
The regulatory role of endoplasmic reticulum chaperone proteins in neurodevelopment

By Hongji Sun, Mengxue Wu, Minxin Wang, Xiaomin Zhang, and Jia Zhu
Excerpt from the article published in Frontiers of Neuroscience, 15 November 2022, Sec. Neurodevelopment, DOI: https://doi.org/10.3389/fnins.2022.1032607
Editor’s Highlights
- Neurodevelopment is a highly complex and precisely regulated process.
- The endoplasmic reticulum (ER) chaperones play key roles in neural development through various processes, such as neuronal migration and localization, neuronal morphogenesis, and synaptic modification.
- Sigma-1 receptor (Sig-1R) is a trans-membrane ER chaperone that regulates neuronal morphogenesis and dendritic axon growth and is mainly localized in the hippocampus.
- Sigma-1 receptor regulates mitochondrion-associated endoplasmic reticulum membrane conduction and affects neural development.
Abstract
The endoplasmic reticulum (ER) is the largest tubular reticular organelle spanning the cell. As the main site of protein synthesis, Ca2+ homeostasis maintenance and lipid metabolism, the ER plays a variety of essential roles in eukaryotic cells, with ER molecular chaperones participate in all these processes. In recent years, it has been reported that the abnormal expression of ER chaperones often leads to a variety of neurodevelopmental disorders (NDDs), including abnormal neuronal migration, neuronal morphogenesis, and synaptic function. Neuronal development is a complex and precisely regulated process. Currently, the mechanism by which neural development is regulated at the ER level remains under investigation. Therefore, in this work, we reviewed the recent advances in the roles of ER chaperones in neural development and developmental disorders caused by the deficiency of these molecular chaperones.
Introduction
Neurodevelopment is a highly complex and precisely regulated process (Bagni and Zukin, 2019). The establishment of the human embryonic central nervous system begins on the 22nd day after fertilization when the neural tube begins to form (Devakumar et al., 2018). As development progresses, the anterior portion of the neural tube expands to form “brain vesicles”, and the brain develops from five small vesicle walls filled with fluid (Jiang and Xu, 2019). The initial vesicle wall has only two layers: the compartment layer and the marginal layer (Bostan et al., 2018; Moon and Xiong, 2022). Neural precursor cells (NPCs) are located in the inner layer of vesicles, and cells in the ventricular layer proliferate. Some of the newly generated cells remain in the ventricular layer where they to continue to divide and differentiate. Finally, these cells proliferate and form different brain regional structures, such as the cerebral cortex, thalamus, and hippocampus (Trujillo et al., 2005; Turrero García and Harwell, 2017). Another group of daughter cells located furthest from the ventricle side begin to migrate outward. After these cells reach various regions of the brain and occupy specific locations, they lose their ability to divide, undergo cell differentiation, and finally form the brain (Crelin, 1974; Li and Pleasure, 2014; Belmonte-Mateos and Pujades, 2022).
At the cellular level, NPCs proliferate and differentiate into immature neurons (Riquelme et al., 2008). Then, immature neurons undergo a series of morphological changes, including neuronal axon and dendrite development (Omotade et al., 2018; Prigge and Kay, 2018), axonal myelination, and dendritic spine formation (Nimchinsky et al., 2002), and gradually become mature neurons. Neurons are interconnected by synapses formed by both axonal endings and dendritic spines, forming complex neural networks (Omotade et al., 2018; Kadoyama et al., 2019). Abnormalities in any of these processes may impair neural development, resulting in brain dysfunction and subsequent leading to brain diseases (Guerrini and Dobyns, 2014).
The endoplasmic reticulum (ER) is the largest tubular reticular organelle in eukaryotic cells (Rapoport, 2007; Pobre et al., 2019) and plays several essential roles, including Ca2+ storage and release, lipid synthesis, intracellular signaling, and protein synthesis (Phillips and Voeltz, 2016; Schwarz and Blower, 2016). ER molecular chaperones are a class of proteins ubiquitous in living organisms and are widely found in prokaryotes and eukaryotes (Doyle et al., 2013; Bose and Chakrabarti, 2017). These molecular chaperones are mainly responsible for assisting in correct protein folding, assembly, trans-location, degradation of misfolded proteins and inhibition of protein aggregation to maintain normal protein homeostasis (Carver et al., 2018; Freilich et al., 2018; Takeuchi, 2018; Ma et al., 2020).
Studies have found that abnormal expression of molecular chaperones often leads to abnormal neurodevelopment and even causes a variety of different neurodevelopmental diseases (Fatemi et al., 2005; Lammert et al., 2017). ER chaperones involved in neural development are mainly divided into the following categories: heat shock family proteins including heat shock protein 70 (HSP70) and heat shock protein 90 (HSP90) (Miller and Fort, 2018), glycan-binding lectin chaperones including calnexin (CNX) and calreticulin (CRT) (Ni and Lee, 2007), protein disulfide isomerase (PDI) (Turano et al., 2002), sigma-1 receptor (Sig-1R) (Su et al., 2010; Zhemkov et al., 2021; Dalwadi et al., 2022) and prefoldin (PFDN) (Siegert et al., 2000).
HSP70 and HSP90, members of the heat shock protein family, are highly conserved and ubiquitous molecular chaperones (Tsuboyama et al., 2018). The reversible interaction between HSP70 and peptides plays an important role in protein folding, transport, degradation of misfolded peptides and maintenance of cellular homeostasis (Fernández-Fernández and Valpuesta, 2018). HSP90 and its co-chaperones coordinate key physiological processes, such as cell survival, cell cycle control, hormone signaling, and apoptosis (Hoter et al., 2018). Binding immunoglobulin protein (Bip) and glucose-regulated protein 94 (GRP94) are the main HSP70 and HSP90 proteins in the ER. Bip cannot function alone, and the binding and separation between Bip and nascent proteins require the participation of other auxiliary proteins, such as the cochaperone SIL1, the main function of which is to serve as a nucleotide exchange factor (NEF) to assist Bip protein in binding nascent peptide chains and mediating their folding and assembly (Otero et al., 2010). However, GRP94 expression in the ER is mostly related to tumor cancer and other related diseases (Gewirth, 2016). CRT and CNX bind Ca2+ and act as molecular chaperones in the transition of proteins from the ER to the extracellular membrane and regulate Ca2+ balance in the ER (Tjoelker et al., 1994). In addition to assisting protein folding consistent with the Sig-1R and PFDN protein, PDI is also involved in the formation and isomerization of disulfide bonds to prevent intermolecular aggregation (Wilkinson and Gilbert, 2004).
In addition, in the ER, chaperones not only exert their respective functions individually but also interact with each other to maintain ER homeostasis and protein homeostasis. For example, glycan-binding lectin chaperones form the glycosylation-deglucosylation cycle. As a reticulin, CRT is involved in a quality control system for newly synthesized proteins and glycoproteins that relies on multiple additional chaperons, including CNX and protein disulfide isomerase family A member 3 (PDIA3) (Fucikova et al., 2021). This system is often referred to as the CNX/CRT cycle. The CNX/CRT cycle specifically recognizes glycoproteins linked by N2 glycosidic bonds and is an important monitoring mechanism for protein folding and assembly in eukaryotic cells as well as regulating Ca2+ homeostasis and Ca2+signaling processes (Insert a) in the ER (Helenius and Aebi, 2001). As a highly conserved molecular chaperone, Bip assists in a wide range of folding processes through its two domains: the nucleotide binding domain (NBD) and the substrate binding domain (SBD) (Mayer and Gierasch, 2019). When adenosine diphosphate (ADP) binds the NBD, the SBD conformation changes, and the substrate binding affinity of Bip increases. The SBD lid closes on the bound substrate, limiting the conformational freedom of the substrate and protecting the bound substrate from premature folding or aggregation (Hendershot et al., 1996). The NEF of Bip can break the hydrogen bond contact and detach the bound ADP molecule (Tyson and Stirling, 2000) so that NBD can bind to the new adenosine triphosphate (ATP) molecule, and the substrate binding affinity is reduced. SBD docks on the NBD and opens the lid to release the bound polypeptide substrate (Insert b). At present, SIL1 and glucose-regulated protein (GRP170) are the two co-chaperones that can bind Bip to promote NEF activity (Yan et al., 2011). Bip is also a regulator of Ca2+ homeostasis in the ER, maintaining Ca2+ balance in the ER (Lièvremont et al., 1997). In addition, when unfolded/misfolded proteins in the ER exceed the capacity of the protein folding mechanism, Bip can initiate the unfolded protein response (UPR), assist in ER-associated degradation (ERAD), reduce the unfolded/misfolded protein load, and induce autophagy (Wang J. et al., 2017).
Recent studies have shown that many ER chaperone proteins participate in neural development. Therefore, in this work, we review the regulatory roles of ER chaperones in various stages of central nervous system development as well as recent research progress on related neurodevelopmental diseases.
The role of binding immunoglobulin protein in neurodevelopment
Heat shock proteins are a large family of evolutionarily conserved molecular chaperones that play key roles in cell survival and development, and their expression is induced by different factors, including heat shock, nutrient deficiency, hypoxia, and intoxication (Tsuboyama et al., 2018). HSP70 family plays an essential role in protein folding, transport, degradation of misfolded polypeptides, and maintenance of cellular homeostasis (Fernández-Fernández and Valpuesta, 2018). Among them, Bip [also known as glucose-regulated protein 78 (GRP78) or heat shock protein A5 (HSPA5)] with a molecular weight of 78 kDa, mainly exists in the ER and is responsible for binding the nascent peptide chain and mediating its folding and assembly (Wang et al., 2009). Bip cannot perform its functions alone, but other auxiliary proteins are required for its binding and separation from nascent proteins (Otero et al., 2010). The effects of Bip on neurodevelopmental processes are mainly reflected in embryonic development, cortical neuron migration, and coordination of developmental processes in conjunction with some ER cochaperone proteins.
Binding immunoglobulin protein regulates the migration and localization of developing cortical neurons
Mutant mice with Bip deficiency die during the embryonic period (Luo et al., 2006; Jin et al., 2017), and the main cause of death is abnormal expression of the pulmonary surfactant protein surfactant protein-A (SP-A) and prominently surfactant protein-C (ProSP-C), which subsequently triggers respiratory insufficiency in neonatal or newborn mice and leads to death. In addition to respiratory failure, Bip mutant mice exhibit abnormal neuronal migration and dysplasia. Normal cortical neurogenesis in the ventricular zone (VZ), and newborn neurons migrate to different layers of the cerebral cortex through radial migration (Nadarajah and Parnavelas, 2002; Marín et al., 2010). Neurons generated earlier migrate to the deep cortex, whereas neurons generated later migration to shallow layers (Ghashghaei et al., 2007; Kwan et al., 2012). The subplate (SP), cortical plate (CP), and marginal zone (MZ) are formed in an inside-out migration pattern during development (Caviness, 1982). However, newborn neurons in Bip mutant mice are the first to migrate to the MZ and remain there, whereas later-born neurons fail to migrate to the upper layers. The mutant brain exhibits an outside-in pattern during neocortical layer formation. This phenotype is observed because loss of Bip affects the expression of reelin, a protein that plays an essential role in neuronal migration. In wild-type mice, reelin-secreting Cajal-Retzius (CR) cells are located in the superficial layer of the cortex. In contrast, CR cells in mutant Bip mice are scattered around the upper layer of the neocortical primordium, and CR cells in Bip mutants do not secrete reelin. Therefore, Bip mutant mice exhibit a phenotype similar to that of reelin knockout mice (Mimura et al., 2008).
Binding immunoglobulin protein regulates motor neuron development
Heterozygous mutant Bip mice exhibit no significant difference in lifespan compared with wild-type mice, but mutant Bip mice develop renal tubular-interstitial lesions as they age (Kimura et al., 2008). Some mutant Bip mice develop paralysis and tremors after 12 months. These mice exhibit motor nerve injury symptoms, such as loss of righting reflex, and suffer from paralysis (Jin et al., 2014). In addition, studies have shown that Bip deletion is associated with the neurodegenerative disease amyotrophic lateral sclerosis (ALS). ALS is a fatal neurodegenerative disease characterized by progressive degeneration of upper and lower motor neurons (Beghi et al., 2011; Filareti et al., 2017). In recent years, impaired protein homeostasis has been found to be a key factor in the pathogenesis of ALS (Zhao et al., 2008; Ruegsegger and Saxena, 2016), and Bip plays an important role in this process. Studies have found that Bip-deficient mice show more severe neurological decline and spinal motor neuron loss symptoms than normal mice. Gómez-Almería et al. created mutant superoxide dismutase 1 (mSOD1) and Bip double mutant (mSOD1/Bip+/-) mice to study the role of Bip in a mouse model of the neurodegenerative disease ALS (Gómez-Almería et al., 2021). They found that knockout of Bip show more intense neurological decline than mSOD1 single knockout mice. Then, to determine the relationship between the Bip protein and the pathogenesis of ALS, they also discussed the potential relationship between the Bip protein and the type-1 cannabinoid (CB1) receptor that mediates neuroprotection. CB1 receptor is mainly located in neurons of the central nervous system (Chiarlone et al., 2014). Numerous studies have reported that CB1 receptor-mediated neuroprotection is related to ALS (Abood et al., 2001; Rossi et al., 2010). They found that in mSOD1 mice with partial Bip gene deletion, the level of the CB1 receptor was significantly decreased, which indirectly indicated that Bip may play a neuroprotective role in ALS (Gómez-Almería et al., 2021). Similarly, Apolloni et al. found that increasing Bip and Hsp70 protein levels in the spinal cord and cortex of ALS model mice could rescue the loss of motor neuron dendritic spines (Apolloni et al., 2019). These studies suggest that Bip plays vital roles in motor neuron development.
Binding immunoglobulin protein regulates the plasticity of the central nervous system
Studies have shown that the interaction between Bip and N-methyl-D-aspartate (NMDA) receptors may affect synaptic transmission. NMDA receptors play essential roles in brain development, plasticity, and pathology. Functional NMDA receptors are tetramers mainly composed of two GluN1 subunits and two GluN2 regulatory subunits (Cull-Candy et al., 2001; Yashiro and Philpot, 2008). The number and composition of GluN2A and GluN2B containing NMDA receptors at synapses are dynamically regulated during development and neuronal activation, which is thought to control synaptic plasticity (Nase et al., 1999; Lopez de Armentia and Sah, 2003; Liu et al., 2004). Using immunoprecipitation, the authors examined the distribution of Bip in neurons and found that Bip is strongly expressed in both cortical and hippocampal neuronal cell bodies and neurites. Furthermore, Bip co-localizes with GluN2A in cultured cortical neuron dendrites, and preventing the binding of Bip to GluN2A reduces synaptic transmission and impairs memory formation (Zhang et al., 2015).
Binding immunoglobulin protein and SIL1 jointly regulate neural development
SIL1 is a 54-kDa ER protein composed of 461 amino acids and is a NEF for Bip (Wang J. et al., 2017). As an auxiliary protein of Bip, the synergistic effect of SIL1 and Bip has consistently been a research hotspot. The central nervous system of mice with SIL1 deficiency exhibits developmental defects, including abnormal neuronal morphological development, cortical neuron migration, and localization (Inaguma et al., 2014).
SIL1-binding immunoglobulin protein coordinately regulates neuronal migration
SIL1 plays a role in neuronal migration in the neocortex. Inaguma et al. constructed a SIL1 knockout plasmid, transfected it into the neural progenitor cells of the VZ of the embryonic mouse brain through in utero electroporation (Tabata and Nakajima, 2001; Nishimura et al., 2012) and observed the localization of the transfected cells at the post-natal day 0 (P0). In the control group, neurons normally migrated to the superficial layers (layers II-IV) of the CP, whereas the SIL1-deficient neurons remained in the middle or lower layers of the CP. Then, the authors performed immunostaining to assess the differentiation status of the abnormally localized cells and found that the abnormally localized cells remained in the immature neuronal state (NPCs or basal progenitor cells) (Inaguma et al., 2014).
Considering that SIL1 acts as a chaperone for Bip and regulates its function, Bip may also be involved in neuronal migration. Therefore, Inaguma et al. also examined the role of Bip in neuronal migration during cortical formation and compared it with SIL1. They found that a large fraction of Bip-deficient (through RNA interference) neural progenitor cells, which remain in the lower part of CP, intermediate zone (IZ), and subventricular zone (SVZ)/VZ at P0, exhibit a phenotype similar to that of SIL1-deficient cells. Bip-deficient neurons showed delayed migration at embryonic day 17 (E17) but localized in normal positions at post-natal day 7 (P7). However, after inhibiting the SIL1-Bip interaction, neuronal migration was significantly inhibited. In conclusion, the coordination function of SIL1 and Bip plays a key role in neuronal migration (Inaguma et al., 2014).
SIL1 and binding immunoglobulin protein jointly regulate reelin protein and affect development
In addition to regulating neuronal migration during cortical development, Reelin is currently believed to be involved in neuronal dendrite development and changes in brain plasticity (Niu et al., 2004; Jossin and Goffinet, 2007). The distribution of cortical neurons in heterozygous reeler (Reelin knockout) mice did not appear disordered, but their dendritic development was affected. Dendritic spine density decreased on the dendrites of pyramidal neurons in the CA1 region of the mouse hippocampus at 21 and 32 days after birth, whereas more mature neurons showed a lower density of dendritic spines accompanied by decreased expression levels of post-synaptic density protein 95 (PSD95) and NMDA receptors (Niu et al., 2008). These findings indicate that Reelin promotes dendritic development and the generation and maintenance of dendritic spines during post-natal development. The loss of Bip is one of the reasons for the down-regulation of Reelin protein, indicating that Bip may affect the development of dendrites and dendritic spines by affecting Reelin protein expression. Loss of SIL1, Bip, or Reelin function leads to similar abnormal phenotypes of neuronal migration (Leemhuis and Bock, 2011; Stranahan et al., 2013), and the loss of function of the ER molecular chaperone Bip inhibits normal Reelin expression. Thus, the relationship among these three proteins during neural development and whether the role played by Reelin protein in development is co-regulated by the coordination of Bip and SIL1 represent questions that deserve to be explored.
The role of calnexin during neurodevelopment
Calnexin is a 90-kDa ER integral protein with a long amino-terminal domain (460 amino acids) located in the lumen of the ER, a hydrophobic trans-membrane domain and a short acidic cytoplasmic domain (91 amino acids). CNX binds to Ca2+ and acts as a chaperone in the transition of proteins from the ER to the outer membrane (Tjoelker et al., 1994). CNX and CRT constitute the CNX/CRT cycle and are responsible for the folding and quality control of newly synthesized proteins (Li et al., 2011).
Many researchers have confirmed the role of CNX as a molecular chaperone in neurodevelopment. The CANX gene (coding CNX) is activated in neuronal tissue early in embryonic development (Coe et al., 2008). Immunohistochemical staining of mouse brain tissue sections showed that CNX exists in hippocampal neurons and is expressed at high levels in soma, dendrites, and synapses (Itakura et al., 2013). Additional studies have shown that in mice, CNX deficiency leads to a phenotype of peripheral axonal demyelination and reduced peripheral nerve conduction velocity, demonstrating that CNX may play an essential role in neuron growth and development (Jung et al., 2018).
Calnexin regulates dendrite and synapse development
Denzel et al. found that approximately 50% of CNX knockout mice died within 48 h after birth, most of the remaining mice died within 4 weeks, and only a few mice survived to 3 months (Denzel et al., 2002). CNX gene knockout (cnx–/–) mice exhibited significant movement impairment, including unsteady gait, inactive walking, and obvious trunk ataxia. The authors then examined the sciatic nerve in the gene-deficient mice through electron microscopy and found that most of the mice exhibited a reduced number of medullary nerve fibers and a reduced sciatic nerve diameter (Denzel et al., 2002). However, the cause of the short-lived death of cnx–/– mice has not yet been discovered but might be related to the coordinating role of CNX proteins in development. This hypothesis deserves further exploration.
Recent studies assessing the localization of CNX and NMDA receptors on neurons based on specific immunostaining and biotin chemical reagent labeling revealed that a considerable portion of CNX was localized on the cell surface of cultured neurons, and this localization was regulated by NMDA receptors (Itakura et al., 2013). These results suggest that CNX may play an essential role in NMDA receptor-mediated neuronal function. Other studies demonstrated that α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptors interact with CNX and Bip in neuronal cell bodies and hippocampal pyramidal neuron dendrites (Rubio and Wenthold, 1999; Gerrow and Triller, 2010). In addition, the AMPA receptor is a tetramer composed of four subunits, GluA1, GluA2, GluA3, and GluA4. This receptor is expressed in both neurons and glial cells (Pick and Ziff, 2018). It is well known that NMDA receptors and AMPA receptors play essential roles in central nervous system development. NMDA receptors are involved in experience-dependent synapse and dendritic development (Nadarajah and Parnavelas, 2002; Luo et al., 2006), whereas AMPA receptor expression directly affects neuronal dendritic complexity, synaptic maturation, and neural network formation (Buffington et al., 2014). Defects in AMPA receptor expression are the underlying causes of developmental diseases, degenerative diseases, and cognitive dysfunction (Kessels and Malinow, 2009; Henley and Wilkinson, 2016; Guo and Ma, 2021). However, the specific effects of CNX loss on NMDA receptor or AMPA receptor membrane expression or function remain to be investigated. Moreover, studies have shown that CNX can be attached to membranes through NMDA receptor-mediated pathways (Itakura et al., 2013). Therefore, CNX may regulate central development by participating in receptor function at the cell membrane. In addition, CNX regulates calcium levels in the ER (Roderick et al., 2000), which subsequently regulates receptor transport to influence synaptic strength (Turrigiano, 2008). Therefore, CNX may directly affect synaptic strength and synaptic development through this mechanism. However, the direct correlation between CNX and synaptic development and function needs to be expanded.
Calnexin cooperates with the cytoskeleton-associated protein to regulate neural plasticity
Activity-regulated cytoskeleton-associated (Arc) protein is a master regulator of long-term synaptic plasticity and memory formation (Bramham et al., 2010; Korb and Finkbeiner, 2011; Shepherd and Bear, 2011; Nikolaienko et al., 2017). Arc proteins control synaptic strength by promoting AMPA receptor endocytosis (DaSilva et al., 2016) and activity-dependent long-term changes in synaptic strength (Ehlers, 2000; Newpher and Ehlers, 2008). Arc overexpression decreased the density of GluA1 subunits of AMPA receptors on the cell surface and AMPA receptor-mediated miniature excitatory post-synaptic currents (mEPSCs). Arc mutations that prevent clathrin-adaptor protein 2 (AP-2) interaction reduce Arc-mediated endocytosis of GluA1 and abrogate AMPA receptor-mediated mEPSCs reduction (DaSilva et al., 2016). Endocytosis is the process by which cells internalize substances through the plasma membrane. Endocytosis plays an important role in different stages of the development of the nervous system, such as the formation of continuous synaptic transmission (Sudhof, 2004), the entry of drugs into the central nervous system (Smith et al., 2008), and the internalization of receptor-ligand complexes to nerve endings for intracellular signaling (Gloor et al., 2001). CNX is also involved in clathrin-mediated endocytosis of neurons (Li et al., 2011). Kraus et al. performed an electron microscopy analysis of the cerebellum of 7-day-old wild-type and CNX-null mice to assess whether CNX is involved in neuronal endocytosis. The results showed that the number of synaptic vesicles was significantly increased in the cerebellum of CNX-deficient mice compared with wild-type mice, demonstrating that CNX deficiency leads to increased endocytic activity in the mouse neuronal system (Kraus et al., 2010). Recent studies have found a direct interaction between Arc and CNX in neurons, and the interaction between recombinantly expressed glutathione S-transferase (GST)-tagged Arc and endogenous CNX has also been found in HEK293, SH-SY5Y neuroblastoma, and PC12 cells (Myrum et al., 2017). Given that both Arc and CNX mediate clathrin-dependent endocytosis and that both are expressed in excitatory synapses, it is possible that Arc and CNX cooperate in regulating endocytosis. Therefore, whether CNX is involved in the synaptic effect is also a mechanism that needs further exploration.
Calreticulin regulates neurodevelopment
Calreticulin is a multifunctional protein with a molecular weight of 46 kDa. CRT is expressed in almost all eukaryotic cells and is preferentially located in the ER. Depending on the sublocalization of CRT in cells, CRT has different physiological functions, including chaperone function and lectin binding. CRT was initially considered to be a high-affinity calcium-binding protein (Rauch et al., 2000), and early studies focused on its role as a calcium storage molecule in the ER (Michalak et al., 1992), including regulating calcium channel activation and maintaining intracellular calcium homeostasis (Chen et al., 2015). Later, CRT was considered a multifunctional protein that interacts with other proteins and participates in various processes (Michalak et al., 1999). In recent years, the focus of CRT research has been refocused to its role as an ER chaperone that aids in glycoprotein folding (Pacheco et al., 2020). The role of CRT in neurodevelopment mainly involves regulating embryonic development, neural differentiation, and axonal growth in the peripheral nervous system.
Calreticulin regulates embryonic development of the central nervous system
To study the role of CRT during development in vivo, Rauch et al. generated CRT-deficient mice by targeted inactivation of the CRT gene. The results demonstrated that CRT heterozygous mice were viable and fertile, whereas crt–/– mice were embryonic lethal and exhibited multiple disease phenotypes, including cardiomyopathy, anencephaly, and omphalocele (Rauch et al., 2000). In addition, CRT is also important for cranial neural tube closure and is highly expressed in the developing brain during neurogenesis (Mesaeli et al., 1999). Neural tube formation is a complex process that requires the interaction of neuroepithelial cells and the underlying mesenchyme (Lynch et al., 2012; Correll et al., 2019). Disruption of neural tube closure was found in several CRT knockout mice, in which proteins involved in neuronal migration and calcium signaling were inactivated (Solheim et al., 1997; Rauch et al., 2000). These results demonstrate that CRT is important for embryonic brain development, especially the neural tube development. However, it is currently unclear which proteins interact with CRT to regulate embryonic neural development.
Calreticulin regulates neuronal differentiation
Neurotrophic factors, including nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophic factor-3 (NT-3), and neurotrophic factor-4/5 (NT-4/5), are key tissue factors that control nervous system development (Chao, 2003). The NGF/tyrosine receptor kinase A (TrkA) signaling pathway is necessary for neural development, and abnormalities in this pathway can lead to abnormal neuronal differentiation. Shih et al. found that in NGF-stimulated PC12 cell differentiation, CRT levels were increased by activation of mitogen-activated protein kinase (MAPK) mediated by extracellular signal-regulated kinase (ERK). CRT deficiency significantly reduced NGF-induced neuronal differentiation. Furthermore, CRT overexpression enhanced neuronal differentiation through simultaneous activation of the ERK-dependent MAPK pathway (Shih et al., 2012). The Ca2+regulation ability of CRT is also essential for NGF-triggered neuronal differentiation (Pilquil et al., 2020). In addition, studies have shown that CRT is a novel oncogenic N-MYC (MYCN) inhibitor that can down-regulate MYCN promoter activity and protein expression to regulate neuronal differentiation. This study also found that CRT-mediated MYCN inhibition resulted in increased neurite length (Lee et al., 2019).
Calreticulin regulates axonal growth in the peripheral nervous system
Calreticulin localizes to axons of cultured dorsal root ganglion neurons in vitro and peripheral nervous system axons in vivo (Willis et al., 2005; Vuppalanchi et al., 2010), and automatic axonal regeneration occurs after injury to adult peripheral neurons (Bradke et al., 2012). The intrinsic growth capacity of peripheral nervous system axons after injury is greater than that of central nervous system axons. Pacheco et al. constructed in vitro and in vivo neuronal axonal injury models to study the relationship between CRT and axonal regeneration after axonal injury. Intra-axonal CRT levels were elevated after peripheral nerve injury in vivo. Unilateral sciatic nerve crush injury was performed in the mid-thigh of male SD rats, and then sciatic nerve sections were collected and stained 1 h, 6 h, 18 h, and 7 days after the crush injury. Compared with the control sciatic nerve, axonal CRT levels were significantly increased 6 h after injury and reached a maximum at 18 h after injury. The authors then performed axotomy on dorsal root ganglion neurons to study the effect of CRT deletion on the post-operative axonal growth process. The results showed that the shRNA-mediated CRT deletion group exhibited exacerbated post-axotomy contractions. In contrast, axon-targeted expression of unrestricted CRT mRNA reduces contraction and promotes axon regeneration after axotomy in vitro (Pacheco et al., 2020). In conclusion, the overexpression of axon-targeted CRT promotes axon regeneration after axonal injury, indicating that CRT plays an important role in axonal growth.
The role of protein disulfide isomerase in neurodevelopment
Protein disulfide isomerase is a class of multifunctional proteins present in the ER lumen, including endoplasmic reticulum protein 57 (ERp57, also known as PDIA3), endoplasmic reticulum protein 29 (ERp29), protein disulfide isomerase-1 (PDI-1), endoplasmic reticulum protein 72 (ERp72), protein disulfide isomerase A1 (PDIA1), and protein disulfide isomerase p (PDI p) (Turano et al., 2002). As a molecular chaperone, PDI binds to the stretched or partially folded peptide chain through its polypeptide binding site to form an intermediate to prevent incorrect protein folding (Wilkinson and Gilbert, 2004).
Protein disulfide isomerase-1 regulates neuronal migration
Protein disulfide isomerase-1 controls neuronal migration by regulating Wnt secretion. PDI is a family of protein chaperones that reside in the ER and are capable of catalyzing the formation (oxidation), cleavage (reduction), and rearrangement (isomerization) of disulfide bonds (Wilkinson and Gilbert, 2004). The formation of a proper Wnt protein structure requires the formation of disulfide bonds, which are critical for Wnt secretion and signaling (Zhang X. et al., 2012; MacDonald et al., 2014). Wnt proteins play essential roles in neuronal migration and axon-dendritic guidance in vertebrates (Yoshikawa et al., 2003; Bocchi et al., 2017), and they also control neural developmental processes in C. elegans, including neuronal migration, polarity, and axon guidance (Coudreuse et al., 2006; Hilliard and Bargmann, 2006; Pan et al., 2006). Therefore, abnormal Wnt expression or function significantly impairs neural development.
PDI-1 controls the secretion of EGL-20, the Wnt homolog in C. elegans, and therefore affects neuron migration (Torpe et al., 2019). EGL-20/Wnt is expressed and secreted in subcutaneous (epidermal) and muscle cell subsets of C. elegans (Whangbo and Kenyon, 1999). EGL-20 function is critical for the migration of hermaphroditism-specific neuron (HSN) (Desai et al., 1988). Torpe et al. used HSN development as a readout of EGL-20 function to identify molecules that control Wnt maturation and secretion. They hypothesized that PDI is important for EGL-20-directed HSN development. The expression of five PDI-encoding genes (pdi-1, pdi-2, pdi-3, pdi-6, and C14B9.2) in C. elegans was reduced using RNA interference, and the development of HSN was analyzed. This study found a specific requirement for PDI-1 during HSN development with approximately 20% of PDI-1-silenced animals exhibiting a phenotype deficient in HSN migration. Subsequently, to determine whether PDI-1 also controls EGL-20-dependent post-embryonic neuron development, the authors performed post-embryonic neuronal migration assays in PDI-1-deficient animals and found that PDI-1 knockout animals exhibited migration defects (Torpe et al., 2019). PDI inhibitors reduce Wnt3a secretion in human cells. Most mammalian genomes contain 19 Wnts and greater than 20 PDI proteins, which exhibit distinct expression domains and biological functions (Ellgaard and Ruddock, 2005; Galligan and Petersen, 2012; Willert and Nusse, 2012). Mammalian Wnt3a secretion was abolished when a single cysteine residue was replaced by alanine (MacDonald et al., 2014), suggesting that Wnt secretion is influenced by disulfide coordination. These results suggest that PDI can affect neuronal migration by regulating the secretion of Wnt and its homolog EGL-20.
The role of the Wnt signaling pathway in early cortical development is not limited to the migratory localization of neurons. It is also involved in neurogenesis, neuronal differentiation, and axon-dendritic guidance (Freese et al., 2010; Munji et al., 2011). In addition, the atypical Wnt signaling pathway is involved in the assembly of neural circuits (Hinata et al., 2007; Wang et al., 2011), especially dendrite development and synaptogenesis in cultured pyramidal neurons (Salinas and Zou, 2008). Wnt pathway dys-regulation is closely related to human neurological diseases (Castelo-Branco et al., 2003). However, the mechanism by which PDI further affects other developmental processes by affecting the Wnt signaling pathway is unclear and deserves further exploration.
The role of sigma-1 receptor in neurodevelopment
Sigma-1 receptor is a 223-amino acid long trans-membrane ER chaperone with a molecular weight of 25.3 kDa (Hanner et al., 1996; Dalwadi et al., 2022). Most Sig-1R proteins are located in the ER, especially in the mitochondria-associated ER membrane, and are critical for regulating energy balance and calcium homeostasis (Hayashi and Su, 2007). Sig-1R is highly expressed in the central nervous system, especially in the cortex, basal ganglia, spinal cord, and brainstem. It is also vital to maintain the proper functioning of motor neurons (Su et al., 2010). Sig-1R has recently attracted extensive attention as a potential drug target for neurological diseases and cancer (Merlos et al., 2017; Penke et al., 2018).
Recently, Sig-1R was shown to be important in neurodevelopmental research as an ER chaperone and is known to be involved in learning and memory. Within the nervous system, Sig-1R is predominantly localized in the gray matter of neurons and multiple glial cell types (Peviani et al., 2014; Robson et al., 2014). In addition to its role in regulating intercellular signaling (Su et al., 2010), neuroprotection (Ruscher et al., 2011), neural recovery (Ruscher et al., 2012), neuroplasticity (Kourrich et al., 2012), and neurotransmitter release (Zheng, 2009), Sig-1R also regulates various neural developmental processes, such as hippocampal neuronal dendritic morphogenesis, dendritic spine maturation, and neuroplasticity.
Sigma-1 receptor regulates neuronal morphogenesis
Sigma-1 receptor regulates neurite outgrowth (Ishima et al., 2014) as well as primary neuron morphogenesis (Ishima and Hashimoto, 2012). Dendritic spines in hippocampal neurons play important roles in neuroplasticity and memory formation. siRNAs were used to silence the expression of Sig-1R to study their role in the morphogenesis of primary hippocampal neurons in vitro. The results showed impaired dendrite extension and branching after Sig-1R gene silencing. Moreover, Sig-1Rs also have a major impact on the formation and maturation of dendritic spines in later stages (Tsai et al., 2009). In rat hippocampal primary neurons, reducing the expression of Sig-1Rs by siRNA resulted in defects in dendritic spine formation. Tsai et al. observed and analyzed the dendritic spine morphology and maturity of cultured hippocampal neurons. On DIV 16, the control group neurons formed filamentous protrusions on the dendrites. On DIV 22, control neurons formed clusters of short, thick mushroom-like spine heads without filopodia, indicating spine maturation. In contrast, Sig-1R-siRNA-transfected (siSig-1R-tf) neurons formed elongated protrusions with a few spine heads at the tips. The longer the culture time, the more pronounced the abnormal phenotype. This finding suggests that Sig-1Rs are endogenous regulators of hippocampal dendritic spine formation (Tsai et al., 2009).
Sigma-1 receptor regulates synaptic function
Sigma-1 receptor also plays an important role in the development of synaptic function (Ryskamp et al., 2019). Active synapses express specific proteins and receptors for proper function. Studies have shown that Sig-1R knockdown inhibits functional synapse formation. GluA2, GluA3, and PSD-95 staining revealed a strong immune response on dendritic spines in control neurons. However, this response was not observed in filopodia protrusions of siSig-1R-tf neurons, whereas these proteins were still present in dendritic shafts. The axonal terminals of siSig-1R-tf neurons also showed significantly reduced immune responses to synaptophysin. Neurons labeled with FM4-64, a membrane-selective red fluorescent dye, were depolarized with KCl to measure synaptic activity. The results showed that siSig-1R-tf neurons did not form functional synapses. These results suggest that Sig-1Rs have essential effects on synapse formation and synaptic function (Ryskamp et al., 2019).
Earlier, we mentioned that the NMDA receptor is associated with learning and memory. A study found that changes in Sig-1R can cause changes in NMDA receptor expression. Sig-1R was overexpressed by intraperitoneal injection of the Sig-1R activation agonist SKF10,047 in SD rats, and subsequent immunofluorescence staining revealed increased GluN2A, GluN2B, and PSD95 expression (de Montigny et al., 1992; Monnet et al., 1994, 1996). This effect was abolished after the addition of the Sig-1R antagonist BD1063 (Matsumoto et al., 1995; McCracken et al., 1999). In addition, they found increased interactions between Sig-1R and GluN2 subunits as well as increased surface levels of NMDA receptor upon Sig-1R activation, which suggests that Sig-1R regulates the expression of NMDA receptor and related neural function (Pabba et al., 2014).
Sigma-1 receptor regulates mitochondrion-associated endoplasmic reticulum membrane conduction and affects neural development
The membrane of the ER of a cell forms contacts directly with mitochondria, and the contact is referred to as the mitochondrion-associated ER membrane (MAM) (Mori et al., 2013). The MAM is a subdomain of the ER that engages in a direct physical association with mitochondria (Rizzuto et al., 2004; Csordás et al., 2006; Szabadkai et al., 2006). The MAM integrates many signaling pathways and is important for cellular survival because it serves as the “tunnel” for lipid transport and Ca2+ signaling between the ER and mitochondria (Cárdenas et al., 2010). Sig-1Rs are ER chaperones that localize specifically at the MAM and regulate a variety of cellular functions including Ca2+ signaling between ER and mitochondria, neuronal differentiation, ion channel activities, and notably cellular survival (Aydar et al., 2002; Fontanilla et al., 2009; Fukunaga and Moriguchi, 2017). However, exactly how Sig-1R regulates the neuronal differentiation process at the MAM is not fully clarified. Neurons are highly differentiated cells and require great amounts of ATP for the maintenance of cell membrane ionic gradients and neurotransmission (Xavier et al., 2016). Mitochondria, the main intracellular energy transducers, play a key role in neural development by producing ATP and biosynthetic substrates, regulating Ca2+ homeostasis, and initiating apoptosis (Kann and Kovács, 2007). Given the vital role of Sig-1R in the MAM and the importance of energy transport for neural development, exploring the energy transduction mechanism of Sig-1R in the MAM may be key to studying the developmental process.
The function of prefoldin during neurodevelopment
Prefoldin is a heterohexameric protein complex (Glover and Clark, 2015) consisting of two α subunits (PFDN3 and PFDN5) and four β subunits (PFDN1, PFDN2, PFDN4, and PFDN6). PFDN interacts with nascent polypeptide chains and can act as a substitute for Hsp70 in vitro to facilitate protein folding and prevent intermolecular aggregation (Siegert et al., 2000). In recent years, studies have found that PFDN regulates the toxicity of misfolded proteins, including those that lead to neurodegenerative diseases. For example, PFDN can protect neuronal cells from polyglutamine toxicity by preventing aggregate formation or maintaining proteostasis by unfolding misfolded proteins in physiopathological conditions (Tashiro et al., 2013; Tahmaz et al., 2022). PFDN-deficient mice exhibit phenotypic features of defective cytoskeletal function, including ciliary dyskinesia, neuronal loss, and defects in B- and T-cell development and function. As ubiquitous components of the cytoskeleton, actin and tubulin play essential roles in multiple stages of neural development, including dendritic axon growth, neuronal polarization, neuronal migration, and synaptic signal transduction (Cao et al., 2008; Bertling and Hotulainen, 2017), whereas efficient synthesis of actin and tubulin requires the participation of the protein complex formed by PFDN and t-complex protein 1 ring complex (TRiC)/chaperonin-containing t-complex protein 1 (CCT). As an ER chaperone, PFDN promotes the post-translational folding of actin and other cytoskeletal proteins through the t-complex protein 1 (TCP1)-containing loop complex chaperone TRiC (Cao et al., 2008). PFDN and TCP1 play essential roles in metazoan development. In C. elegans, RNA interference screens revealed that knockdown of a single PFDN or TCP1 subunit resulted in morphological abnormalities and distinct penetrating embryonic lethal phenotypes (Delgehyr et al., 2012), suggesting that these proteins are essential in multiple tissue development and early embryonic development (Monzo et al., 2010). In contrast, PFDN1-deficient mice develop defective cytoskeletal function, loss of neural tracts, hydrocephalus, neuromuscular defects, abnormal lymphocyte development and function, and shortened lifespan (Lee et al., 2011). Furthermore, abnormal actin and/or tubulin cytoskeleton assembly and microtubule formation may underlie the Pfdn5nmf5a(encoding PNDF5) disease phenotype, including photoreceptor degeneration (Kwon et al., 2019), central nervous system abnormalities, and male sterility (Lee et al., 2011). Although PFDN5 is expressed in various tissues, defects in PFDN5 homozygotes appear to be limited to neuronal cells. Genetic disruption of the mouse Pfdn5 gene results in reduced formation of microtubules and microfilaments, leading to progressive neurodegeneration, hydrocephalus, and reproductive abnormalities. These results suggest that PFDN5 is required for normal sensory and neuronal development (Lee et al., 2011).
Endoplasmic reticulum molecular chaperones and neurodevelopmental disorders
Neurodevelopmental disorders (NDDs) refer to the abnormal development of the nervous system caused by inherited or acquired diseases, resulting in brain dysfunction. NDDs include intellectual disability (ID), autism spectrum disorder (ASD), attention-deficit hyperactivity disorder (ADHD), and bipolar disorder (BD) (Rutter et al., 2006; Thapar et al., 2017; Ismail and Shapiro, 2019; Morris-Rosendahl and Crocq, 2020). At present, the global prevalence of unexplained ID is 2–3% (Olusanya et al., 2020), the prevalence of ASD is 1–2% (Baxter et al., 2015; Zablotsky et al., 2015), the prevalence of ADHD is 5–7.2% (Polanczyk et al., 2007, 2014; Sokolova et al., 2017; Wang T. et al., 2017), and the prevalence of BD is 1–4% (Merikangas et al., 2011). Numerous studies have shown that children with NDDs have a high incidence of mental health problems compared with normal children (Arim et al., 2015; Eyre et al., 2019). NDDs bring heavy economic and mental burdens to families and society, seriously affect the quality of life of patients and their families and represent a public health problem of high concern worldwide. The pathogenesis of NDDs is complex. Environmental risk factors, such as the inflammatory response, immune disorders and metabolic disorders, and genetic factors, such as neuronal dysfunction and chromosome deletion, are involved in the occurrence of NDDs (Bădescu et al., 2016). The abnormal expression of ER chaperone proteins can lead to the dys-regulation of neurodevelopmental processes and participate in the occurrence of many neurodevelopmental diseases.
Endoplasmic reticulum molecular chaperones and autism spectrum disorder
Autism spectrum disorder is a brain developmental disorder characterized by language disorders, social difficulties, and repetitive and stereotyped behaviors (Lai et al., 2014).
Calreticulin and autism spectrum disorder
Current studies have reported copy number variations (CNVs) in specific genes, and the ASD-related literature has cited the involvement of ER chaperone proteins. However, the molecular mechanisms are not well understood. For example, when studying the relationship among CD47 overexpression, brain overgrowth and 16p11.2 deletion syndrome, Li et al. found that ER chaperone CRT-mediated phagocytosis was related to the pathogenesis of ASD (Li et al., 2021). CNV at 16p11.2 has been implicated in neuropsychiatric disorders, such as ASD and schizophrenia (Kumar et al., 2008; Weiss et al., 2008; McCarthy et al., 2009; Zarrei et al., 2019). Carriers with 16p11.2 deletions tend to have macrocephaly (or enlarged brain), whereas carriers with 16p11.2 duplications often have microcephaly (Bartholomeusz et al., 2002; Hazlett et al., 2011; Maillard et al., 2015; Sacco et al., 2015). Human induced pluripotent stem cells (hiPSCs) from controls and subjects with 16p11.2 deletions and duplications were employed to understand the underlying mechanisms regulating brain overgrowth. CD47 (the “do not eat me” signal) was found to be overexpressed in cells with the 16p11.2 deletion vector, and CRT was also highly expressed. Cells with high CRT expression should be eliminated by immunophagocytosis, whereas cells escape immunophagocytosis due to high expression of CD47 (Oldenborg et al., 2000). The phagocytosis and CRT expression of hiPSCs with 16p11.2 deletion were restored to the control level when anti-CD47, a CD47 blocking agent, was applied (Li et al., 2021). This finding indicates that the normal phagocytosis mediated by CRT is affected in 16p11.2 null cells, and this mechanism may be involved in the pathogenesis of ASD.
Protein disulfide isomerase A1 and autism spectrum disorder
Lammert et al. found that increased PDIA1 was associated with ASD (Lammert et al., 2017). Studies have shown that RELN gene mutation can lead to ASD complications (Fatemi et al., 2005). In the ASD mouse model based on the RELN R2290C (Reelin mutant gene) (Iossifov et al., 2014; Wang et al., 2014; Lammert and Howell, 2016) mutation, PDIA1 expression was increased in the neurospheres of RELN R2290C heterozygous (+/-) as well as in the cerebellum of RELN Orleans (Orl) +/- mice. Reelin protein is highly expressed in the developing cerebral cortex and cerebellum and plays an important role in neuronal migration. As an ER-resident chaperone, PDIA1 can ensure the formation of correct disulfide bonds in nascent proteins (Parakh and Atkin, 2015), but its overexpression may cause neuropathological diseases (Perri et al., 2016; Zeeshan et al., 2016). This finding indicates that the increased expression of PDIA1 may be associated with the risk of ASD.
Sigma-1 receptor and autism spectrum disorder
Fragile X syndrome (FXS) is a disorder of synaptic development and dysfunction (Hagerman et al., 2009) and is the most prevalent genetic form of ID, ASD, and ADHD (Boyle and Kaufmann, 2010; Budimirovic and Kaufmann, 2011; Kaufmann et al., 2017). FXS mouse models recapitulate anxiety phenotypes similar to those observed in the clinic. Blarcamesine is a Sig-1R agonist (Villard et al., 2011), and the important role of Sig-1R in calcium homeostasis and synaptic function (Su et al., 2016; Schmidt and Kruse, 2019) makes blarcamesine a potential candidate for FXS. Blarcamesine has shown preliminary efficacy in patients with Alzheimer’s disease, Rett syndrome (RTT), synaptic neurodegeneration and NDDs (Hampel et al., 2020). Reyes et al. conducted research on the role of blarcamesine in FXS (Reyes et al., 2021). These researchers used the Fmr1 knockout FXS mouse model (Bakker et al., 1994; Mientjes et al., 2006; Dahlhaus, 2018) to evaluate the effect of blarcamesine on key cognitive and behavioral aspects that represent the FXS phenotype. When FXS mice were administered blarcamesine, two key neurobehavioral phenotypes, the open field test (hyperactivity) and contextual fear conditioning (associative learning), returned to normal.
Kaufmann et al. conducted further studies on the role of blarcamesine in RTT (Kaufmann et al., 2019). RTT is a progressive, non-inherited, X-linked neurodevelopmental disorder with an incidence of approximately 1 in 10,000 female births (Laurvick et al., 2006; Neul et al., 2010). Most patients carry a mutation in the methyl-CpG-binding protein 2 (MECP2) gene, which encodes a rich transcriptional regulator in the brain (Amir et al., 1999; Neul et al., 2014). The disorder is characterized by a variety of neurological impairments, particularly affecting cognition (i.e., developmental delay, ID, communication deficits), behavior, and motor and autonomic dysfunction (Neul et al., 2010; Kaufmann et al., 2016). No specific effective treatment is available for RTT, and treatment is mainly symptomatic. Using female Mecp2 mutant mice (Samaco et al., 2013; Lombardi et al., 2015; Tan and Zoghbi, 2019) as the research object, the authors and others administered the Sig-1R agonist blarcamesine and found that motor defects, acoustic starvation defects and visual defects were significantly improved. These data suggest that Sig-1Rs play an important role in FXS and in neurological disorders that are genetically characterized as FXS.
Endoplasmic reticulum molecular chaperones and intellectual disability
Intellectual disability (ID) is a common disorder of nervous system development that leads to intellectual disabilities. Various factors can cause ID, including loss of important synaptic function, abnormal neuronal morphological development, and neuronal migration and localization disorders (Kaufman et al., 2010; Ropers, 2010; Mehregan et al., 2016).
SIL1 and intellectual disability
Studies have shown that SIL1 gene mutations causes Marinesco-Sjögren syndrome (MSS), and it is the only gene that has been found to cause MSS. MSS is a rare autosomal recessive genetic disease. Clinical studies have found that approximately 90% of patients with MSS caused by SIL1 mutation present with moderate to severe ID, motor delay, congenital cataract, cerebellar atrophy, and other NDDs (Roos et al., 2014; Osborn et al., 2017). It has been well established that the decreased interaction between Bip and SIL1 or the loss of SIL1 function alone leads to the abnormal migration and localization of cortical neurons, representing a possible cause of ID (Krieger et al., 2013; Inaguma et al., 2014).
Calnexin and intellectual disability
In a study of mucopolysaccharidosis type II (MPS II), Osaki et al. demonstrated that CNX expression directly affects intellectual deficits caused by iduronate 2-sulfatase (IDS) mutations (Osaki et al., 2019). MPS II is one of the most common mucopolysaccharidoses (Wraith et al., 1987; Stapleton et al., 2018) and is caused by mutations in the gene encoding IDS (Wilson et al., 1990). The loss of IDS function leads to the accumulation of heparan sulfate and dermatan sulfate of glycosaminoglycans throughout the body, resulting in skeletal deformities, retardation, rigid joints, and thick skin (Ficicioglu et al., 2018). It was found that mutant IDS retained in the ER requires binding to CNX to achieve folding. Thus, CNX knockdown reduces the transport of mutant IDS from the ER to lysosome and its enzymatic activity. These studies indicate that proper folding by interaction with CNX ensures its functional activity. These findings reveal the existence of a role between IDS and CNX for the treatment of ID caused by MPSII and IDS mutations (Osaki et al., 2019).
Sigma-1 receptor and intellectual disability
α-thalassemia X-linked intellectual disability (ATR-X) syndrome is caused by mutations in ATRX (Gibbons et al., 1992, 1995). ATR-X syndrome is characterized by a variety of clinical manifestations, including severe ID, facial dysmorphism, genital abnormalities, and seizures (Gibbons et al., 2008). Similarly, ATR-X model mice lacking Atrx exon 2 (Nogami et al., 2011) show phenotypes similar to the symptoms of ID noted in humans, cognitive deficits (Kaufmann and Moser, 2000; Levenga and Willemsen, 2012), and abnormal dendritic formation (Shioda et al., 2011). Yamaguchi et al. investigated whether Sig-1R promotes the increased activity of neurotrophic factors, such as BDNF, to induce neuroprotection and nerve regeneration (Yamaguchi et al., 2018). They found that treatment with the Sig-1R activator SA4503 (Matsuno et al., 1996) reversed axonal development and dendritic spine abnormalities in primary cultured cortical neurons of ATR-X model mice. In addition, SA4503 treatment rescued the cognitive deficits noted in ATR-X model mice.
Protein disulfide isomerase family A member 3 and intellectual disability
Recessive gene mutations underlie many developmental disorders and often lead to disabling neurological problems (Martin et al., 2018). Bilches et al. conducted clinical and genetic studies on large close relatives with ID (Bilches Medinas et al., 2022). These researchers isolated a homozygous mutant of PDIA3, c.170G > A (p. Cys57Tyr or C57Y), from the genes of clinical patients with ID and determined that this disease was associated with PDIA3. PDIA3 is an oxidoreductase containing a thioredoxin-like domain that catalyzes the formation and isomerization of disulfide bonds in the ER and is associated with syndromic ID (Ellgaard and Ruddock, 2005). Experiments in zebrafish embryos demonstrated that the PDIA3C57Y mutation is pathogenic and causes developmental defects, such as axonal disorganization and skeletal abnormalities. Abnormal PDIA3C57Y expression in the hippocampus leads to impaired synaptic plasticity and memory consolidation (Bilches Medinas et al., 2022).
Endoplasmic reticulum molecular chaperones and bipolar disorder
Bipolar disorder is a common mental disorder characterized by recurrent episodes of mania, hypomania, and depression. BD is characterized by mood swings between elevated and depressed moods (Rosso et al., 2007). The exact mechanism of these mood swings remains unclear and needs to be further elucidated.
Binding immunoglobulin protein and bipolar disorder
There is increasing evidence from previous in vitro studies that the ER, as a protein folding factory, plays a major role in BD (So et al., 2007; Hayashi et al., 2009; Pfaffenseller et al., 2014). The role of the ER molecular chaperone Bip in BD diseases is more reflected in ER stress. ER stress activates the UPR (Zhang and Kaufman, 2008; van Schadewijk et al., 2012), which is partially impaired in BD (Pfaffenseller et al., 2014). Therefore, Bengesser et al. analyzed Bip and C/EBP homologous protein (CHOP) gene expression and X-box binding protein 1 (XBP1) splicing in the peripheral blood of BD patients and control study participants (Bengesser et al., 2018). They isolated RNA from fasting blood of BD patients and control study participants, reverse transcribed it into cDNA, and analyzed Bip and CHOPgene expression. The results significantly increased Bip gene expression in BD samples. In addition, valproate, an atypical antipsychotic drug, has been shown to play a neuroprotective role by increasing the expression of chaperone proteins that assist ER protein folding, such as CRT and Bip (Chen et al., 2000; Bown et al., 2002; Kim et al., 2005). This neuroprotective effect has been suggested to be potentially related to the pathophysiology of neuropsychiatric disorders, such as schizophrenia and BD (Aghajani et al., 2006). However, the role of the specific pathophysiology of this neurological disorder remains to be elucidated.
Endoplasmic reticulum molecular chaperones and attention-deficit hyperactivity disorder
Attention-deficit hyperactivity disorder is a syndrome characterized by inattention, hyperactivity, emotional impulsivity, and learning difficulties (Tripp and Wickens, 2009).
At present, there are few studies on the role of molecular chaperones in ADHD, and more relevant studies exist at the level of drug treatment targets. Methylphenidate (MPH) is a commonly used stimulant drug for ADHD (Capp et al., 2005; Arnsten, 2006). The symptoms of ADHD patients are mostly consistent with dysfunction in the pre-frontal cortex (PFC) (Barkley et al., 1992), a highly functional region that directs and organizes attention, thoughts, and emotions (Arnsten and Li, 2005). It has been demonstrated that low-dose MPH injection into the PFC can improve working memory performance (Arnsten and Dudley, 2005). High MPH doses can cause behavioral sensitization and a high risk of addiction (Amini et al., 2004). Sig-1R receptor proteins are highly distributed in the PFC, striatum and hippocampus. At the cellular level, Sig-1R is post-synaptic (Hayashi and Su, 2004) and is an excitatory target for the treatment of ADHD (e.g., cocaine) (Matsumoto et al., 2001; Maurice et al., 2002). MPH was found to increase NMDA receptor-mediated synaptic transmission, which was mediated by Sig-1R. Under the same dose of MPH stimulation, the NMDA-induced post-synaptic strength in the hippocampus of mice supplemented with the Sig-1R agonist PRE-084 was significantly higher than that of the control group (Motawe et al., 2020). Similarly, pretreatment with the Sig-1R antagonist BD1063 effectively prevented hyperactivity caused by MPH hyperactivity (Matsumoto et al., 1995), confirming the importance of Sig-1R receptors in excitatory synaptic transmission (Zhang C. L. et al., 2012). Therefore, the active use of Sig-1R receptor ligands in the treatment of ADHD and prevention of MPH-induced addiction may have unexpected effects.
Conclusion
A considerable amount of evidence indicates that ER chaperones play key roles in neural development through various processes, such as neuronal migration and localization, neuronal morphogenesis, and synaptic modification. There are two main mechanisms by which ER chaperones participate in the regulation of neural development: 1. ER chaperones directly act on specific developmental periods to regulate developmental stages. 2. ER chaperones indirectly regulate development by regulating the spatial and temporal expression of secreted proteins or central development-related receptor proteins. Of these methods, the indirect regulation mode mainly affects the secretory protein Reelin and membrane receptor proteins AMPA receptor, NMDA receptor and Wnt receptor. Bip protein maintains protein homeostasis under ER stress. In addition, the key effects of these partners in neurodevelopmental diseases also deserve our attention (Table 1). Bip, SIL1, and PDI-1 regulate neuronal migration. Bip, CNX, CRT and PFDN regulate early embryonic development. CNX, CRT and Sig-1R regulate neuronal morphogenesis and dendritic axon growth. Bip, CNX, and Sig-1R regulate synaptic function (Figure 1). These direct effects occur through membrane receptor-independent mechanism and are related to the regulation of ER calcium levels by ER chaperones. However, the direct mechanisms of action remain unclear and need to be further studied.
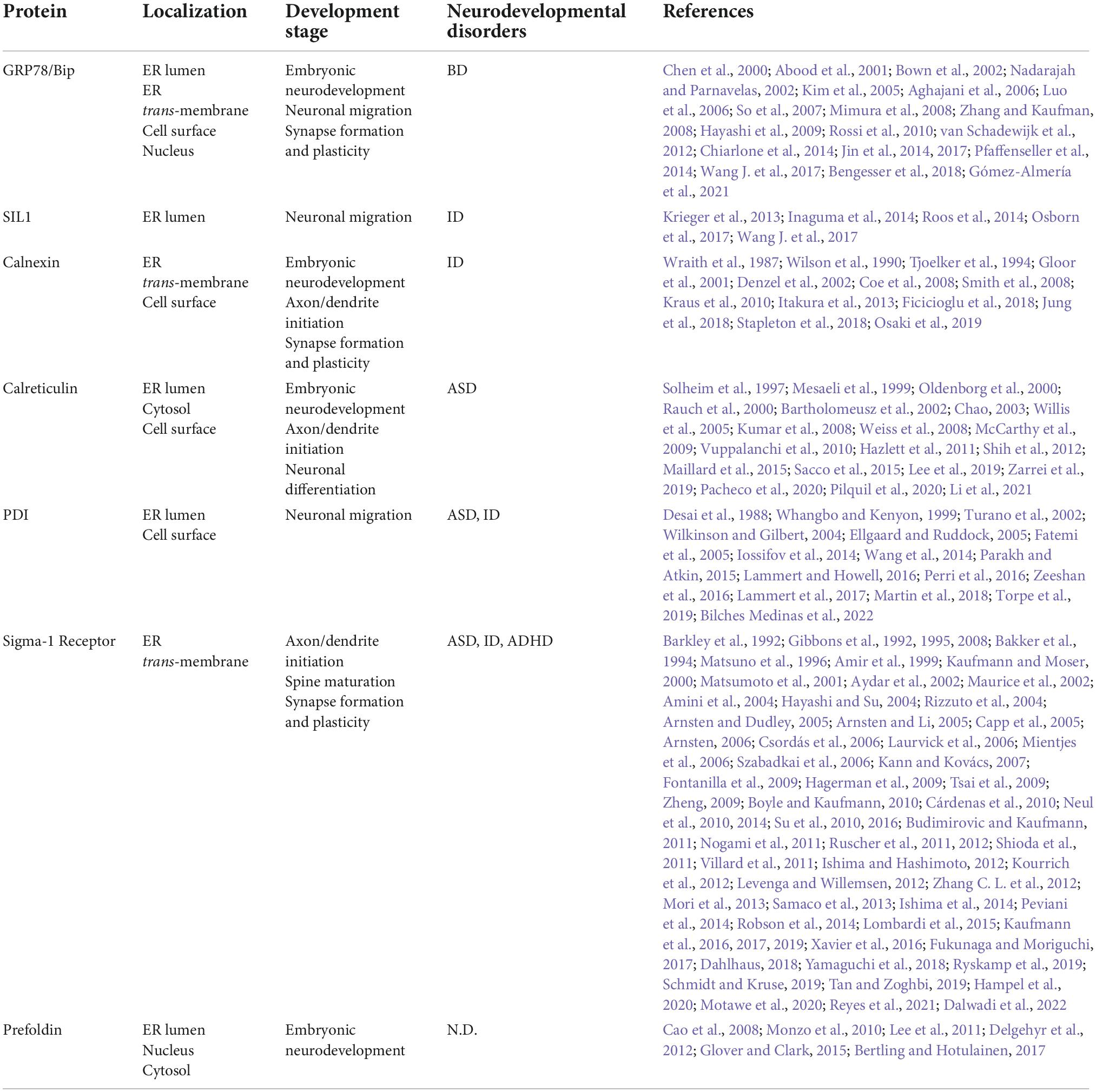
The role of Endoplasmic reticulum (ER) chaperones in neural development.

Endoplasmic reticulum (ER) molecular chaperones involved in neurodevelopment. ER molecular chaperones regulate central nervous development through two mechanisms: 1. ER chaperones directly act on specific developmental periods and regulate developmental stages. 2. ER chaperones indirectly act on secretory proteins or receptor proteins and regulate ER stress by ensuring protein homeostasis. CRT and Sig-1R regulate neuronal morphogenesis and dendritic axon growth (A); Bip, SIL1, and PDI-1 regulate cortical neuron migration (C); CRT regulates Ca2+ to guide NGF differentiation into mature neurons (D); Bip, CNX, and Sig-1R regulate synaptic function (B). Indirect regulation mainly affects the secreted protein Reelin and AMPA and NMDA membrane receptors (C), which act on the post-synaptic membrane and nerve migration. Bip proteins are involved in endoplasmic quality control and transport misfolded proteins for degradation, thereby maintaining protein homeostasis under ER stress conditions. Insert (a) CRT cooperates with CNX and PDIA3 to form the CNX/CRT cycle that controls protein folding within the ER. The CNX/CRT cycle can specifically recognize glycoproteins linked by N2 glycosidic bonds and regulate Ca2+ homeostasis and Ca2+ signaling processes in the ER. Insert (b) Bip is involved in a variety of ER functions dependent on ATP-induced conformational changes. The adenosine triphosphatase (ATPase) cycle of Bip is regulated by its co-chaperones of two families, namely, the ER-localized DnaJ-like proteins (ERdjs) and the nucleotide exchange factors (NEFs). ERdjs enables Bip to stably associate with its target protein. The NEF SIL1 then mediates the release of the target protein from the upper Bip and further folds to form the mature protein. Modified from Fucikova et al. (2021), Ichhaporia and Hendershot (2021).
In summary, considerable research on the function of ER molecular chaperones has been performed, and some progress has been made. However, some limitations remain. 1. ER molecular chaperones have specific expression sites in the early stage of neural development, and specific spatial and temporal patterns coincide with different stages of neural development. For example, Bip is involved in early embryonic development and is closely related to cortical neuronal migration. Sig-1R regulates neuronal morphogenesis and dendritic axon growth and are mainly localized in the hippocampus. Do these findings suggest that the functions of these molecular chaperones are focused at specific developmental sites at specific developmental times? 2. The mechanism by which ER chaperones affect central nervous system development is still not well defined. For example, Bip and SIL1 coregulate the coordination mechanism during central development. CNX regulates the specific regulatory processes of the NMDA receptor and AMPA receptor. Whether PDI-mediated Wnt protein-dependent disulfide bond formation affects other developmental processes in addition to migration remains unclear. As well characterized molecular ER chaperone, the role of GRP94 in protein folding and assembly as well as in cancer and related diseases has been widely studied (Fu and Lee, 2006; Pan et al., 2009). However, there are few studies on GRP94 in neural development, and further exploration is needed. 3. At present, studies on the role of molecular chaperones in developmental function are insufficient, and further studies on the functions of these molecular chaperones in different brain regions are needed. Finally, studies on the key role and induction mechanism of molecular chaperones in neurodevelopmental diseases are limited.
At present, neurodevelopmental diseases have become a problem of great concern. The complex etiology and specific pathogenesis have affected the health of numerous individuals. Determining whether it is possible to repair developmental defects by controlling the expression of chaperone proteins and subsequently treat developmental diseases is the focus of our research.