
Sigma-1 receptor maintains ATAD3A as a monomer to inhibit mitochondrial fragmentation at the mitochondria-associated membrane in amyotrophic lateral sclerosis
By Seiji Watanabe, Mai Horiuchi, Yuri Murata, Okiru Komine, Noe Kawade, Akira Sobue, and Koji Yamanaka
Excerpt from the article published in Neurobiology of Disease, 2 February 2023, 106031, ISSN 0969-9961, DOI: https://doi.org/10.1016/j.nbd.2023.106031
Editor’s Highlights
- Sigma-1 receptor (σ1R) is a mitochondria-associated membranes (MAM)-specific chaperone protein.
- σ1R is essential for MAM integrity and that σ1R deficiency is closely associated with amyotrophic lateral sclerosis (ALS) pathogenesis.
- MAM-associated protein, AAA ATPase domain-containing protein A3 (ATAD3A), is involved in the pathogenesis of neurological diseases.
- ATAD3A dimer/monomer state is involved in ALS pathology and is associated with the loss of σ1R function that causes inherited ALS.
- Both σ1R and ATAD3A are critical factors of the MAM to maintain mitochondrial homeostasis, which are impaired in neurological diseases.
- Targeting σ1R–ATAD3A axis at the MAM to prevent mitochondrial dysfunction would lead to development of a novel therapeutic strategy for neurodegenerative diseases.
Autors’ Highlights
- σ1R maintains the MAM with the help of ATAD3A.
- ATAD3A localizes to the MAM via its N-terminal two coiled-coil domains.
- σ1R maintains ATAD3A as a monomer, which is associated with the inhibition of mitochondrial fragmentation.
- σ1R–ATAD3A interaction is disrupted and involved in mitochondrial fragmentation in ALS model mice.
Abstract
Organelle contact sites are multifunctional platforms for maintaining cellular homeostasis. Alternations of the mitochondria-associated membranes (MAM), one of the organelle contact sites where the endoplasmic reticulum (ER) is tethered to the mitochondria, have been involved in the pathogenesis of neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS). However, the detailed mechanisms through which MAM integrity is disrupted in ALS have not been fully elucidated. Here, we examined whether AAA ATPase domain-containing protein 3A (ATAD3A), a mitochondrial membrane AAA ATPase accumulating at the MAM, is involved in ALS. We found that sigma-1 receptor (σ1R), an ER-resident MAM protein causative for inherited juvenile ALS, required ATAD3A to maintain the MAM. In addition, σ1R retained ATAD3A as a monomer, which is associated with an inhibition of mitochondrial fragmentation. ATAD3A dimerization and mitochondrial fragmentation were significantly induced in σ1R-deficient or SOD1-linked ALS mouse spinal cords. Overall, these observations indicate that MAM induction by σ1R depends on ATAD3A and that σ1R maintains ATAD3A as a monomer to inhibit mitochondrial fragmentation. Our findings suggest that targeting σ1R–ATAD3A axis would be promising for a novel therapeutic strategy to treat mitochondrial dysfunction in neurological disorders, including ALS.
1. Introduction
Organelle contacts between the endoplasmic reticulum (ER) and mitochondria, known as the mitochondria-associated membranes (MAMs), are multifunctional platforms for maintaining cellular homeostasis, including cholesterol synthesis, Ca2+ transfer and autophagy induction (Fujimoto and Hayashi, 2011; Markovinovic et al., 2022; Vance, 2014). MAM integrity is generally compromised in amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative disease characterized by a selective loss of motor neurons (Paillusson et al., 2016). We previously reported that MAM disruption is a general pathomechanism in ALS based on studies employing ALS model mice and the MAM reporter system named MAMtrackers (Sakai et al., 2021; Watanabe et al., 2016). Impaired MAM integrity is involved in other neurological disorders, such as Alzheimer’s disease (AD) (Area-Gomez and Schon, 2017), Parkinson’s disease (Gomez-Suaga et al., 2018), and Huntington’s disease (HD) (Maity et al., 2022). Accumulating evidence has revealed that MAM integrity is essential for mitochondrial homeostasis, especially in mitochondrial dynamics such as fission and fusion. Various proteins associated with mitochondrial dynamics are accumulated at the MAM. For instance, mitofusin-2 (MFN2), which tethers the ER to mitochondrial outer membranes, interacts with apoptosis regulator protein Bax to promote mitochondrial fusion (Cerqua et al., 2010). Syntaxin-17 (STX17), a SNARE protein crucial for the fusion of autophagosomes and lysosomes, regulates mitochondrial division with dynamin-related protein 1 (Drp1) at the MAM (Arasaki et al., 2015). Moreover, mitochondrial fission protein 1 (Fis1), an essential factor of mitochondrial fission, is also accumulated at the MAM to form the ER–mitochondria tethering complex with B-cell receptor-associated protein 31 (BAP31) (Iwasawa et al., 2011). Mitochondrial fragmentation associated with mitochondrial dysfunction is observed in the various neurodegenerative diseases (Knott et al., 2008); thus, targeting MAM is a promising strategy to prevent mitochondrial dysfunction. (See Fig. 1, Fig. 2, Fig. 3, Fig. 4, Fig. 5, Fig.
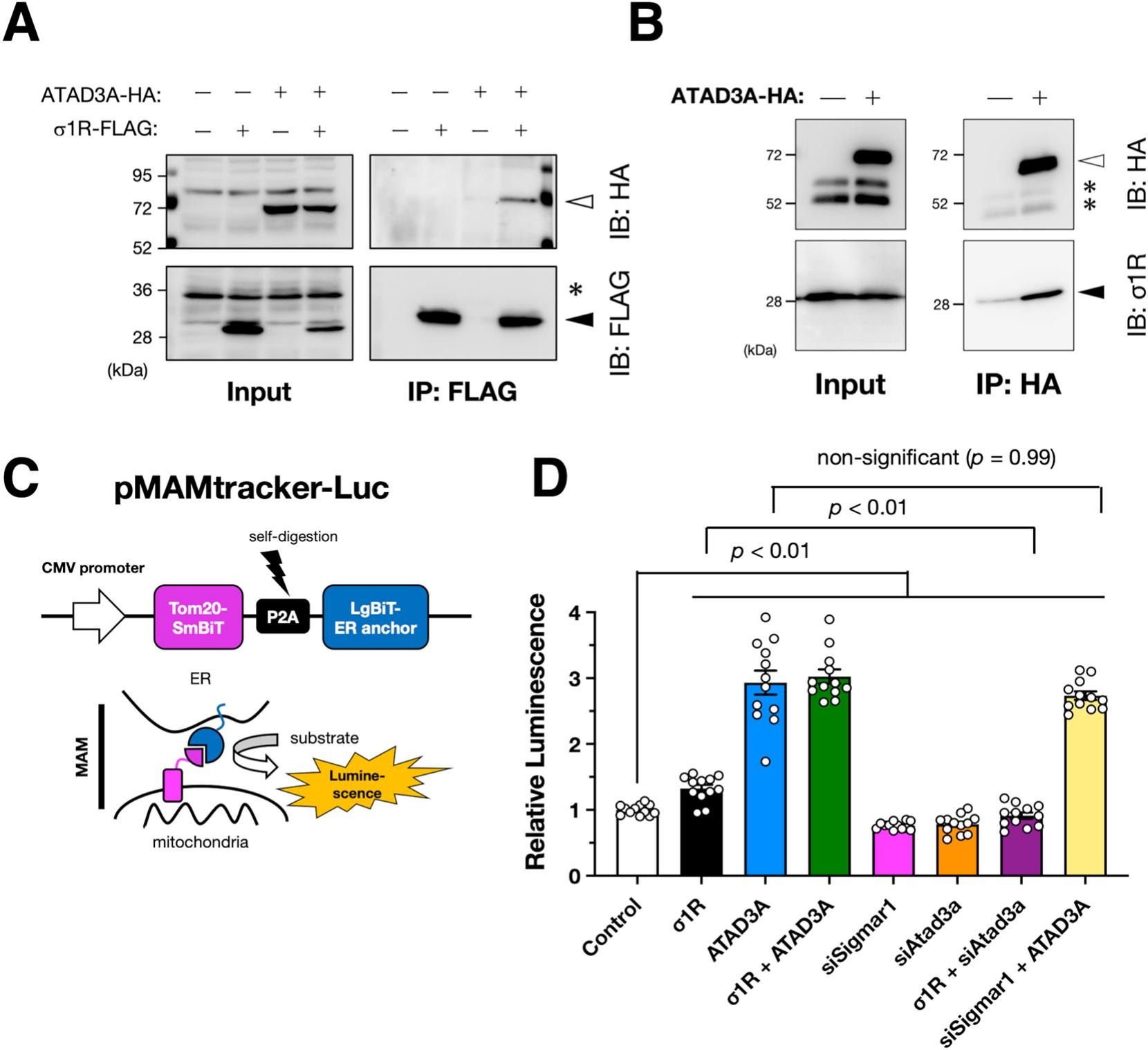
Sigma-1 receptor (σ1R) requires AAA ATPase domain-containing protein A3 (ATAD3A) to induce the mitochondria-associated membranes (MAM).
A and B) ATAD3A interacts with σ1R. HA-tagged human ATAD3A was co-immunoprecipitated with FLAG-tagged (A) or endogenous (B) human σ1R in human cervical carcinoma HeLa cells. Representative immunoblotting images are shown. Asterisks represent non-specific bands. Open and filled triangles indicate HA-tagged ATAD3A and σ1R, respectively. (C) Schematic illustration of our MAM reporter, MAMtracker-Luc. Bicistronically expressed compartments of Nanobit luciferase, smbit, and Lgbit, reconstitute its enzymatic activity at the MAM. (D) Relative normalized luminescence was measured using mouse neuroblastoma Neuro2a cells stably expressing MAMtracker-Luc (N2a・MAMLuc). The cells were transfected with small interfering RNAs targeting murine endogenous σ1R (siSigmar1) or ATAD3A (siAtad3a) or plasmids encoding FLAG-tagged human σ1R (σ1R) or HA-tagged human ATAD3A (ATAD3A). After transfection, the cells were incubated overnight before the measurement. Data from four independent experiments performed in triplicate are expressed as mean ± standard error of the mean (SEM) with p-values, analyzed by one-way ANOVA followed by multiple comparison tests with Sidak’s correction.
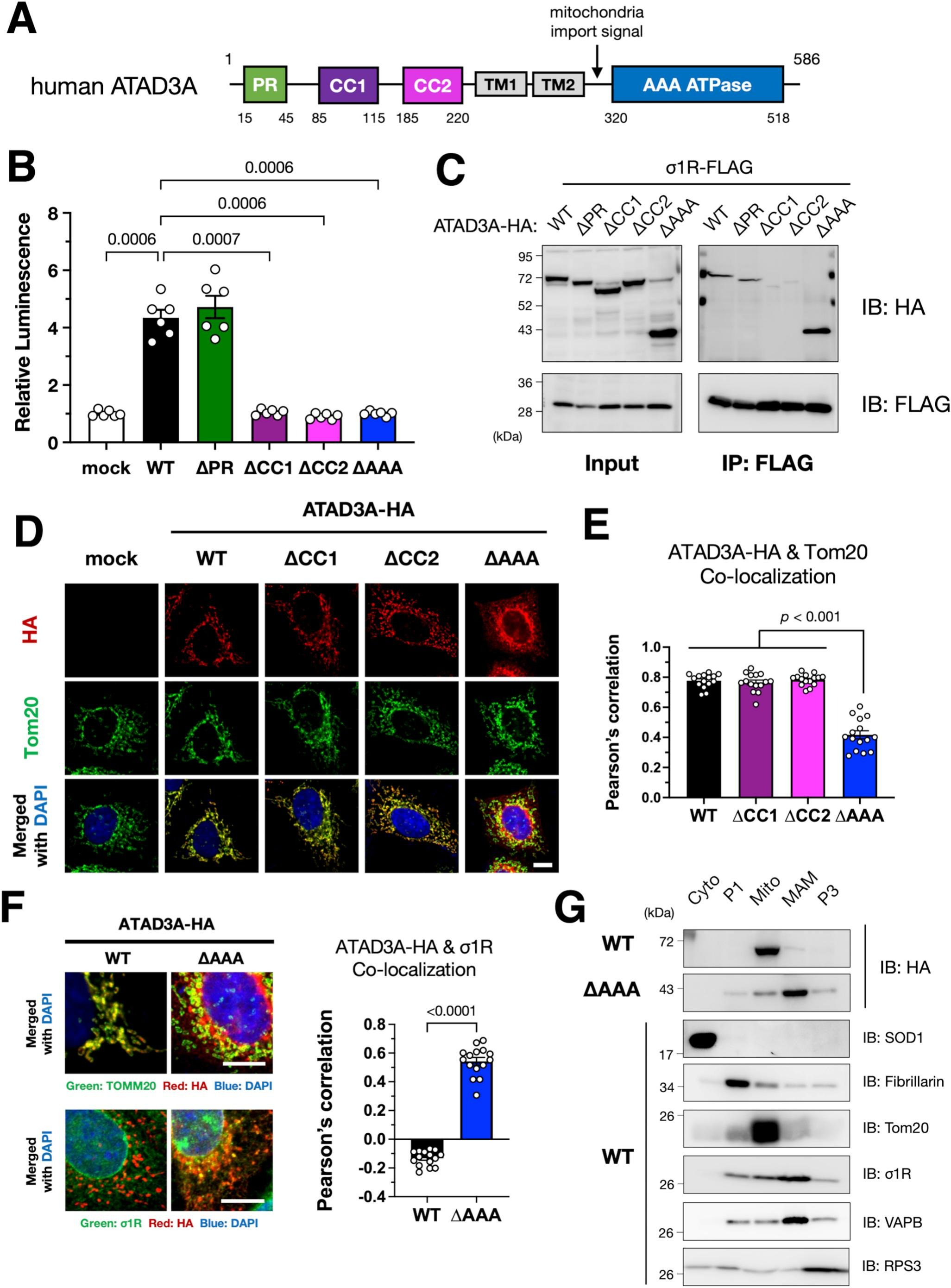
Identification of crucial domains of ATAD3A for the σ1R–ATAD3A interaction.
(A) Schematic illustration of human ATAD3A domains. Predicted functional domains, a proline-rich region (PR), coiled-coil domains (CC1/2), transmembrane domains (TM1/2), and an AAA ATPase domain (AAA) are indicated by colored rectangles. (B) CC1/2 and AAA domains are crucial for MAM induction by ATAD3A. Wild-type (WT) or mutants lacking each domain of ATAD3A were transiently expressed in N2a・MAMLuc cells. The relative luminescence was determined by normalizing to that in mock-transfected cells. Three independent experiments were performed in duplicate. Data are expressed as mean ± SEM with p-values, analyzed by one-way ANOVA followed by multiple comparison tests with Sidak’s correction. (C) CC1/2 domains of ATAD3A were crucial for the interaction with σ1R. Each HA-tagged ATAD3A deletion mutant was co-immunoprecipitated with FLAG-tagged σ1R from co-transfected Neuro2a cell lysates. The lysates and immunoprecipitates were analyzed by immunoblotting using the indicated antibodies. (D–G) AAA is essential for ATAD3A localization to the mitochondria in HeLa cells. ∆AAA mutants were not localized to the mitochondria in immunocytochemistry (D–F) and subcellular fractionation (G) but localized to the MAM (F and G). HeLa cells expressing WT or mutant ATAD3A were immunostained using the indicated antibodies. Representative confocal images (D and F, left panel) and Pearson’s correlation co-efficiency (E and F, right panel) were shown. HeLa cells expressing WT or ∆AAA ATAD3A were fractionated as described in the Methods section (Cyto, cytoplasm; P1, nuclei, and debris; Mito, mitochondria; MAM, mitochondria-associated membrane; P3, microsomal fraction). Each fraction was subject to immunoblotting (G). Scale bars: 10 μm.
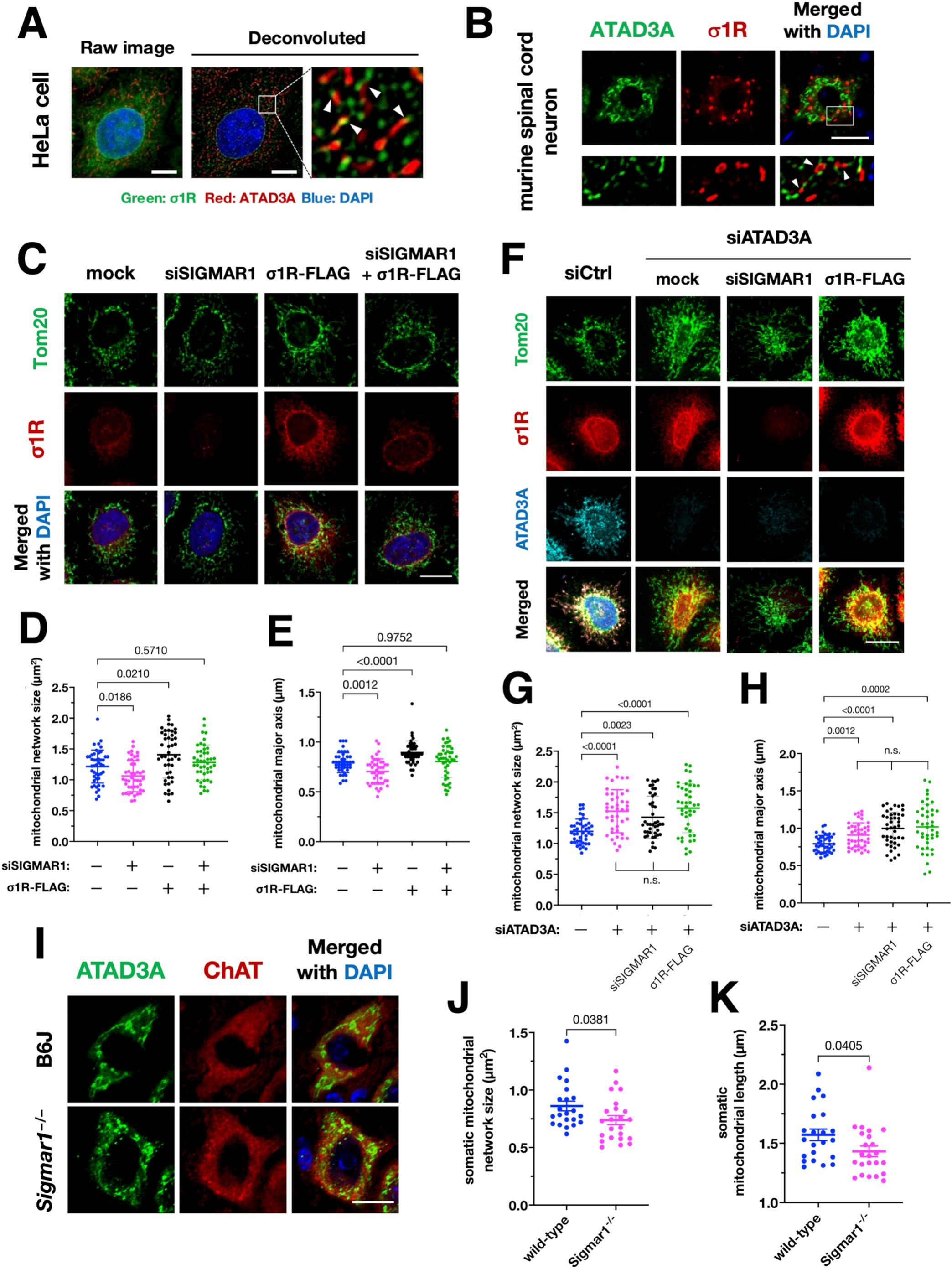
Impairment of the σ1R–ATAD3A interaction is associated with mitochondrial fragmentation.
(A and B) σ1R localized at edges of the mitochondria in HeLa cells (A) or mouse spinal cord neurons (B). Representative deconvoluted (A, right) or confocal (B) images of HeLa cells or mouse neurons stained with antibodies against ATAD3A and σ1R are shown, respectively. The mitochondria were visualized by endogenous ATAD3A. The lower panels in (B) show enlarged images of the area surrounded by the white rectangle in the top panel. Arrowheads indicate σ1R localization at the edges of the mitochondria. Scale bars: 5 μm (A) and 20 μm (B). (C–E) σ1R promoted mitochondrial elongation in HeLa cells. Representative confocal images of HeLa cells stained with the antibodies against TOMM20 are shown (C). The size (D) and major axis length (E) of the mitochondria were measured to quantify mitochondrial morphology. Scale bar: 10 μm. (F–H) σ1R-dependent mitochondrial elongation required ATAD3A in HeLa cells. Representative confocal images of HeLa cells stained with the antibodies against TOMM20, σ1R, and ATAD3A are shown in (F). The size (G) and major axis length (H) of the mitochondria were measured to quantify mitochondrial morphology. Note that neither σ1R depletion nor overexpression affected mitochondrial morphology when ATAD3A was depleted. Scale bar: 10 μm. (I–K) σ1R deficiency was associated with mitochondrial fragmentation in the motor neurons in 6 months-old mouse spinal cords. Representative confocal images of mitochondria in the ChAT-positive lumbar motor neurons of wild-type (B6J) and Sigmar1−/−− mice visualized by endogenous ATAD3A (I). The size (J) and major axis length (K) of mitochondria were measured to quantify mitochondrial morphology. All the data are expressed as mean ± SEM with p-values, analyzed by one-way ANOVA followed by multiple comparison tests with Sidak’s correction for three or more groups and Welch’s t-tests for two groups. Scale bar: 20 μm.
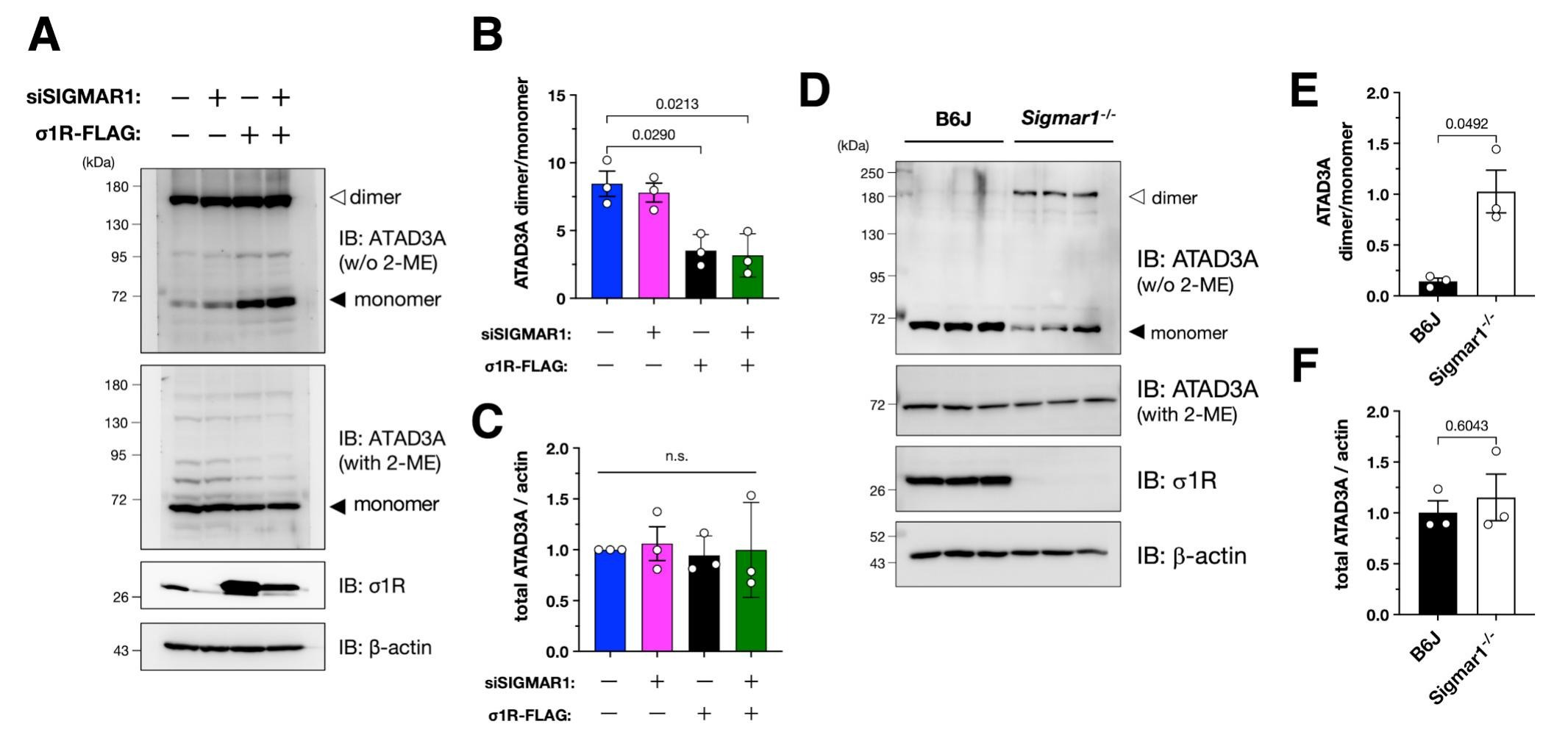
σ1R reduces the levels of ATAD3A oxidative dimerization.
(A–C) σ1R promoted ATAD3A monomerization in HeLa cells. Representative immunoblotting images are shown in (A). The normalized ATAD3A dimer per monomer (B) or total ATAD3A (C) levels were quantified. Data from three independent experiments are expressed as mean ± SEM with p-values, analyzed by one-way ANOVA followed by multiple comparison tests with Sidak’s correction. (D–F) σ1R deficiency induced ATAD3A oxidized dimerization in mouse spinal cords. Representative immunoblotting images of 6 months-old wild-type or Sigmar1−/− mouse spinal cord lysates are shown in (D). Relative ATAD3A dimer/monomer ratio (E) and total ATAD3A levels (F) were quantified. Data are expressed as mean ± SEM with p-values (n = 3 per group), analyzed by one-way ANOVA followed by Welch’s t-tests.
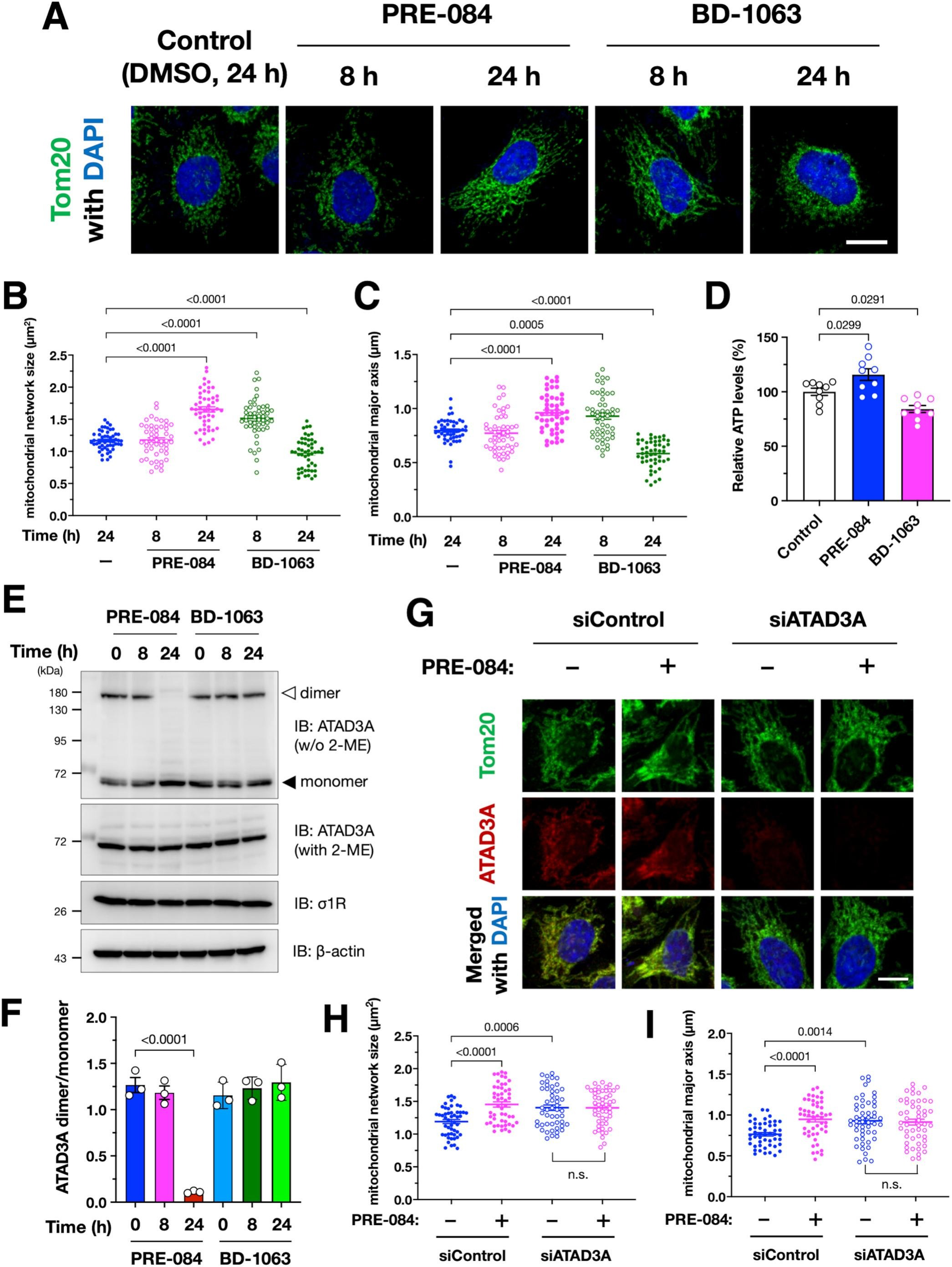
Pharmacological activation of σ1R induces mitochondrial elongation dependent on ATAD3A.
(A–C) Pharmacological activation or inactivation of σ1R using its specific agonist, PRE-084, or antagonist, BD-1063, alters mitochondrial morphology. Representative immunofluorescent images of HeLa cells are shown in (A). Mitochondrial size or major axis length was quantified in a total of 50 cells from three independent experiments are shown in (B) and (C), respectively. (D) Intracellular ATP levels were increased by PRE-084 treatment but decreased by BD-1063 treatment. Relative intracellular ATP levels were quantified using a cellular ATP measurement kit (TOYO Benet). (E and F) PRE-084 reduced ATAD3A dimer/monomer ratio, but BD-1063 did not affect the ATAD3A dimer/monomer state. Representative immunoblotting images are shown in (E) with arrowheads indicating ATAD3A dimer (open triangle) and monomer (closed triangle). ATAD3A dimer/monomer ratio quantified in three independent experiments is shown in (F). (G–H) ATAD3A is required for PRE-084-induced mitochondrial elongation. Representative immunofluorescent images of Hela cells are shown in (G). The cells were incubated in the presence or absence of 10 μM PRE-084 for 24 h. Mitochondrial size or major axis length was quantified in a total of 50 cells from three independent experiments are shown in (H) and (I), respectively. Note that PRE-084 failed to induce mitochondrial elongation when ATAD3A was suppressed by siRNA. All the data are expressed as mean ± SEM with p-values, analyzed by one-way ANOVA followed by Welch’s t-tests. n.s.: non-significant (p > 0.05). Scale bars: 5 μm.
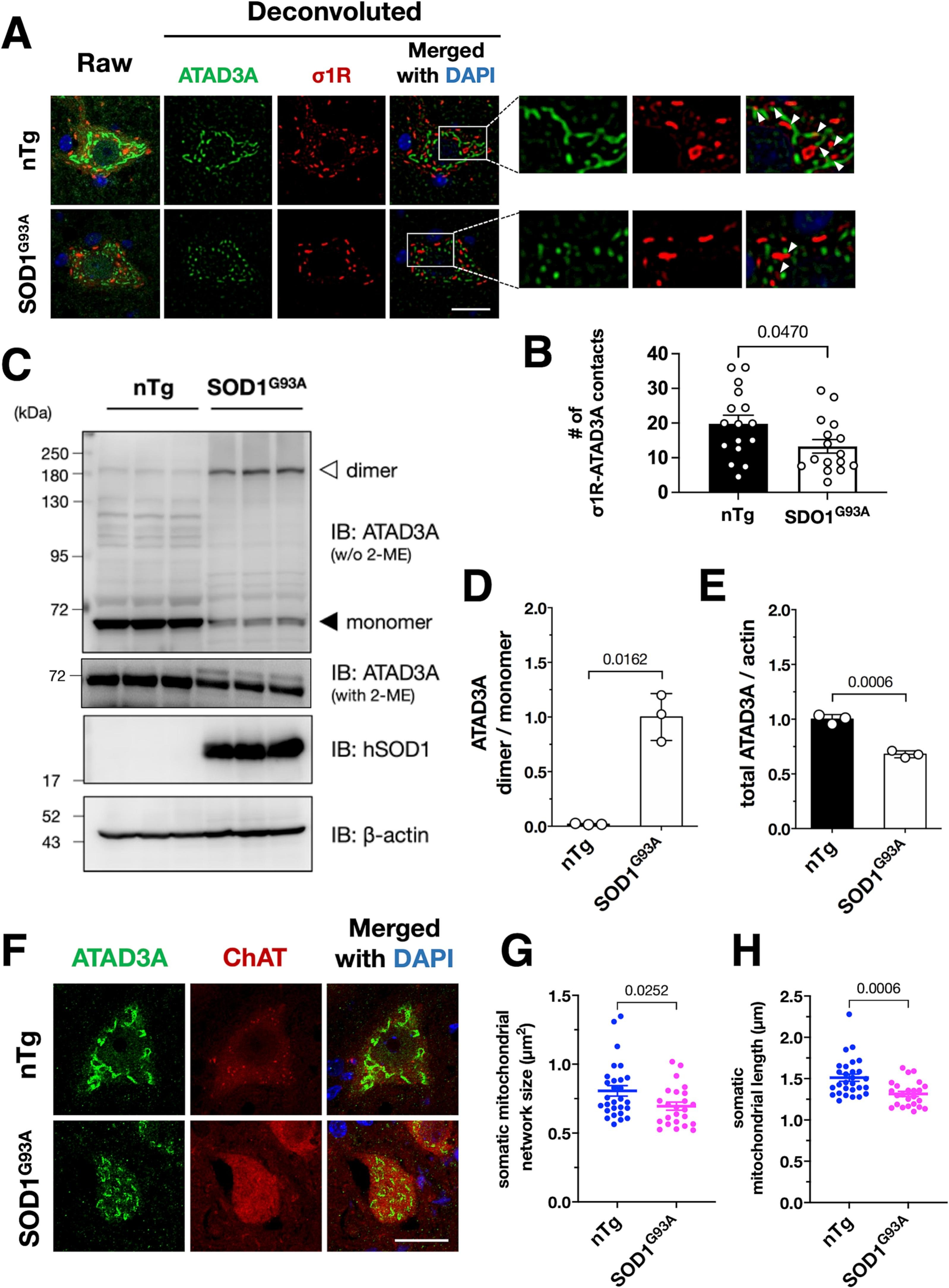
ATAD3A oxidative dimerization and mitochondrial fragmentation are induced in SOD1G93A transgenic mice.
(A and B) The σ1R–ATAD3A interaction was disrupted in SOD1G93A mouse spinal cord neurons at the end-stage. Five months-old wild-type mice (nTg) were used as the control. The right panels in (A) show enlarged images surrounded by the white rectangles in the left panel. Arrowheads indicate contacts of σ1R and ATAD3A, which were disrupted in SOD1G93A mice. The number of σ1R–ATAD3A contacts in at least five motor neurons from three mice per genotype were quantified and plotted as the mean ± SEM with p-value, analyzed by one-way ANOVA followed by Welch’s t-tests (B). The number of contacts were normalized by total mitochondrial network size indicated by ATAD3A in each neuron. (C–E) ATAD3A oxidative dimers were induced in SOD1G93A mouse spinal cords. Representative immunoblotting images of 5 months-old wild-type or end-stage SOD1G93A mouse spinal cord lysates are shown in (C). The normalized ATAD3A dimer/monomer ratio (D) or total ATAD3A levels (E) were quantified. Data from three mice are expressed as mean ± SEM with p-values, analyzed by one-way ANOVA followed by Welch’s t-tests. (F–H) Mitochondrial fragmentation was induced in the lumbar motor neurons of SOD1G93A mice. Representative confocal images of mitochondria in the ChAT-positive lumbar motor neurons of nTg and SOD1G93A mice visualized by ATAD3A staining (F). The size (G) and major axis length (H) of mitochondria were measured to quantify mitochondrial morphology. Data are expressed as mean ± SEM with p-values, analyzed by one-way ANOVA followed by multiple comparison tests with Welch’s t-tests. Scale bar: 20 μm.
Sigma-1 receptor (σ1R) is a MAM-specific chaperone protein. Σ1R is best known as a regulator of Ca2+ transfer from the ER to the mitochondria, which is mediated by inositol triphosphate receptor type 3 (IP3R3) (Hayashi and Su, 2007). It has been reported, including ours, that the loss of σ1R function is causative of an inherited form of juvenile ALS (ALS16) (Al-Saif et al., 2011; Watanabe et al., 2016). Specifically, we found that σ1R deficiency induced mislocalization of IP3R3 from the MAM, resulting in reduced Ca2+supply into mitochondria and overflow of cytoplasmic Ca2+ to accelerate disease onset in ALS model mice (Watanabe et al., 2016). In addition, σ1R aggregation, leading to the loss-of-function of σ1R, was observed in the motor neurons of sporadic ALS cases (Prause et al., 2013). Furthermore, σ1R agonist administration extended the survival times of ALS model mice (Mancuso et al., 2012; Ono et al., 2013). These studies demonstrate that σ1R is essential for MAM integrity and that σ1R deficiency is closely associated with ALS pathogenesis.
On the other hand, recent studies revealed that another MAM-associated protein, AAA ATPase domain-containing protein A3 (ATAD3A), is involved in the pathogenesis of neurological diseases such as AD (Zhao et al., 2022) and HD (Zhao et al., 2019). ATAD3A is a nuclear-encoded mitochondrial membrane protein that accumulates at the MAM (Gilquin et al., 2010). ATAD3A reportedly contributes to maintaining mitochondrial DNA stability and supports mitochondrial protein synthesis (He et al., 2012; He et al., 2007; Peralta et al., 2018). Moreover, ATAD3A scaffolds mitochondrial inner membrane structures, including OXPHOS complexes (Arguello et al., 2021). An increased level of oxidative ATAD3A dimer induces Drp1 translocation, leading to mitochondrial fission, and inhibition of Drp1 binding to ATAD3A dimer ameliorates mitochondrial dysfunction in HD model cells or mice (Zhao et al., 2019). Similar observations were also confirmed in AD (Zhao et al., 2022). In these neurodegenerative diseases, mitochondrial fragmentation related to mitochondrial dysfunction was associated with oxidative ATAD3A dimerization. Preventing mitochondrial fragmentation induced by the ATAD3A–Drp1 axis is a potential therapeutic strategy for AD and HD. However, the role of ATAD3A in ALS pathogenesis has yet to be uncovered.
Thus, in this study, we aimed to determine whether the deregulation of ATAD3A dimer/monomer state is involved in ALS pathology and is associated with the loss of σ1R function that causes inherited ALS. Using our MAM reporter system, MAMtracker-Luc, we identified ATAD3A as a novel inducer of the MAM that interacts with σ1R. Indeed, ATAD3A was crucial for MAM induction by σ1R. Moreover, σ1R retained ATAD3A as a monomer, which was associated with mitochondrial fusion in cultured cells. Conversely, in Sigmar1 (a gene coding σ1R)-knockout (Sigmar1−/−) or ALS-linked mutant SOD1 transgenic mice, oxidative ATAD3A dimerization was markedly induced with mitochondrial fragmentation in the spinal cord. These findings indicate that σ1R tethers the ER to the mitochondria by interacting with ATAD3A and maintains mitochondrial function by preventing oxidative ATAD3A dimerization, suggesting the role of σ1R–ATAD3A axis at the MAM in maintaining homeostasis in motor neurons.
…
3. Results
3.1. ATAD3A is crucial for σ1R-mediated MAM induction
First, to examine whether σ1R and ATAD3A interact with each other, we performed a co-immunoprecipitation assay using mouse neuroblastoma Neuro2a cells. ATAD3A bound to both overexpressed and endogenous σ1R (Fig. 1 A & B), suggesting that σ1R and ATAD3A contribute tethering of the ER and mitochondria, which leads to MAM formation. We also tried to examine the interaction between endogenous σ1R and ATAD3A; however, we were not able to immunoprecipitate enough amount of target proteins by anti-σ1R antibody used in this study (Supplementary Fig. S3), possibly due to its low efficiency of the immunoprecipitation. In our previous study, we developed a split luciferase-based MAM reporter, MAMtracker-Luc, which enables high-throughput quantification of the MAM in living cells (Fig. 1C) (Sakai et al., 2021). The two components of luciferase are anchored to mitochondrial or ER membranes, respectively, and complimented when the ER and mitochondria are in proximity (< 200 nm) at the MAM. Thus, using Neuro2a cells stably expressing MAMtracker-Luc, we examined whether σ1R and ATAD3A require each other for MAM induction. As shown in Fig. 1D, σ1R overexpression induced the MAM, which was consistent with previous studies, including ours (Prause et al., 2013; Sakai et al., 2021; Watanabe et al., 2016). Similarly, ATAD3A overexpression induced the MAM. However, the impact on MAM induction was much greater for ATAD3A [1.32 (σ1R) vs. 2.93 (ATAD3A), relative to mock control]. Conversely, depletion of either σ1R or ATAD3A resulted in MAM reduction [0.762 (siSigmar1) and 0.783 (siAtad3a), respectively]. As expected, depletion of ATAD3A prevented MAM induction by σ1R overexpression. However, ATAD3A overexpression induced the MAM even when σ1R was depleted. Moreover, overexpression of both σ1R and ATAD3A did not further induction of the MAM. These findings indicate that ATAD3A can form MAM independent of σ1R. Indeed, ATAD3A reportedly interacts with another MAM-associated ER membrane protein, Wiskott-Aldrich syndrome protein family member 3 (WASF3) (Teng et al., 2016); therefore, for MAM induction, σ1R requires ATAD3A, but ATAD3A does not require σ1R.
3.2. Coiled-coil domains of ATAD3A are essential for MAM induction and interaction with σ1R
ATAD3A is composed of four putative functional domains, two transmembrane regions, and a mitochondrial targeting signal (Fig. 2A) (Gilquin et al., 2010; Zhao et al., 2019). To determine which ATAD3A domain is crucial for MAM induction, we prepared ATAD3A domain deletion mutants. Except for deletion of proline-rich region (PR), deletion of either three other domains, namely coiled-coil domain 1 (CC1), coiled-coil domain 2 (CC2), or AAA ATPase domain (AAA), failed to increase the MAM (Fig. 2B). CC1 or CC2 deletion mutant, i.e., ∆CC1 or ∆CC2, was not co-immunoprecipitated with σ1R (Fig. 2C), indicating that CC1 and CC2 are essential for interacting with σ1R. On the other hand, AAA lacking (∆AAA) mutant maintained its ability to interact with σ1R, indicating that mitochondrial localization of ATAD3A is not necessary for the interaction with σ1R. We confirmed that ∆AAA mutant was not able to localize at the mitochondria as previously reported (Gilquin et al., 2010), whereas ∆CC1 and ∆CC2 mutants retained their mitochondrial localization (Fig. 2D and E). Consistent with our co-immunoprecipitation results, ∆AAA mutant colocalized with σ1R (Fig. 2F). Furthermore, ∆AAA mutant was mainly fractionated into the MAM fraction, whereas wild-type ATAD3A was predominantly fractionated into the mitochondrial fraction (Fig. 2G). These observations suggest that ∆AAA mutant is unable to induce the MAM, at least partially, due to a loss of its proper localization at mitochondrial membranes.
3.3. σ1R and ATAD3A are associated with mitochondrial fragmentation in vitro and in vivo
ATAD3A did not require σ1R for MAM induction, implying that the σ1R and ATAD3A may play another physiological role. In human cervical carcinoma HeLa cells, σ1R partially colocalized with ATAD3A at edges of the mitochondria (Fig. 3A). Similarly, σ1R made contact with the edges of the mitochondria, indicated by ATAD3A, in murine spinal cord neurons (Fig. 3B). VAPB, another MAM-related protein (De Vos et al., 2012), also showed close contact with ATAD3A (Supplementary Fig. S4). These findings led us to hypothesize that the σ1R and ATAD3A regulates mitochondrial fission and/or fusion. Thus, in HeLa cells with depletion or overexpression of σ1R, we quantified the size and major axis of the mitochondria. The size and major axis of the mitochondria were significantly decreased in σ1R-depleted HeLa cells (Fig. 3C–E). Conversely, σ1R overexpression increased the size and major axis of the mitochondria. In HeLa cells depleted ATAD3A, the size and major axis of the mitochondria were increased. However, under the ATAD3A depleted conditions, altering the level of σ1R did not affect mitochondrial morphology (Fig. 3F–H). Mitochondrial fragmentation determined by a decrease in the size and length of somatic mitochondria was also observed in the spinal motor neurons of Sigmar1−/−mice (Fig. 3I–K). Collectively, these results indicate that σ1R and ATAD3A interaction is associated with mitochondrial fragmentation.
3.4. σ1R prevents ATAD3A oxidative dimerization to inhibit mitochondrial fragmentation
Given that oxidative ATAD3A dimerization reportedly induces mitochondrial fragmentation via Drp1 recruitment into the mitochondria (Zhao et al., 2022; Zhao et al., 2019), we hypothesized that σ1R prevents ATAD3A dimerization to inhibit mitochondrial fragmentation. To test this hypothesis, we analyzed the dimerization state of ATAD3A using immunoblotting, as previously reported (Zhao et al., 2019). As shown in Fig. 4A and B, ATAD3A monomer was induced in σ1R-overexpressed HeLa cells. Because the oxidative ATAD3A dimer was dominant in HeLa cells, no further increase of the oxidative ATAD3A dimer by σ1R depletion was observed. In contrast to our observations in HeLa cells, oxidative ATAD3A dimerization was almost absent in mouse spinal cord lysates (Fig. 4D), and σ1R deficiency markedly induced oxidative ATAD3A dimerization (Fig. 4E). Total ATAD3A levels were not altered in either HeLa cells or mouse spinal cord lysates (Fig. 4C and F), suggesting that σ1R did not affect total levels of ATAD3A. Taken together, these results indicate that σ1R maintains ATAD3A as a monomer to inhibit mitochondrial fragmentation.
3.5. Pharmacological activation of σ1R maintained ATAD3A as a monomer, which is associated with mitochondrial elongation
Next, we examined whether the pharmacological activation or inactivation of σ1R affects the ATAD3A dimerization state and mitochondrial morphology. PRE-084, an agonist of σ1R, induced mitochondrial elongation after 24 h of the treatment (Fig.5A–C). On the other hand, BD-1063, an antagonist of σ1R, temporarily elongated the mitochondrial length, which is consistent with the previous report (Bernard-Marissal et al., 2015), but then resulted in mitochondrial fragmentation (Fig.5A–C). Moreover, PRE-084 increased intracellular ATP levels, whereas BD-1063 decreased (Fig. 5D). Although PRE-084 also affects glycolysis (Motawe et al., 2020), this observation implies the possibility that PRE-084-induced mitochondrial elongation is associated with increased ATP synthesis in the mitochondria. ATAD3A dimer was apparently decreased after 24 h of the PRE-084 treatment, whereas BD-1063 did not affect the ATAD3A dimer/monomer ratio at all (Fig.. 5E and F). Consistent with the above observations, ATAD3A knock-down prevented the mitochondrial elongation induced by PRE-084 (Fig. 5G–I), suggesting that the pharmacological activation of σ1R leads to mitochondrial elongation via ATAD3A. In contrast, BD-1063-induced mitochondrial fragmentation was independent of ATAD3A (Supplementary Fig. S5). Thus, the pharmacological activation of σ1R maintained ATAD3A as a monomer, which is associated with mitochondrial elongation, whereas the pharmacological inactivation of σ1R led to fragmentation of the mitochondria independent of ATAD3A.
3.6. ATAD3A monomerization and localization in mitochondria are decreased in SOD1G93A mice
In our previous studies, we revealed that the MAM is generally disrupted in inherited ALS (Sakai et al., 2021; Watanabe et al., 2016). Therefore, we investigated whether the disrupted interaction between σ1R and ATAD3A was involved in mitochondrial alternation observed in ALS. In the spinal motor neurons of SOD1G93A ALS model mice, we found that the number of σ1R and ATAD3A contacts was decreased (Fig. 6A and B). The levels of the oxidative ATAD3A dimer were also increased (Fig. 6C and D). Simultaneously, the total level of ATAD3A was decreased in SOD1G93A mouse motor neurons (Fig. 6A, C and E). Similar to the Sigmar1−/− mice, the mitochondria, indicated by ATAD3A, were highly fragmented (Fig. 6F–H) in SOD1G93A mice. These findings are well consistent with our observations in Sigmar1−/− mice, suggesting that ATAD3A dysregulation is associated with mitochondrial fragmentation in ALS.
4. Discussion
In the present study, we identified ATAD3A as a novel interactor of σ1R, which is essential for σ1R-dependent MAM induction. Although ATAD3A induced the MAM without σ1R, a deficit of the σ1R–ATAD3A interaction was associated with mitochondrial fragmentation and oxidative ATAD3A dimer formation. Collectively, our findings indicate that σ1R contributes to the homeostasis of the MAM and mitochondria via its interaction with ATAD3A.
σ1R is reportedly crucial for MAM integrity (Fujimoto et al., 2012; Su et al., 2010; Weng et al., 2017). Our findings indicate that σ1R induces tethering of the ER to the mitochondria with the help of ATAD3A; however, because of the limitation of our co-immunoprecipitation studies, we cannot address whether σ1R directly binds to ATAD3A. Recent studies have proposed that σ1R has a topology oriented toward the ER lumen (Lee et al., 2016; Sharma et al., 2021). Thus, it seems reasonable that σ1R and ATAD3A do not directly interact with each other. Because σ1R binds to IP3R3 and stabilizes the complex formed with mitochondrial voltage-dependent anion channel 1 (VDAC1) and glucose-regulated protein 75 (GRP-75) (Szabadkai et al., 2006), stabilizing the complex including ATAD3A to tether the ER and mitochondria would be a possible mechanism of the σ1R-mediated MAM induction. Proteomic analyses would help to identify the molecule on the ER that is responsible for forming σ1R–ATAD3A complex in the future. Despite such a limitation of our study, the σ1R–ATAD3A interaction is an interesting mechanism underlying MAM induction. As we have previously reported, in the central nervous system, IP3R3 expression is strictly limited to motor neurons in the brain stem and spinal cord (Watanabe et al., 2016). In contrast, σ1R and ATAD3A are expressed throughout the brain. Thus, the σ1R may also contribute to the maintenance of the MAM in neurons lacking IP3R3 via ATAD3A.
To date, various sets of proteins are known to tether the ER and mitochondria, including vesicle-associated membrane protein-associated protein B (VAPB)–regulator of microtubule dynamics protein 3 (RMDN3/PTPIP51) (Stoica et al., 2014), mitofusin-2 (Mfn2) (de Brito and Scorrano, 2009), B-cell receptor-associated protein 31 (BAP31)–mitochondrial fission protein 1 (FIS1) (Iwasawa et al., 2011), and PDZ domain-containing protein 8 (Hirabayashi et al., 2017). The σ1R and ATAD3A form a novel set of proteins comprising a MAM tethering complex. Interestingly, the loss of either σ1R or ATAD3A affects only about 30% of the total MAM, suggesting that the MAM is an extremely diverse intracellular structure. This notion is supported by our previous electron microscopy observations, which revealed that the MAM is only partially reduced in Sigmar1−/− or ALS model mice (Watanabe et al., 2016). Future studies focused on the MAM diversity might contribute to revealing specific physiological roles of the MAM dependent on each tethering complex.
Previously, the unfolded PR domain of ATAD3A was predicted to be responsible for interaction at the MAM (Baudier, 2018); however, we found that CC1/2 domains rather than PR domain were required for interaction with the ER and MAM induction. It is still unclear whether the CC1/2 domains of ATAD3A are really located on mitochondrial outer membranes. According to some previous studies, ATAD3A N-terminal domains are localized in intermembrane spaces (Arguello et al., 2021; Baudier, 2018); however, our finding that ∆AAA mutant is localized at the MAM suggests that ATAD3A N-terminal domains can directly bind to ER proteins. Given that ATAD3A N-terminal domain locates in intermembrane spaces when ATAD3A forms an oxidative dimer (Zhao et al., 2019), one possible interpretation is that ATAD3A alters its conformations and/or localization depending on its oligomerization status or interacting partners.
Members of the AAA ATPase family are known to form hexameric ring complexes (Miller and Enemark, 2016) and ATAD3A has been shown to oligomerize without any oxidation, which is crucial for the physiological function of ATAD3A (Miller and Enemark, 2016; Peralta et al., 2018). Conversely, oxidative ATAD3A dimerization is reportedly involved in HD (Zhao et al., 2019) and AD (Zhao et al., 2022), which leads to mitochondrial dysfunction. Mitochondrial impairment has also been documented in various ALS models (Smith et al., 2019). Indeed, in our previous study, MAM disruption decreased ATP supply from mitochondria (Watanabe et al., 2016). Mitochondrial fragmentation is also a pathological feature observed in ALS motor neurons (Magrané et al., 2014; Vinsant et al., 2013). Moreover, mitochondrial fragmentation is observed in various neurodegenerative diseases, including AD and HD (Knott et al., 2008). Therefore, the prevention of mitochondrial fragmentation is a possible protective mechanism.
Although the MAM is important for mitochondrial homeostasis, especially in mitochondrial dynamics of fission and fusion, and various proteins associated with the mitochondrial division are accumulated at the MAM, it is unclear how MAM alteration affects mitochondrial function and morphology. In this study, we found that MAM induction by σ1R–ATAD3A axis prevented mitochondrial fragmentation, and σ1R deficit led to MAM deficiency and was associated with mitochondrial fragmentation, which is observed in ALS models. On the other hand, mitochondria are also reportedly fragmented in AD models (Medala et al., 2021), in which the MAM is excessively induced (Area-Gomez et al., 2012). One possible interpretation is that the impact of MAM induction on mitochondrial morphology is dependent on the molecules involved. For example, BAP31 prevents Fis1-dependent Drp1 recruitment (Iwasawa et al., 2011); thus, Fis1 and BAP31-mediated MAM formation prevents mitochondrial fission. On the other hand, STX17-associated MAM promotes mitochondrial fission by recruiting Drp1 (Arasaki et al., 2015). In the case of ATAD3A, ATAD3A dimer induces Drp1 recruitment (Zhao et al., 2019); that is, ATAD3A dimer at the MAM promotes mitochondrial fragmentation. In our observations, σ1R, which induced the MAM, decreased the dimer/monomer ratio of ATAD3A; thus, MAM induction by σ1R–ATAD3A axis promoted mitochondrial elongation rather than fragmentation. Therefore, it is likely that mitochondrial fragmentation in various neurodegenerative diseases is differently associated with MAM alternations specific to each disease.
Another question is how σ1R prevents ATAD3A dimerization. Because the MAM, including σ1R-containing MAM, regulates Ca2+ transfer into the mitochondria (Raeisossadati and Ferrari, 2020; Rizzuto et al., 2012), the production of reactive oxygen species (ROS) due to excess Ca2+ loading may cause ATAD3A oxidation. Indeed, in AD models, ATAD3A dimerization was reportedly increased (Zhao et al., 2022). Pharmacological inhibition of σ1R increases mitochondrial size possibly due to decreased Ca2+ loading to the mitochondria (Bernard-Marissal et al., 2015). We confirmed that the mitochondrial size was transiently increased by σ1R inhibition using BD-1063. Since BD-1063 did not affect the dimer/monomer state of ATAD3A, the transient mitochondrial elongation by σ1R inhibition is independent of ATAD3A dimerization. On the other hand, σ1R activation, which increases Ca2+ loading to the mitochondria (Watanabe et al., 2016), was associated with ATAD3A monomerization in the present study. Although it remains to be elucidated how σ1R retains ATAD3A as a monomer against the increased ROS, interaction of ATAD3A with unidentified ER proteins mediated by σ1R may block the target site, K135 in CC1 domain (Zhao et al., 2019), of ATAD3A oxidation from ROS.
Finally, accumulating evidence, including ours, shows that both σ1R and ATAD3A are critical factors of the MAM to maintain mitochondrial homeostasis, which are impaired in neurological diseases. Targeting σ1R–ATAD3A axis at the MAM to prevent mitochondrial dysfunction would lead to development of a novel therapeutic strategy for neurodegenerative diseases.