
BS148 Reduces the Aggressiveness of Metastatic Melanoma via Sigma-2 Receptor Targeting
By Claudia Sorbi, Silvia Belluti, Claudio Giacinto Atene, Federica Marocchi, Pasquale Linciano, Neena Roy, Elia Paradiso, Livio Casarini, Simone Ronsisvalle, Tommaso Zanocco-Marani, Livio Brasili, Luisa Lanfrancone, Carol Imbriano, Giulia Di Rocco, and Silvia Franchini
Excerpt from the article published in the International Journal of Molecular Sciences. 2 June 2023; 24(11):9684. DOI: https://doi.org/10.3390/ijms24119684
Editor’s Highlights
- Cutaneous melanoma is considered one of the most serious because of the high potential for metastasis and mortality worldwide.
- By 2040, the number of new cases of cutaneous melanoma per year will increase by more than 50%.
- Binding to sigma-2 receptor (S2R) decreases the expression of genes participating in the cholesterol signaling pathway also associated with the activation of the ER stress response, resulting in decreased proliferation of metastatic melanoma cells.
- A decrease in S2R expression, in Melanoma Patient-Derived Xenograft (PDX) models, reduces cell viability and migration properties, representing a promising preclinical candidate to be further developed for the treatment of metastatic melanoma.
Abstract
The management of advanced-stage melanoma is clinically challenging, mainly because of its resistance to the currently available therapies. Therefore, it is important to develop alternative therapeutic strategies. The sigma-2 receptor (S2R) is overexpressed in proliferating tumor cells and represents a promising vulnerability to target. Indeed, we have recently identified a potent S2R modulator (BS148) that is effective in melanoma. To elucidate its mechanism of action, we designed and synthesized a BS148 fluorescent probe that enters SK-MEL-2 melanoma cells as assessed using confocal microscopy analysis. We show that S2R knockdown significantly reduces the anti-proliferative effect induced by BS148 administration, indicating the engagement of S2R in BS148-mediated cytotoxicity. Interestingly, BS148 treatment showed similar molecular effects to S2R RNA interference-mediated knockdown. We demonstrate that BS148 administration activates the endoplasmic reticulum stress response through the upregulation of protein kinase R-like ER kinase (PERK), activating transcription factor 4 (ATF4) genes, and C/EBP homologous protein (CHOP). Furthermore, we show that BS148 treatment downregulates genes related to the cholesterol pathway and activates the MAPK signaling pathway. Finally, we translate our results into patient-derived xenograft (PDX) cells, proving that BS148 treatment reduces melanoma cell viability and migration. These results demonstrate that BS148 is able to inhibit metastatic melanoma cell proliferation and migration through its interaction with the S2R and confirm its role as a promising target to treat cancer.
1. Introduction
Among all types of cancer, cutaneous melanoma is considered one of the most serious because of the high potential for metastasis and mortality worldwide. According to estimates, by 2040, the number of new cases of cutaneous melanoma per year will increase by more than 50% [1]. Although our understanding of the biology of melanoma and the development of targeted and immune therapies have advanced significantly over the past five years, a large fraction of patients do not respond to therapy or rapidly acquire drug resistance [2]. To effectively address this issue and guide future therapy development, a deeper knowledge of the actionable molecular pathways in metastatic melanoma is required and innovative treatments urgently needed. Sigma receptors play an important role in many physiological and pathological conditions, such as brain plasticity, learning and memory processes, CNS disorders, drug abuse, and neuropathic pain [3,4,5,6]. In addition, a growing body of evidence supports their function in tumor development, growth, and migration due to their upregulation in various human cancer cells compared with healthy tissue [7]. Since the identification of the sigma receptors in the mid-1970s, efforts have been made to characterize their molecular structure and function [8,9]. Sigma receptors were historically divided into two distinct subtypes, namely, sigma-1 receptor (S1R) and sigma-2 receptor (S2R), the latter coded by the TMEM97 gene [10,11]. Crystallization of both S1R and S2R has brought this receptor family into the new era of drug discovery, enabling a more efficient toolbox for the identification of selective molecular probes and new drugs [12,13]. Some representative S2R ligands under investigation for the diagnosis or treatment of cancer are shown in Figure 1. Sigma-1 antagonists and sigma-2 agonists have been reported to induce cell death in several human cancer cells, although there are no sigma-receptor-targeting drugs approved for and currently applied as anti-cancer therapies in clinics [14,15]. Recently, a highly selective sigma-2 radioligand, [18F]ISO-1 (Figure 1), has been validated as a PET imaging biomarker to assess the proliferative status of tumors and as a predictor of therapy response using noninvasive techniques [16].

Representative sigma-2 ligands involved in cancer.
Our research group has been active in the field of sigma-receptor ligands [17]. In our previous works, a large library of substituted benzylpiperidines and benzylpiperazines was synthesized, and the effect of modifications to the aralkyl moiety has been studied systematically [18]. Among them, BS148 exhibited nanomolar affinity and good preference for S2R over S1R (pKi S1R = 6.27 (Ki 540 nM); pKi S2R = 7.71 (Ki 20 nM); S2R/S1R = 28). Functionally, BS148 treatment induced cell death in SK-MEL-2 metastatic malignant melanoma cells with a GI50 of 22 μm, which was comparable to the reference S2R agonist siramesine. In the tail-flick test, BS148 decreased the analgesic effect of morphine, supporting an S1R agonist profile and thus excluding the involvement of the S1R in cell death, as S1R agonists show a pro-survival action [14]. Therefore, we assumed that the anti-proliferative effect of BS148 was possibly mediated by the modulation of the S2R, suggesting an S2R agonist profile. Although S2R selective compounds with excellent antitumor activity have been identified, it is still unclear whether their mechanism of action is mediated by this receptor, as opposite results have been obtained [19,20].
To this aim, in this work, we elucidate the molecular basis of BS148 anti-cancer activity by designing and synthesizing a BS148 fluorescent probe for confocal microscopy study. Through siRNA-mediated knockdown of TMEM97, coding for S2R, we validated the BS148 target in melanoma cells. In addition, we investigated the effects induced by S2R after treatment with BS148 at the molecular level using Western blotting and RT-qPCRs. Finally, we assessed the anti-proliferative and anti-migration activity of BS148 in metastatic melanoma PDX-derived cells, which represent an important platform for elucidating new treatments in oncology.
2. Results
2.1. BS148 Enters Melanoma Cells and Localizes into the Cytoplasm
2.1.1. Design and Synthesis of BS148 Fluorescent Probe
To shed light on the mechanism of action of BS148, we first assessed its ability to enter melanoma cells and investigated its subcellular localization using confocal microscopy analysis. To this aim, a BS148 fluorescent probe conjugate (BS148-fluo) was synthesized. A preliminary investigation of the binding mode of BS148 at the S2R binding site was performed with docking calculation to identify the portion of the BS148 molecule more prone to being derivatized with the insertion of the fluorescent probe without significantly affecting the binding of the conjugate to the receptor. The structure of S2R remained a mystery until very recently, when the four X-ray crystallographic structures of bovine S2R in complex with S2R ligands, namely, PB28 (PDB ID: 7M93), roluperidone (PDB ID: 7M94), Z1241145220 (PDB ID: 7M95), and Z4857158944 (Ki S2R = 4 nM; PDB ID: 7M96), were successfully resolved [13]. In this work, the binary complex S2R-roluperidone (PDB ID 7M94) was used for docking calculation because it is the one with the highest resolution (2.41 Å) among the currently available X-ray crystallographic structures. Molecular docking was performed with AutoDock 4.2.6. The docking model was first validated by redocking the cognate ligand roluperidone into the parent crystal structure. The predicted binding mode for roluperidone closely matched the crystallographic pose with a root-mean-square deviation (RMSD) of 0.95 Å, thus confirming the suitability of the docking protocol for effectively predicting the binding mode of the ligand with S2R. The predicted binding pose for BS148 is reported in Figure 2A. BS148 properly accommodated within the S2R binding site. The binding is primarily driven by the salt bridge between the protonated nitrogen atom of the benzylpiperidine moiety and the key residue Asp29. This electrostatic interaction is known to be crucial for the formation of the biologically active conformation of proteins [21]. Moreover, two additional π-cation interactions between the protonated amine and Tyr147 and Tyr150 contribute to reinforcing the binding of the ligand with the receptor and its positioning within the binding site. Indeed, these polar interactions guide the orientation of the two main hydrophobic moieties of BS148 that flanks the tertiary amine. Specifically, the 1,4-dithiaspiro[4.5]decane moiety is located in the deepest and highly hydrophobic portion of the binding pocket formed by Phe66, Phe69, Leu70, and Tyr150; these hydrophobic interactions contribute to stabilizing the compounds in the binding site. Conversely, the benzyl moiety is oriented toward the entrance of the binding site which is lined with hydrophobic and aromatic residues and opens into the lipidic bilayers of the membrane where S2R is anchored rather than the aqueous environment of the cytoplasm. Interestingly, no significative differences in the binding mode or the docking score were observed for either R- or S-enantiomers (docking score −8.439 and −8.415 for (S)-BS148 and (R)-BS148, respectively; Figure 2B).
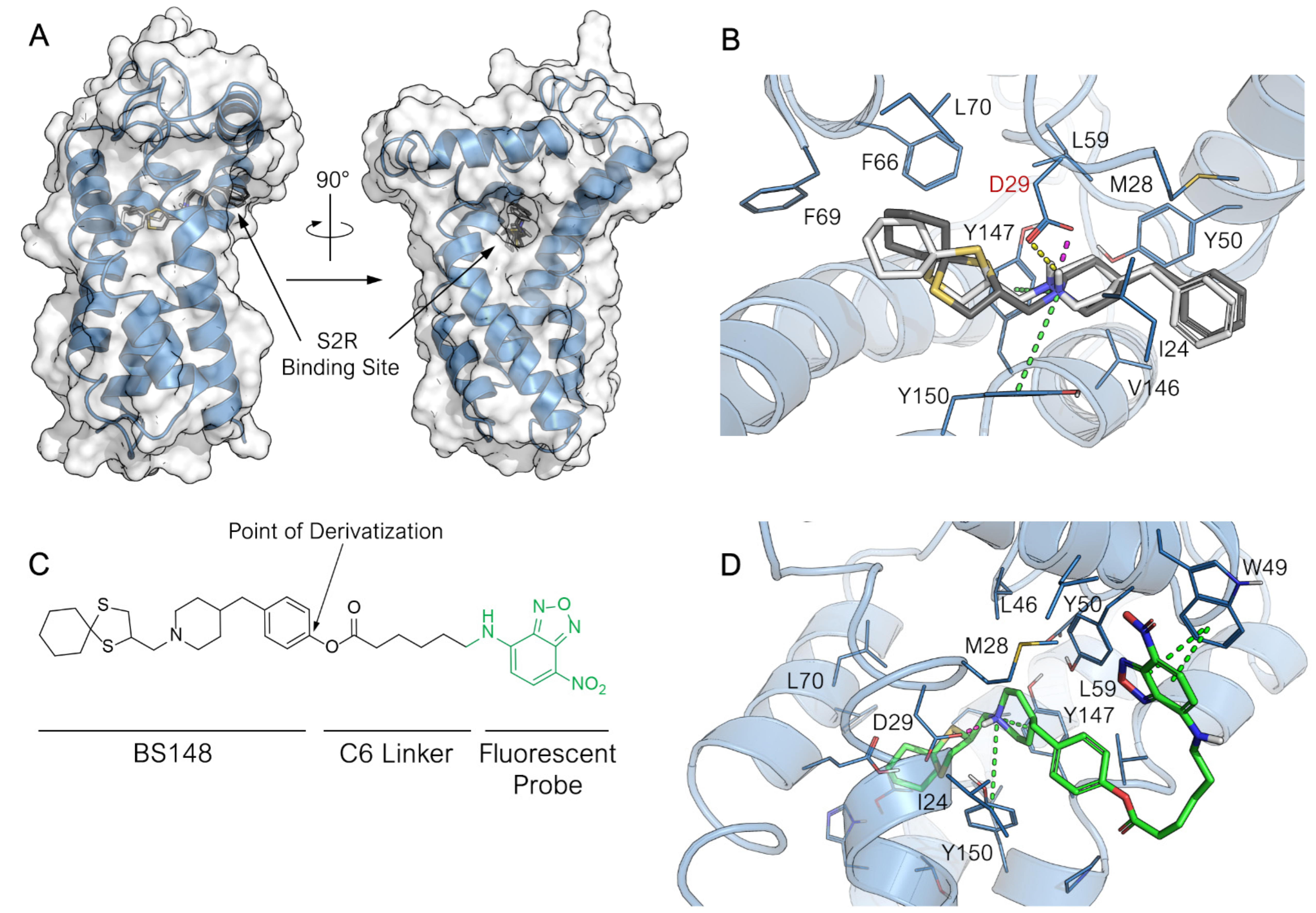
Predicted binding mode of BS148 and BS148-fluo at the S2R binding site. (A) Overall structure of S2R in complex with highest-score binding pose of BS148 and view of the entrance to the binding pocket. The tertiary structure of the protein (PDB ID: 7M94) is represented as blue cartoon. The surface of the protein is displayed in transparency. (B) Focus on the binding mode of (S)-BS148 (in white stick carbons) and (R)-BS148 (in grey stick carbon) within the S2R binding pocket. (C) Chemical structure of BS148-fluo. The point of derivatization of BS148 for appending the probe is highlighted. (D) Binding mode of BS148-fluo (in green stick carbons) with a particular focus on the orientation of the probe over the S2R surface. The 3D structure of S2R is represented in blue cartoon, and the key amino acid residues interacting with the ligand are represented in lines. The heteroatoms are color-coded: oxygen in red, nitrogen in blue, and sulfur in yellow. The H-bonding, salt bridge, π-cation, and π–π stacking are represented in yellow, pink, green, and blue dotted lines, respectively.
Based on the predicted binding mode of BS148, we chose the benzyl moiety as the anchor point for the introduction of the fluorescent probe, as it is located in a wider region outside the binding pocket and therefore should not adversely affect the binding of the conjugate to S2R (Figure 2C). We chose the green-emitting 7-nitrobenzofurazan as a fluorogenic moiety previously used in confocal microscopy study for selective S2R fluorescent probes [22]. This fluorescent probe was connected to the functionalized BS148 through a six-carbon spacer. Thus, the binding of BS148-fluo at S2R was preliminarily investigated with docking calculation. No significative differences between BS148-fluo and the parent compound in the binding mode for the core 1-((1,4-dithiaspiro[4.5]decan-2-yl)methyl)-4-benzylpiperidine scaffold were observed. As for BS148, the chirality seemed to also not affect the binding of BS148-fluo (docking score −10.142 and −9.859 for (S)-BS148-fluo and (R)-BS148-fluo, respectively). As expected, the fluorescent 7-nitrobenzofurazan moiety is completely positioned outside the binding pocket. This results in several predicted orientations for the 6-(7-nitrobenzofuran-4-ylamino)hexanoate moiety. To assess the binding stability of the BS148-fluo binding pose and investigate the preferred and most recurring orientation of the 6-(7-nitrobenzofuran-4-ylamino)hexanoate tail, molecular dynamic simulation was performed. To reproduce a more realistic environment, the S2R was embedded within a membrane bilayer. The positioning of S2R within the membrane was retrieved from the OPM (Orientation of Proteins in Membrane) database. The prepared system was subjected to a 50 ns MD run. To obtain insight into the dynamic behavior and stability of the ligand complexes, the backbone Root-Mean-Square Deviation (RMSD) was computed. The RMSD of the system sharply increased during the initial equilibration phase because of the change in simulation condition but rapidly converged at 1.4 ± 0.2 Å after 2 ns. The protein RMSD does not fluctuate significantly throughout the duration of the simulation (Figure S1A), thus indicating the dynamic stability of the system upon BS148-fluo binding to the receptor. The ligand RMSD was measured as well to assess the stability of the ligand with respect to the protein and its binding pocket. The RMSD of the ligand increased to 3.159 ± 0.445 Å during the initial equilibration phase as a result of the settling of the ligand from its docked position and then underwent a further sharp increase at around 5.2 ns of simulation. The RMSD of the ligand fluctuated around 6.000 ± 1.000 Å for the next 15 ns and then converged at 22 ns and settled to 5.863 ± 0.221 Å for the entire duration of the simulation. To understand the reasons for this change in the RMSD of the ligand, the Ligand Root-Mean-Square Fluctuation (L-RMSF), which expresses the changes in the ligand atom positions, was computed. As displayed in the Ligand RMSF plot reported in Figure S1B, the BS148 core of the conjugate molecule is firmly anchored within the S2R binding site, and it does not show remarkable fluctuation during the entire simulation with an RMSF of 0.895 ± 0.200 Å (atoms 1–25, Figure S1B). Conversely, the 6-(7-nitrobenzofuran-4-ylamino)hexanoate moiety (atoms 26–45) had the highest RMSF of 3.286 ± 0.544 Å, indicating a greater fluctuation for this membrane-exposed portion of the molecule. Considering the average conformation assumed by BS148-fluo during the latest converged 25 ns (Figure 2D), the 7-nitrobenzofurazan moiety folded over the surface of S2R, and it was accommodated within a minor hydrophobic groove on the surface of the protein delimited by helix-1 and -2 and outlined by the hydrophobic amino acid side chains such as Ile 24, Leu27, Met28, Leu43, and Leu46, where in addition to the hydrophobic interactions, an almost conserved π–π interaction was established between the aromatic ring of the benzofurazan moiety and the side chain of Trp49. Summation of the overall frequency of contacts to the binding site of S2R supports the docking results and confirms the capability of BS148-fluo to form stable interactions with the receptor.
The synthetic pathway used to obtain the BS148-fluo is depicted in Scheme 1. We planned to insert a hydroxyl group at the para position of the benzyl moiety of BS148 to facilitate bond formation with the selected fluorescent probe 6-(7-nitrobenzofuran-4-ylamino)hexanoic acid. Briefly, the previously synthesized 2-(chloromethyl)-1,4-dithiaspiro[4.5]decane was reacted with piperidin-4-ylmethylphenol (2 eq.) to give the key intermediate 4-((1-((1,4-dithiaspiro[4.5]decane-2-yl)methyl)piperidin-4-yl)methyl)phenol (1) [23]. Two microwave (MW) cycles of 3.5 h each were performed at 130–140 °C, affording the desired product in low yield (14%) due to the low nucleophilicity of the amine, commercially available as hydrochloride salt. Compound 1 was then reacted with the 6-(7-nitrobenzofuran-4-ylamino)hexanoic acid using the conventional EDC coupling chemistry (EDC, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) in anhydrous methylene chloride (MC) under argon atmosphere in the presence of a catalytic amount of triethylamine, affording the desired compound in 35% yield. BS148-fluo was then converted into the corresponding oxalate salt in order to obtain a water-soluble probe required for the biological tests.
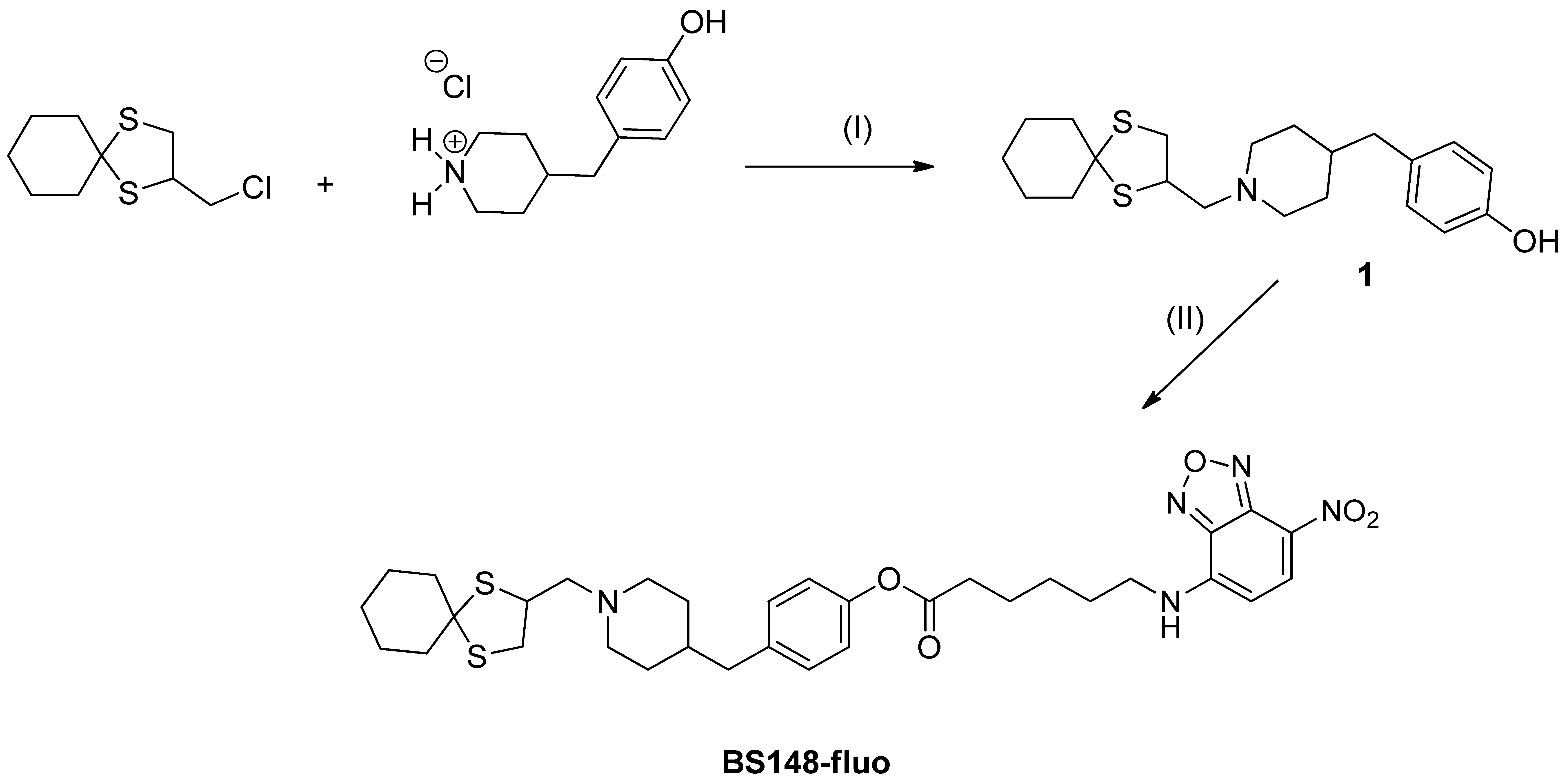
BS148-fluo. Reagents and conditions: (I) anhydrous acetonitrile, K2CO3 (2 eq.), catalytic KI, 135 °C, MW; (II) 6-(7-nitrobenzofuran-4-ylamino)hexanoic acid, methylene chloride, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), Ar, triethylamine (TEA), r.t.
The dose-response curve for BS148-fluo at S2R is reported in Figure S2. As shown in Table 1, the S2R binding affinity of the BS148-fluo is comparable to that of the parent compound BS148. This result agrees with the docking studies.
| Compound | pKi S2R a ± SD |
|---|---|
| BS148-fluo | 7.00 ± 0.11 |
| BS148 * | 7.71 b |
Affinity constants (pKi) of BS148-fluo and the reference compound at S2R.
a Values represent the mean of at least two separate experiments performed in duplicate. * See [18]. b SD is within ±20% [18].
2.1.2. Confocal Microscopy Studies
BS148-fluo was used to assess BS148 cellular localization using confocal microscopy. SK-MEL-2 cells were grown to sub confluence and treated with BS148-fluo (100 nM), and the fluorescent intensity was analyzed with confocal microscopy. MCF7 cells were used as a positive control, as they express S2R at high levels but low S1R [24]. The results demonstrate the internalization of BS148-fluo in SK-MEL-2 and MCF7 cells (Figure 3). The localization of the probe in the perinuclear region and cytoplasm and the absence of uptake in the nucleus are consistent with the localization of S2R in the endoplasmic reticulum [25].
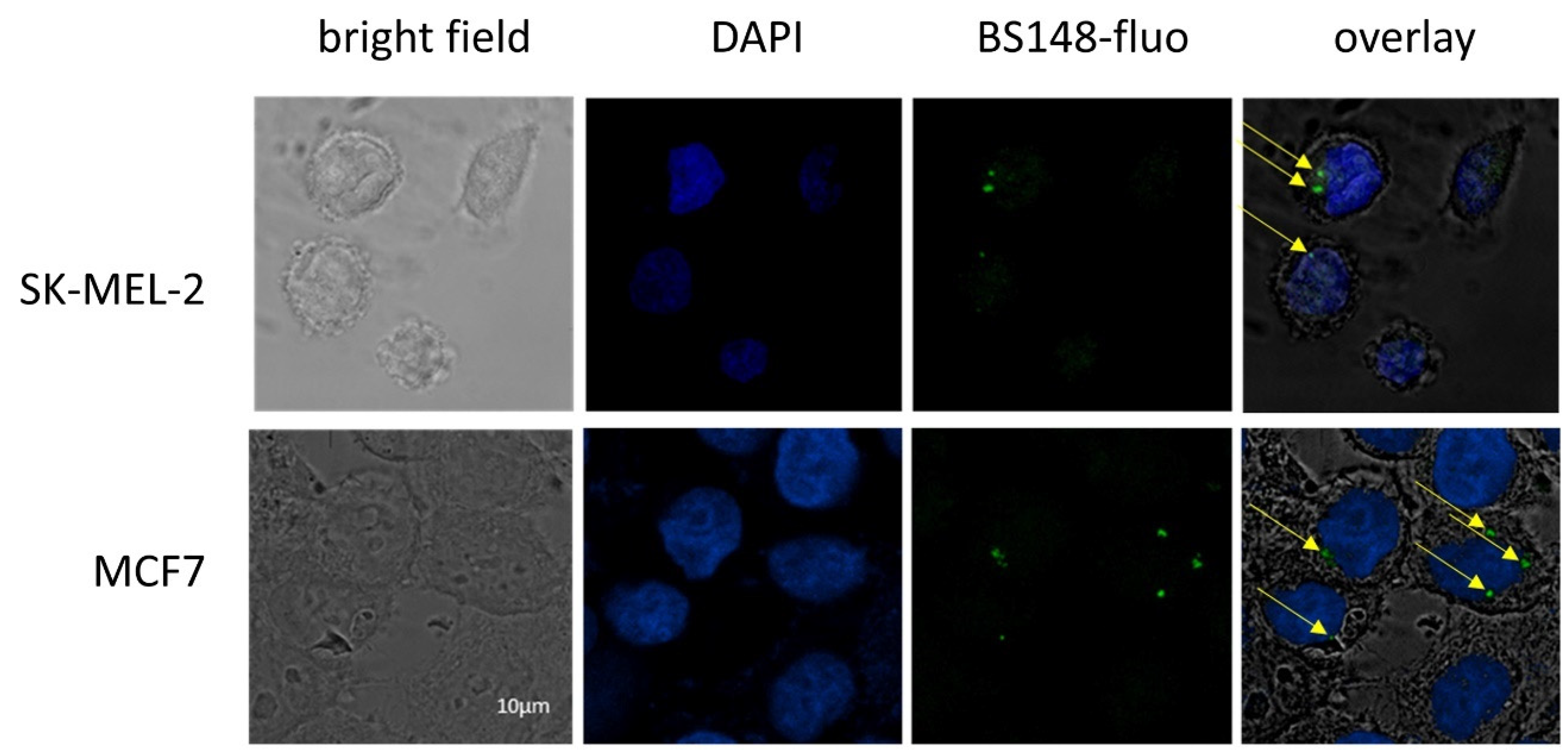
Confocal microscopy analyses of SK-MEL-2 cells labeled with BS148 fluorescent probe. MCF7 cells were used as a positive control. Bright field (first column); DAPI staining (blue, second column); staining with BS148-fluo (100 nM, green, third column); overlay (fourth column). Arrows indicate the localization of BS148-fluo within the cells. Images are representative of one experiment out of three.
2.2. S2R Is Involved in BS148-Induced Decrease in Cell Viability
The analysis of the expression of sigma receptors in SK-MEL-2 cells, using RT-qPCR, highlighted higher transcript levels of S2R compared to S1R (Figure 4A). We then evaluated whether the anti-proliferative effects induced by BS148 can be ascribed to specific S2R targeting. To this purpose, we knocked down TMEM97 mRNA transcripts with siRNA delivery in SK-MEL-2 cells and evaluated viability following the administration of increasing concentrations of BS148 for 24 h and 48 h. Western blot analysis of the total cellular extracts confirmed the decrease in S2R protein expression by RNA interference (Figure 4B). The comparison between the growth curves of TMEM97-silenced and control (mock-transfected) cells showed significant protection from cell death by TMEM97 siRNA upon 60–100 µM administration of BS148 (Figure 4C), with the GI50 being higher than 100 µM compared to 56.6 ± 22.7 µM in the mock-transfected cells.
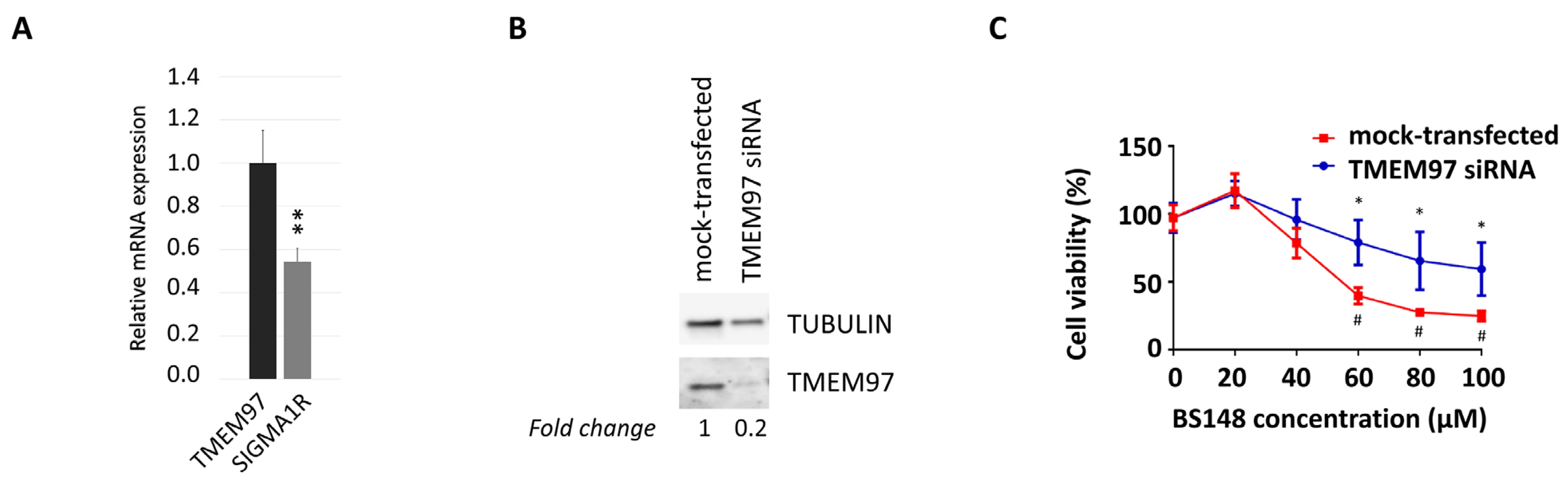
TMEM97-mediated decrease in cell viability induced by BS148. (A) qRT-PCR analysis of TMEM97 and S1R transcripts in SK-MEL-2 cells. Normalized S1R mRNA levels are reported as fold change (2-dCT) vs. TMEM97, arbitrarily set at 1 (mean ± SD; n = 3); ttest was used to calculate statistical significance (** p < 0.01). (B) Western blot analysis of S2R protein expression in control or TMEM97 knocked down SK-MEL-2 cells. Tubulin served as a loading control. TMEM97 protein levels, derived from densitometry analysis, were normalized to tubulin and expressed as fold change versus mock-transfected control cells. (C) The viability of control and knocked-down cells was evaluated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay after 48 h of treatment with 20–100 µM BS148. Cells transfected with control siRNAs served as a positive control (mock). Absorbance data were collected and represented as the percentage (%) of viable cells (means ± SD). * = significantly different versus control in the same treatment condition; # = significantly different versus the untreated mock-transfected condition (two-way ANOVA; p ≤ 0.05; n = 8).
2.3. BS148 Affects the Expression of Genes Involved in Cholesterol Biosynthesis and Activates the ER Stress Response in Melanoma Cells
The membrane-bound S2R localizes in multiple subcellular organelles, including the endoplasmic reticulum (ER), and several lines of evidence highlight the crucial role of S2R in cholesterol homeostasis [26]. With accelerated cholesterol and lipid metabolism being the hallmarks of tumor cells, we hypothesized that BS148 could modulate the expression of genes participating in cholesterol biosynthesis that sustain the proliferation and migration of cancer cells. RT-qPCR analysis on the total RNA extracted from the control and BS148-treated cells at 40 µM and 80 µM doses showed a dose-dependent decrease in the expression of Acetyl-CoA acetyltransferase (Acat2), Farnesyl-diphosphate farnesyltransferase 1 (Fdft1), and Farnesyl diphosphate synthase (Fdps), all genes related to the cholesterol pathway (Figure 5A). Taking into consideration that ER stress regulates cholesterol metabolism and homeostasis, we analyzed the expression of genes related to the ER stress response as well. The transcriptional activation of activating transcription factor 4 (ATF4), C/EBP homologous protein (CHOP), and the short splice variant of the IRE-1α/X-box binding protein 1 (Xbp1) gene support the hypothesis that S2R targeting by BS148 induces ER stress. Differently, the expression of the protein-folding ER chaperone BiP/Grp78/HspA5 that improves the protein-folding function of the ER was not increased. Unexpectedly, the analysis of mRNA levels in the SK-MEL-2 cells knocked down for TMEM97 by RNAi perfectly overlapped with the transcriptional effects induced by BS148 administration. To shed light on this result, we quantified the TMEM97 mRNA levels following the administration of BS148. RT-qPCR showed a striking decrease in TMEM97 transcription that was further validated with Western blot protein analysis of the same samples. As shown in Figure 5B, the protein levels were consistent with the mRNA levels. The increased expression of ER stress genes was coherent with the activation of a critical process known as unfolded protein response (UPR) which is usually associated with ER disturbance. At first, the UPR promotes an adaptive mechanism to restore ER homeostasis, but if the stress is prolonged or the adaptive response fails, the cells undergo apoptotic cell death. The analysis of the expression levels of γH2AX and cleaved PARP1 suggests that BS148 administration did not trigger DNA damage and caspase-dependent apoptosis, but rather it could inhibit cell proliferation or caspase-independent cell death (Figure 5B). We further explored ER stress by investigating the related signaling pathways (Figure 5C). The p38 MAPK promotes ER stress-induced cellular processes and may phosphorylate CHOP to activate its transcription through ATF6 [27]. Western blot on the total protein extracts from the BS148-treated cells showed a dose-dependent increase in p38 MAPK phosphorylation. ER stress and UPR were also coupled with the MEK/ERK signaling pathway. Similarly, we observed the phosphorylation of ERK1/2, which could contribute to non-caspase-dependent pathways of injury after ER stress [28].
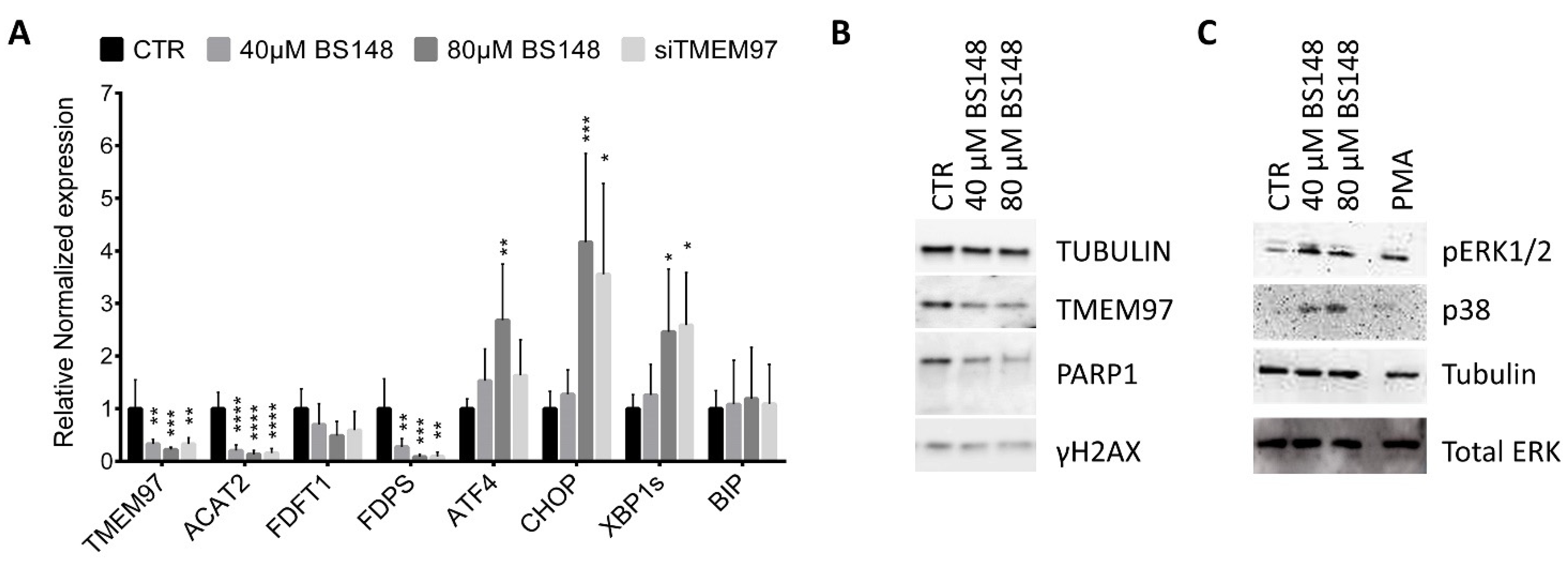
Effects of BS148 treatment in SK-MEL-2 cells. (A) RT-qPCR analysis of the indicated transcripts in SK-MEL-2 cells treated with BS148 for 48 h or siRNA against TMEM97 transcripts. RPS20 and GAPDH were used as reference genes. Normalized mRNA levels are reported as fold change vs. control cells (CTR), arbitrarily set at 1. Data represent mean ± SD, n = 6 for BS148 treatments and n = 3 for siTMEM97 transfections; one-way ANOVA was used to calculate statistical significance (* p < 0.05, ** p < 0.01, *** p< 0.001, **** p < 0.0001) vs. CTR. (B) Western blot of total extracts from SK-MEL-2 cells treated 24 h with BS148 with the indicated antibodies. Tubulin has been used as loading control. (C) Representative images of the phosphorylation of ERK1/2 and p38 MAPK using Western blotting. Cells were treated 4 h with 40 µM and 80 µM BS148. Phorbol-12-Myristate-13-Acetate (PMA) was used as a positive control. Total ERK and tubulin were used as normalizer for pERK1/2 and p38 MAPK, respectively.
Overall, these results suggest that the inhibition of the cholesterol pathway associated with the activation of the ER stress response could be responsible for the anti-proliferative activity induced by BS148.
2.4. BS148 Reduces Viability and Migratory Capacity of Metastatic Melanoma PDX Cells
The expression of S2R in metastatic melanoma PDX-derived cells (MM13, MM2, MM27, MM16) [29] was investigated using Western blot analysis and compared to that in SK-MEL-2 and SK-MEL-28 (Figure 6). While at different levels, all the PDX cells show S2R expression, regardless of their genetic background (MM13/MM16 are NRAS-mutant and MM2/MM27 BRAF-mutant melanomas) (Figure 6A). Then, we investigated the anti-proliferative activity of BS148 in PDX cells, showing that increasing doses of BS148 significantly reduced cell viability in all four melanomas (Figure 6B).
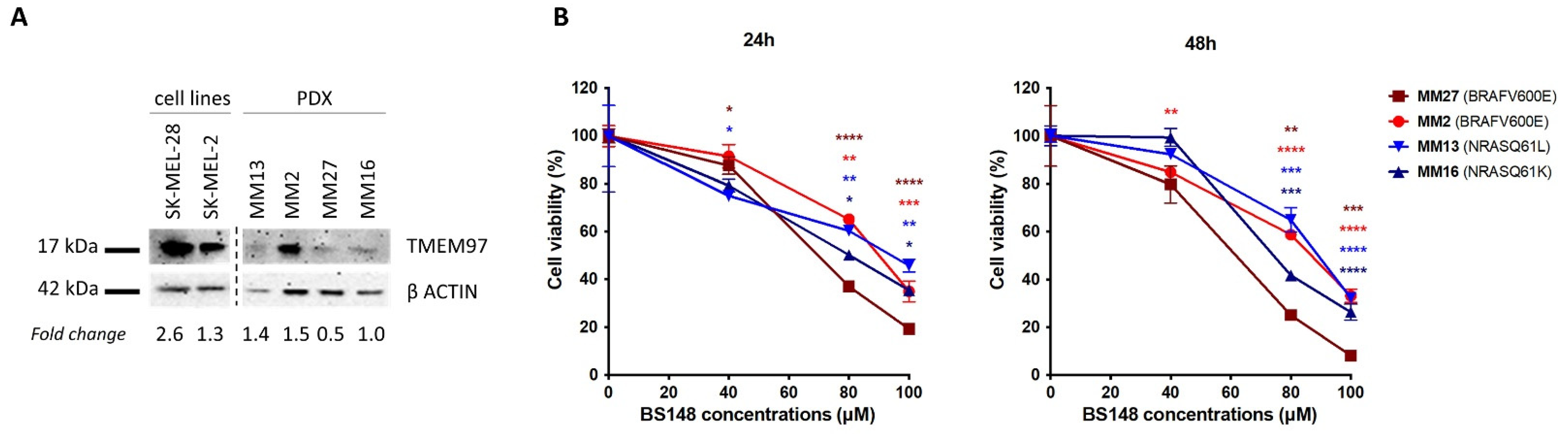
Effects of BS148 treatment in PDX cells. (A) Expression of S2R in SK-MEL-2, SK-MEL-28, and metastatic melanoma PDX-derived cells with Western blotting. TMEM97 protein levels, derived from densitometry analysis, were normalized to β-actin and expressed as fold change versus MM16. (B) Cell Titer Glo Assay for viability assessment upon BS148 treatment for 24 h (left) and 48 h (right). Cell viability is reduced by increasing concentrations of BS148 in the four PDXs. Viability was normalized to CTR (DMSO-treated cells); triplicate experiment (n = 3); one-way ANOVA was used to calculate statistical significance (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001) vs. CTR.
Moreover, BS148 strongly reduces the migratory features of three PDXs (Figure 7).
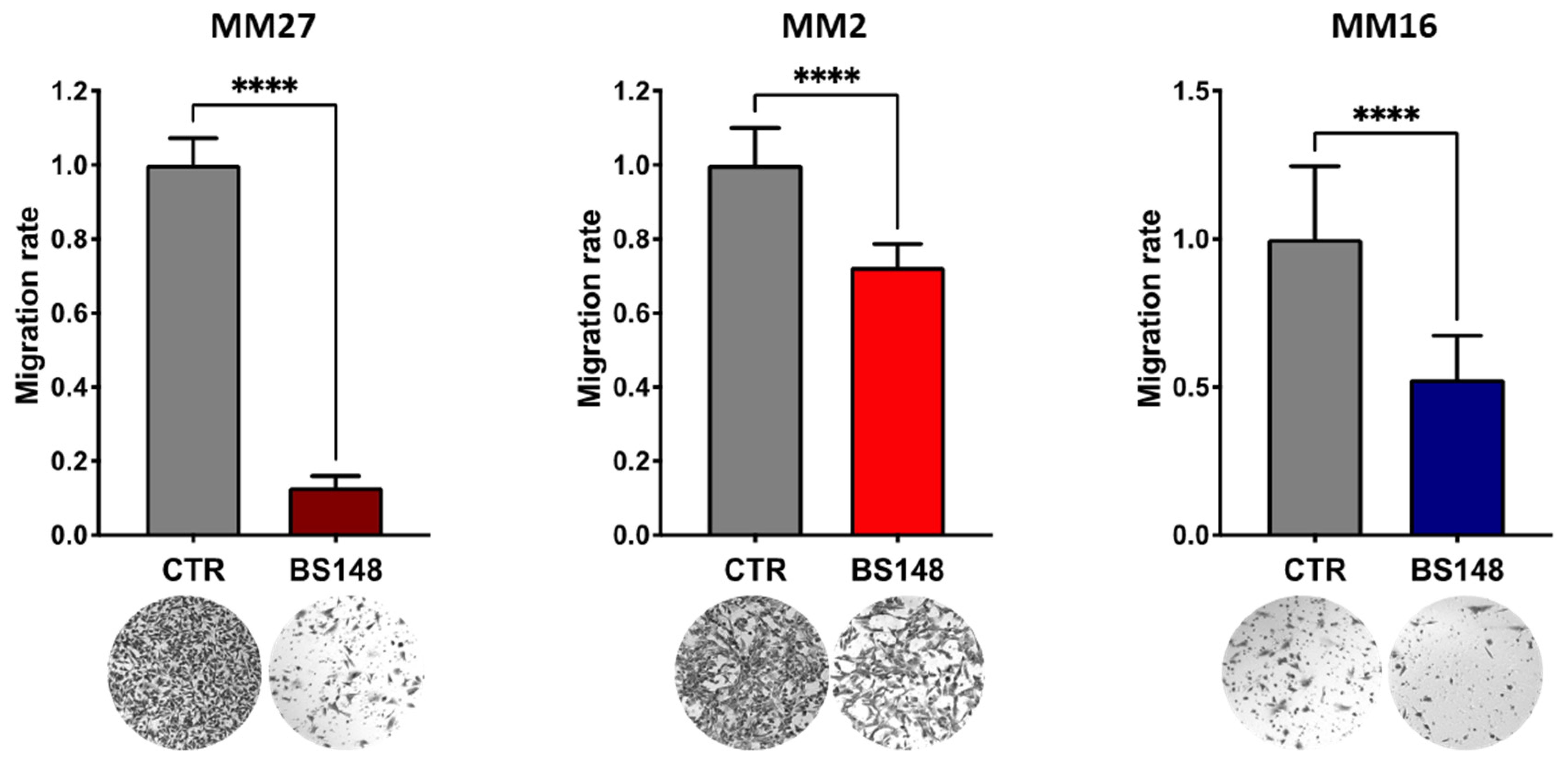
BS148 reduces the migratory properties of PDX cells. PDX cells were pre-treated with 50 μM BS148 for 24 h and then plated in modified Boyden chambers. Cell migration was assessed after 36 h and normalized to CTR (DMSO-treated cells); triplicate experiment (n = 3). t test was used to calculate statistical significance (**** p < 0.0001) vs. CTR (DMSO-treated).
These findings prompted us to evaluate the potential therapeutic relevance of S2R in the management of melanoma patients. Hence, we analyzed RNAseq data from the TCGA (The Cancer Genome Atlas) and GTEx projects to determine the transcript levels of TMEM97 in patients with melanoma compared to those in healthy patients (Figure 8). TMEM97 mRNA levels are significantly increased in tumor vs. healthy tissues, supporting the rationale of TMEM97-targeting drugs. Moreover, high levels of TMEM97 expression are associated with patients with poor overall survival probability, hinting that it may be used as a prognostic marker of survival in patients affected by skin cutaneous melanoma (SKCM).
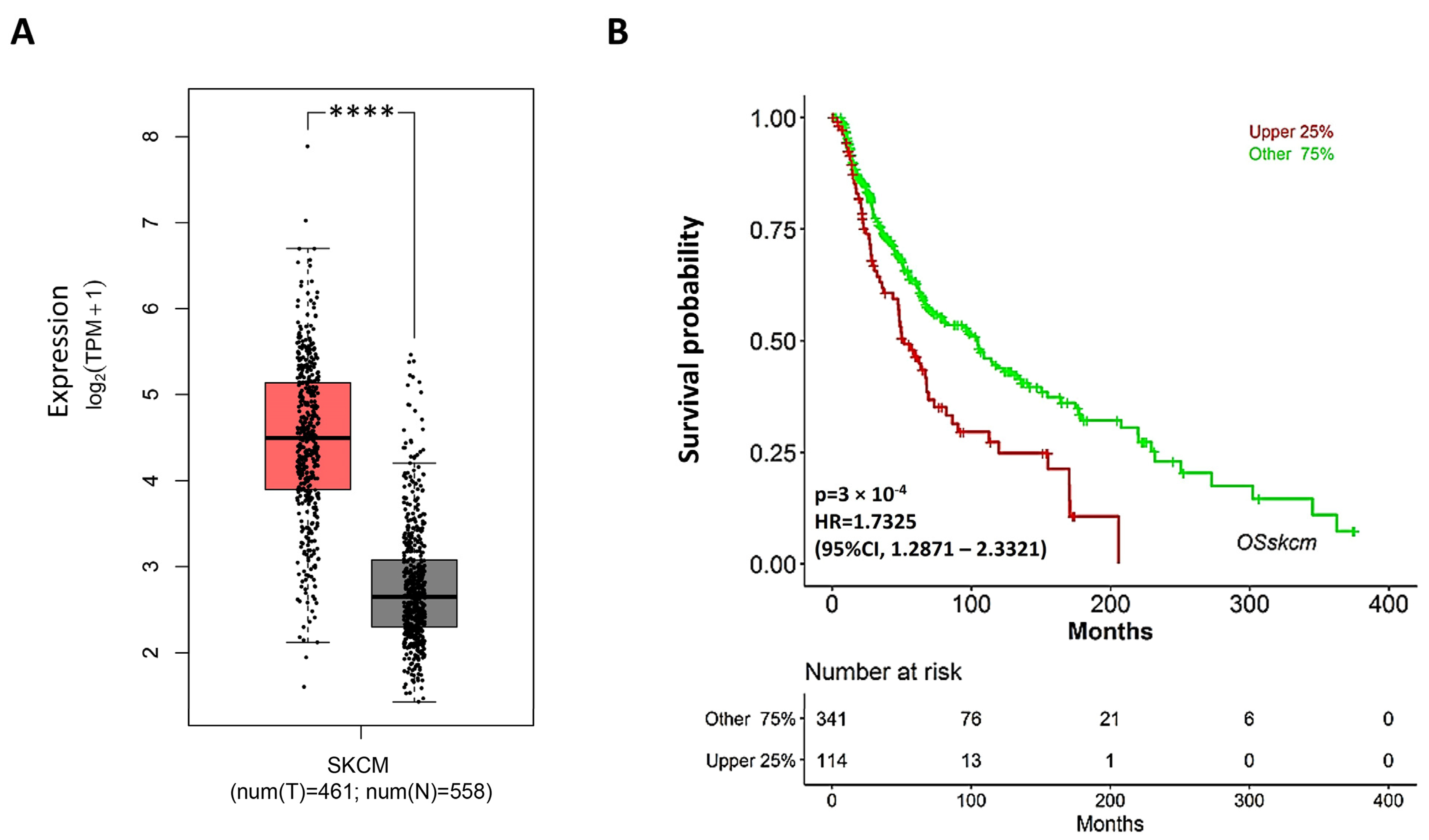
TMEM97 expression increases in SKCM patients, and its higher levels are associated with poor prognosis. (A) Expression levels of TMEM97 measured as transcripts per million (TPM) in skin cutaneous melanoma (SKCM) patients (red bar, T) compared to normal ones (grey bar, N). **** p < 0.0001. (B) Overall survival curve of cutaneous melanoma patients based on SKCM TCGA data. Upper 25%: cases ranked in top 25% higher expression level for TMEM97 gene; other 75%: cases ranked in bottom 75% lower expression level of TMEM97 gene.
3. Discussion
The S2R is an endoplasmic-reticulum-resident transmembrane protein overexpressed in various proliferative tumors. Its expression is positively correlated with tumor progression and recurrence and poor survival [7,11]. Here, we show that TMEM97 transcript levels rise significantly in tissues from SKCM patients compared to those from healthy ones, and high levels could represent a prognostic marker for the survival prediction of patients. S2R is an interesting pharmacological target because of its involvement in cholesterol homeostasis. Melanoma cells are characterized by an impressive metabolic plasticity, including the lipid metabolic network that confers cancer aggressiveness. The analysis of TCGA data from SKCM patients demonstrates that enhanced expression of cholesterol synthesis genes is associated with decreased melanoma patient survival [30]. These data support the rationale for developing S2R-targeting molecules that could be further developed as anti-cancer drugs. Novel ligands were recently developed to induce cancer-specific cytotoxic activity through the receptor [20], although opposite results were also achieved [19]. In fact, ligands inducing receptor coupling to death pathways are limited, while others mediate effects consistent with cancer cell survival [31]. These diverging effects are due to conformational issues of the receptor in the active state, resulting in intracellular signaling bias and ligand-specific, preferential activation of metabolically stimulative or inhibiting effects.
Our research team has previously identified a sigma ligand, BS148, which exhibited nanomolar affinity and good preference (25-fold) for S2R over S1R, as assessed with receptor-binding assay [18]. This compound showed a marked cytotoxic effect on SK-MEL-2 cells and was classified as a putative S2R agonist on the basis of the commonly accepted definition [32]. In this work, we translated our results into patient-derived xenograft (PDX) cells, proving that BS148 exerts anti-proliferative effects not only in SK-MEL-2 cells but also in melanoma PDX-derived cells. In these cellular models, we also observed inhibitory activity on the migration properties following BS148 treatment. Parallel studies of cell viability in control and knocked-down metastatic melanoma cells demonstrated that the anti-proliferative effects observed following BS148 administration are mediated by S2R targeting, although we cannot exclude that additional molecular targets may be involved. Overall, our results strengthen the concept that S2R could be a candidate for pharmacological anti-cancer treatments [33], including against melanoma [34].
Through the analysis of protein extracts and RNA, we investigated the possible molecular effects at the base of BS148 anti-proliferative activity. With cholesterol homeostasis being one of the main activities ascribed to S2R, we focused on the expression of genes representative of the cholesterol pathway. The expression of the selected genes was previously shown to correlate with S2R activity and expression [35]. The decrease in the transcription of cholesterol genes in BS148-treated cells comparable to TMEM97 knocked-down cells prompted us to investigate TMEM97 expression. mRNA and protein levels were similarly reduced in BS148-treated cells, a phenomenon previously described at the protein level with other S2R ligands [20]. Interestingly, the decrease in TMEM97 expression exerted by BS148 was similar to that observed for S2R RNA interference-mediated knockdown, which has been already shown to inhibit both proliferation and migration of cancer cells [36]. While compounds that mimic S1R knockdown have been historically classified as S1R antagonists, in the case of S2R ligands, the definition of agonist or antagonist activity is still controversial and appears to be context-dependent. Therefore, the negative correlation between BS148’s anti-proliferative effect and TMEM97 expression levels does not hamper the classification of BS148 as an S2R agonist, as was recently shown for PB28, another well-known S2R agonist [37].
The observed decrease in TMEM97 expression induced by BS148 can even represent an advantage in terms of therapeutic application, as small molecules are extremely stable, easier to administer, and less expensive compared to genetic manipulation. Various mechanisms could be involved in BS148-mediated TMEM97 transcriptional regulation. For example, TMEM97 has been identified as a target of the oncosuppressor p53 [38] or of c-Jun NH(2)-terminal kinase 2 (JNK) [39] and TGF-ß1 [40]. Further molecular analysis would better identify whether BS148 can affect the expression of these regulators that, in turn, could modulate TMEM97 transcription. Moreover, because TMEM97 itself is regulated by cholesterol-related signals, such as sterol depletion or SREBP expression levels [26], it could be that BS148 activates a feedback loop that impairs the expression of cholesterol proteins and consequently affects TMEM97gene expression. Finally, we cannot exclude that BS148 triggers a desensitization-like mechanism that leads to the decrease in TMEM gene expression. Cholesterol homeostasis and ER stress are highly connected, and altered cholesterol metabolism is a well-documented inducer of ER stress [41]. Correspondingly, we observed a concomitant overexpression of ER stress genes that hinted at the activation of the IRE1 and PERK pathways [42]. Further, Western blot analysis displayed both pERK1/2 and p38 MAPK activation as the main intracellular signaling signature. The activation of pERK1/2 is even slightly enhanced upon cell treatment with 40 µM BS148, while it is not at higher concentrations, suggesting a biphasic behavior of the compound. Moreover, we described for the first time the link between S2R and the p38 MAPK pathway, which occurs under treatment with BS148 and extends the phosphorylation landscape of the receptor to a new partner. The role of p38 MAPK was previously investigated in melanoma, where this kinase may exert inhibitory effects on cancer cell growth and could be targeted to block its proliferation [43,44,45,46,47,48,49,50,51]. Opposite results were also found [52,53,54], demonstrating that the analysis of one single signaling pathway could not provide a comprehensive explanation of the whole metabolic pattern of the cell and that further investigations are required to achieve solid conclusions. For instance, opposite effects on melanoma cell metabolism could be supported by different p38 MAPK isoforms not evaluated herein [55]. In any case, the activation of this kinase may upregulate p53, plausibly via reactive oxygen species (ROS) generation, leading to melanoma cell apoptosis [55,56,57,58]. Interestingly, the progression of melanoma cell growth may be inhibited upon downregulation of both p38 MAPK and pERK1/2 [59], suggesting that tumor cell growth occurs as a balance between the contribution of different pathways exerting opposite metabolic effects.
…
5. Conclusions
In conclusion, in this work, we demonstrated that BS148-induced cytotoxicity is mediated by S2R targeting. BS148 binding to S2R decreases the expression of genes participating in the cholesterol signaling pathway also associated with the activation of the ER stress response, resulting in decreased proliferation of metastatic melanoma cells in vitro. Interestingly, BS148 treatment induces a decrease in S2R expression, producing the same effect as S2R RNA interference-mediated knockdown. In PDX models, BS148 reduces cell viability and migration properties, representing a promising preclinical candidate to be further developed for the treatment of metastatic melanoma.