
Sigma-1 receptor-regulated efferocytosis by infiltrating circulating macrophages/microglial cells protects against neuronal impairments and promotes functional recovery in cerebral ischemic stroke
By Gufang Zhang, Qi Li, Weijie Tao, Pingping Qin, Jiali Chen, Huicui Yang, Jiaojiao Chen, Hua Liu, Qijun Dai, and Xuechu Zhen
Excerpt from the article published in Theranostics. 2023 Jan 1;13(2):543-559. DOI: 10.7150/thno.77088. PMID: 36632219; PMCID: PMC9830433.
Editor’s Highlights
- Removing the dead neurons or debris of apoptotic cells, is one of the key defense mechanisms for neuronal survival following a ischemic stroke
- Sig-1R is a pivotal molecule that mediates efferocytosis by macrophages, which contributes to inflammation resolution and functional recovery after cerebral ischemia
- Modulation of Sig-1R/Rac1 signaling pathway-mediated phagocytic activity may be a potent therapeutic approach for ischemic stroke
Abstract
Background: Efferocytosis of apoptotic neurons by macrophages is essential for the resolution of inflammation and for neuronal protection from secondary damage. It is known that alteration of the Sigma-1 receptor (Sig-1R) is involved in the pathological development of some neurological diseases, including ischemic stroke. The present study aimed to investigate whether and how Sig-1R regulates the phagocytic activity of macrophages/microglia and its significance in neuroprotection and neurological function in stroke.
Methods: The roles of Sig-1R in the efferocytosis activity of microglia/macrophages using bone marrow-derived macrophages (BMDMs) or using Sig-1R knockout mice subjected to transient middle artery occlusion (tMCAO)-induced stroke were investigated. The molecular mechanism of Sig-1R in the regulation of efferocytosis was also explored. Adoptive transfer of Sig-1R intact macrophages to recipient Sig-1R knockout mice with tMCAO was developed to observe its effect on apoptotic neuron clearance and stroke outcomes.
Results: Depletion of Sig-1R greatly impaired the phagocytic activity of macrophages/microglia, accordingly with worsened brain damage and neurological defects in Sig-1R knockout mice subjected to tMCAO. Adoptive transfer of Sig-1R intact bone marrow-derived macrophages (BMDMs) to Sig-1R knockout mice restored the clearance activity of dead/dying neurons, reduced infarct area and neuroinflammation, and improved long-term functional recovery after cerebral ischemia. Mechanistically, Sig-1R-mediated efferocytosis was dependent on Rac1 activation in macrophages, and a few key sites of Rac1 in its binding pocket responsible for the interaction with Sig-1R were identified.
Conclusion: Our data provide the first evidence of the pivotal role of Sig-1R in macrophage/microglia-mediated efferocytosis and elucidate a novel mechanism for the neuroprotection of Sig-1R in ischemic stroke.
Introduction
Ischemic stroke is a devastating disease, and effective therapeutics against its neurological consequences remain a challenge 1. Accumulating evidence indicates that immune cells, especially brain-resident microglia and infiltrated circulating macrophages, are intimately involved in the pathological development of ischemic stroke 2, 3. At an early stage, blocking nutrition and oxygen supply in response to brain ischemia activates various cell death processes, including apoptosis, autophagy and necrosis 4. A large amount of damage-associated molecular patterns (DAMPs) released by all types of dead cells trigger inflammatory responses by activating microglial cells or macrophages 5. Excessive neuroinflammation further promotes neuronal and glial cell damage and impairs brain repair and functional recovery. Thus, cell death is an integral part of DAMP-induced inflammatory responses activated by ischemia and reperfusion.
Efferocytosis is a process by which dead cells or debris are rapidly and efficiently cleared by professional phagocytes such as macrophages, which helps to inhibit DAMP release and subsequent reactive inflammation 6, 7. Inflammation and tissue repair are impaired owing to failure to activate efferocytosis 8, 9. Defective efferocytosis leads to the accumulation of dead cells and debris, which exacerbates inflammation, ultimately inducing progressive brain damage with poor outcomes of ischemic stroke 10, 11. Brain resident microglia and infiltrated monocyte-derived macrophages are the essential elements of the innate immune responses during the acute phase following brain ischemic insult. Inflammatory M1 macrophages/microglia exacerbate disease by releasing detrimental mediators such as a cascade of proinflammatory cytokines, hydrolyzed proteases and peroxides, while resolving M2 macrophages favors the maintenance of tissue homeostasis by efficiently clearing dead/dying cells or debris and reducing the production of proinflammatory cytokines 12–14. Given the importance of efferocytosis, targeting efferocytosis regulation may be an effective pharmacological approach as a therapeutic strategy for neuronal damage-related diseases such as stroke.
Sigma-1 receptor (Sig-1R) is a chaperone protein that modulates multiple aspects of cellular functions, such as cell death, autophagy/apoptosis, neuronal differentiation, and inflammation. Alterations in Sig-1R functions have been implicated in a few neurological and psychiatric diseases, such as depression, addiction, schizophrenia and Parkinson’s disease 15–19. In an ischemic stroke mouse model with permanent MCAO, Sig-1R knockout mice displayed significantly increased infarct volumes compared to littermate wild-type controls 20. The Sig-1R agonist Comp-AD was reported to produce a neuroprotective effect after transient middle cerebral artery occlusion in mice associated with ER stress 21. We have previously demonstrated that activating Sig-1R significantly suppresses LPS-induced proinflammatory cytokine release by microglia 18. In addition to regulating neuroinflammation, it was also shown that Sig-1R is involved in the regulation of autophagy and apoptosis of neuronal cells, including in stroke 22–24. It is known that macrophage-mediated efferocytosis plays an important role in cellular protection and inflammation regulation; however, little is known about how Sig-1R regulates efferocytosis and its contribution to the protective effects of Sig-1R in cerebral ischemia.
In the present study, we focused on the effects of Sig-1R on the phagocytic modulation of macrophages post ischemic stroke. We provide the first evidence that Sig-1R is an important regulator of efferocytosis in macrophages. We found that the phagocytic activity of apoptotic cells depended on the activation of Sig-1R. We further demonstrated that Sig-1R physically interacts with Rac1 and that Rac1 activity is critical for Sig-1R-stimulated efferocytosis in macrophages. Moreover, our data revealed that Sig-1R-mediated efferocytosis in circulating macrophages/microglia cells contributed largely to the protection both in brain tissue impairments and neurological functions in ischemic stroke.
Results
Activating the sigma-1 (Sig-1R) receptor promotes macrophage efferocytosis
To explore whether Sig-1R is involved in the regulation of efferocytosis, we investigated how Sig-1R activation regulates the clearance of dead cells in macrophages, which is a vital process for tissue injury and inflammation development after ischemic stroke. We employed HT-22 neurons as donors of dead cells (late apoptosis/necrosis) in which the cells were prelabeled with CMFDA (5-chloromethylfluorescein diacetate) and then incubated with hydrogen peroxide to induce up to 70% cell death (Figure S1). Bone marrow-derived macrophages (BMDMs) were preincubated with the Sig-1R agonist PRE-084 for 30 min and cocultured with dead/dying HT-22 cells at a 1:5 ratio as described in the figure legend. Confocal imaging analysis showed the enhanced uptake activity on dead cells by selective Sig-1R agonist PRE-084-treated macrophages compared with that of the control. Preincubation with the Sig-1R antagonist BD1047 abolished the effect of PRE-084 (Figure (Figure1A-B),1A-B), revealing a Sig-1R-mediated event. Consistent with the results, PRE-084 application to macrophages also enhanced the expression of biomarkers of the M2 phenotype, such as Cd206, Ym1 and Tgf-β1, which was also blocked by BD1047 pretreatment after exposure to dead cells (Figure (Figure1C-E).1C-E). These results suggested that Sig-1R activation promoted macrophage uptake of dying/dead cells and facilitated macrophage switching to an anti-inflammatory phenotype.
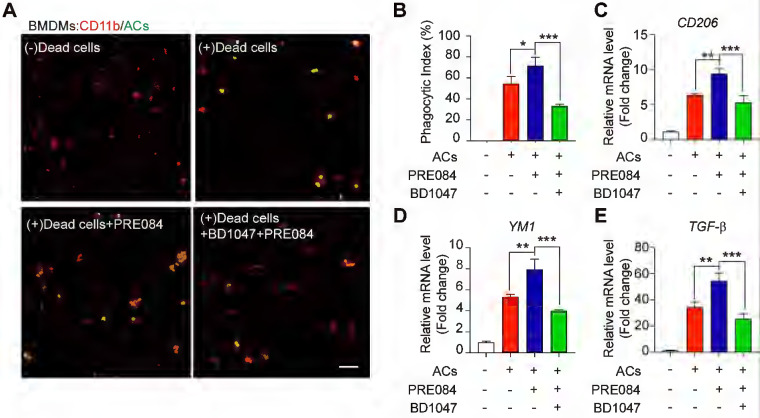
Sig-1R activation promotes efferocytosis and M2 phenotype polarization of macrophages.
(A) Bone marrow -derived macrophages (BMDM) pre-treated with PRE-084 (10 μM) or BD1047 (10 μM) followed incubation with dead HT-22 neuronal cells which was prepared as described in Method for 30 min. Dead cells were labeled with CMFDA (Green), macrophages were marked with CD11b (Red). Scale bar: 50 μm. (B) Statistical analysis of phagocytotic index of macrophages. (C) mRNA level of CD206, (D) YM1 and (E) TGF-β was determined 24 h in macrophages after incubation with dead cells. Data are presented as means ± SD, and were analyzed using one-way ANOVA followed by Dunnett’s post-hoc tests, n = 3. *P < 0.05; **P < 0.01, ***P < 0.001.
Engulfment of dead cells by macrophages/microglia is dependent on the sigma-1 receptor
To further verify the effect of Sig-1R in regulating the engulfment of dead cells, we compared the phagocytic ability between WT and Sig-1R-deficient macrophages from Sig-1R knockout mice. Dying/dead HT-22 neuronal cells were labeled with CMFDA (green) and then cocultured with macrophages at a 1:5 (macrophages: dead cells) ratio as described. The percentage of engulfed dead cells (green) was analyzed by flow cytometry (Figure (Figure2A).2A). In relation to the fact that 25% of macrophages from WT mice were CMFDA positive, the proportion, however, was markedly decreased to approximately 8% in Sig-1R knockout macrophages (Figure (Figure2B).2B). Consistent with the results, we also observed a significant decrease in engulfed dead cells in Sig-1R knockout macrophages compared to WT macrophages (Figure (Figure2C).2C). M2 phenotypic markers such as Ym1 and CD206 were significantly higher (mRNA expression) in WT macrophages than in Sig-1R-/- macrophages. The basal expression of the two markers was not altered between WT and Sig-1R-/- macrophages (Figure S2). The protein expression of MerTK and CD36, both of which are critical molecules for recognizing dead cells, was not altered in WT and Sig-1R-/- macrophages in response to dead cell treatment (Figure S3). In support of this hypothesis, we demonstrated that Sig-1R knockout disrupted phagocytic activity in primary microglia from Sig-1R knockout mice (Figure (Figure2D).2D). Interestingly, we noticed that macrophages appeared to have a stronger efferocytosis capacity than microglia, since we observed that CMFDA-positive macrophages engulfed more dead cells than microglial cells. In addition, we demonstrated that WT macrophages exhibited significantly stronger phagocytic activity toward E. Coli and zymosan bioparticles than Sig-1R-/- macrophages (Figure S4). Our data thus clearly revealed that Sig-1R is involved in wide spectrum of efferocytosis by macrophages or microglia.
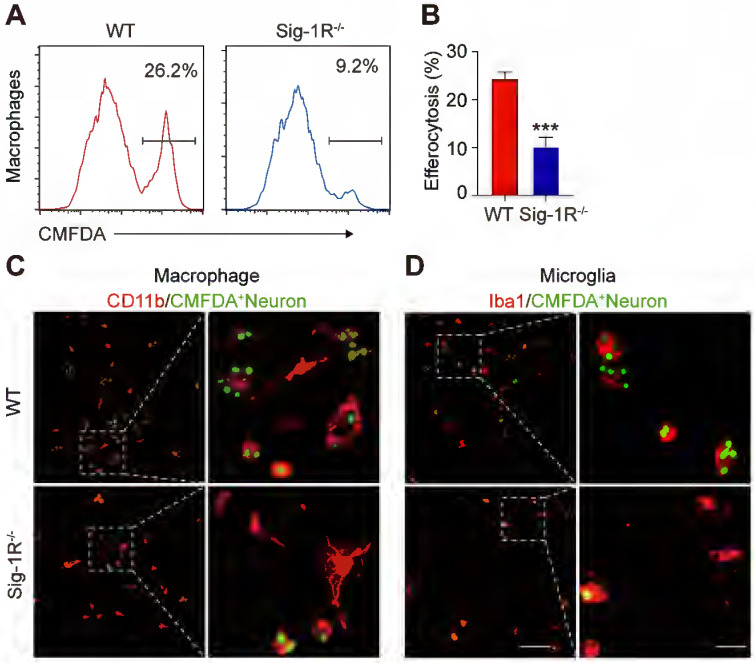
Depletion of Sig-1R abrogates efferocytosis activity of macrophages.
Bone marrow derived macrophages (BMDMs) from either wildtype (WT) or Sig-1R knockout (Sig-1R-/-) mice were incubated with dead cells for 30 min. (A) Flow cytometry analysis of engulfed macrophage or microglia (CMFDA+ cells) was conducted as described. Presentative images of wildtype (WT) macrophages (Red line) and Sig-1R-/- macrophages (blue lines) were exhibited. (B) Phagocytic index of macrophages was calculated as CMFDA+ cells among total cells. (C) WT or Sig-1R-/- BMDMs were incubated with dead cells for 30 min at 1:5 ratio. Dead cells were labeled with CMFDA (Green) and macrophages were marked with CD11b (Red). Scale bar: 50 μm. (D) WT or Sig-1R-/- primary cortical microglia were incubated with dead cells for 30 min at 1:5 ratio. Dead cells were labeled with CMFDA (Green) and macrophages were marked with Iba1 (Red). Scale bar: 50 μm. Data are presented as means ± SD and were analyzed using student t – test, n = 3. ***P < 0.001.
Sigma-1 receptor knockout impairs clearance of apoptotic cells in response to cerebral ischemia
To elucidate the role of Sig-1R in efferocytosis activity in ischemic stroke, we first explored the functional implication of Sig-1R in the acute phase in brain ischemia. Cerebral ischemia was induced by transient middle artery occlusion (tMCAO) and reperfusion in mice as described. As shown in Figure Figure3A&B,3A&B, the brain infarct volume in Sig-1R-/- mice was significantly larger than that in WT mice. In agreement with previous reports, we also confirmed that Sig-1R deletion exacerbated neurological deficits in response to ischemic stroke in tMCAO mice (Figure (Figure3C).3C). To observe the effect of Sig-1R on efferocytosis activity of macrophages/microglia in vivo, macrophages/microglia were labeled with CD68, which is predominantly expressed in late endosomes and lysosomes of macrophages 30; NeuN and TUNEL double-positive cells were marked as apoptotic neurons at the infarct striatum; CD68, NeuN and TUNEL triple-positive cells were apoptotic cells engulfed by macrophages/microglia. The high-power 3-D images clearly indicated phagocytic macrophages/microglia in the infarct striatum (Figure (Figure3D).3D). Consistent with the exacerbation of brain infarction, there were significantly more dead/dying neurons (NeuN+TUNEL+ cells) in the infarct striatum at 5 d after stroke in Sig-1R-/- mice than in WT mice (Figure (Figure3E).3E). The number of macrophages/microglia that phagocytosed dead/dying cells (CD68+NeuN+TUNEL+) was significantly reduced in Sig-1R-/-mice (Figure (Figure3F).3F). In support of this hypothesis, the phagocytic index, which reveals the efferocytosis ability of macrophages/microglia, was markedly reduced in Sig-1R-/- mice (Figure (Figure3G).3G). In addition to removing dead and dying neurons, microglia can also engulf live neurons 31. We also noted that a few live neurons (NeuN+TUNEL–) were engulfed by microglia/macrophages among all CD68 and NeuN double-positive cells (yellow arrow in Figure Figure3D).3D). Interestingly, Sig-1R knockout mice exhibited a markedly higher percentage of engulfed live neurons in microglia/macrophages than WT mice (Figure (Figure3H),3H), and whether Sig-1R knockout contributes to exacerbated impairment in Sig-1R-/- mice in response to ischemic stroke remains unclear. Taken together, these results suggest that Sig-1R plays a critical role in the regulation of the phagocyte function of macrophages/microglia that may contribute to brain injury following ischemic insult.
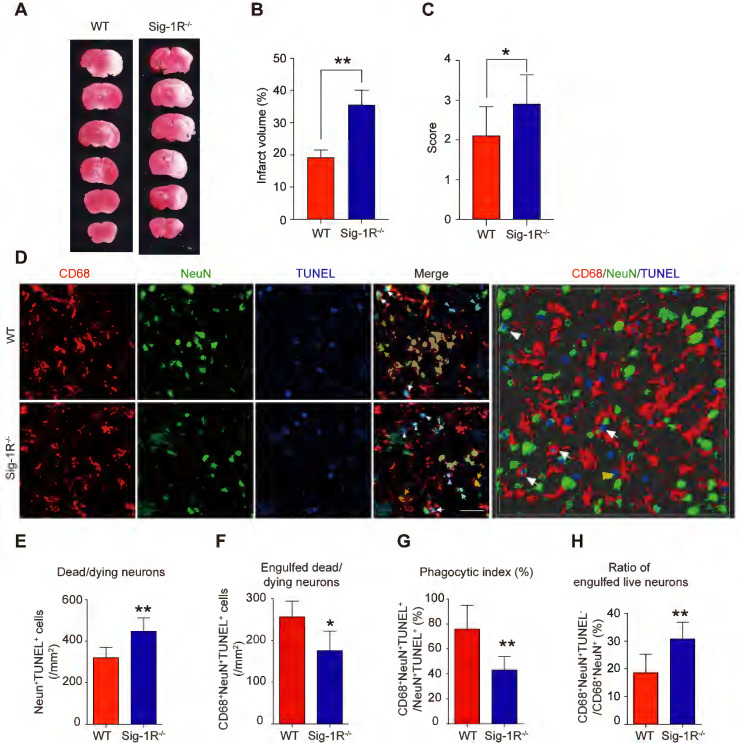
Depletion of Sig-1R sensitized the brain injure and neurological defects companied with impaired phagocytic activity of macrophage/microglia.
WT or Sig-1R-/- mice were subjected to tMCAO as described. Mice were monitored with neurological scores before sacrificed on day 5 after reperfusion. n = 7 mice / group. (A) Representative image of brain infarct area detected with TTC assay for each treatment. (B) Statistical analysis of infarct volume. (C) Neurological score for animals was assessed according to Material and Methods. (D) Representative images for CD68 (macrophage marker, Red) and NeuN (neuronal marker, Green) which was co-labeled with TUNEL (Blue) in infarct areas. Scale bar: 50μm. High-power 3-D image generated from D (right panel). White arrows indicate microglia / macrophages that engulfed dead / dying neurons (CD68+NeuN+TUNEL+). White arrowheads indicate dead / dying neurons that were not engulfed by microglia / macrophages (CD68–NeuN+TUNEL+). Yellow arrows indicate live neurons were engulfed by microglia / macrophages (CD68+NeuN+TUNEL–). Yellow arrowhead indicates a TUNEL– neuron touched by an CD68+ cell (CD68+NeuN+TUNEL–). Scale bar: 50 μm. (E) The number of total dead / dying neurons in the ischemic striatum of mice. (F) The number of CD68+NeuN+TUNEL+ cells (microglia / macrophages with engulfed dead / dying neurons) in the ischemic striatum of mice. (G) Phagocytic index, as quantified the percentage of dead / dying neurons engulfed by microglia / macrophages ([number of CD68+NeuN+TUNEL+ cells / number of NeuN+TUNEL+cells] × 100%), in WT and Sig-1R-/- mice brains. n = 6 mice per group. (H) Percentage of live neurons were engulfed by macrophages / microglia among all engulfed neurons ([number of CD68+NeuN+TUNEL– cells / number of CD68+NeuN+ cells] × 100%) in WT and Sig-1R-/- mice brains. n = 6 mice per group. Data are presented as means ± SD and were analyzed using student t-test. *P < 0.05; **P < 0.01.
Sigma-1 receptor deletion exacerbates inflammation after brain ischemia
The phenotypic shift of macrophages/microglia in the acute phase of brain ischemia is an essential factor in the outcomes after stroke. To understand the roles of Sig-1R knockout on the polarization of macrophages/microglia after stroke, we first compared the pro-inflammatory phenotypic marker CD16 (Figure (Figure4A)4A) and anti-inflammatory marker CD206 (Figure (Figure4C)4C) expression in macrophages/microglia from the ischemic striatum at 5 d after reperfusion between WT and Sig-1R knockout mice. As shown in Figure Figure4B,4B, the number of ionized calcium binding adaptor molecule 1 (Iba1) and CD16 double-positive cells was significantly higher in Sig-1R knockout mice. In contrast, the number of Iba1 and CD206 double-positive cells was reduced (Figure (Figure4D).4D). To further confirm the role of Sig-1R in the phenotypic polarization of infiltrated circulating macrophages after stroke, ischemic brains from WT or Sig-1R knockout mice 5 d after tMCAO were assessed and subjected to flow cytometry analysis. CD45highCD11b+Ly6G–macrophages (sorting strategy shown in Figure S5) displayed a stronger MFI of the proinflammatory marker CD86 (Figure (Figure4E-F)4E-F) and a weaker MFI of the anti-inflammatory marker CD206 (Figure (Figure4G-H)4G-H) in Sig-1R knockout mice than in the same subset of macrophages from WT mice subjected to tMCAO.
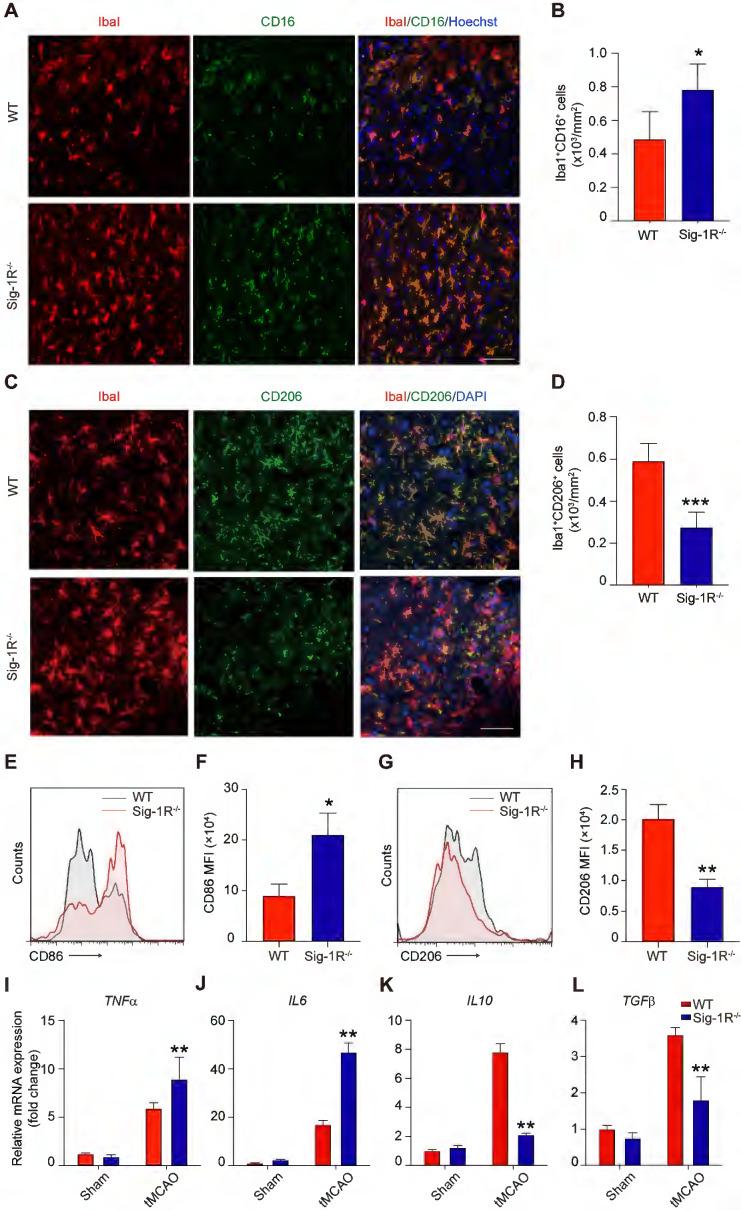
Sig-1R deletion promotes macrophages/microglia activation and its tMCAO.
Brains of WT or Sig-1R-/- mice were subjected to tMCAO as described, and brains were collected on day 5 for further experiments. (A) Representative images of microglia/macrophage marker Iba1 (Red) and inflammatory phenotype marker CD16 (Green) double-staining. Scale bar: 50 μm. (B) Quantification of Iba1+CD16+ proinflammatory macrophages/microglia in ischemic areas. n = 5 mice per group. (C)Representative images of Iba1 (Red) and CD206 (Green) double staining. Scale bar: 50 μm. (D) Quantification of Iba1+ / CD206+ macrophages/microglia in ischemic areas. n = 5 mice per group. Data are presented as means ± SD and were analyzed using student t -test. *P < 0.05; ***P < 0.001. (E) Flow cytometry analysis of CD86 expression in CD45+CD11b+Ly6G– macrophages in the brain, mean fluorescent intensity (MFI) was calculated (F). (G) Flow cytometry analysis of CD206 level in CD45+CD11b+Ly6G– macrophages in the brain, mean fluorescent intensity (MFI) was calculated(H). mRNA expression of proinflammatory cytokines TNF-α (I) and IL-6 (J), resolving cytokines IL-10 (K) and TGF-β (L)were measured with RT-PCR in the ipsilateral hemisphere. n = 5 mice per group. Data are presented as means ± SD and analyzed with one-way ANOVA followed by Dunnett’s post-hoc tests. **P < 0.01.
Consistent with the phenotypic switch to a proinflammatory phenotype, Sig-1R deficiency led to augmented local inflammation. The mRNA expression of proinflammatory cytokines, including TNF-α(Figure (Figure4I)4I) and IL-6 (Figure (Figure4J),4J), was significantly elevated at 5 d after stroke in Sig-1R-/- mice. In contrast, the mRNA expression of the anti-inflammatory cytokines IL-10 (Figure (Figure4K)4K) and TGF-β1 (Figure (Figure4L)4L) was significantly decreased in Sig-1R -/- mice compared with WT mice. Taken together, these results indicated that Sig-1R defects promoted macrophage polarization into the M1 type and infiltration into the ischemic area, consequently sensitizing the neuroinflammation.
Transferring intact Sig-1R macrophages rescues the acute neurological defect in Sig-1R knockout mice with tCAMO
It is known that circulating macrophages infiltrate injured brain regions as early as 3 hours after reperfusion 32. These macrophages effectively clear dead cells or debris, which is critical for the pathological development and outcomes of ischemic stroke 10. To further elucidate the importance of Sig-1R in macrophages on neurological functions in response to cerebral ischemia, we transferred bone marrow-derived macrophages (BMDMs) obtained from either WT or Sig-1R-/- mice to Sig-1R knockout ischemic mice 2 h after reperfusion (Figure (Figure5A).5A). BMDMs were transfected with lentivirus containing GFP elements prior to injection into recipient mice. Five days after injection, GFP-positive cells colocalized with CD68 accumulated in the infarct area (Figure (Figure5B),5B), and these BMDMs were able to engulf apoptotic cells (GFP+CD68+NeuN+TUNEL+) (Figure (Figure5C).5C). The basal number of GFP+CD68+ cells exhibited no significant difference between WT and Sig-1R-/- mice (Figure (Figure3D),3D), while adoptive transduction of BMDMs from WT mice markedly enhanced efferocytosis activity compared with that of Sig-1R-/- BMDMs (Figure (Figure5E).5E). Consistent with this observation, BMDMs transferred from WT mice greatly decreased the infarct volume of Sig-1R-/- recipient mice, as did neurological functional impairment. In contrast, BMDMs transferred from Sig-1R-depleted mice did not produce any significant effects on those parameters (Figure (Figure55F-H).
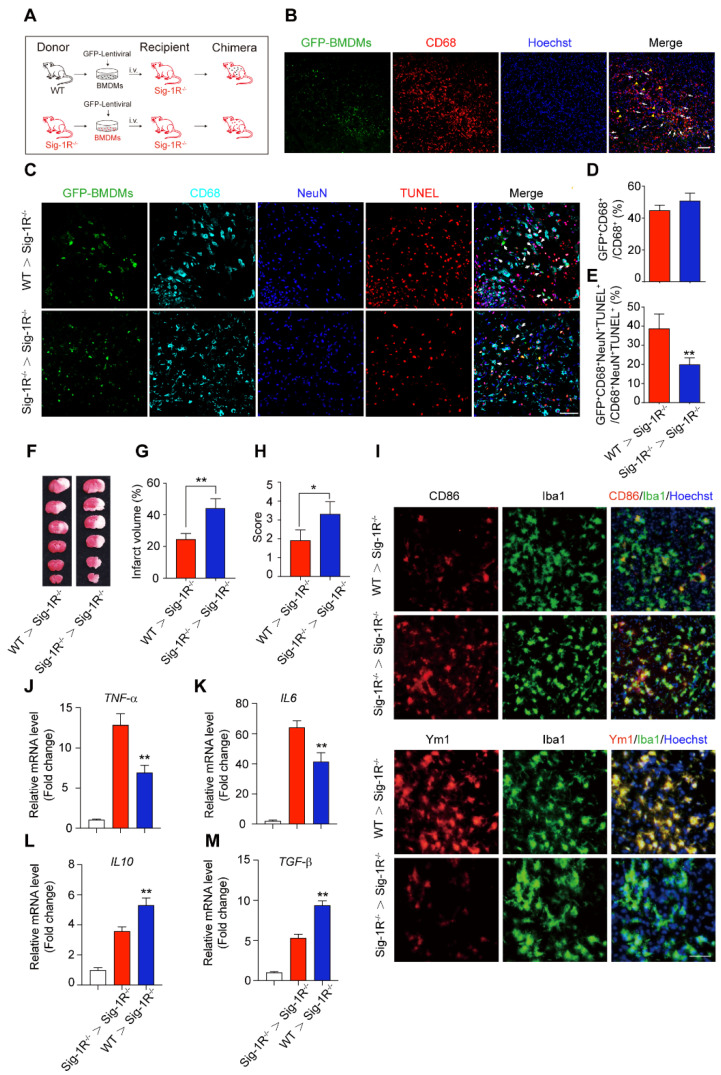
Circulating macrophages attribute to Sig-1R-afforded protection against ischemic stroke.
(A) Age- and weight-matched Sig-1R-/- mice as recipients were subjected tMCAO, BMDMs were prepared as depicted from either WT or Sig-1R-/- mice. Chimeric mice were constructed by transferring WT or Sig-1R-/- macrophages through tail vein injection to recipient Sig-1R-/- mice immediately after reperfusion. (B) Reprehensive images indicating GFP+ BMDMs were accumulated at the brain infarct area, scale bar: 50 μm. (C) Reprehensive images depicting transferring BMDMs effectively phagocyte apoptotic neurons (white arrow, GFP+CD68+NeuN+TUNEL+), scale bar: 50 μm. (D) Percentage of transferred BMDMs (GFP+CD68+) among all CD68+ microglia/macrophages. (n = 5 mice per group) (E) Percentage of transferred BMDMs contribute to the efferocytosis among all the effectively phagocytes (GFP+CD68+NeuN+TUNEL+ / CD68+NeuN+TUNEL+). (n = 5 mice per group). **P < 0.01, one-way ANOVA. (F) Representative image of infarct area of for each treatment on day 5 after tMCAO. (G) The statistical analysis of infarction area and (H) neuronal score in each group was performed on day 5 after tMCAO. n = 7 mice / group. (I) Brains were collected from recipient mice and microglia/macrophage phenotypes were analyzed. Representative images for Iba1 (Green) and CD86 (Red) double staining and Iba1 with Ym1 (Red) double staining. Scale bar: 50 μm. mRNA expression of proinflammatory cytokines TNF-α (J) and IL-6 (K), IL-10 (L) and TGF-β (M) were measured with RT-PCR in the ipsilateral hemisphere. n = 5 mice / group. **P < 0.01, data were analyzed with one-way ANOVA.
We also elucidated the roles of intact Sig-1R BMDMs in macrophage/microglia polarization in the ischemic area 5 days after ischemic insult. As indicated in Figure Figure5I,5I, the expression of CD86 (red) in Iba1-positive (green) macrophages/microglia was significantly reduced in mice that accepted BMDMs from Sig-1R-intact WT mice but not Sig-1R-knockout mice. In contrast, the expression of Ym1 (red) in Iba1-positive (green) macrophages/microglia was significantly elevated (Figure (Figure5I).5I). Accordingly, we detected a significant decrease in TNF-α (Figure (Figure5J)5J) and IL-6 (Figure (Figure5K)5K) mRNA expression. However, the expression of IL-10 (Figure (Figure5L)5L) and TGF-β (Figure (Figure5M)5M) was significantly increased in mice treated with Sig-1R-intact BMDMs from WT mice compared to Sig-1R knockout mice. These data revealed that the transfer of sigma-1 receptor-intact macrophages was sufficient to polarize M1 macrophages into M2 phenotypes, leading to the inhibition of neuroinflammation. Taken together, the results indicated that Sig-1R-regulated macrophages/microglia play a critical role in acute brain damage and neurobiological functions in response to stroke.
To further evaluate the role of Sig-1R-modulated macrophages in long-term functional recovery after brain ischemia, neurological functions, including sensorimotor gating and cognitive ability, in recipients with BMDM transplantation from either WT or Sig-1R KO mice were assessed over a period of four weeks following tMCAO. Transferring macrophages from WT mice significantly ameliorated neurological deficits in Sig-1R KO mice compared to that of Sig-1R KO macrophage transplantation (Figure (Figure6A),6A), as did the rotarod test (Figure (Figure6B)6B) and corner test (Figure (Figure6C).6C). In addition, transferring macrophages from WT mice improved the general activity of animals and anxiety as measured by travel distance (Figure (Figure6D)6D) and the time spent in the center (Figure (Figure6E),6E), respectively, in the open-field test. This result indicated that transplantation with macrophages from WT mice to Sig-1R-depleted mice with tMCAO improved long-term neurological deficits. Taken together, the results indicated that macrophage Sig-1R-mediated protection is critical for decreasing brain damage and improving neurobiological functions after ischemic stroke.
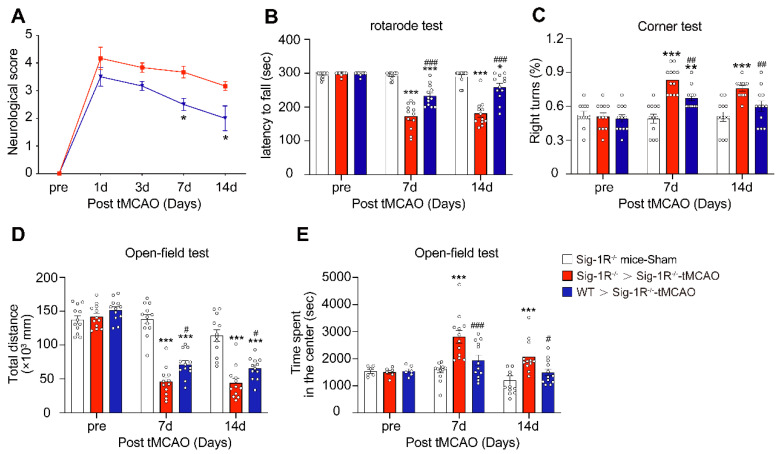
Circulating WT macrophages alleviate neuronal deficits after ischemic stroke.
Sig-1R knockout mice were used as recipient mice to accepting WT or Sig-1R knockout BMDMs immediately after cerebral ischemia. Neurological function of recipient mice was observed each week. (A) Neurological scores were summarized (n = 6 mice per group). (B) The summary for rotarod test, (C) corner test and (D and E) open-field test were assessed to evaluate the motor and sensory function of recipient mice. n = 12 mice / group. Data are presented as means ± SEM. Two-way ANOVA followed by Dunnett’s post-hoc tests were applied. *P < 0.05; **P < 0.01, ***P < 0.001 compared with sham group; #P < 0.05; ##P < 0.01, ###P < 0.001 compared with sham group.
The sigma-1 receptor promotes efferocytosis through Rac1 signaling in macrophages
Thus far, we have discovered that Sig-1R plays a critical role in the regulation of efferocytosis. Depletion of Sig-1R resulted in impaired efferocytosis, which led to enhanced neuroinflammation and tissue damage, ultimately aggravating neurological defects. We next explored the potential mechanism for Sig-1R-regulated phagocytic activity of microglia/macrophages. It is known that the internalization of dead cells by phagocytes depends on actin polymerization, which is mediated by small GTPases, especially Rac1. Therefore, we first examined Rac1 activation in WT and Sig-1R-/- macrophages. After incubation with dead cells, the activation of Rac1 was significantly decreased in Sig-1R-/- macrophages compared to WT macrophages (Figure (Figure7A-B).7A-B). Furthermore, activation of Sig-1R by the specific agonist PRE-084 promoted phagocytic activity of macrophages, which was abrogated by EHop-016 (Figure (Figure7C-D),7C-D), a Rac1 inhibitor that has been proven to effectively inhibit Rac1 activity in the brain without affecting basal behavior through intraperitoneal injection 33. To further confirm the effect of EHop-016 on Sig-1R activation-induced clearance of dead neurons, ischemic mice were treated with EHop-016 (i.v., 20 mg/kg) via the tail vein 1 h before intraperitoneal administration of PRE-084 (i.p., 10 mg/kg). Five days post drug treatment, consistent with the in vitro results, PRE-084 treatment enhanced the clearance of apoptotic neurons (CD68+NeuN+TUNEL+) by microglia/macrophages compared to that of vehicle treatment. As expected, EHop-016 pretreatment markedly attenuated the Sig-1R activation-stimulated efferocytosis (Figure (Figure7E-F).7E-F). Interestingly, we observed that EHop-016 treatment also inhibited microglia/macrophage recruitment to the infarct area (Figure (Figure7E).7E). Taken together, these results indicated that Sig-1R-regulated efferocytosis of macrophages depends on Rac1 activation.
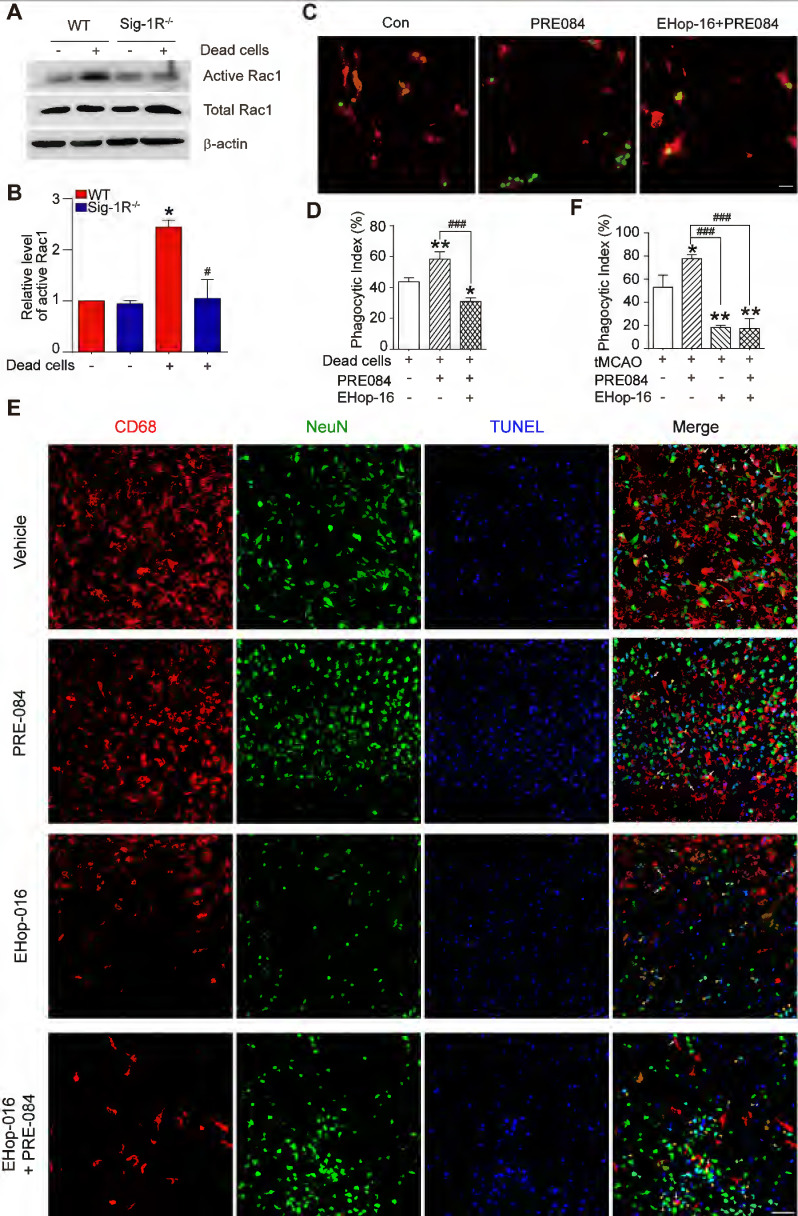
Sig-1R regulate macrophage efferocytosis through Rac1.
(A) WT or Sig-1R-/- BMDMs were incubated with dead cells for 1 h, Rac1 activity was determined. (B) Summary of Rac1 activity in WT and Sig-1R-/- macrophages. n = 3, data are presented as means ± SD and analyzed with one-way ANOVA followed by Dunnett’s post-hoc tests. *P < 0.05 versus WT without dead cells group. #P < 0.05 versus WT with dead cells group. (C) Macrophages were pre-treated with Rac1 inhibitor EHop-016 (1 μM) for 30 min followed by Sig-1R agonist PRE084 (10 μM) prior to incubation with dead cells. Dead cells were labeled with CMFDA (Green) and macrophages were marked with CD11b (Red). Scale bar: 20 µM. (D) Phagocytotic index was determined as described. n = 3, data are presented as means ± SD and analyzed with one-way ANOVA followed by Dunnett’s post-hoc tests. *P < 0.05, **P < 0.01 versus control group; ###P < 0.001 versus PRE-084-treated group. (E) Effects of Rac1 inhibitor EHop-016 on Sig-1R agonist PRE-084 induced efferocytosis in the mice brain infarct area after cerebral ischemia. Representative Images of CD68 (Red), NeuN (Green), and TUNEL (blue) triple-staining cells in each group. Scale bar: 50 μm. White arrows indicate apoptotic neurons were phagocyted by microglia/macrophages which resented as CD68+NeuN+TUNEL+ cells, yellow arrowheads indicate apoptotic neurons were not effectively phagocyted by microglia/macrophages which presented as CD68–NeuN+TUNEL+ cells. (F) Statistical analysis of phagocyte index in each group, which calculated as the percentage of CD68+NeuN+TUNEL+ among all NeuN+TUNEL+ cells. n = 5, data are presented as means ± SD and analyzed with one-way ANOVA followed by Dunnett’s post-hoc tests. *P < 0.05, **P < 0.01 versus vehicle group; ###P < 0.001 versus PRE-084-treated group.
Due to the chaperone nature of Sig-1R, we next investigated whether Sig-1R directly interacts with Rac1. As shown in Figure Figure8A,8A, we detected enhanced colocalization between Rac1 and Sig-1R in macrophages in response to incubation with dead ells. This was further supported by the coimmunoprecipitation assays, which showed an elevated association between Rac1 and Sig-1R in macrophages that were preincubated with dead cells. Interestingly, we found that PRE-084 application enhanced the association, which was blocked by the Sig-1R-specific antagonist BD1047 (Figure (Figure8B).8B). We further confirmed the association in vitro using co-expressed Myc-tagged Rac1 and Sig-1R in HEK293T cells (Figure (Figure8C).8C). Interestingly, we found that after transfection with inactive Rac1 (T17N mutant), we detected a weak interaction between mutant Rac1 and Sig-1R, while constitutively active Rac1 (Q61 L mutant) enhanced its interaction with Sig-1R (Figure S6). To identify the structural basis for the interaction between Rac1 and Sig-1R, we used docking analysis to predict the interaction amino site (Table S1) based on the protein structure of Sig-1R and Rac1. Eight Rac1 residues with the highest interaction score were selected, and a single-mutated construct was generated and expressed in HEK293 cells, which included I33A, T40S (Switch I domain, GDP/GTP exchange), L67S, L70A (Switch Ⅱ domain, GDP/GTP exchange), I126A, T138S, P140A and L143A (Insert domain, membrane ruffling and actin organization). Coimmunoprecipitation assays showed that I33A, I126A, T138S or P140A mutations of Rac1 impaired the interaction with Sig-1R (Figure (Figure88D).
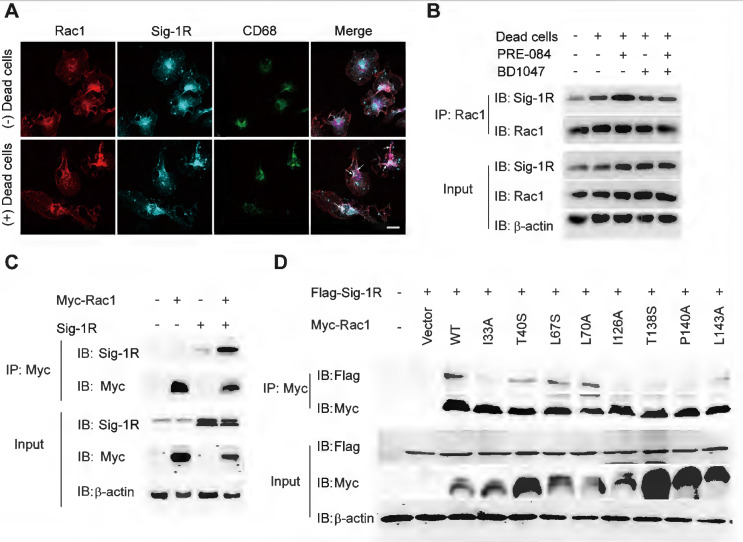
Sig-1R activates and interacts with Rac1 to regulate macrophage efferocytosis.
(A) Macrophages were incubated with dead cells for 30 min before processed for immunostaining. Representative images of Rac1 (Red), Sig-1R (Cyan) and CD68 (Green) triple staining were shown from at least three repeated experiments with similar results. Scale bar: 20 μm. (B)Macrophages were pre-treated with BD1047 (10 μM) for 30 min before stimulation with PRE084 (10 μM) for 30 min. The cells were then incubated with dead cells for additional 60 min before collection for further assays. Co-immunoprecipitation (Co-IP) analysis for Rac1 and Sig-1R interaction was shown. (C) HEK293T cells were co-transfected with Myc-Rac1 and Sig-1R plasmids. the interaction between Rac1 and Sig-1R was monitored by co-immunoprecipitation (Co-IP) with anti-Myc beads, followed by SDS-PAGE separation and detected by respective antibody. (D) Single mutation of Rac1 plasmid was prepared as described in Method section, HEK293T cells were transfected with Flag-Sig-1R and wild type Myc-Rac1 or its single mutation, as indicated. After Co-IP with anti-Myc beads, the IP and input samples were separated by SDS-PAGE and probed with indicated antibodies.
Discussion
It is well established that efferocytosis maintains tissue homeostasis through the removal of dead cells, which is believed to be a critical process in tissue damage and repair, inflammation modulation and neurological functions. The present study identified, for the first time, that Sig-1R plays a pivotal role in the regulation of macrophage/microglia efferocytosis, which contributes to receptor activation-promoted neuronal protection, tissue repair and neurological functional recovery in response to cerebral ischemic insults. The major discoveries of the present study are as follows: (1) Sig-1R is a critical regulator of efferocytosis activity in the clearance of dead cells by macrophages. Depletion of Sig-1R impaired the phagocytic ability of macrophages, consequently promoting neuroinflammation and tissue injury and sensitizing neurological defects in the ischemic brain. (2) Adoptive transfer of intact Sig-1R macrophages significantly attenuated tissue injury and neurological defects in Sig-1R knockdown mice subjected to ischemic stroke. (3) Sig-1R-mediated phagocytic activity depended on activation and interaction of the small GTPase protein Rac1.
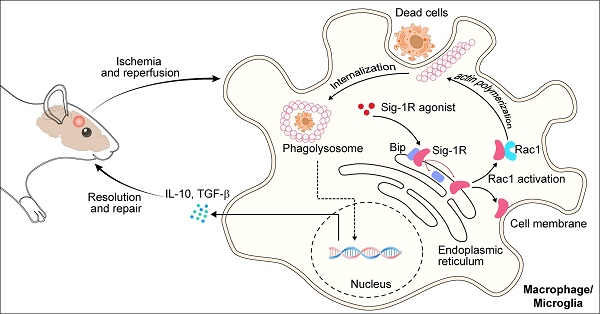
Sig-1R is localized at mitochondria-associated membranes (MAMs) and acts as a chaperone protein. Upon activation with its agonist, such as PRE-084, the receptor dissociates from Bip and translocates to the plasma membrane or other sub-organelles to interact with other molecules to elicit multiple biological responses. PRE-084 and BD1047 is widely used as Sig-1R agonist and antagonist, respectively, which bind to the receptor to activate or inhibit the activation of the receptor. The neuroprotective effects of Sig-1R are generally believed to be associated with receptor activation-regulated autophagy, neuroinflammation and mitochondrial functions 17, 22. In addition, there were also studies shown that Sig-1R activation could induce GNDF/BDNF production 18, 34, 35, and blocking Sig-1R by BD-1047 markedly alleviated ethanol-induced neurotoxicity 36. Thus, targeting the Sig-1R receptor has been an important approach in drug discovery for a number of CNS diseases, such as AD, PD, depression, and stroke 19, 37. The present work, for the first time, revealed that Sig-1R is a critical regulator of efferocytosis that was associated with the ischemic brain damage, therefore, revealing a novel mechanism for Sig-1R-mediated neuroprotection.
Removing the dead/dying neurons or debris of apoptotic cells, i.e., efferocytosis is one of the key defense mechanisms for neuronal survival following neuronal damage, such as ischemic stroke, which is believed to be conducted by professional phagocytes, such as microglia, macrophages, neutrophils and dendritic cells. Effective clearance of dead cells in a timely manner is in favor of maintaining the brain homeostatic status by preventing DAMP release and subsequent excessive inflammation 38. Furthermore, engulfed dead cells promote microglia and macrophages to reprogram into the M2 phenotype, which contributes to inflammation resolution by producing anti-inflammatory cytokines, trophic factors and bioactive lipids and consequently accelerates tissue repair 6, 10, 39. The present data clearly demonstrated that Sig-1R is a key modulator of the phagocytic activity of macrophages, as supported by the following facts: activation of Sig-1R enhanced the phagocytic activity of macrophages on dead / dying cells, which was blocked by a Sig-1R antagonist; Sig-1R knockout macrophages greatly impaired its engulfment of dead cells (Figure (Figure1A,1A, Figure Figure2A2A – B). Thus, the present data provide the first evidence to reveal that Sig-1R plays a critical role in the regulation efferocytosis by macrophages or microglia. In support of this hypothesis, we found that activation of Sig-1R inhibited macrophage/microglial activation and promoted an M2 phenotype shift (Figure (Figure1C-E),1C-E), while depletion of the receptor sensitized the inflammation by stimulating the proinflammatory M1 phenotypes (Figure S2) 40. In addition, we found that depletion of Sig-1R also impaired phagocytic activity toward E. Coli and zymosan (Figure S4), indicating that loss of Sig-1R leads to a broader defect in phagocytosis.
We further investigated the functional roles of Sig-1R-regulated phagocytic activity in ischemic stroke. Dead astrocytes/neurons were observed within an hour after acute cerebral ischemia 10, DAMPs released from the dead cells induced microglial activation and peripheral macrophage infiltration into the injured region. It was reported that brain resident microglial cells are the immune cells that react the earliest to ischemic injury, and circulating macrophages enter the brain and accumulate in the infarct area within 24 h and peak at 3 days 10. These macrophages/microglia engulf dead neurons or apoptotic cells in response to “eat-me” signals 41–44. On the other hand, studies have shown that cell phagocytosis may also target living cells and lead to tissue damage 45. These results indicate that phagocytosis is a critical regulatory mechanism for neuronal damage and repair. Therefore, targeting phagocytic activity may represent a potential approach for ischemic stroke treatment 45. Indeed, it was shown that depletion of circulating monocytes during the first three days after stroke impairs long-term neuronal function recovery 46; transferred peripheral macrophages to mice reduced their infarct volume and ameliorated the accumulation of dead cells in the ischemic territory 47. Our data showed that there was a remarkable reduction in the phagocytic index at the infarct sites in Sig-1R knockout mice compared with WT mice subjected to tMCAO (Figure (Figure3G),3G), indicating an impairment of phagocytic activity in Sig-1R-deficient macrophages. It is also noted that we observed that live neurons were also engulfed by microglia/macrophages in the ischemic area, and Sig-1R-/- mice appeared to have more live neurons engulfed than WT mice (Figure (Figure3H).3H). Whether it indicates an additional protective mechanism for Sig-1R remains to be studied. In agreement with previous reports 20, 21, we also confirmed that Sig-1R deletion exacerbated the infarction area and neurological deficits (Figure (Figure3A-C)3A-C) and promoted microglia/macrophage activation (Figure (Figure4)4) in response to ischemic stroke in tMCAO mice. Consistently, transferring Sig-1R intact macrophages from WT mice improved acute and long-term neurological defects and brain damage in Sig-1R knockout mice with tMCAO (Figure (Figure55 and and6),6), which may be attributed to the clearance of apoptotic neurons by transferred BMDMs (Figure (Figure5B-C).5B-C). Our data thus revealed that Sig-1R-regulated efferocytosis contributes to the protective effects of Sig-1R activation on brain injury and functional recovery in response to ischemic stroke. In addition, it is known that the polarization of microglia/macrophages plays a critical role in the process of tissue damage and functional recovery after ischemic stroke 10, 48. In agreement with previous reports 10, 49, 50, we also found that CD45highCD11b-positive macrophages in the ischemic hemisphere of Sig-1R-/- mice expressed significantly higher CD86 but lower CD206 than those of WT mice (Figure (Figure4D),4D), which exhibited a proinflammatory phenotype. The M1 phenotype, however, was switched to the M2 phenotype, while ischemic Sig-1R-/- mice were adoptively transferred with intact sig-1R macrophages (Figure (Figure5I).5I). This confirms the important role of Sig-1R in macrophage/microglia polarization. This may also contribute to the beneficial effects of Sig-1R activation in brain damage and functional recovery after ischemic stroke 51, 52, which has been proved in our study that transfering WT BMDMs to Sig-1R-/- mice subjected to tMCAO partly rescue impaired neurological functions (Figure (Figure6).6). Another important question is what mechanisms underlie Sig-1R-/--mediated macrophage efferocytosis. It is known that efferocytosis is a complex process, and any defect can cause alterations in macrophage engulfment and degradation 13. During the process of dead cells engulfment, GTPase, especially Rac1, is a crucial regulator in mediating actin remodeling, which results in the formation of apoptotic bodies 11. Interestingly, it was reported that Sig-1R physically interacts with Rac1 in mitochondria 53. We discovered that Sig-1R activation-mediated macrophage/microglia efferocytosis appears to depend on Rac1: (1) Rac1 activation was aborted in Sig-1R-depleted macrophages during efferocytosis (Figure (Figure7A-B).7A-B). (2) The Sig-1R activation-promoted phagocytic activity of macrophages was abolished by the Rac1-specific inhibitor EHop-016 in vitro and in vivo (Figure (Figure7C7C and E). We further revealed that Rac1 activation promoted the interaction between Sig-1R and Rac1 (Figure (Figure8,8, Figure S6). Furthermore, we mapped the structural basis for the interaction by conducting multiple mutations and identified that residues 33, 126, 138 and 140 of Rac1 appeared to be the key amino sites for the binding pocket (Figure (Figure8D).8D). However, whether these single alleles are decisive sites to mediate rac1-mediated efferocytosis in response to Sig-1R activation requires further investigation. On the other hand, how Rac1 interaction alters the translocation or functions of Sig-1R remains to be elucidated.
In summary, our study identifies Sig-1R as a pivotal molecule that mediates efferocytosis by macrophages, which contributes to inflammation resolution and functional recovery after cerebral ischemia. We also demonstrated that Sig-1R activation promotes phagocytic activity mainly through interaction with Rac1 in macrophages. The data revealed that modulation of Sig-1R/Rac1 signaling pathway-mediated phagocytic activity may be a potent therapeutic approach for ischemic stroke. It is also noted that although we provided sufficient evidence to demonstrate the importance of Sig-1R in the regulation of efferocytosis by macrophages/microglia, loss-of-function studies were performed using global Sig-1R knockout mice to develop macrophage-specific Sig-1R knockout mice, which may be a more precise approach to further elucidate the role of the receptor in phagocytic activity.