
Sigma receptor ligands are potent anti-prion compounds that act independently of sigma receptor binding
By Robert C. C. Mercer, Nhat T. T. Le, Mei C. Q. Houser, Aaron B. Beeler, and David A. Harris
Excerpt from the article published in bioRxiv 2023.11.28.569035; November 29, 2023. DOI: https://doi.org/10.1101/2023.11.28.569035
Editor’s Highlights
- Prion diseases are invariably fatal neurodegenerative diseases of humans and other animals.
- The most common human prion disease, Creutzfeldt-jakob disease (CJD), has a lifetime risk of about 1:5000.
- Sigma receptor agonists are excellent candidates for preclinical studies for the treatment of prion disease.
- σ1R and σ2R are not the direct molecular targets responsible for the anti-prion effects of these compounds.
- The ability of the sigma receptor ligands to block this cascade could result from their effects on PrPC or PrPSc at the neuronal plasma membrane, or on subsequent steps of the synaptotoxic signaling pathway.
Abstract
Prion diseases are invariably fatal neurodegenerative diseases of humans and other animals for which there are no treatment options. Previous work from our laboratory identified phenethyl piperidines as novel class of anti-prion compounds. While working to identify the molecular target(s) of these molecules, we unexpectedly discovered ten novel anti-prion compounds based on their known ability to bind to the sigma receptors, σ1R and 2R, which are currently being tested as therapeutic or diagnostic targets for cancer and neuropsychiatric disorders. Surprisingly, however, knockout of the respective genes encoding σ1R and σ2R (Sigmar1 and Tmem97), in prion infected N2a cells did not alter the anti-prion activity of these compounds, demonstrating that these receptors are not the direct targets responsible the anti-prion effects of their ligands. Further investigation of the most potent molecules established that they are efficacious against multiple prion strains and protect against downstream prion-mediated synaptotoxicity. While the precise details of the mechanism of action of these molecules remains to be determined, the present work forms the basis for further investigations of these compounds in pre-clinical studies. Given the therapeutic utility of several of the tested compounds, including rimcazole and haloperidol for neuropsychiatric conditions, (+)-pentazocine for neuropathic pain, and the ongoing clinical trials of SA 4503 and ANAVEX2-73 for ischemic stroke and Alzheimer’s disease, respectively, this work has immediate implications for the treatment of human prion disease.
Introduction
Prion diseases are invariably fatal neurodegenerative diseases of humans and other animals (1). The most common human prion disease, Creutzfeldt-jakob disease (CJD), accounts for ∼95% of cases; it has a sporadic etiology with a worldwide incidence of 1-2 cases per million people per year, which translates to a lifetime risk of about 1:5000. A smaller number of cases (5-10%) are due to germline mutations in PRNP (Prnp in mouse gene nomenclature), the gene encoding the prion protein (PrPC) (2,3). Fewer still are due to infection as a result of exposure to contaminated tissue, either by ingestion or nosocomial means (4).
The central event of prion disease is the self-templated structural rearrangement of the primarily α- helical PrPC to its β-sheet enriched, disease-associated conformer, PrPSc (5,6). This three-dimensional change imparts biochemical features upon PrPSc that differentiate it from PrPC, including resistance to proteolytic digestion and insolubility in detergents. Following protracted and clinically silent incubation periods, accumulation of PrPSc in the central nervous system (CNS) results in the loss of neurons and eventual death of the host (1). Divergent PrPScatomic structures with identical PrPC sequences are the basis of prion strains, which cause diverse clinical and pathological outcomes by currently unknown mechanisms (5,7,8).
Extensive efforts have been devoted to identifying effective therapeutics for prion disease. Most anti- prion compounds have been discovered by exposing prion infected mouse neuroblastoma cells (ScN2a) to compound libraries and assessing changes in the levels of proteinase K (PK) resistant PrP. Molecules discovered in this manner have been shown to prolong the disease course of mice infected with mouse prions but none have yet been effective in transgenic mice expressing human PrP infected with CJD prions, or in patients (9,10). Anti-prion compounds have also been shown to have strain-specific efficacy both in vitro and in vivo (11–13), complicating efforts to develop therapeutics. It has been demonstrated that some of these molecules prevent prion propagation through a direct interaction with PrPC; by stabilizing its structure or by sterically occluding its interactions with PrPSc which are critical for conversion (14–16). Others are known to inhibit the conversion process by direct interaction with PrPSc(17,18). The majority of anti-prion compounds, however, do not interact with either PrP conformer and, presumably, target other molecules (19). These compounds present a more attractive pharmaceutical target as they could potentially be used to combat multiple prion strains (5,7,8).
Our laboratory recently identified phenethyl piperidines as a novel class of anti-prion compound (20). This discovery was made through application of the Drug Based Cellular Assay (DBCA) in a high throughput screen for molecules that suppress the increased sensitivity to antibiotics induced by the expression of an internally deleted form of PrP (ΔCR) (21). These compounds were refined through structure-activity relationship studies, and the most potent derivative, JZ107, can permanently cure ScN2a cells of infection by multiple prion strains. JZ107 also rescues hippocampal neurons from prion- induced synaptotoxicity in vitro, an important secondary assay performed on cultured hippocampal neurons (20,22,23). Phenethyl piperidine molecules do not appear to bind to PrP at concentrations used in these assays, making the identity of their interaction partners of significant interest.
In the present paper, we describe our efforts to identify the molecular target of JZ107. Through this, we discovered that ligands of the sigma receptors (σ1R and σ2R) can potently reduce the levels of PK resistant PrP, a biochemical indicator of infection, in ScN2a cells and can also prevent prion-induced retraction of dendritic spines on hippocampal neurons, a measure of synaptotoxicity. Surprisingly, however, we find that these effects are independent of direct interaction of the compounds with the sigma receptors. Because these molecules are known to cross the blood brain barrier, and some are already in clinical use for other diseases, they make excellent candidates for preclinical therapeutic studies.
Results
Discovery of novel anti-prion compounds
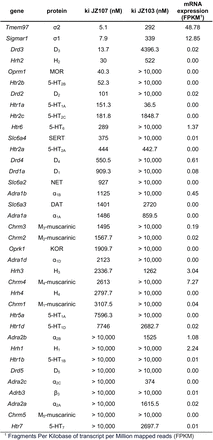
We previously described the ability of the phenethyl piperidine, JZ107, to reduce the levels of PrPSc in ScN2a cells infected with two prion strains (RML and 22L) at low micromolar concentrations (20). In an effort to identify potential non-PrP molecular targets of JZ107, we utilized the National Institute of Mental Health Psychoactive Drug Screening Program (NIMH PDSP), which assays the binding affinity of small molecules to a panel of central nervous system channels, receptors, and transporters (24). By performing radioligand binding competition assays, a list of inhibition constants (Ki) was obtained for JZ107 and an inactive analogue, JZ103, against 37 potential target proteins (Table 1). We found that the two targets in the PDSP panel with the highest affinity for JZ107 were the sigma-1 and sigma- 2 receptors (σ1R and σ2R), which bound JZ107 with Ki values of 7.9 and 5.1 nM, respectively. In contrast, the Ki values for JZ103 binding were 339 nM (σ1R) and 292 nM (σ2R), 43x and 57x higher, respectively, than JZ107. Transcriptomic data obtained through RNAseq was used as an additional filter of this dataset, with 0.5 FPKM chosen as the lower limit for expression of each gene. Surprisingly, expression of the Sigmar1 (σ1R) and Tmem97 (σ2R) genes in N2a cells was found to be higher than that of any other proteins assayed in the PDSP (Table 1).
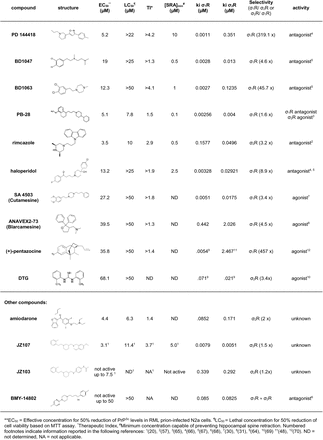
These results led us to test several known sigma receptor ligands for their ability to reduce the levels of PK resistant PrP. RML infected ScN2a cells were cultured in the presence of increasing concentrations of each compound or DMSO vehicle for a total of seven days, with passaging on the third day. Cell lysates were then exposed to 10 µg/ml of PK before western blotting while MTT assays were performed in parallel as a measure of cell viability. Using this approach, we identified ten novel anti-prion compounds: PD 144418 (25) (EC50 = 5.2 µM) (Figure 1A); BD1047 (26) (EC50 = 19 µM) (Figure 1B); BD1063 (26) (EC50 = 12.3 µM) (Figure 1C); PB-28 (27) (EC50 = 5.1 µM) (Figure 1D); rimcazole (BW234U) (28) (EC50 = 3.5 µM) (Figure 1E); haloperidol (29) (EC50 = 13.2 µM) (Figure 1F); SA4503 (Cutamesine) (30) (EC50= 27.2 µM) (Figure 1G), ANAVEX2-73 (Blarcamesine) (31) (EC50 = 39.5 µM) (Figure 1H), (+)-pentazocine (32) (EC50 = 35.8 µM) (Figure 1I), and ditolylguanidine (DTG) (33) (EC50 = 68.1 µM) (Figure 1J). BMY-14802 (34) was found to be ineffective in lowering the levels of PK resistant PrP at concentrations up to 50 µM, and PrPSc reduction at higher concentrations paralleled a loss of cell viability (Figure 1K). For ease of visual comparison, dose response curves are plotted together (Figure 1L), and all data, including compound structures, are compiled in Table 2 along with corresponding data for JZ107 and JZ013 (20). Based on MTT assays of cellular toxicity (Figure 1, dashed lines), molecules with anti-prion activity exhibited LC50 values ranging from 7.8 – >50 µM, with corresponding therapeutic indices (TI = LC50/EC50) of 1.5 – >4.2 (Table 2).
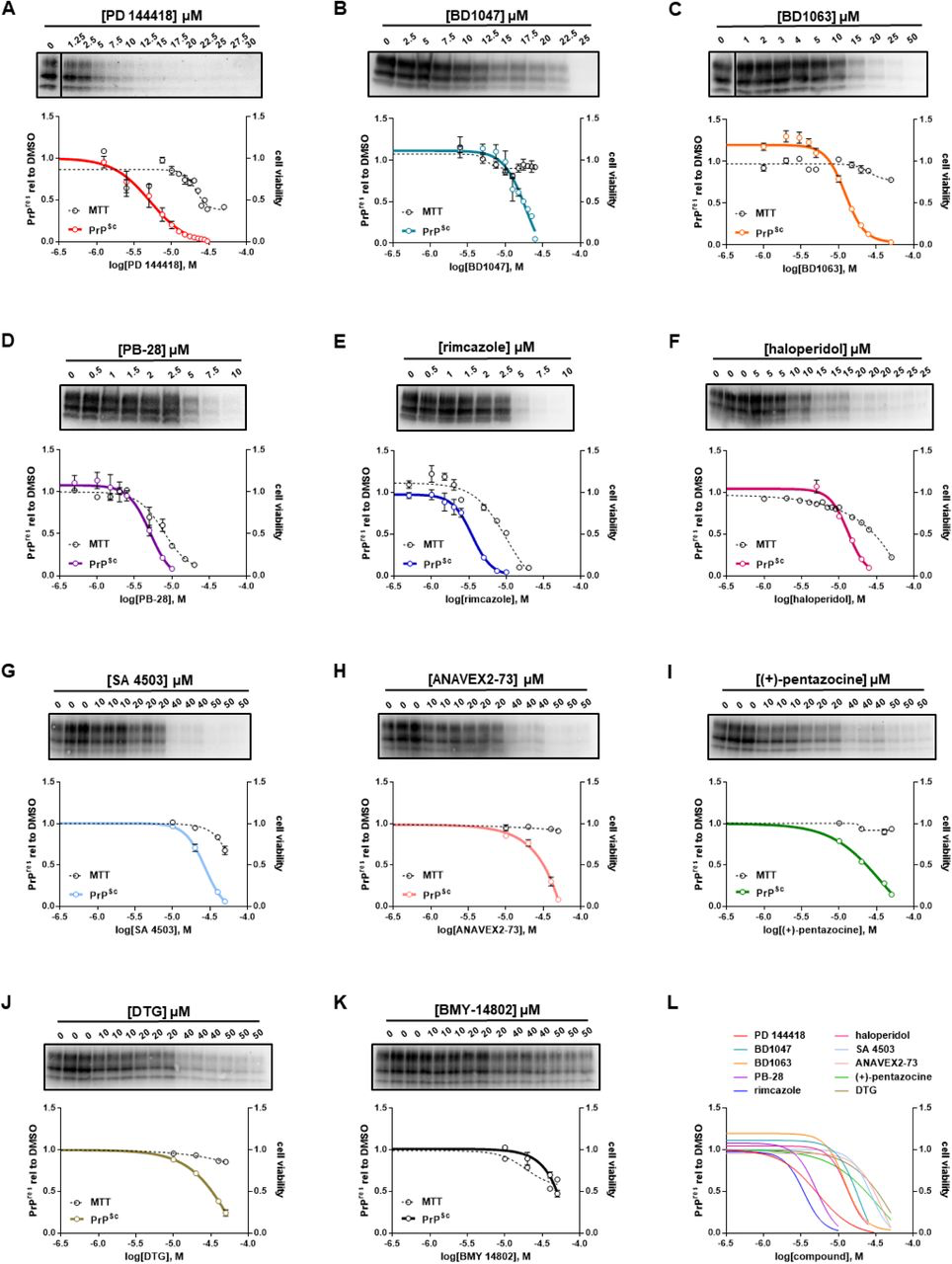
Sigma receptor ligands reduce the levels of PK resistant PrP in ScN2a-RML cellsN2a cells chronically infected with RML prions were incubated with the indicated concentrations of compound for a total of seven days, before being lysed for PK digestion and analysis by western blot. Cultures treated in parallel were subjected to MTT assay. All data points represent three independent replicates. A) PD 144418; B) BD1047; C) BD1063; D) PB-28; E) rimcazole; F)haloperidol; G) SA 4503; H) ANAVEX2-73; I) (+)-pentazocine; J) DTG; K) BMY 14802; L) EC50 curves of A-K plotted together. All curves were fit by least squares regression using GraphPad software, n = 3 replicates.
Sigma receptor binding characteristics of anti-prion compounds
Ki values reported in the literature for sigma receptor ligands are often difficult to compare, as they have been obtained using different assay methods and/or source tissues. We therefore submitted our set of anti-prion compounds to the PDSP to experimentally verify their sigma receptor binding characteristics. The PDSP assays the ability of test compounds to compete with binding of [3H]-(+)- pentazocine and [3H]-DTG to membrane preparations of receptor-expressing HEK293T cells to determine Ki values for σ1R and σ2R, respectively. We observed that, irrespective of anti-prion activity, all compounds bound to both σ1R and σ2R; most with sub-micromolar affinity (Table 2). Of the ligands with anti-prion activity, PB-28 and JZ107 had similar affinities for σ1R and σ2R (1.6X and 1.5X selectivity for σ1R and σ2R, respectively). Showing slight selectivity for σ1R were BD1047 (4.6x), haloperidol (8.9x), SA 4503 (3.4x), and ANAVEX2-73 (4.5x), while rimcazole and DTG displayed the opposite preference (3.2X and 3.4X for σ2R, respectively). Finally, three ligands were highly selective for σ1R: PD 144418 (319.1x), BD1063 (45.7x), and (+)-pentazocine (457x).
We attempted to correlate the observed anti-prion effects with the categorization of these compounds as agonists or antagonists of the sigma receptors. This categorization must be regarded cautiously, since there is no agreed upon biochemical or physiological activity of either receptor to serve as a criterion for assigning the pharmacological effects of its ligands (Table 2). Although most compounds with anti-prion activity have been classified as antagonists in published literature, some (SA 4503 and ANAVEX2-73) have been classified as agonists, and one (PB28) as an antagonist of σ1R and an agonist of σ2R. We also assessed the relationship between the observed anti-prion activities of these molecules with their binding affinity for the two sigma receptors. When we plotted the EC50 values determined using ScN2a cells against Ki values for σ1R and σ2R determined using the PDSP, we observed no correlation between these two parameters for either receptor (Figure 2). Taken together, these results fail to confirm a correlation between the anti-prion potency of the ten ligands and their affinity for or purported pharmacological action on σ1R and σ2R.
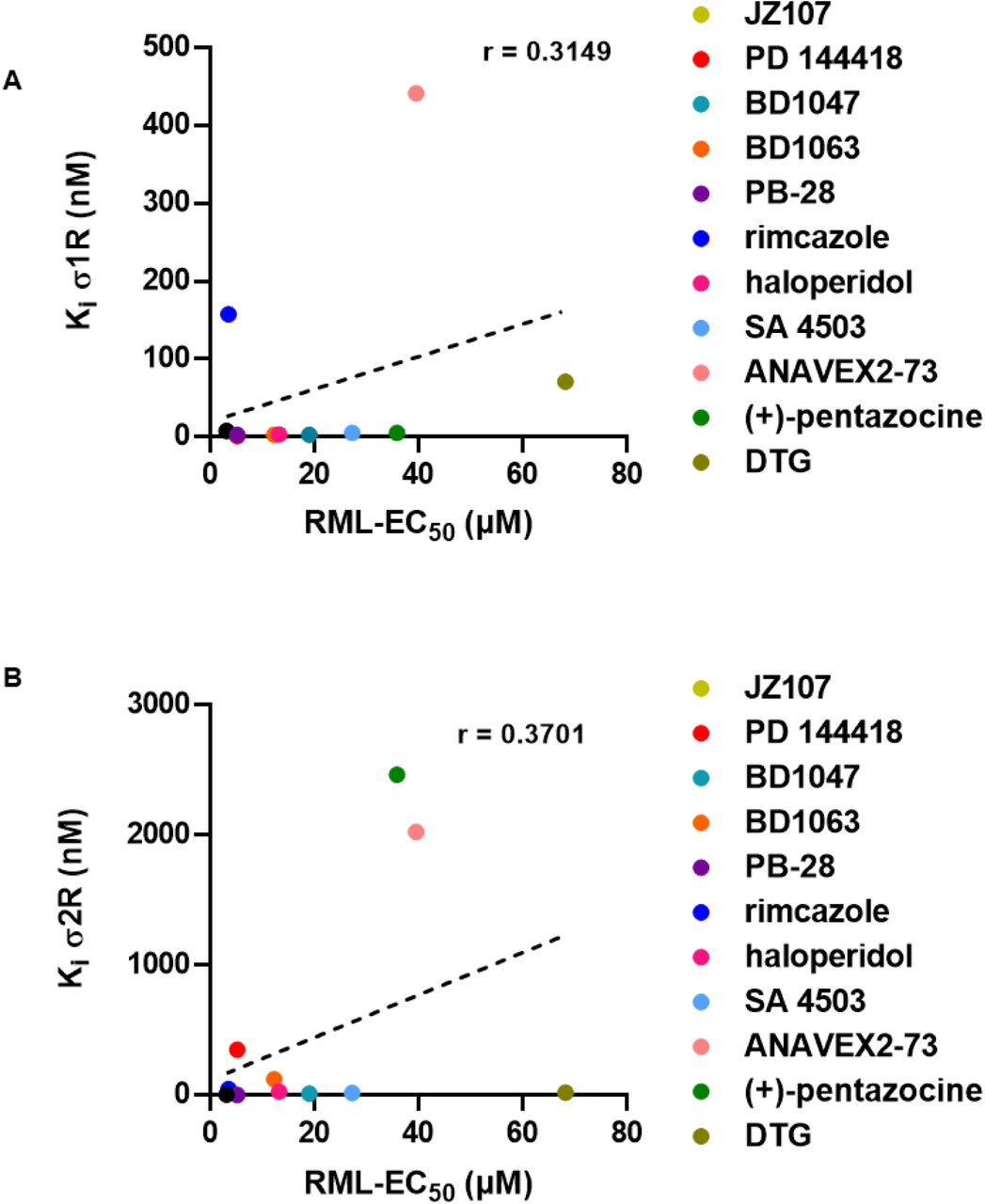
Correlation of sigma receptor binding affinity and anti-prion activityThe corresponding Ki for each compound for σ1R or σ2R as determined by the PDSP is plotted against the EC50 determined against RML prions. PDSP derived Ki values of (+)-pentazocine and DTG were determined by another group (48,64). A) σ1R Kivs RML EC50: r = 0.3149, p = 0.3455. B) σ2R Ki vs RML EC50: r = 0.3701, p = 0.2626. Correlation analysis was performed using GraphPad software.
Sigma receptor knockout does not abrogate anti-prion effects
To investigate the role of σ1R and σ2R in prion propagation and in the anti-prion effects of their ligands, we edited both the Sigmar1 and Tmem97 genes in ScN2a cells using CRISPR/Cas9 to disrupt the respective open reading frames. Since N2a cells are well known to display clonal variations in prion infection susceptibility and propagation (35), we utilized a CRISPR/Cas9 platform capable of high- efficiency gene editing without the need for cloning (see Experimental Procedures). As a control, we used a sgRNA targeting the Rosa26 safe harbor locus. Editing efficiency was determined by Sanger sequencing of PCR amplicons and deconvolution using the Inference of CRISPR Edits (ICE) tool (36). The editing efficiency (percentage of open reading frames with indels), and the knock-out score (percentage of edits producing a frameshift) was > 94% for both Sigmar1 and Tmem97 (Figure 3A). Quantitative RT-PCR analysis demonstrated that Sigmar1 and Tmem97 transcripts underwent incomplete nonsense mediated decay, with transcript levels of Sigmar1 and Tmem97 decreasing relative to Rosa26 targeted cells (Figure 3B). Confirming nucleotide analysis, σ1R and σ2R could not be detected in Sigmar1 + Tmem97 knockout cells by western blot (Figure 3C). These genetic manipulations had no effect on either the transcript level of Prnp, (Figure 3B), or upon the level of total or PK resistant PrP (Figure 3C, D). These results indicate that genetic reduction of σ1R and σ2R expression in ScN2a cells has no significant effect on the basal levels of PrPSc in the absence of sigma receptor ligands.
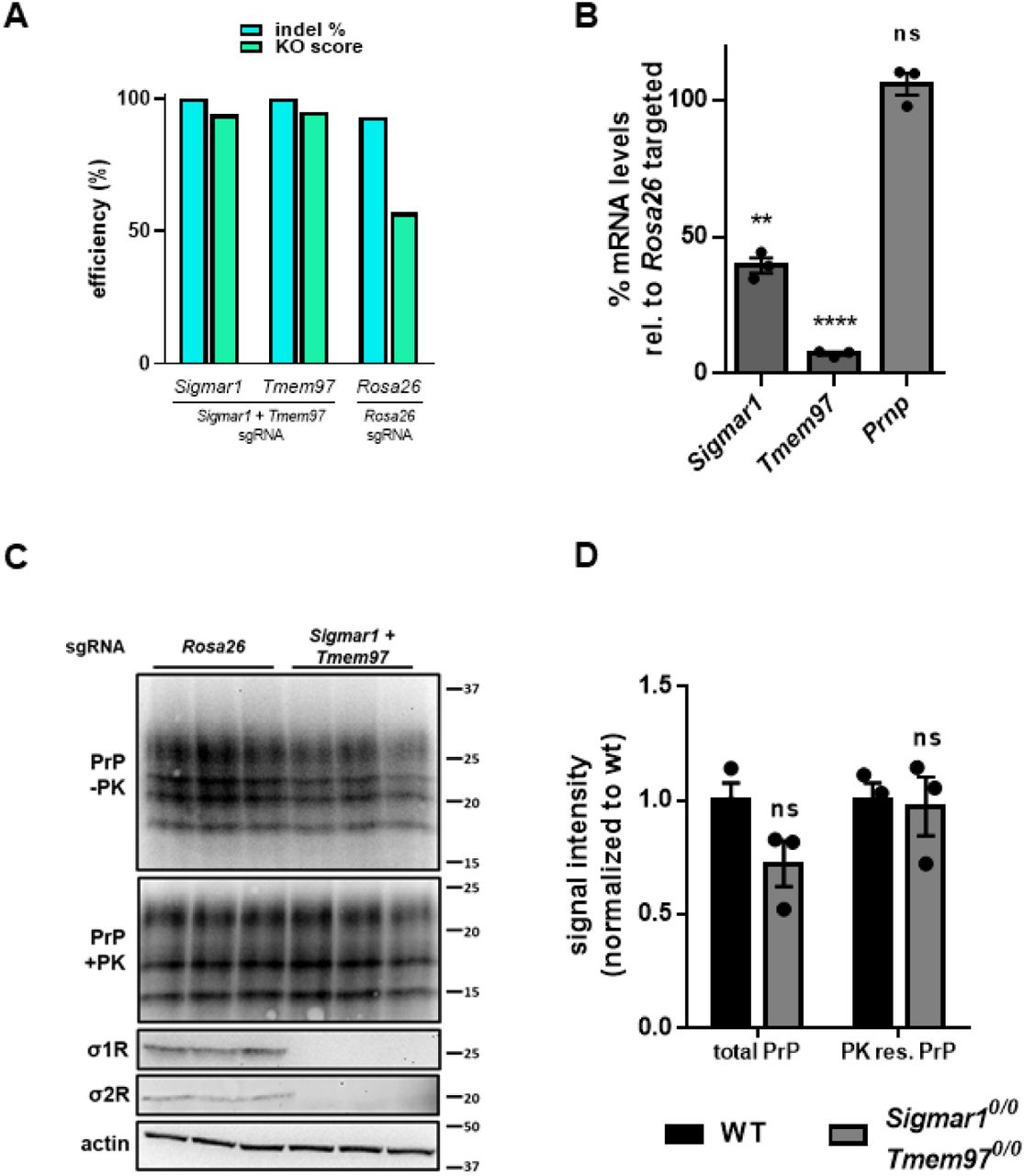
Combined CRISPR/Cas9 mediated editing of the σ1R and σ2R genes has no effect on basal levels of PrPC or PrPSc>A) Inference of CRISPR Edits (ICE) analysis of Sigmar1 and Tmem97 gene disruption. Following PCR amplification and cleanup of targeted loci, ICE analysis allows for the deconvolution of Sanger sequencing data to provide indel % (editing efficiency) and a knockout score (KO: proportion of edits that result in a frame shift). The Rosa26 safe harbor locus is targeted with a single guide RNA as a control. B) Quantitative RT-PCR analysis using the ΔΔCt method. C) High knockout efficiency is demonstrated by undetectable levels of σ1R and σ2R protein in double targeted cells by western blot. Blotting for PrP with and without PK demonstrates that levels are unaffected by the ablation of Sigmar1 and Tmem97. D) Quantification of PrP blots in C. For all experiments n = 3 replicates. p < 0.0001 = ****; p < 0.001 = ***; p < 0.01 = **; p < 0.05 = *; ns = not significant using a two-tailed students t-test.
We next sought to determine if knockout of σ1R and σ2R influenced the ability of sigma receptor ligands to reduce PrPSc levels. For these experiments, we subjected these cells to our standard drug treatment regimen using the determined EC50 of each compound against RML prions in unedited cells (Figure 1; Table 2). Surprisingly, we found that dual knockout of Sigmar1 and Tmem97 did not affect the ability of any of the compounds to reduce the levels of PK resistant PrP (Figure 4). These data demonstrate that, despite being sigma receptor ligands, the anti-prion activity of these compounds does not require expression of σ1R or σ2R.
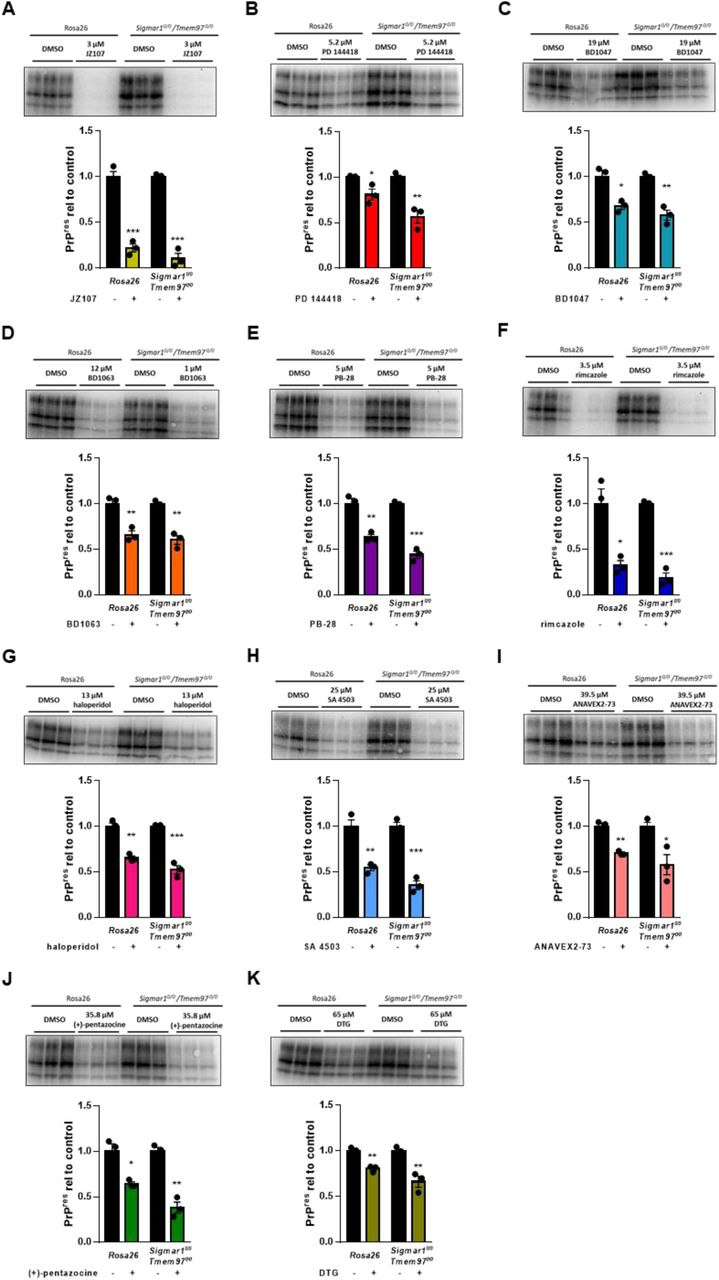
Elimination of σ1R and σ2R expression does not alter the anti-prion effects of sigma receptor ligandsCompound efficacy against RML prions in chronically infected Sigmar1 + Tmem97 KO cells. For each compound, the EC50determined against RML prions in N2a cells was used. Levels of PK resistant PrP relative to DMSO treated controls are plotted (mean ± SEM). A) JZ107; B) PD 144418; C) BD1047; D) BD1063; E) PB-28; F) rimcazole; G) haloperidol; H) SA 4503; I)ANAVEX2-73; J) (+)- pentazocine; K) DTG. For all experiments n = 3 replicates. p < 0.0001 = ****; p < 0.001 = ***; p < 0.01= **; p < 0.05 = *; ns = not significant using the Holm-Sidak test for multiple comparisons.
Other binding partners are not responsible for anti-prion effects
While σ1R or σ2R were the targets identified by the PDSP that were most highly expressed in N2a cells and that had the highest affinity for the test compound JZ107, (Table 1), other proteins that interact with one or more of sigma receptor ligands are expressed at lower levels (Figure 5A; Table 1; Supplementary Table 1). Using the same strategy employed for Sigmar1and Tmem97, we disrupted the open reading frames of Htr6, Drd4, Hrh1, Hrh3, and Chrm4. Attempts to detect the corresponding proteins by western blot were unsuccessful, likely due to their low levels of expression in N2a cells (Table 1). However, disruption of the open reading frames observed through ICE analysis gives us confidence that these genes are no longer functional (Figure 5B). Again using each compound at its previously determined EC50, we were unable to demonstrate the requirement for any of these proteins for the observed anti-prion effects of the tested compounds (Figure 5C-N).
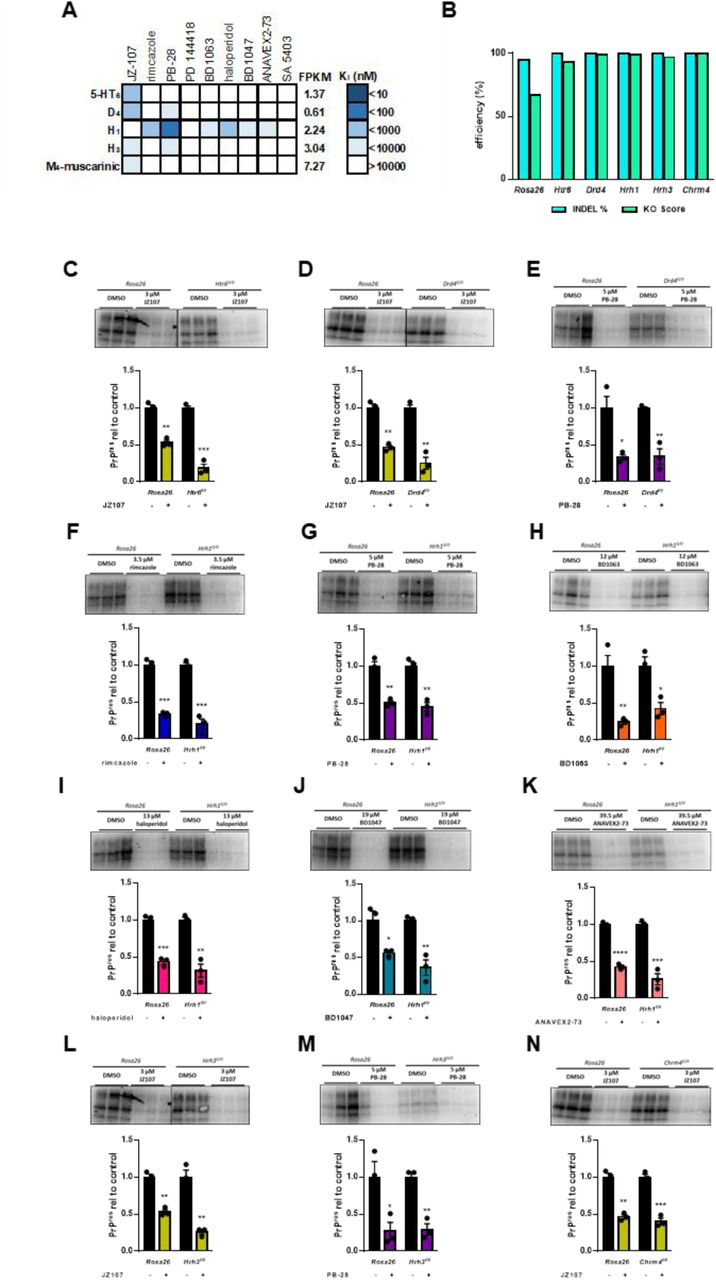
Other receptors identified in the PDSP do not mediate the observed anti-prion effectsA) Graphical representation of active compound Ki values for other receptors known to be expressed in N2a cells (FPKM > 0.5). B) Inference of CRISPR Edits (ICE) analysis of gene disruption. The Rosa26 safe harbor locus is targeted with a single guide RNA as a control. C-N) Compound efficacy against RML prions in chronically infected KO cells. For each compound, the EC50determined against RML prions in N2a cells was used. Levels of PK resistant PrP are expressed relative to DMSO treated controls in Rosa26 targeted cells vs KO cells ± SEM : C) JZ107 against Htr60/0 cells; D) JZ107 against Drd40/0 cells; E) PB-28 against Drd40/0 cells; F) rimcazole against Hrh10/0cells; G) PB-28 against Hrh10/0 cells; H) BD1063 against Hrh10/0 cells; I)haloperidol against Hrh10/0 cells; J) BD1047 against Hrh10/0 cells; K) ANAVEX2-73 against Hrh10/0 cells; L) JZ107 against Hrh30/0cells; M) PB-28 against Hrh30/0 cells; N) JZ107 against Chrm40/0 cells. Assays shown in C and L, and in D and N were done using the same western blot, necessitating the use of the same Rosa26 control images. For all experiments n = 3 replicates. p < 0.0001 = ****; p < 0.001 = ***; p < 0.01 = **; p < 0.05 = *; ns = not significant using the Holm-Sidak test for multiple comparisons.
Phospholipidosis as a potential anti-prion mechanism
σ1R and σ2R were recently identified in a screen for host molecules that interact with SARS-CoV-2 proteins and it was demonstrated that known sigma receptor ligands, including some under investigation here, possessed antiviral activity (37). However, a subsequent study found that these effects were not correlated with binding affinity for σ1R or σ2R, but instead depended on the ability of these molecules to induce phospholipidosis (38). A phenomenon that often confounds drug discovery efforts, phospholipidosis occurs due to the capacity of cationic, amphiphilic molecules to accumulate in endosomes and lysosomes, resulting in disruptions of lipid processing and alterations in the morphology of these compartments (39). Because lysosomal function is known to be important for PrPSc degradation, and some of the molecules under investigation here are reported inducers of phospholipidosis, we next explored this as a potential mechanism of action of these anti-prion compounds (38,40).
Interestingly, in addition to being a potent inducer of phospholipidosis (38), the antiarrhythmic medication amiodarone has been previously identified as an anti-prion compound (41) and sigma receptor ligand (42). In our hands, amiodarone has an EC50 of 4.4 µM and a LC50 of 6.3 µM (Figure 6A), and Ki values of 85.3 nM and 171 nM for σ1R and σ2R, respectively (Table 2). Here, we used amiodarone as a positive control in phospholipidosis assays, which were monitored using LipidTox, a fluorescent reagent that stains intracellular lipid accumulations (43). Similar results were obtained with an alternative reporter, NBD-PE (not shown, (38)). Of the most potent compounds, only rimcazole induced phospholipidosis at levels > 50% that of amiodarone after incubation with N2a cells at 10 µM for 24 hours (Figure 6B, C). Further, the level of phospholipidosis induced by these compounds did not correlate with their anti-prion activity in ScN2a cells (r = −0.4436, p = 0.2709) (Figure 6D). These results indicate that phospholipidosis itself is unlikely to be a major mechanism underlying the anti- prion effects of the sigma receptor ligands analyzed here, although we cannot rule out that other alterations in lysosomal or autophagosomal pathways could be a contributing factor to the action of some of the compounds.
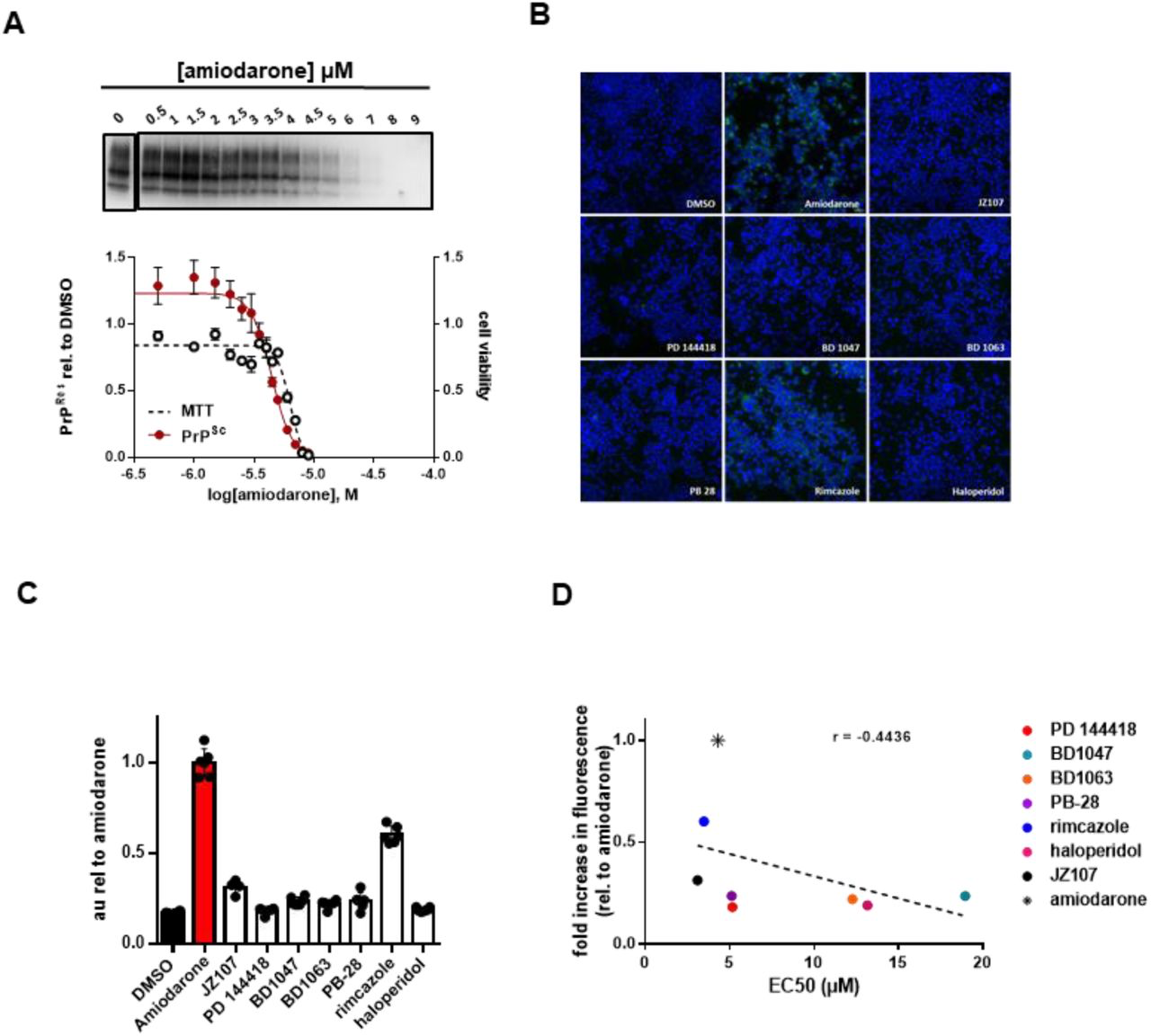
Phospholipidosis induction is not correlated with anti-prion activityA) N2a cells chronically infected with RML prions were incubated with the indicated concentrations of amiodarone for a total of seven days, before being lysed for PK digestion and analysis by western blot. Cultures treated in parallel were subjected to MTT assay. All data points represent three independent replicates. B) Uninfected N2a cells were incubated in 10 µM of the indicated compound with LipidTox for 24 hours. Green fluorescence represents LipidTox staining, blue represents Hoechst nuclei staining. C) Total LipidTox fluorescence was normalized to signal derived from Hoechest. D) For each compound, the level of phospholipidosis induction relative to DMSO is plotted against the EC50 determined against RML prions. r = −0.4436, p = 0.2709. Correlation analysis was performed using GraphPad software.
Sigma receptor ligands do not alter the levels or sub-cellular localization of PrPC
To further probe the mechanism of action of the sigma receptor ligands, we tested their effects on the total levels and sub-cellular localization of PrPC, factors that could potentially influence cellular content of PrPSc. Focusing on compounds with an EC50 of < 20 µM, we found that none significantly altered the total levels of PrPC assessed by western blotting when tested at their EC50 values for anti-prion activity (Figure 7A; Supplementary Figure 1). The compounds also had no observable effect on the sub-cellular localization of PrPC based on immunofluorescence staining (Figure 7B).
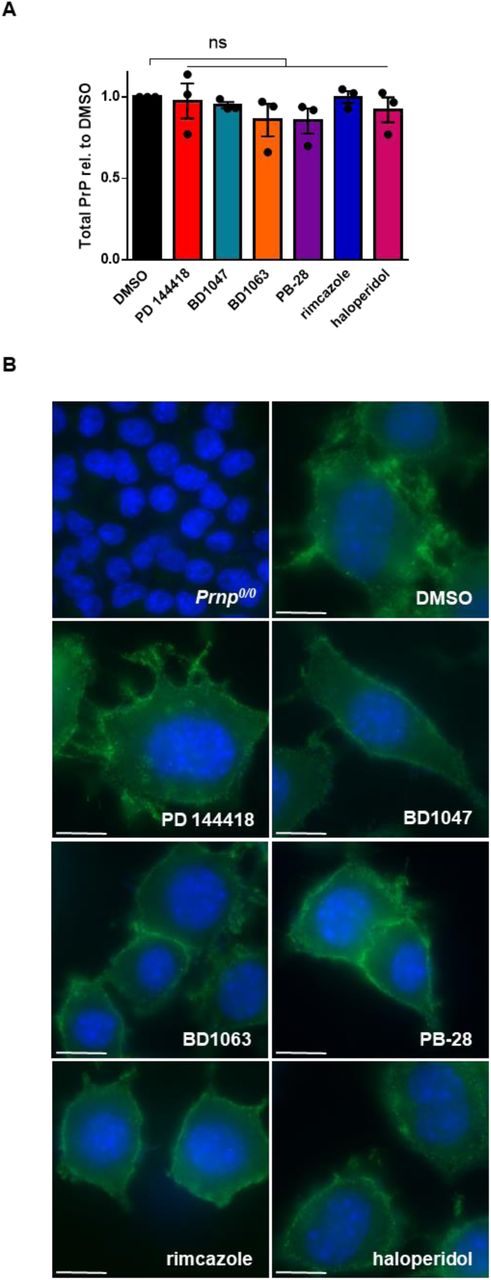
Anti-prion compounds do not alter PrPC levels or sub-cellular localizationA) Effect of compounds on total PrP levels in uninfected N2a cells. For each compound, the EC50 determined against RML prions in Figure 2 was used. Following incubation with each compound for seven days, levels of total PrP, compared to DMSO treatment, were plotted as mean ± SEM. B) Compounds do not alter the cell-surface localization of PrPC. For each compound, the EC50 determined against RML prions was used. PrPC was imaged by immunofluorescence staining using D18. DAPI is used as a nuclear counter stain. Scale bar = 10 μm. For all experiments, n = 3 replicates. p < 0.0001 = ****; p < 0.001 = ***; p < 0.01 = **; p < 0.05 = *; ns = not significant using a two-tailed students t-test.
Prion strain specificity of sigma receptor ligands
The efficacy of some anti-prion compounds has been shown to be strain dependent (11,44). To examine the potential for strain specificity, we used N2a cells chronically infected with a second strain of prions (22L), again focusing on compounds that displayed an EC50 of < 20 µM against RML prions. We observed that three of these compounds have efficacies against 22L prions that are comparable to those against RML prions: When tested at their EC50 values for RML prions, BD1063 reduced PK resistant PrP to 63.3 ± 1.2% (p = < 0.0001), PB-28 by 52.6 ± 2.6% (p = < 0.0001), and haloperidol by 43.0 ± 8.6% (p = 0.0007) of that observed in vehicle treated cells (Figure 8A, Supplementary Figure 2C, D, F). Two molecules were more effective against 22L prions than RML prions, resulting in reductions in PK resistant PrP by BD1047 to 19.1 ± 10.5% (p = 0.0004) and by rimcazole to 13.5 ± 7.8% (p = < 0.0001) of that seen in controls (Figure 8A, Supplementary Figure 2B, E). PD 144418, however, was ineffective against 22L prions at the EC50 determined against RML prions (Figure 8A; Supplementary Figure 2A). However, a dose response curve using 22L infected ScN2a cells revealed that PD 144418 is effective at higher concentrations, with an EC50 of 12.6 µM against this strain compared to an EC50 of 5.2 µM against RML prions (Figure 8B).
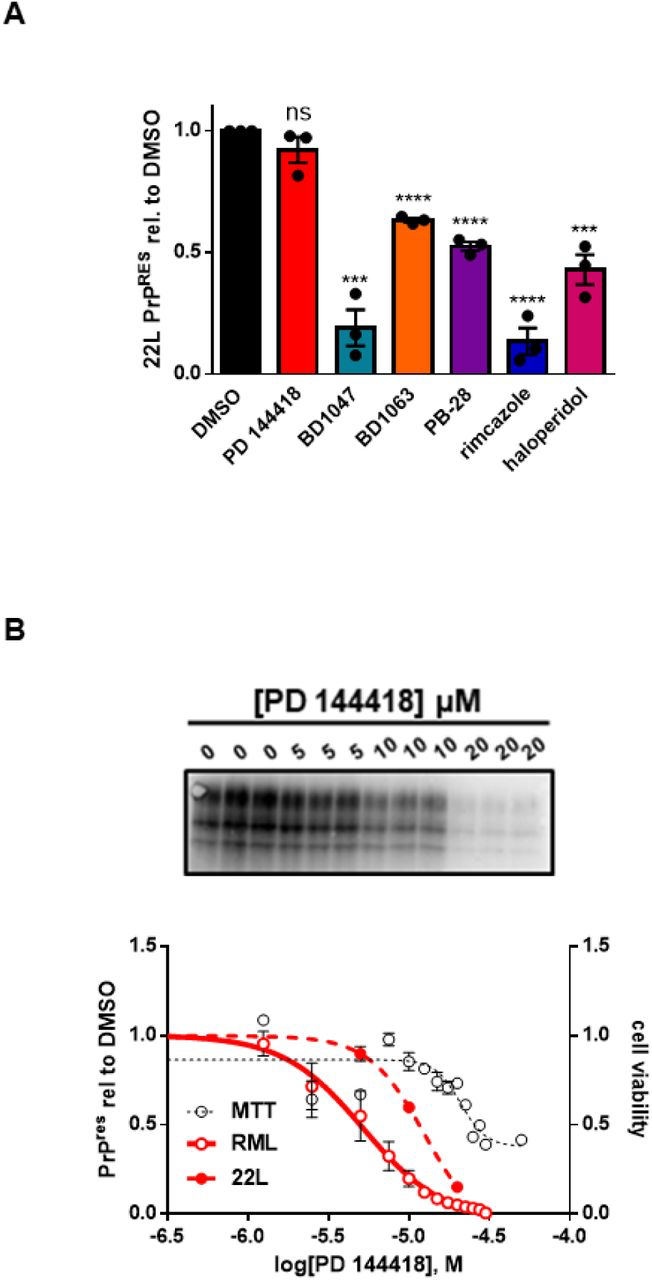
Anti-prion compounds do not exhibit strain specificityA) Compound efficacy against 22L prions in chronically infected N2a cells. For each compound, the EC50 determined against RML prions was used. Levels of PK resistant PrP, compared to DMSO treatment, were plotted as mean ± SEM. B) Dose response curves of PK resistant PrP remaining after incubation with increasing concentrations of PD 144418. RML and MTT values are replotted from Figure 1A. For all experiments, n = 3 replicates. p < 0.0001 = ****; p < 0.001 = ***; p < 0.01 = **; p < 0.05 = *; ns = not significant using a two-tailed students t-test.
Sigma receptor ligands prevent prion-induced synaptotoxicity
We previously demonstrated that exposure of cultured hippocampal neurons to prion infected brain homogenates or purified PrPSc causes a rapid (<12 hours) and dramatic retraction of dendritic spines (22,45,46). This phenomenon, which we have used as an assay of the synaptotoxic activity of prions, is dependent on the presence of cell-surface PrPC and is blocked by NMDA receptor antagonists and p38 MAPK inhibitors. Our results suggest that rapid conversion of PrPC to PrPSc on the cell surface triggers an intracellular signaling cascade that ultimately leads to collapse of the actin cytoskeleton within dendritic spines. In this scenario, compounds that inhibit the conversion of PrPC to PrPSc would be predicted to prevent spine collapse upon prion exposure. To test this prediction, hippocampal neurons (21 DIV) were pre-incubated with one of the six most potent compounds or DMSO vehicle for 2 hours before treatment with either purified RML prions or mock-purified material from uninfected, age-matched control brains (Supplementary Figure 3). Following 24 hours of prion exposure in the continued presence of the test compound, spine density was determined by immunofluorescence staining for F-actin. As expected, the application of purified prions to these cultures in the absence of test compound caused dendritic spine collapse, reducing spine density to less than 40% of that observed in cultures treated with mock-purified material (Figure 9; mock vs prion). Strikingly, all six sigma receptor ligands tested in this assay were able to significantly prevent spine loss (Figure 9). Further, PD 144418 (Figure 9A), PB-28 (Figure 9D), and haloperidol (Figure 9F) restored spine density to levels that were not statistically different from those of neurons treated with mock-purified material.
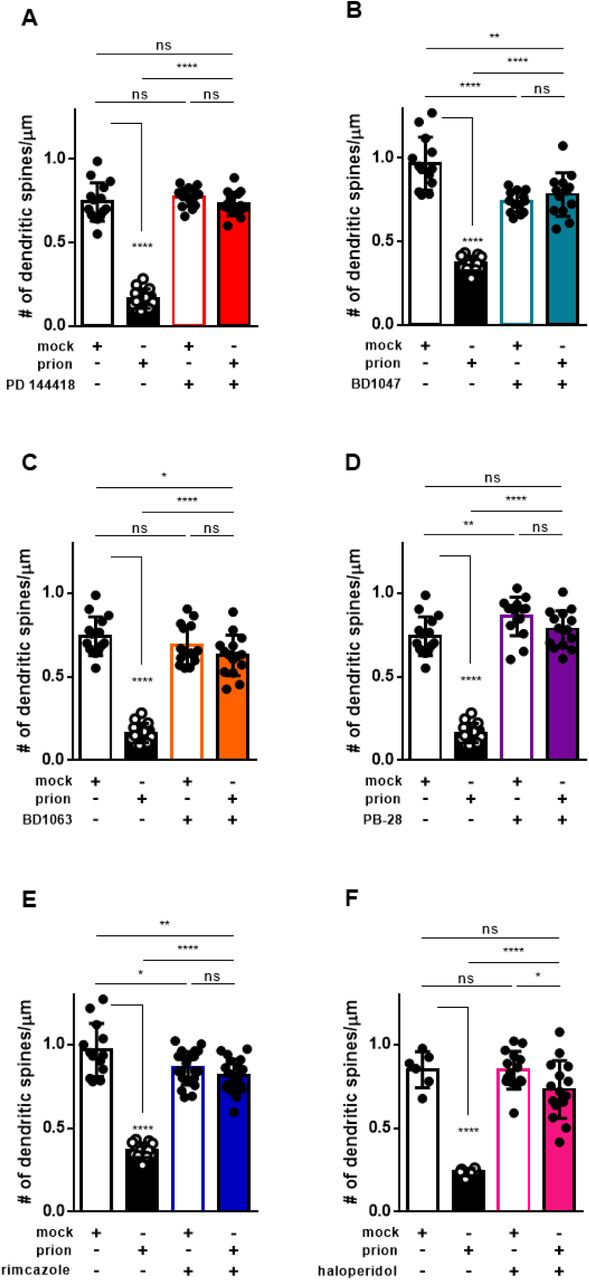
Sigma receptor ligands prevent prion induced synaptotoxicityFollowing a 2 hour pre-incubation with the indicated compound, or DMSO vehicle, hippocampal neurons were exposed to PrPScpurified from the brains of RML-infected mice (prion), or to mock- purified material from uninfected brains (mock). Dendritic spine number was assayed 24 h later by imaging F-actin in spines using Alexa Fluor 488 conjugated phalloidin. Mean number of dendritic spines/μm ± SEM is plotted for the following treatments: A) 10 μM PD 144418; B) 0.5 μM BD1047; C) 1 μM BD1063; D) 0.1 μM PB-28; E) 0.5 μM Rimcazole; F) 2.5 μM haloperidol. All numerical values can be found in Supplementary Table 2. p < 0.0001 = ****; p < 0.001 = ***; p < 0.01 = **; p < 0.05 = *; ns = not significant using a two-tailed student’s t-test.
Discussion
Identification of sigma receptor ligands as anti-prion agents
Using the DBCA, we previously identified phenethyl piperidines as a novel class of anti-prion compound, capable of reducing the levels of PK resistant PrP in RML and 22L-infected ScN2a cells at low micromolar concentrations (20,21). To identify the target of these molecules relevant to their anti-prion effects we submitted JZ107, a structurally optimized phenethyl piperidine, and JZ103, an inactive analogue, to the PDSP. In this screen, the highest affinity receptors for JZ107 were σ1R and σ2R, both of which were highly expressed in N2a cells (Table 1). Based on this result, we tested known ligands for σ1R and σ2R, and identified ten novel anti-prion compounds: PD 144418, BD1047, BD1063, PB-28, rimcazole, haloperidol, SA 4503, ANAVEX2-73, (+)-pentazocine, and DTG (Figure 1; Table 2). Each of these molecules were found to significantly reduce PrPSc levels in ScN2a cells and to bind σ1R and σ2R with Kivalues of 1.1 nM – 2.5 µM (Table 2).
Exploration of the mechanism of action of sigma receptor ligands
σ1R and σ2R, originally thought to be a novel class of opioid receptors, were eventually shown to be a pharmacologically and molecularly distinct pair of receptors that reside primarily in ER membrane domains. Their endogenous ligands and physiological functions are uncertain (47–49). There is evidence that σ1R functions as a ligand-gated chaperone for other receptors, and that σ2R plays a role in cholesterol exchange between the ER and lysosomes; both receptors have an evolutionary relationship to sterol isomerases. Sigma receptors are being actively investigated as therapeutic targets for treatment of cancer, as well as neurological disorders, including neuropathic pain, amyotrophic lateral sclerosis, Alzheimer’s disease, and Parkinson’s disease (50).
Given their localization in the ER and their involvement in sterol metabolism, it seemed plausible that σ1R and σ2R could play a role in the formation or degradation of PrPSc in ScN2a cells, and thereby represent the molecular targets responsible for the anti-prion effects of the compounds identified in this study. However, a comparison of the affinity for σ1R or σ2R and the EC50 of these compounds against RML prions reveals that these parameters are not correlated (Figure 2). Further, dual knockout of the genes encoding σ1R and σ2R in ScN2a cells did not alter either basal levels of PK resistant PrP (Figure 3) or blunt the susceptibility of the cells to the anti-prion effects of the compounds (Figure 4). These results conclusively demonstrate that σ1R and σ2R are not the direct molecular targets responsible for the anti-prion effects of these compounds. This was surprising, given that we chose to test the anti-prion activity of these compounds based on the fact that they are well known ligands for the sigma receptors. However, σ1R and σ2R are known to be promiscuous in their ligand binding profiles, and binding to the sigma receptors does not rule out the possibility that these ligands may act via other molecular targets. To explore the possibility that these compounds may be acting though other, distinct, interaction partners, we applied a similar experimental approach to test five other target proteins identified by the PDSP (Htr6, Drd4, Hrh1, Hrh3, and Chrm4). However, similarly to what we observed with the sigma receptors, these additional target knockouts did not diminish the observed anti-prion effects (Figure 5).
Other potential mechanisms of anti-prion activity
During the course of this study, the sigma receptors and their ligands were thrust into the spotlight for a potential role in the pathogenesis and treatment of SARS-CoV-2 infection (37). However, the affinity of these compounds for the sigma receptors did not correlate with their anti-viral activity (38). It was subsequently determined that the anti-viral effects were due to the ability of these compounds to induce phospholipidosis, a poorly understood phenomenon that often confounds drug discovery efforts. Because these findings included some of the compounds under examination in the present work, and because similar mechanisms could plausibly disrupt prion propagation, we next investigated phospholipidosis as a potential mechanism of action for these anti-prion compounds (Figure 6). While we found no correlation between phospholipidosis induction and the anti-prion effects of the molecules currently under investigation, we cannot rule out the possibility that phospholipidosis is a mechanism whereby some anti-prion compounds exert their effects, such as amiodarone, or that other kinds of disruptions of lysosomal or autophagosomal pathways contribute to reductions in PrPSc.
We determined that none of the six most potent sigma receptor ligands identified here had a significant effect on the total levels or cellular localization of PrPC (Figure 7; Supplementary Figure 1). This observation argues against an inhibitory mechanism dependent on reducing or redistributing the PrPC substrate, as has been proposed for some anti-prion compounds (16,51). Although there were some variations in the potency of the compounds when tested on ScN2a cells infected with the RML and 22L strains, all of them significantly reduced PrPSclevels in cells infected with either strain, suggesting lack of a strong preference for particular PrPSc conformations (Figure 8).
A striking observation that may provide mechanistic insight is that all six of the most potent sigma receptor ligands, like JZ107 (20), significantly reduced PrPSc-induced retraction of dendritic spines on cultured hippocampal neurons (Figure 9). We have previously demonstrated that spine retraction and decrements in synaptic transmission occur rapidly (< 12 hrs) after exposure to PrPSc, and these events are entirely dependent on the expression of full length PrPC by target neurons (22,45,52). We have also shown that the synaptotoxic effects of PrPSc depend on activation of an NMDA receptor/p38 MAPK-mediated signal transduction cascade, likely initiated by formation of PrPSc at the cell surface. The ability of the sigma receptor ligands to block this cascade could result from their effects on PrPC or PrPSc at the neuronal plasma membrane, or on subsequent steps of the synaptotoxic signaling pathway.
Therapeutic implications
The compounds identified here have several properties that make them attractive candidates as therapeutics for prion disease. In addition to their ability to inhibit prion propagation in ScN2a cells infected with multiple prion strains, the most potent compounds prevent prion-induced retraction of hippocampal neuron dendritic spines. This event is one of the earliest pathologies observed over the course of prion disease (53). Further, these compounds are all known to be blood brain barrier penetrant, and five of them, rimcazole, haloperidol, SA 4503, ANAVEX2-73, and (+)-pentazocine have a history of use in humans (49,54–62). The preclinical molecules SA 4503 and ANAVEX2-73, known respectively as cutamesine and blarcamesine, were well tolerated in phase II trials: SA 4503 for ischemic stroke (60), and ANAVEX2-73 for Alzheimer’s and Parkinson’s diseases (61). Despite the negative side effects associated with high and/or prolonged dosing with rimcazole, haloperidol, and (+)-pentazocine (57–59), all five molecules make excellent candidates for preclinical studies for the treatment of prion disease.