
Sigma-1 receptor regulates Tau phosphorylation and axon extension by shaping p35 turnover via myristic acid
By Shang-Yi A. Tsai, Michael J. Pokrass, Neal R. Klauer, Hiroshi Nohara, and Tsung-Ping Su
Excerpt from the article published in PNAS May 11, 2015, 112(21) 6742-6747 DOI: https://doi.org/10.1073/pnas.1422001112
Editor’s Highlights
- Malfunction of axonal maintenance, regeneration, and target recognition contribute to CNS disorders such as Parkinson’s disease (PD).
- Cyclin-dependent kinase (Cdk5) is a ubiquitously expressed enzyme; however, its activator, p35, is almost exclusively expressed in neurons. Cdk5 signaling supports neurite projection and proper neuronal migration. Dysregulation of cdk5 activity leads to hyperphosphorylation of several substrates, including NF proteins and tau, which causes the formation of neurofibrillary tangles.
- Because of this lipid-binding property of the sigma-1 receptor (Sig-1Rs) and because the myristoylation of p35 contributes to its degradation, due to its ability to bind myristic acid.
Significance
Neurodegeneration is tightly linked to tauopathy as a result of the overactivated cyclin-dependent kinase (cdk) 5 in the form of cdk5/p25. P35, another activator of cdk5, which is short-lived and myristoylated, is cleaved by calpain to the degradation-resistant activator p25. One way to tune down the aberrant, overreactive cdk/p25 is to reduce available p35. The sigma-1 receptor (Sig-1R) is an endoplasmic reticulum (ER) chaperone of unknown relation to tauopathy. Here we found that Sig-1Rs bind myristic acid and promote the degradation of p35. Further, the aberrant tau hyperphosphorylation and stunting of axon elongation in Sig-1R–KO neurons are mitigated by exogenously added myristic acid. Our results establish the role of ER Sig-1Rs in tauopathy and implicate the importance of myristic acid as a substance to fight against neurodegeneration.
Abstract
Dysregulation of cyclin-dependent kinase 5 (cdk5) per relative concentrations of its activators p35 and p25 is implicated in neurodegenerative diseases. P35 has a short t½and undergoes rapid proteasomal degradation in its membrane-bound myristoylated form. P35 is converted by calpain to p25, which, along with an extended t½, promotes aberrant activation of cdk5 and causes abnormal hyperphosphorylation of tau, thus leading to the formation of neurofibrillary tangles. The sigma-1 receptor (Sig-1R) is an endoplasmic reticulum chaperone that is implicated in neuronal survival. However, the specific role of the Sig-1R in neurodegeneration is unclear. Here we found that Sig-1Rs regulate proper tau phosphorylation and axon extension by promoting p35 turnover through the receptor’s interaction with myristic acid. In Sig-1R–KO neurons, a greater accumulation of p35 is seen, which results from neither elevated transcription of p35 nor disrupted calpain activity, but rather to the slower degradation of p35. In contrast, Sig-1R overexpression causes a decrease of p35. Sig-1R–KO neurons exhibit shorter axons with lower densities. Myristic acid is found here to bind Sig-1R as an agonist that causes the dissociation of Sig-1R from its cognate partner binding immunoglobulin protein. Remarkably, treatment of Sig-1R–KO neurons with exogenous myristic acid mitigates p35 accumulation, diminishes tau phosphorylation, and restores axon elongation. Our results define the involvement of Sig-1Rs in neurodegeneration and provide a mechanistic explanation that Sig-1Rs help maintain proper tau phosphorylation by potentially carrying and providing myristic acid to p35 for enhanced p35 degradation to circumvent the formation of overreactive cdk5/p25.
Axons are structurally and functionally distinct protrusions of neurons that modulate neurotransmitter release and neural function. Malfunction of axonal maintenance, regeneration, and target recognition contribute to CNS disorders such as Alzheimer’s disease (AD), Parkinson’s disease, stroke, and spinal cord injuries (1–3). Cyclin-dependent kinase (Cdk) 5 activities within the axon play a significant role in the cytoskeletal dynamics of microtubules and actin neurofilaments (NFs), which determine axonal path and length. Specifically, cdk5 active complexes phosphorylate proteins that contribute to the stabilization or destabilization of microtubules, formation of neurofibrillary tangles, and axonal pathfinding (4–6).
Cdk5 is a ubiquitously expressed enzyme; however, its activator, p35, is almost exclusively expressed in neurons (7, 8). Cdk5 signaling supports neurite projection and proper neuronal migration (9–11). Dysregulation of cdk5 activity leads to hyperphosphorylation of several substrates, including NF proteins and tau, which causes the formation of neurofibrillary tangles (6, 12). Although the kinetics of cdk5 activity when in complex with p35 or p25 are the same, cdk5/p25 complexes are proposed to be responsible for neurodegenerative pathophysiology because of their longer duration of activity in neurons (13, 14). Cdk5 activity is largely dependent on the availability of its activators, p35 and p39 (8, 15–17). Transcription of p35 is under the control of early growth response protein 1 (EGR-1) (18–20). p35 is degraded by two main pathways: the ubiquitin–proteasome pathway is the predominant form of degradation in fetal neurons, whereas, in adult neurons, p35 is preferentially cleaved to p25 by the calcium-dependent protease calpain (21–23). P35 is myristoylated at an N-terminal glycine residue, and this modification determines the membrane localization of cdk5/p35 complexes as well as p35’s vulnerability to proteasomal degradation (24, 25). When p35 is cleaved to p25, the N-terminal glycine is on a separate p10 fragment and the remaining cdk5/p25 complex is free to diffuse into the cytosol (14, 22).
The sigma-1 receptor (Sig-1R) is an endoplasmic reticulum (ER) chaperone that resides at the ER/mitochondrion interface referred to as the mitochondrion-associated ER membrane (26–28) that has been implicated in neurodegenerative disorders (29–31). The exact molecular mechanism underlying the neuroprotective property of Sig-1Rs remains to be totally clarified. Although we previously reported that Sig-1Rs promote dendritic arborization (28), whether Sig-1Rs are involved in the regulation of axon elongation remains unknown. A recent report demonstrated the efficacy of a Sig-1R ligand in attenuating the β-amyloid–induced neurotoxicity via the ligand’s action in blocking the glycogen synthase kinase (GSK) activity (32) that also causes tau phosphorylation. We decided to examine here if a potential relation between Sig-1R and cdk5 may exist. In particular, the Sig-1R has been shown to bind cholesterol as well as sphingolipids, ceramides, progesterone, and other endogenous lipids (26, 33–36). Thus, because of this lipid-binding property of Sig-1Rs and because the myristoylation of p35 contributes to its degradation, we set forth to examine if the action of Sig-1Rs in tauopathy, if any, may relate to Sig-1R’s potential ability to bind myristic acid. The results are presented in this report.
Results
Sig-1Rs Regulate Axonal Length and Density.
We previously reported that rat hippocampal neurons transfected with Sig-1R siRNA (siSig-1R) displayed reduced dendritic spine formation compared with control siRNA (siCon) cells (28). This led us to further examine the role of Sig-1Rs in neuronal morphology. Rat hippocampal neurons were transfected with siCon or siSig-1R in combination with EGFP on the first day in vitro (div). Neurons were stained on div 3 and div 7 with Tau antibody to visualize axonal length. Neurons transfected with siSig-1R exhibited diminished axonal length in comparison with the control group. Significant differences in axonal elongation were evident as early as div 3 and persisted to div 7 (Fig. 1A). Cortical brain slices from Sig-1R–KO mice corroborated the role of Sig-1Rs in axonal development (Fig. 1B). Cortical brain slices were immunostained with heavy-chain NF (NF-H) to visualize NF proteins, and with DAPI in Sig-1R WT and KO mice. The cortex of KO animals shows diminished axonal density compared with WT cortices (Fig. 1B).
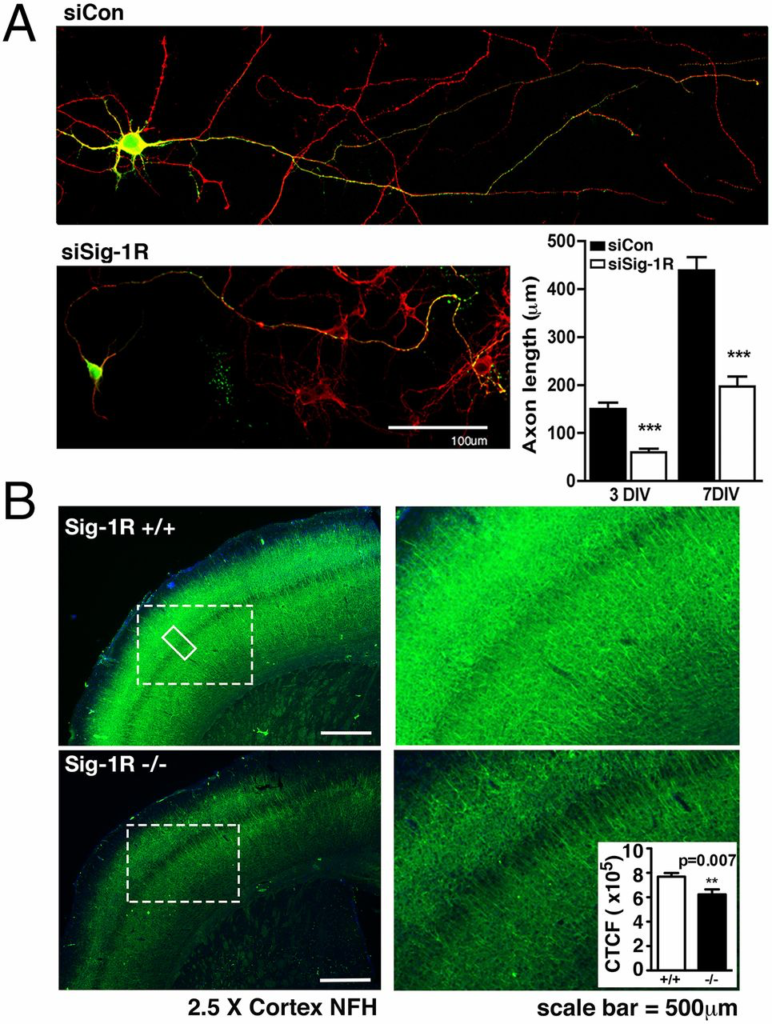
Sig-1Rs affect axonal extension. (A) Confocal microscopic images of neurons stained for tau proteins (red) and expressing EGFP as evidence of siRNA expression. Neurons expressing siSig-1R exhibit shorter axons in comparison with neurons expressing siCon when observed on div 3 and div 7. Axon length was estimated by using free-hand tracing on approximately 20 axons (WT and KO each) with five sparse neurons on each coverslip. Error bar indicates SEM. (B) Cortical brain slices were collected from Sig-1R WT and KO mice and stained for NF proteins. Note the diminished axonal density in the Sig-1R KO brain slices. NF density was assessed in either of the brain slices from bregma 0.02 mm to bregma −0.46 mm, as shown in the figure, or from bregma −1.06 mm to bregma −2.06 mm. At least five brain slices within the same distance range were used for each animal. Five regions of cortex (as in solid box) from each slice were analyzed by using ImageJ software. NF fluorescence intensity (Inset) was adapted from and calculated by the corrected total cell fluorescence formula: integrated density − (area of selected region × mean fluorescence of background readings). Six each of the age-matched WT and KO mice were used in the study. Error bar indicates SEM.
Sig-1R Regulates Cytoskeleton Protein Phosphorylation and Transport of Mitochondria Toward Nerve Ending.
Neurons with reduced Sig-1R expression exhibit a dramatic increase in hyperphosphorylated tau proteins indicated by paired helical filaments (Fig. 2A). Additionally, Sig-1R–KO neurons accumulate greater levels of phosphorylated NF-Hs (pNF-H) relative to WT neurons (Fig. 2A). Surprisingly, protein phosphatase 2 (PP2) A-C, which plays a major role in dephosphorylating tau (37–39), was increased in KO mice. Sig-1R–KO neurons might have expressed more phosphatases to compensate for the hyperphosphorylated tau (Fig. 2A). At any rate, aberrant hyperphosphorylation of tau proteins may impair tau function, thereby deregulating axonal transport networks (40, 41). We used sucrose gradient fractionation to observe protein compositions in the synaptosomes of siSig-1R cells (Fig. 2B). The synaptosomal marker proteins AMPA receptor subunits 2 and 3 (GluR2/3) and synaptosomal-associated protein 25 (SNAP-25) and the mitochondrial protein cytochrome C were all diminished in the pre- and postsynaptosomal fractions from Sig-1R–knockdown neurons (Fig. 2B). We also analyzed the isolated synaptosomal vesicles by flow cytometry for mitochondrial membrane potential and mass (Fig. 2C). We detected reduced mitochondrial mass as determined by 10-N-nonyl acridine orange (NAO) signal in the isolated synaptosomes from siSig-1R neurons. The reduced mass in mitochondria was accompanied by decreased signal from the mitochondrial membrane potential dyes Rh 123 and MitoTracker Red CMXRos (MT CMXRos; Fig. 2C). Accordingly, live imaging revealed that axonal mitochondrial movements were less dynamic in the Sig-1R–knockdown neurons (Fig. S1).
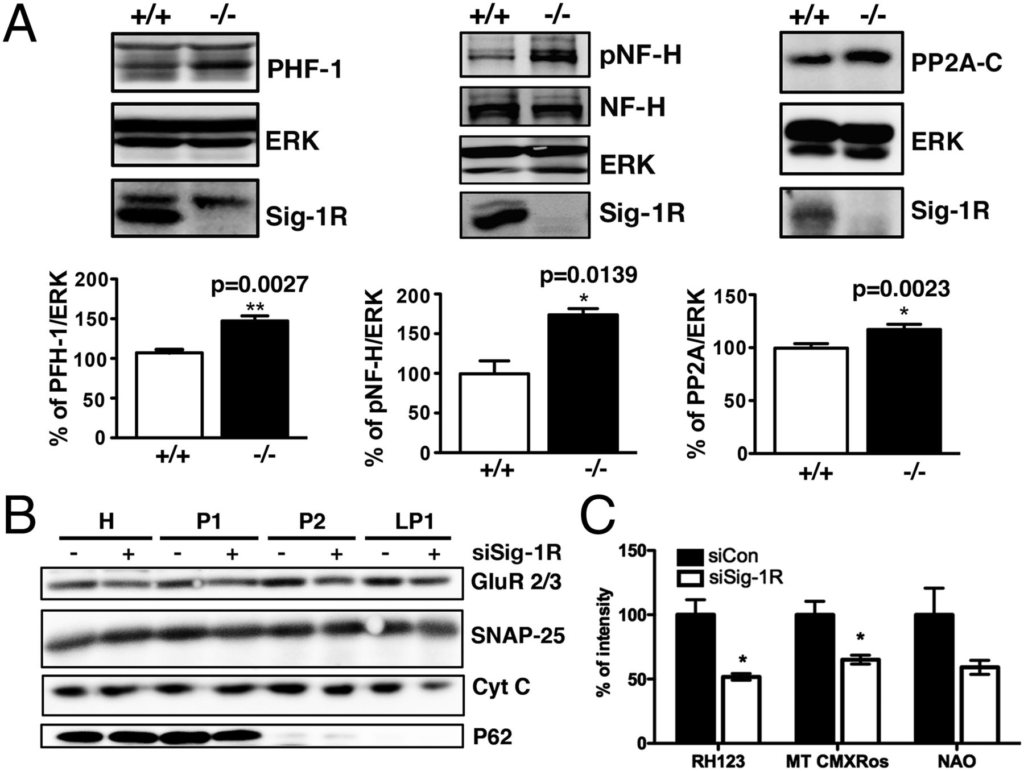
Sig-1Rs affect cytoskeletal proteins and alter numbers of mitochondria at the synaptosome. (A) Elevated basal levels of PHF-1, pNF-H, and PP2A in brains of Sig-1R KO mice (n = 4–6 independent experiments; error bar indicates SEM; *P < 0.05 and **P < 0.01, t test). (B) Sucrose gradient fractionation on synaptosomal proteins. Note the reduced level of GluR2/3, SNAP-25, and cytochrome C in siSig-1R samples, in particular in the synaptosomal membrane fraction (LP1). H, homogenate; P1, nuclei enriched fraction; P2, synaptosomal fraction (n = 3 independent experiments). (C) Reduced mitochondrial membrane potential (labeled by Rh 123) and mass (indicated by MT CMXRos or NAO) seen in isolated synaptosomal vesicles from siCon and siSig-1R neurons as examined by flow cytometric analyses (n = 4 independent experiments; error bar indicates SEM; *P < 0.05, t test).
Sig-1R Controls p35 Degradation Mainly Through the Proteasomal Pathway.
The presence of hyperphosphorylated tau or NFs has been implicated in the pathology of neurodegenerative disorders such as multiple sclerosis, AD, and amyotrophic lateral sclerosis and may be indicative of aberrant cdk5 activity (42–44). We thus investigated cdk5 in complex with its activator, p35, in neurons. Cdk5 activity is dependent on the availability of p35. Interestingly, the p35 levels were found to be elevated in brain tissue from Sig-1R–KO mice (Fig. 3A). Quantitative real-time PCR experiments were conducted to examine the role of the Sig-1R in the transcriptional regulation of p35. No significant difference was found in the amount of p35 or EGR-1 mRNA between Sig-1R WT and KO mice (Fig. 3B). We next studied the degradation of p35 to clarify the effect of Sig-1Rs on cdk5 activity. Cells overexpressing siSig-1R or siCon were treated with protein synthesis inhibitor cycloheximide (CHX) and collected at intervals over a 120-min period. A slower degradation rate of p35 was observed in the siSig-1R cells compared with the siCon-expressing cells (Fig. 3C). Similar degradation trends were seen in Sig-1R–KO cortical neurons and brain slices (Fig. 3 E and F). On the contrary, a faster p35 degradation rate was observed in cells overexpressing Sig-1Rs (Fig. 3D). Cells that were treated with CHX and the proteasome inhibitor lactacystin exhibited accumulated p35, indicating that the degradation observed was primarily caused by the ubiquitin–proteasome pathway (Fig. 3D). To further substantiate the role of Sig-1Rs in the proteasomal degradation of p35, we examined the ubiquitination of p35. Although knocking down of Sig-1Rs increased the total amount of ubiquitinated proteins, the ubiquitinated p35 was reduced in the Sig-1R–knockdown neurons (Fig. S2A). This reduction of ubiquitinated p35 was in agreement with a slower proteasomal degradation of p35 in Sig-1R–knockdown neurons described earlier. However, Sig-1Rs may not be directly involved in the ubiquitination of p35, as we did not observe physical interactions between p35 and Sig-1R (Fig. S2B).
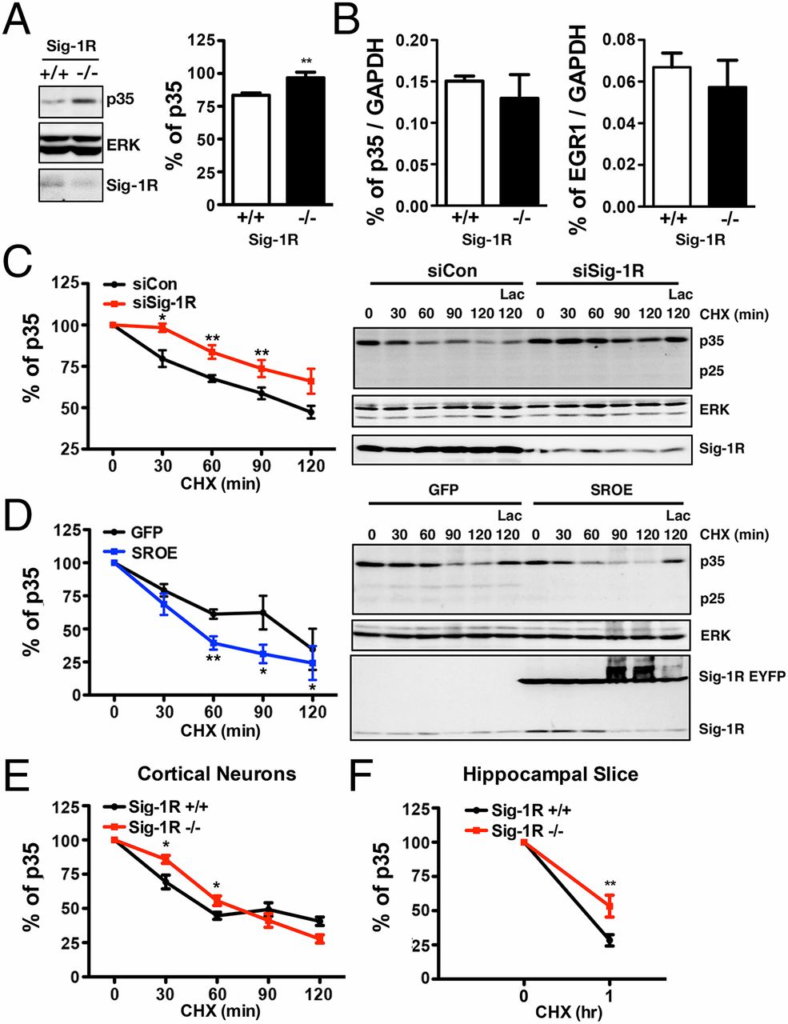
Accumulation of P35 proteins in Sig-1R KO neurons as a result of slower degradation. (A) Up-regulated P35 level in the brain tissue from Sig-1R KO mice (n = 4 independent experiments; error bar indicates SEM; **P= 0.0064, t test). (B) Unaltered level of the mRNA of p35 or its associated transcription factor, EGR-1, in the brain of Sig-1R–KO mice (n = 6 independent determinations; error bar indicates SEM; t test). (C) Slower degradation of p35 in Sig-1R–knockdown CHO cells compared with controls. CHX was used to halt de novo protein synthesis. Cells were transiently transfected with siSig-1R (red curve) or siCon (dark curve) before CHX treatment. (D) Effect of overexpression of Sig-1Rs on the p35 degradation. CHO cells stably overexpressing GFP or enhanced yellow fluorescent protein (EYFP)-tagged Sig-1R (SROE) were treated with CHX. P35 degraded faster in EYFP-tagged Sig-1R cells. (E) Slower degradation of p35 in Sig-1R–KO cortical neurons. (F) Slower p35 degradation in Sig-1R–KO hippocampal brain slices (n = 3 independent experiments in C–F; error bar indicates SEM; *P < 0.05 and **P < 0.01, paired t test).
Sig-1R Effect on p35 Turnover Is Unrelated to Calpain.
We next sought to examine if the Sig-1R–regulated p35 degradation is related to calpain-induced p35 cleavage. We treated crude brain extracts from Sig-1R WT and KO mice with CaCl2to activate calpain and thus observe the conversion of p35 to p25. We found no apparent or dramatic differences between the two sets of brains with regard to the rate of p35 conversion to p25 under conditions without CaCl2 (Fig. 4A, Left) or with CaCl2 (Fig. 4A, Middle). This process was certainly calpain-dependent, as the samples treated with the calpain inhibitor ALLM (N-Acetyl-Leu-Leu-Met-CHO) exhibited negligible p35 cleavage (Fig. 4A, Right). Furthermore, by treating hippocampal neurons with the calcium ionophore A23187 and CaCl2 along with CHX, we observed similar degradation rates of p35 in siSig-1R and siCon cells (Fig. S3A). Those results suggest that the Sig-1R does not regulate the p35 turnover via the calpain-mediated pathway. Moreover, the endogenous subtypes of calpain, as well as the expression level of the endogenous calpain inhibitor calpastatin, did not differ between the Sig-1R–KO animals and the WT animals (Fig. 4B). Sig-1Rs did not interact with calpain subunits calpain 1 (CP1) or calpain 2 (CP2; Fig. S3B). These data together indicate that Sig-1Rs do not affect the calpain conversion of p35 to p25.
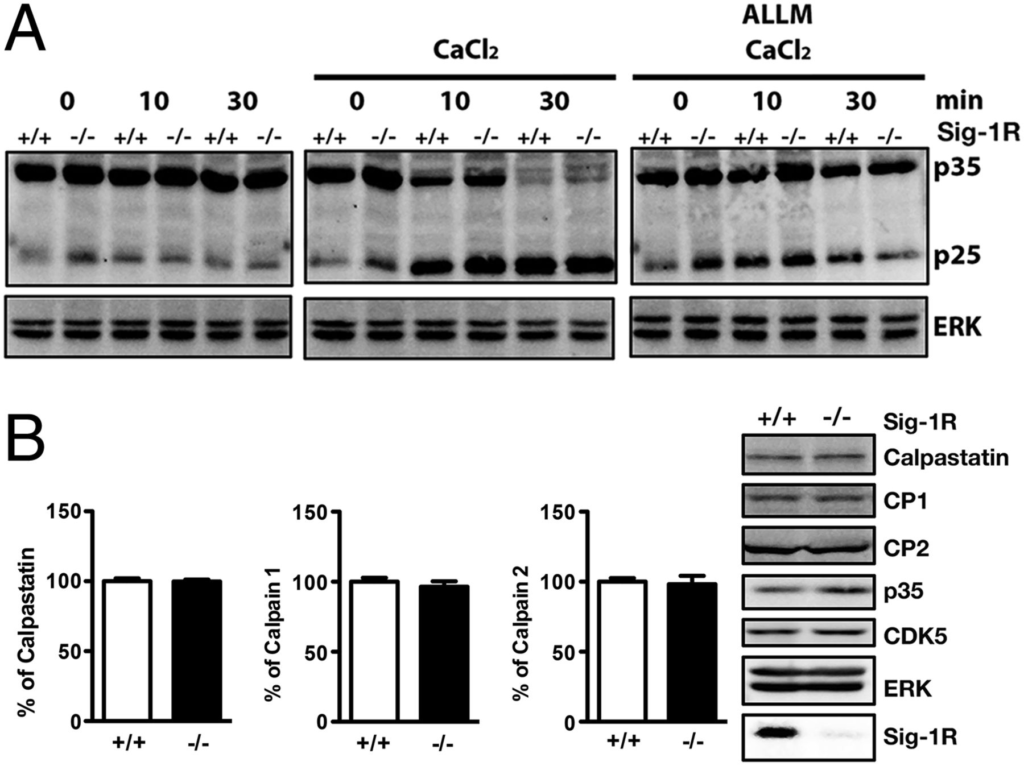
Sig-1R regulation of p35 turnover unrelated to calpain. (A) Brain tissue was harvested from WT and Sig-1R–KO mice and homogenized. ALLM samples were pretreated 1 h before experimentation. Sig-1R WT and KO homogenates exhibited similar rates of p25 accumulation (n = 3 independent experiments). (B) Examination of endogenous calpain subtypes (n = 3 independent experiments) and calpain inhibitor calpastatin (n = 6 independent experiments) protein levels in brain lysates from WT and Sig-1R KO mice. Error bar indicates SEM.
The Sig-1R Binds Myristic Acid and Regulates the Membrane Anchoring of p35.
In this section of the study, we first treated neurons with Sig-1R antagonist BD1063 and found that p35 levels were increased by BD1063 in a time-dependent manner (Fig. 5A). These results were unusual because antagonists do not cause an effect by themselves unless there are already some endogenous agonists at the receptor. It is known that p35 degradation requires its binding to the membrane and that this membrane anchorage is regulated via the N-myristoylation at the second amino acid glycine of p35 (24). To investigate whether the Sig-1R–regulated p35 degradation may relate to the membrane-bound form of p35, we compared the ratio of membrane-bound p35 and soluble p35 first in the Sig-1R antagonist-treated neurons and second in the WT vs. Sig-1R–KO brain samples. Sig-1R antagonist BD1063-treated (60 min) neurons showed a reduced level of membrane-bound p35 and a slightly increased amount of soluble p35 (Fig. 5B), again suggesting that BD1063 was antagonizing some endogenous Sig-1R ligand. Sig-1R–KO mice also showed a reduction of membrane-bound p35 (Fig. 5C). As the N-myristoylation–mediated membrane anchorage of p35 is a critical step to allow for the proteasomal degradation of p35, our data suggested that the Sig-1R may regulate the p35 turnover by affecting the membrane anchoring of p35. However, this regulation may not involve Sig-1R effects on the enzyme that transfers myristic acid to p35, i.e., N-myristoyltransferase 1 (NMT-1). NMT-1 protein levels remained the same in WT and Sig-1R–KO neurons (Fig. 5D). However, it is possible that the enzymatic activity of NMT-1 may differ. We further examined if the myristoylated p35 level might be regulated by Sig-1Rs. By combining the Click reaction (SI Materials and Methods) with immunoprecipitation (IP), we observed a reduced level of myristoylated p35 in the Sig-1R–knockdown cortical neurons (Fig. S4A). Similar results were seen by using the p35 antibody pull-down assay examining the [3H]myristic acid-labeled p35 (Fig. S4B). Those data suggested the possibility that the Sig-1R binds myristic acid and serves to increase the myristoylation of p35 and facilitate the p35 anchoring to the membrane. This idea is not implausible because lipids have been reported to bind to Sig-1Rs (34–36). We therefore examined next whether myristic acid binds to Sig-1Rs. A [3H](+)-pentazocine (PTZ) competition binding assay was performed at 4 °C to determine if the BSA-conjugated myristate (MA-BSA) competes with the selective Sig-1R agonist (+)-PTZ to Sig-1Rs on the plasma membrane. The MA-BSA was chosen to circumvent the solubility problem of myristic acid. The results indicated that myristate competed with the binding of [3H](+)-PTZ to Sig-1Rs and displaced approximately 25% of specific [3H](+)-PTZ (Fig. 5D) binding in the indicated test concentration range of MA-BSA. Higher concentrations of MA-BSA could not be tested because the BSA alone as such increased the [3H](+)-PTZ binding (Fig. S5). Within that workable range of displacement in the binding assay in which BSA did not interfere with the assay, MA-BSA exhibited an apparently high affinity at the Sig-1R (IC50 = 4.54 nM; Fig. 5E). Also, inasmuch as myristic acid was able to displace almost a 25% portion of specific [3H](+)-PTZ binding over a dose range of two log cycles (i.e., 1–100 nM), it is possible that the myristic acid-displaced portion of the [3H](+)-PTZ binding may represent a lipid-specific domain of the Sig-1R binding site. To further substantiate myristate as a Sig-1R ligand and to clarify if myristate may thus function as a Sig-1R agonist or antagonist, we determined the binding nature of myristate to Sig-1Rs by using our previously established method of the Sig-1R–binding immunoglobulin protein (BiP) association/dissociation assay (26). Results showed that myristate at 1 µM caused the dissociation of Sig-1R from BiP only to a slightly lesser extent than caused by 3 µM of (+)-PTZ (Fig. 5F), which is a prototypic Sig-1R agonist (27). Within the limitation of the data as a result of the interference of the BSA control per se, it is plausible to say that myristate is perhaps as potent as (+)-PTZ as a Sig-1R agonist. By using the plasma membrane IP assay on Sig-1Rs, we also found that there is no physical interaction between p35 and Sig-1R (Fig. S6). Results are similar to that seen with the cellular lysate (Fig. S2). The exact mode of action on how Sig-1Rs transfer myristate to p35 awaits further investigation.
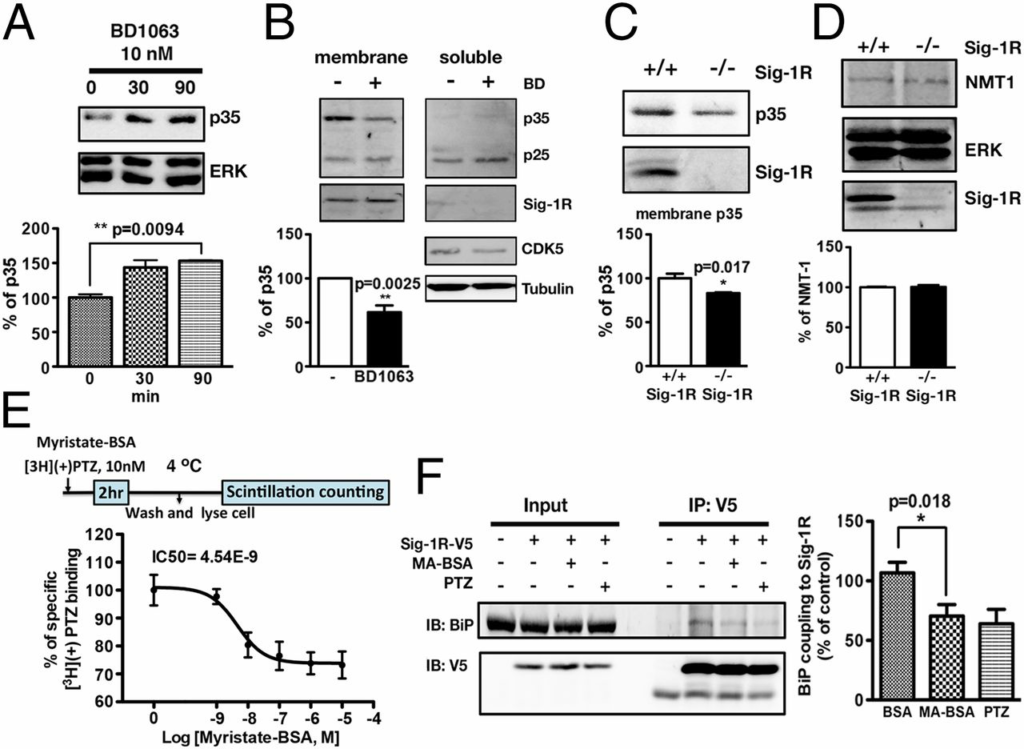
Sig-1Rs regulate p35 degradation by affecting membrane-bound p35 via myristic acid. (A) Effects of Sig-1R antagonist BD1063 on the level of p35 in the whole cellular lysates of neurons. Note the time-dependent increase of p35 by BD1063 (n = 3; error bar indicates SEM; P = 0.0094, t test). (B) Effect of BD1063 on the membrane-bound p35. The cell lysates of neurons treated with BD1063 were centrifuged at 100,000 × g for 1 h at 4 °C to separate the soluble and membrane fractions. The ER chaperone Sig-1R was used as a marker of the membrane fraction. Cdk5 and tubulin were used as markers of the soluble fraction (n = 3; error bar indicates SEM; P = 0.025, t test). (C) Comparison of membrane-bound p35 in WT or Sig-1R–KO brain lysates (n = 3; error bar indicates SEM; P = 0.017, t test). (D) No significant changes of NMT-1 protein levels were found in Sig-1R–KO (−/−) neurons. (E) MA-BSA competes with specific [3H](+)-PTZ for Sig-1R binding. Note: maximum displacement of approximately 25% was reached over two log cycles of the dose range of myristate (i.e., 1–100 nM; n = 4 independent experiments; error bar indicates SEM). (F) MA-BSA as a Sig-1R agonist in causing the dissociation of Sig-1R from BiP. MA-BSA (60 min)-induced dissociation of Sig-1R from BiP is shown. (+)-PTZ was used as prototypic Sig-1R agonist (n = 4; error bar indicates SEM; P = 0.018, t test).
Myristic Acid Enhances p35 Turnover, Reduces Tau Phosphorylation, and Rescues Retarded Axon Elongation in Sig-1R–Knockdown/KO Nervous Systems.
We next examined effects of exogenously added myristate (in the form of MA-BSA) on several neuronal characteristics as they relate to cdk5 signaling. Exogenous myristate abolished the abnormal p35 accumulations in Sig-1R–knockdown neurons and Sig-1R–KO brain tissues (Fig. 6A and Fig. S7). Also, in WT neurons, exogenous myristate remarkably reduced all of the following: hyperphosphorylated tau in the paired helical filaments (PHF-1), pNF-H, and p35 (Fig. 6B, lane 1 vs. lane 2). In Sig-1R–KO neurons, PHF-1, pNF-H, and p35 were also diminished after 24 h of myristate treatment (Fig. 6B, lane 3 vs. lane 4). Further, myristate increased the axon elongation in WT neurons and also rescued the retarded axon elongation seen in KO neurons (Fig. 6C). Those results support the notion that Sig-1Rs, by interacting with endogenous myristic acid, play a pivotal role in the p35 myristoylation and degradation.
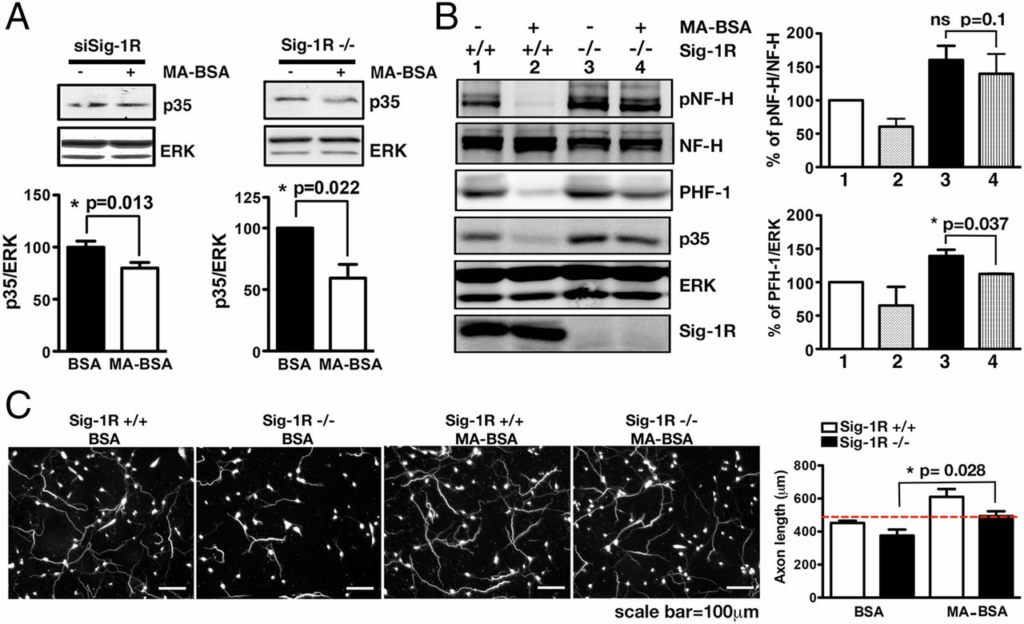
Effects of exogenous myristic acid on p35 and phosphorylated cytoskeletal proteins in WT and Sig-1R–knockdown neurons and associated consequences on axonal lengths. (A) Twenty-four hours of MA-BSA treatment attenuated p35 levels in Sig-1R–knockdown (n = 5) or KO neurons (n = 3). Error bar indicates SEM (*P < 0.05, t test). BSA alone served as controls. (B) Neurons treated with MA-BSA apparently showed the tendency of diminished phospho-NF-H, PHF-1 and, again, p35 in WT and Sig-1R–KO neurons (n = 4 independent experiments; error bar indicates SEM; t test). (C) Effect of BSA alone or MA-BSA on axon lengths in WT and Sig-1R–KO mouse brain. Cortical neurons from WT or Sig-1R–KO mice were treated for 48 h with BSA or MA-BSA. Neurons were stained with phosphorylated NF antibody. Neurons treated with MA developed longer axons relative to those that received BSA alone. Three coverslips of neuron per treatment, at least 10 neurons, were counted in each coverslip. Error bar indicates SEM (P = 0.028, t test).
Discussion
Sig-1Rs have been reported to play a role in the pathogenesis of neurodegenerative disorders including AD (30, 45, 46), x’s disease (47, 48), and motor neuron disorders (49–51). The underlying molecular mechanisms of Sig-1R action remain to be totally clarified. Although we have previously demonstrated several versatile actions of Sig-1Rs as they may affect cellular survival (26–28), other mechanisms of action may exist. Here, we have identified a new action of Sig-1Rs in that Sig-1Rs regulate tau phosphorylation and axon elongation by interacting with myristic acid.
Lipids are receiving considerable attention in the cascades of signal transduction. Specifically, lipids are closely related to neurodegeneration (52, 53). Although Sig-1Rs bind to certain lipids (34–36) and have been shown to partake in lipid biosynthesis (54, 55), the specific physiological role arising from the Sig-1R–lipid interaction remains unknown. Thus, to our knowledge, our present finding constitutes the first report on the physiological consequence of the Sig-1R–lipid interaction. It is noteworthy to again point out that myristic acid is acting as a Sig-1R agonist and is almost as potent as (+)-PTZ in causing the dissociation of Sig-1R from BiP. In this regard, the effects seen with the Sig-1R antagonist BD1063 in the present study can be reckoned as BD1063 blocking the agonistic effect of endogenous myristic acid at the Sig-1R. However, the possibility may be entertained that those data may represent the result of those ligands working competitively on certain yet unknown innate activity of Sig-1Rs. Overall, our data suggest a notion that Sig-1Rs may bind myristic acid and thereby increase the myristoylation of p35. However, the possibility exists that myristic acid may bind Sig-1Rs in the form of an ester or amide. Questions remain concerning the molecular mechanisms involved in this process. For example, do Sig-1Rs bind myristic acid at the ER and then translocate myristic acid to the plasma membrane to affect the p35 myristoylation? Alternatively, do Sig-1Rs transfer myristic acid directly to p35 at the ER? It would be technically challenging to provide answers to those questions. Nevertheless, our results showing that exogenous myristic acid could rescue the axon length in WT and Sig-1R KO neurons (Fig. 6C) suggest that the end role of Sig-1Rs is sending endogenous myristic acid to the plasma membrane. Therefore, the exogenously added myristic acid serves to complete the final goal that Sig-1Rs would have served.
As mentioned in the Introduction, in addition to the cdk5 we examined in this study, GSK3β has also been shown to play a role in the Sig-1R ligand action against β-amyloid–induced neurotoxicity (32). Recent reports have demonstrated cross-talk between the cdk5 and GSK3β pathways (56, 57). The exact relation between Sig-1Rs, cdk5, and GSK3β remains to be clarified.
Our data showing that the knockdown of Sig-1Rs causing a dysregulation of p35 is independent of calpain activity is consistent with our previous report that Sig-1Rs regulate the ER-mitochondrion Ca2+ signaling but do not significantly affect the cytosolic Ca2+ (26). A recent study indicated that overexpression of Sig-1R or addition of Sig-1R agonist increased endogenous calpain inhibitor calpastatin expression in the neuronal PC6.3 cell line (58). However, we found no significant differences in calpain or calpastatin protein levels in Sig-1R KO neurons or when overexpressing Sig-1R in the neuro-2a cells (Fig. 4B).
In summary, we found that Sig-1Rs regulate the turnover of the short-lived cdk activator p35 via myristic acid and thus play important roles in the maintenance of proper tau phosphorylation and axon elongation in the brain.