
Sigma-1 receptor agonism exacerbates immune-driven nociception: Role of TRPV1 + nociceptors

By M. Carmen Ruiz-Cantero, Miguel Á. Huerta, Miguel Á. Tejada, Miriam Santos-Caballero, Eduardo Fernández-Segura, Francisco J. Cañizares, José M. Entrena, José M. Baeyens, and Enrique J. Cobos
Excerpt from the article published in Biomedicine & Pharmacotherapy, Volume 167, November 2023,115534, ISSN 0753-3322, DOI: https://doi.org/10.1016/j.biopha.2023.115534.
Highlights
- Sigma-1 agonism potentiates PGE2-induced hyperalgesia.
- Sigma-1 agonism enhances pain responses after plantar incision–induced inflammation.
- These responses are due to an increase in the proalgesic actions of neutrophils.
- The proalgesic actions of sigma-1 agonists are mediated by TRPV1 + neurons.
- The sigma-1 receptor is present in all mouse and human DRG neurons.
Abstract
The analgesic effects of sigma-1 antagonists are undisputed, but the effects of sigma-1 agonists on pain are not well studied. Here, we used a mouse model to show that the administration of the sigma-1 agonists dextromethorphan (a widely used antitussive drug), PRE-084 (a standard sigma-1 ligand), and pridopidine (a selective drug being investigated in clinical trials for the treatment of neurodegenerative diseases) enhances PGE2-induced mechanical hyperalgesia. Superficial plantar incision induced transient weight-bearing asymmetry at early time points, but the mice appeared to recover at 24 h, despite noticeable edema and infiltration of neutrophils (a well-known cellular source of PGE2) at the injured site. Sigma-1 agonists induced a relapse of weight bearing asymmetry in a manner dependent on the presence of neutrophils. The effects of sigma-1 agonists were all reversed by administration of the sigma-1 antagonist BD-1063 in wild-type mice, and were absent in sigma-1 knockout mice, supporting the selectivity of the effects observed. The proalgesic effects of sigma-1 agonism were also abolished by the TRP antagonist ruthenium red and by in vivo resiniferatoxin ablation of TRPV1 + peripheral sensory neurons. Therefore, sigma-1 agonism exacerbates pain-like responses in mice with a mild inflammatory state through the action of TRPV1 + nociceptors. We also show that sigma-1 receptors are present in most (if not all) mouse and human DRG neurons. If our findings translate to humans, further studies will be needed to investigate potential proalgesic effects induced by sigma-1 agonism in patients treated with sigma-1 agonists.
1. Introduction
The sigma-1 receptor is a Ca2+-sensing and ligand-operated chaperone that modulates several receptors and ion channels [1], [2], [3]. Both sigma-1 agonists and antagonists may have therapeutic utility. Sigma-1 agonists, for instance, have antitussive properties [4]. Dextromethorphan, a classic sigma-1 agonist [5], is a widely used over-the-counter cough suppressant approved by the FDA in 1958 [6]. It has a variety of pharmacological activities in addition to sigma-1 agonism, including NMDA antagonism [6], and it is believed to be a preferentially central-acting cough suppressant [7]. There are other more selective sigma-1 agonists, including PRE-084 and pridopidine. PRE-084 is widely used in preclinical research as a prototypic sigma-1 agonist [5], [8], whereas pridopidine is currently being tested in phase III clinical trials for two central neurodegenerative diseases: Huntington’s disease [9] and amyotrophic lateral sclerosis (ALS) [10].
Prototypic sigma-1 antagonists include BD-1063 and S1RA [1], [5]. S1RA has shown promising results for pain treatment in phase II clinical trials [11]. Early studies (e.g. [12]) showed that sigma-1 antagonism enhances central opioid antinociception, while later studies found it to decrease central sensitization [3], [13] (amplification of neural signaling in the spinal cord), which is of pivotal importance for the development of chronic pain [14]. Several preclinical studies have shown that sigma-1 antagonism decreases sensory hypersensitivity in chronic pain conditions, such as neuropathy, inflammation, and osteoarthritis [1], [3], [13]. Systemic administration of sigma-1 agonists enhances capsaicin-induced mechanical hypersensitivity (a behavioral model of central sensitization), suggesting that sigma-1 agonism might potentiate central pain pathways after priming of the nociceptive system [15]. Sigma-1 receptors have a prominent role in central sensory function, and several studies (e.g. [16] and [17]) have shown that the central (intrathecal) administration of sigma-1 agonists induces sensory hypersensitivity.
Although most studies on the relationship between sigma-1 receptors and pain have focused on central sites, we reported that mice had a much higher density of sigma-1 receptors in the dorsal root ganglion (DRG) (where the somas of peripheral sensory neurons are located) than in the dorsal spinal cord or several pain-related supraspinal areas [18]. These receptors are, in fact, expressed in every single peripheral sensory neuron [19], [20], [21], [22]. Whether they have a similar distribution in human tissue is not known. The role of sigma-1 receptors in peripheral mechanisms of nociception is much less studied. Sigma-1 receptors bind to and modulate the activity of TRPV1 [22], a major transducer for noxious stimuli [23]. We very recently reported that sigma-1 antagonism was able to attenuate hyperalgesia induced by peripheral sensitization, specifically sensory hypersensitivity induced by sensitizers of TRPV1 + neurons, including PGE2 [22]. PGE2 is a major algogenic chemical that is robustly released in pain states involving inflammation such as that occurring during inflammatory responses to tissue injury [24]. PGE2 can be produced by all cell types, but epithelia, fibroblasts, and infiltrating inflammatory cells are the main sources [25]. In short, sigma-1 antagonism has the potential to weaken the connection between the inflammatory environment and TRPV1 + nociceptors. Whether sigma-1 agonism has the opposite effect, that is exacerbation of nociception through TRPVI+ neurons, is unknown.
Taking into account the above considerations, the aims of this study were to test whether sigma-1 agonism enhances PGE2-induced hyperalgesia and pain during inflammation subsequent to plantar incision in mice and to assess the involvement of TRPV1+nociceptors in the effects observed. An additional aim was to study the expression of sigma-1 receptors in human DRG tissue. This research is relevant, as several sigma-1 agonists are already in clinical use or are currently being investigated in clinical trials.
…
3. Results
3.1. Comparison of PGE2-induced mechanical hyperalgesia in wild-type and sigma-1 knockout mice
We first explored the effects of the peripheral sensitizer PGE2 on struggle response latency following mechanical stimulation in wild-type and sigma-1 knockout mice. Responses were tested 10 min after intraplantar injection of PGE2 or its solvent (saline control). Wild-type and sigma-1 knockout mice exhibited a similar (non-significantly different) latency (21.67 ± 1.01 vs. 21.30 ± 1.22). PGE2 (0.125–0.5 nmol) induced a similar dose-dependent decrease in latency (mechanical hyperalgesia) in mice of both genotypes (Fig. 1). Differences in struggle latency between mice administered the low PGE2 dose (0.125 nmol) and those administered solvent were non-significant in both wild-type and sigma-1 knockout mice (Fig. 1). The lack of sensitization observed with the 0.125-nmol dose was not because the evaluation time was too short, as no significant differences in latency were observed between wild-type mice evaluated at 30 and 60 min after PGE2 administration and solvent-treated mice (Fig. S2). The higher PGE2 dose (0.5 nmol), by contrast, induced pronounced, sustained hyperalgesia from 10 min to at least 60 min post-administration, with a struggle latency of approximately 8 s (Fig. S2).
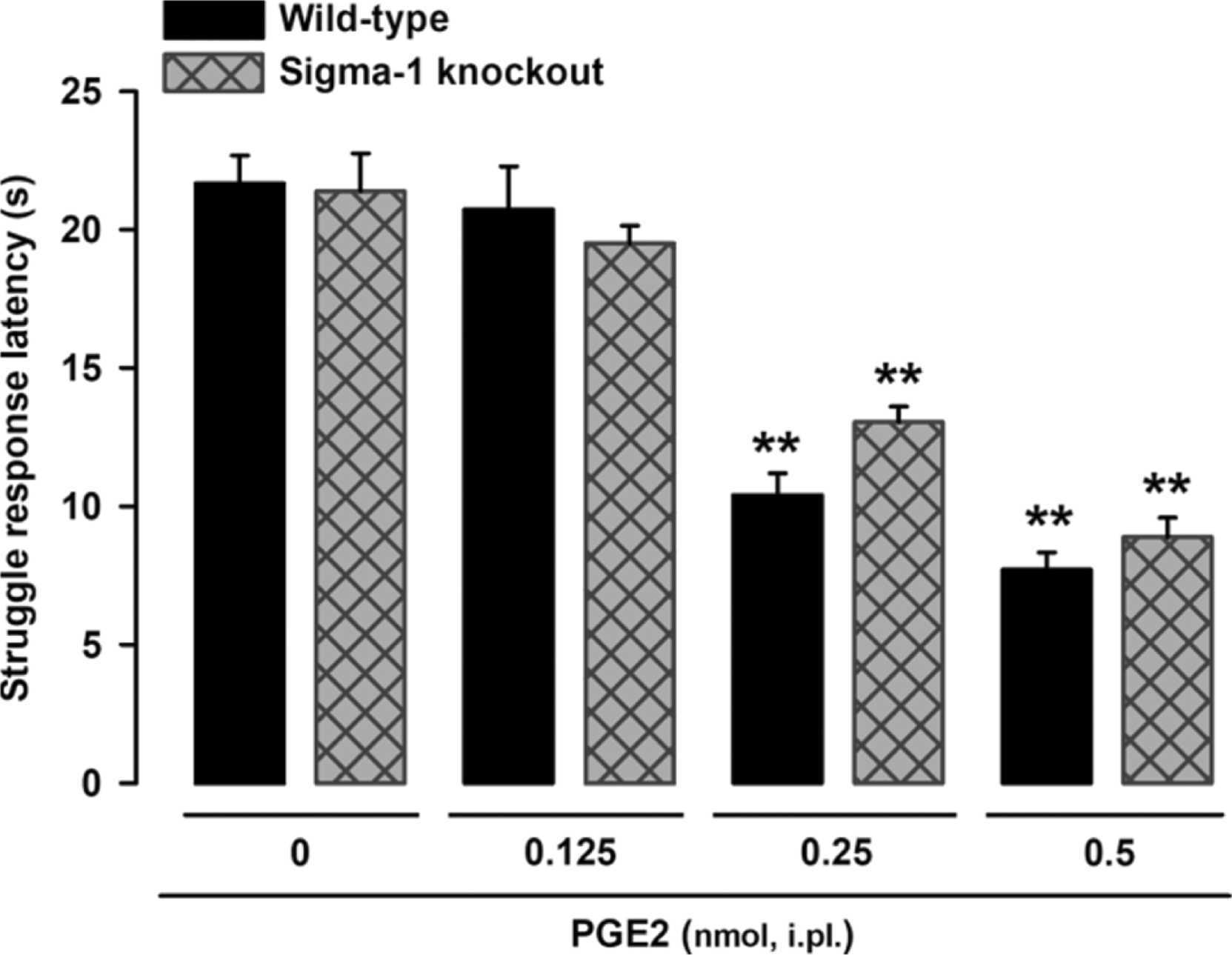
PGE2-induced effects on behavioral responses to mechanical stimulus in wild-type and sigma-1 knockout mice.
The results represent the latency to struggle in response to a mechanical stimulus of 100 g in wild-type and sigma-1 knockout mice intraplantarly (i.pl.) injected with PGE2 (0.125–0.5 nmol) or its solvent (control). Each bar and vertical line represents the mean ± SEM of the values obtained in 7–8 mice. Statistically significant differences between the values obtained in non-sensitized control mice and the other experimental groups (**P < 0.01). No significant differences were found between wild-type and knockout values at any of the PGE2 doses tested (two-way ANOVA followed by Student-Newman-Keuls test).
3.2. Systemic administration of sigma-1 agonists enhances PGE2-induced mechanical hyperalgesia without altering normal mechanical sensitivity
We studied the effects of systemically administered sigma-1 agonists in mice injected with a low dose of i.pl. PGE2 (0.125 nmol), which as shown in the previous section does not induce sensitization to mechanical stimulation. Subcutaneous administration of the nonselective sigma-1 agonist dextromethorphan (8–16 mg/kg) induced a dose-dependent decrease in struggle latency, which was less than 10 s with the 16-mg/kg dose (Fig. 2A). This latency was similar to that observed after sensitization with the much higher dose of PGE2 0.5 nmol (compare Fig. 1 and S2). The effect of PGE2 (dose-dependent decrease in struggle latency) was replicated with the s.c. administration of the prototypic sigma-1 agonist PRE-084 (8–32 mg/kg) and the selective sigma-1 agonist pridopidine (0.125–0.25 mg/kg) (Fig. 2A).
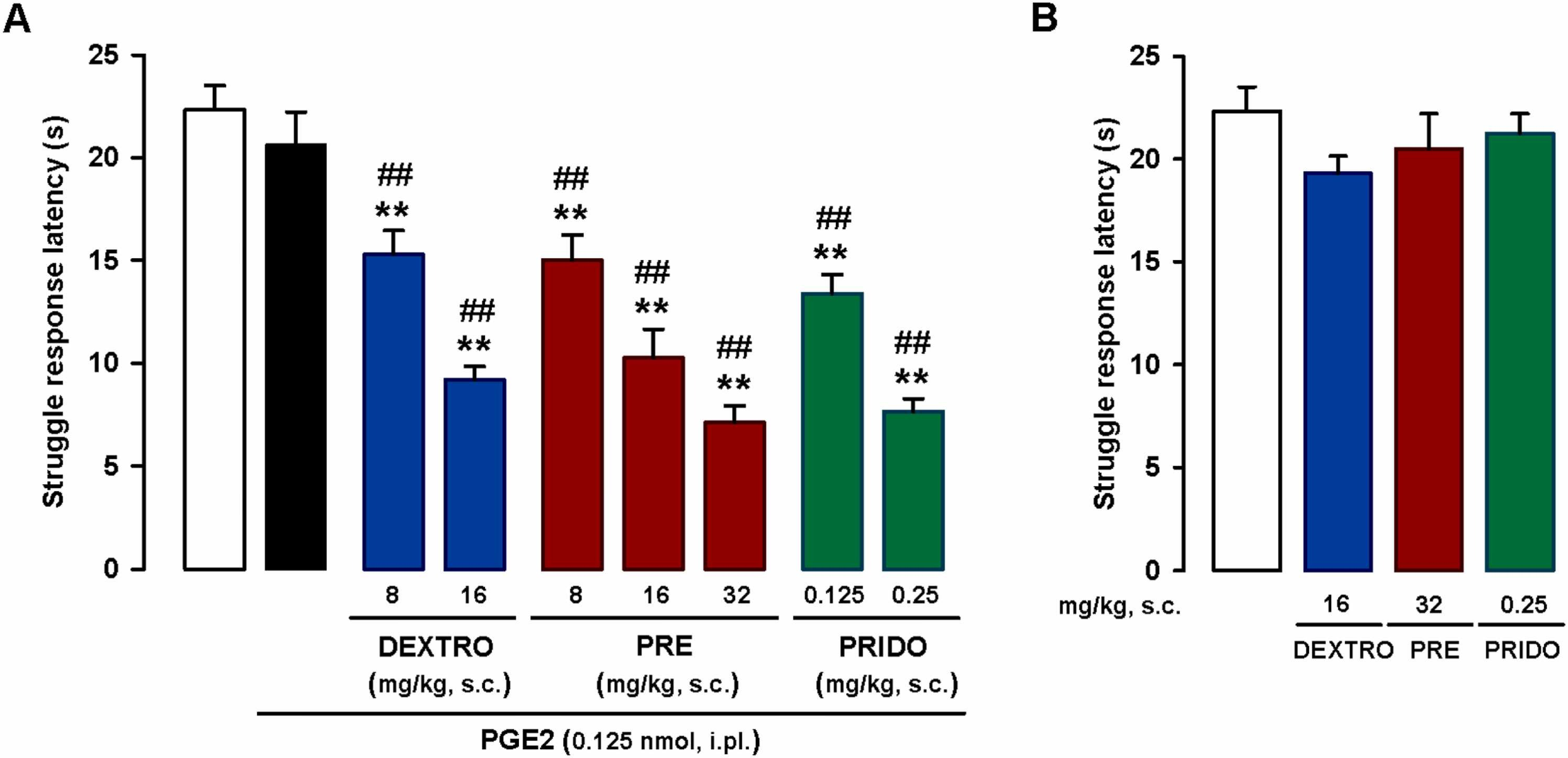
Systemic administration of sigma-1 agonists enhances PGE2-induced mechanical hyperalgesia.
The results represent latency to struggle in response to a mechanical stimulus of 100 g in wild-type mice. (A) Effects of subcutaneous (s.c.) administration of dextromethorphan (DEXTRO), PRE-084 (PRE), pridopidine (PRIDO), or their solvent (saline) in mice sensitized with an intraplantar (i.pl.) injection of a low dose of PGE2 (0.125 nmol). (B) Absence of effect in non-sensitized mice (mice not treated with i.pl. PGE2). (A and B) Each bar and vertical line represents the mean ± SEM of the values obtained in 6–8 mice. (A) Statistically significant differences between the values obtained in non-sensitized control mice (white bar) and the other experimental groups (**P < 0.01) and between values obtained in PGE2-sensitized wild-type mice administered the sigma-1 agonists (blue, red and green bars) or their solvent (black bar) (##P < 0.01) (one-way ANOVA followed by Student-Newman-Keuls test). (B) There were no significant differences between the values obtained in non-sensitized mice treated with the sigma-1 agonists or their solvents (one-way ANOVA followed by Student-Newman-Keuls test).
No changes to struggle latency were observed when the sigma-1 agonists were administered to non-sensitized mice at doses high enough to markedly potentiate PGE2-induced mechanical hyperalgesia (16 mg/kg for dextromethorphan, 32 mg/kg for PRE-084, and 0.25 mg/kg for pridopidine) (Fig. 2B).
These results indicate that systemic sigma-1 agonism is unable to induce sensitization to mechanical stimulus per se, but is able to enhance PGE2-induced mechanical hyperalgesia.
3.3. Hindpaw weight bearing asymmetry following plantar incision: effects of systemically administered sigma-1 agonists and dependence on neutrophil infiltration
As PGE2 is an inflammatory mediator, we next aimed to explore the effects of sigma-1 agonists on a more translational model involving inflammation. Since inflammation is a natural response to tissue damage, we used the hindpaw plantar incision model. Hematoxylin-eosin staining of the incision site showed that the incision had cut through the epidermis and dermis, causing no (or minimal) injury to the fascia or muscle tissue, as seen in the representative images taken 3.5 h after incision (Fig. 3A, middle panels; compare the images for the naïve control in the left panels). Edema and inflammation were still present in the dermis and subcutaneous tissue at 24 h, with a substantial inflammatory infiltrate, even in muscle tissue (Fig. 3A, right panels). FACS showed little neutrophil and macrophage/monocyte recruitment 3.5 h after injury. At 24 h, however, a prominent immune infiltrate composed mainly of neutrophils, with some macrophages/monocytes, was observed (Fig. 3B and C).
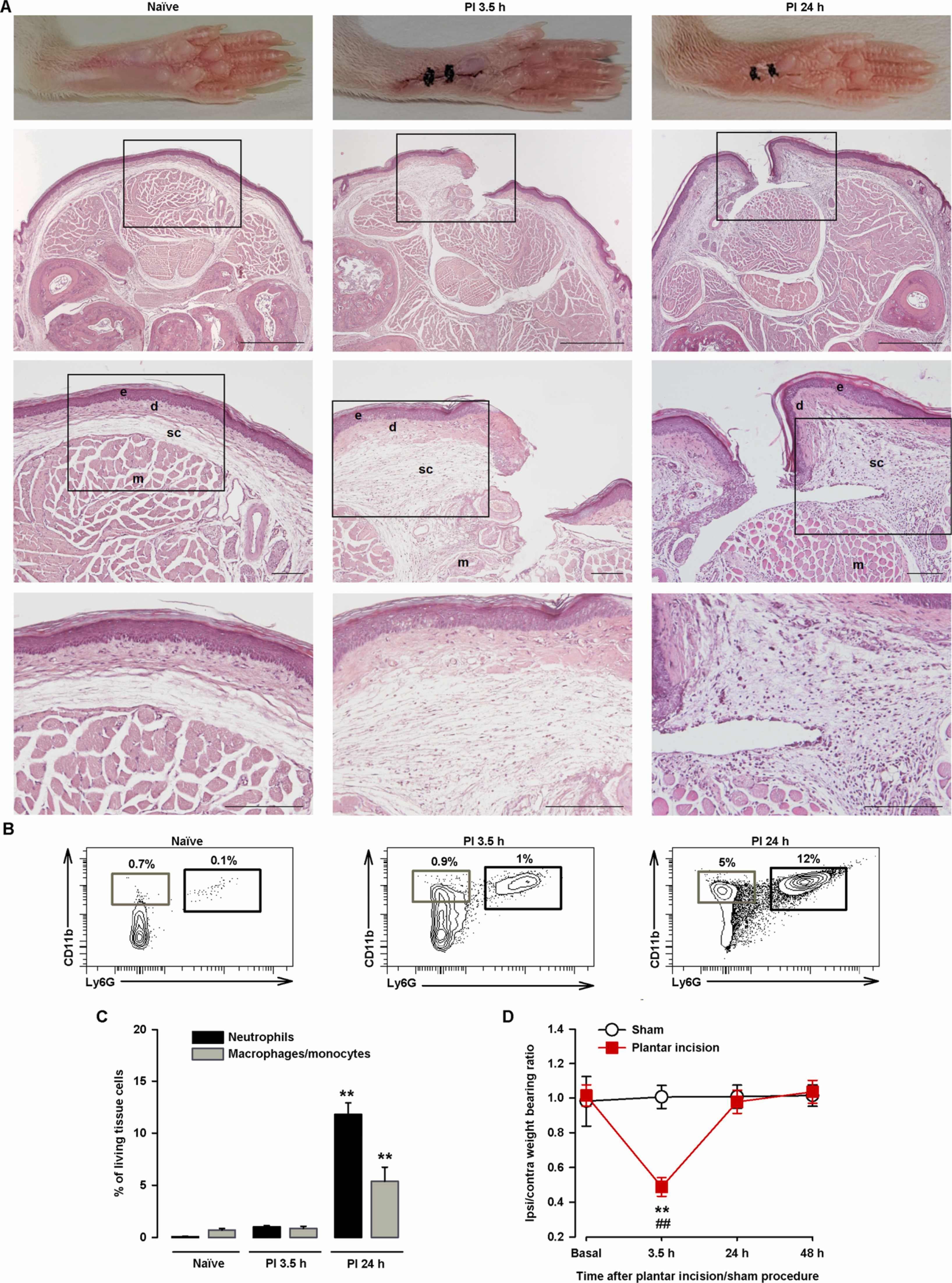
Time course of changes in hindpaw weight bearing ratio, immune cell recruitment, and histological findings after plantar incision.
(A) Representative pictures of paws from uninjured (naïve) mice and injured mice 3.5 h and 24 h after plantar incision and corresponding photomicrographs of hematoxylin- eosin–stained paw sections from the mid-plantar region. Scale bar is 500 µm in the upper panels, 100 µm in the middle panels, and 50 µm in the bottom panels. The middle and bottom panels show details of the boxed areas in the top and middle panels, respectively. The relevant structures are labeled in the middle panels for clarity (e, epidermis; d, dermis; sc, subcutaneous tissue; m, muscle tissue). Note the increase in thickness of d and sc at 3.5 h and 24 h after incision and the inflammatory infiltrate in sc and m at 24 h after incision. (B) Representative FACS diagrams with gating from CD45+ cells showing an increase in neutrophils (CD11b+Ly6G+) and to a lesser extent macrophages/monocytes (CD11b+Ly6G-) in the paw 24 h after incision. Gating for neutrophil and macrophage/monocyte quantification is shown in black and gray rectangles, respectively. (C) Quantification of neutrophils and macrophages/monocytes with respect to number of living cells in paw samples from naïve mice and mice that underwent plantar incision. Each bar and vertical line represents the mean ± SEM of the values obtained in 5 samples per group, with each sample taken from a single animal. Statistically significant differences between the values obtained for each cell type in samples from naïve mice and the other experimental groups (**P < 0.01) (one-way ANOVA followed by Student-Newman-Keuls test). (D) The results represent the ratio between the weight borne by the ipsilateral (ipsi) and the contralateral (contra) paw to the incision before surgery and at 3.5 h, 24 h, and 48 h after incision (or the sham procedure) in wild-type mice. Each point and vertical line represents the mean ± SEM of the values obtained in 7 animals per group. Statistically significant differences between baseline and post-incision values (**P < 0.01) and between the values from sham and injured mice evaluated at the same time points after the procedure (##P < 0.01) (two-way repeated-measures ANOVA followed by Student-Newman-Keuls test).
We next studied nociception after injury by evaluating the ratio of the weight borne by the injured (ipsilateral) hindpaw to that borne by the non-injured (contralateral) hindpaw (weight bearing ratio). The paw pressure prints during the baseline recording (prior to incision) were very similar, with a ratio of close to 1. At 3.5 h after surgery, the mice exhibited significant weight bearing asymmetry, manifested as a significant reduction in the ipsilateral/contralateral hindpaw weight bearing ratio (decreased weight on the ipsilateral paw and increased weight on the contralateral paw). At 24 h, the weight bearing deficits had returned to near-baseline levels (Fig. 3D). Mice in the sham group showed no significant changes in weight bearing ratio at any of the time points tested (Fig. 3D).
Altogether, the above findings show that while tissue injury and inflammation were still observable 24 h after plantar incision, they were not sufficient to induce hindpaw weight bearing asymmetry.
We then tested the effects of sigma-1 agonists and their solvents on hindpaw weight distribution. Evident hindpaw weight bearing asymmetry was observed at 3.5 h after surgical injury in all groups of mice evaluated (Fig. 4A). At 24 h, immediately before the s.c. injection of sigma-1 agonists or their solvents (time 0), the asymmetry had resolved, with a weight bearing ratio of close to 1 (Fig. 4A). The non-selective sigma-1 agonist dextromethorphan (16 mg/kg), the prototypic sigma-1 agonist PRE-084 (32 mg/kg), and the selective sigma-1 agonist pridopidine (0.25 mg/kg) were injected s.c. at doses that had induced sensitization to mechanical stimulation in PGE2-injected mice. All the sigma-1 agonists induced a significant reduction in hindpaw weight bearing ratios after 30–90 min. There was no evidence of asymmetry 48 h after surgical injury (24 h after drug administration) (Fig. 4A). Subcutaneous saline injection had no significant effect on weight bearing ratios during the 24-h test period (Fig. 4A).
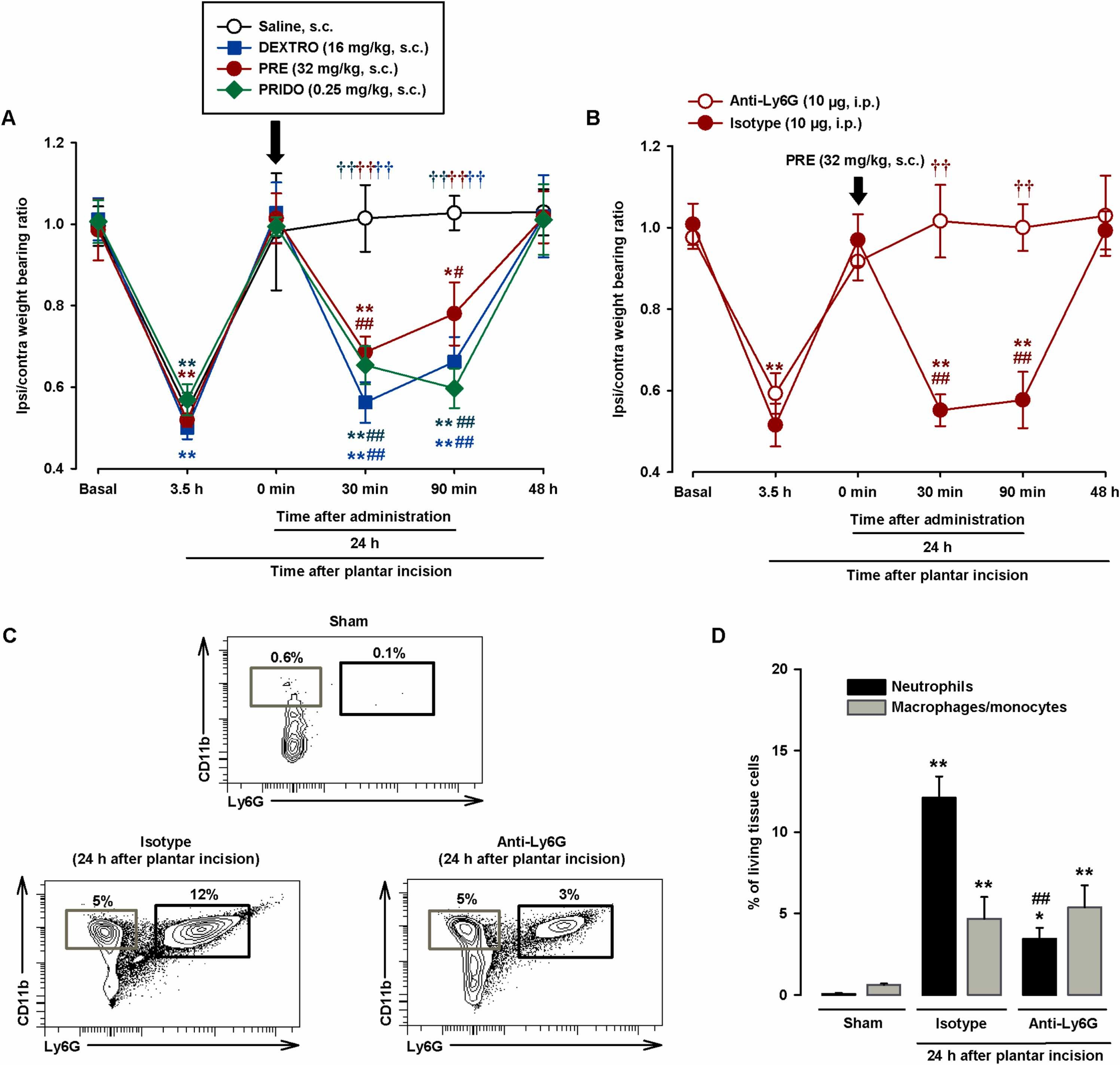
Systemic administration of sigma-1 agonists induces relapse of hindpaw weight bearing asymmetry after plantar incision in wild-type mice through the actions of neutrophils.
(A and B) Weight bearing ratio of injured ipsilateral (ipsi) paw to contralateral (contra) paw in wild-type mice (A) injected subcutaneously (s.c.) with dextromethorphan (DEXTRO), PRE-084 (PRE), pridopidine (PRIDO), or their solvents and (B) injected intraperitoneally (i.p.) with the anti-Ly6G antibody (10 μg) or the isotype control 24 h before plantar incision and injected s.c. with PRE 24 h after incision. Hindpaw weight bearing was recorded before plantar incision (basal) and 3.5 h after injury. The next day, they were evaluated immediately before administration of sigma-1 agonists (0 min) and at 30 min, 90 min, and 24 h after drug injection. (C) Representative FACS diagrams, with gating from CD45 + cells, showing neutrophils (CD11b+Ly6G+) and macrophages/monocytes (CD11b+Ly6G-) in uninjured (sham) mice and mice injected with anti-Ly6G or the isotype control 24 h after plantar incision. Gating for neutrophils and macrophage/monocytes is shown in black and gray rectangles, respectively. (D) Quantification of neutrophils and macrophages/monocytes with respect to number of living cells in the paws of sham mice and anti-Ly6G/isotype control–treated mice at 24 h after incision. (A and B). Each point and vertical line represents the mean ± SEM of the values obtained in 7–9 mice. Statistically significant differences between baseline and other values (*P < 0.05, **P < 0.01), between values obtained before drug administration (0 min) and afterwards (#P < 0.05 ## P < 0.01), between mice treated with the drugs or saline, and between mice treated with anti-Ly6G or the isotype control (††P < 0.01) (two-way repeated-measures ANOVA followed by Student-Newman-Keuls test). (D) Each bar and vertical line represents the mean ± SEM of the values obtained in 5 samples per group, with each sample taken from a single animal. Statistically significant differences between the number of neutrophils and macrophages/monocytes in sham mice and the other experimental groups (*P < 0.05, **P < 0.01) and between the number of neutrophils in mice treated with anti-Ly6G or the isotype control (## P < 0.01). There were no statistical differences between the number of macrophages/monocytes in mice treated with anti-Ly6G or the isotype control (one-way ANOVA followed by Student-Newman-Keuls test).
Considering that immune cells are one of the main sources of PGE2 at inflamed sites (see Introduction for references) and that we observed obvious neutrophil infiltration in our experiments, we administered an anti-Ly6G antibody to test the influence of neutrophil depletion on the proalgesic effect of PRE-084. Mice injected with i.p. anti-Ly6G 10 μg and control mice administered a non-reactive isotype antibody showed similar weight bearing asymmetry 3.5 h after plantar incision, but recovered near-baseline values at 24 h. Subsequent administration of s.c. PRE-084 (32 mg/kg) induced a relapse in weight bearing asymmetry in the control mice (30–90 min after injection of the isotope antibody), but not in the mice treated with anti-Ly6G (Fig. 4B). We also tested the effects of both antibodies on immune cell recruitment after plantar incision. The anti-Ly6G antibody induced a 72% decrease in neutrophil infiltration compared to the isotype control. The differences in macrophage/monocyte recruitment were non-significant (Fig. 4C and D), demonstrating the selectivity of the neutrophil depletion strategy.
In summary, the sigma-1 agonists dextromethorphan, PRE-084, and pridopidine were able to trigger pain-like behaviors after apparent resolution, when post-injury inflammation and immune cell infiltration were still present. The presence of neutrophils at the incision site is essential for the proalgesic effect induced by sigma-1 agonism.
3.4. Selectivity of the pronociceptive effects induced by systemically administered sigma-1 agonists
We also tested the selectivity of the effects induced by the s.c. administered sigma-1 agonists dextromethorphan, PRE-084, and pridopidine on PGE2-induced mechanical hyperalgesia and hindpaw weight bearing asymmetry after surgical injury.
We first tested the effects of combined treatment with these agonists and the sigma-1 antagonist BD-1063 (32 mg/kg, s.c.) on PGE2-induced hyperalgesia. BD-1063 alone did not modify struggle latency in the paw pressure test in non-sensitized mice (Fig. 5A), but when it was administered in association with the sigma-1 agonists at doses capable of markedly enhancing the hyperalgesia induced by low-dose PGE2 (0.125 nmol) (16 mg/kg for dextromethorphan, 32 mg/kg for PRE-084, and 0.25 mg/kg for pridopidine), it was able to fully reverse the sensitizing effect of all sigma-1 agonists tested, increasing struggle latencies to values similar to those observed in non-sensitized control mice (Fig. 5A).
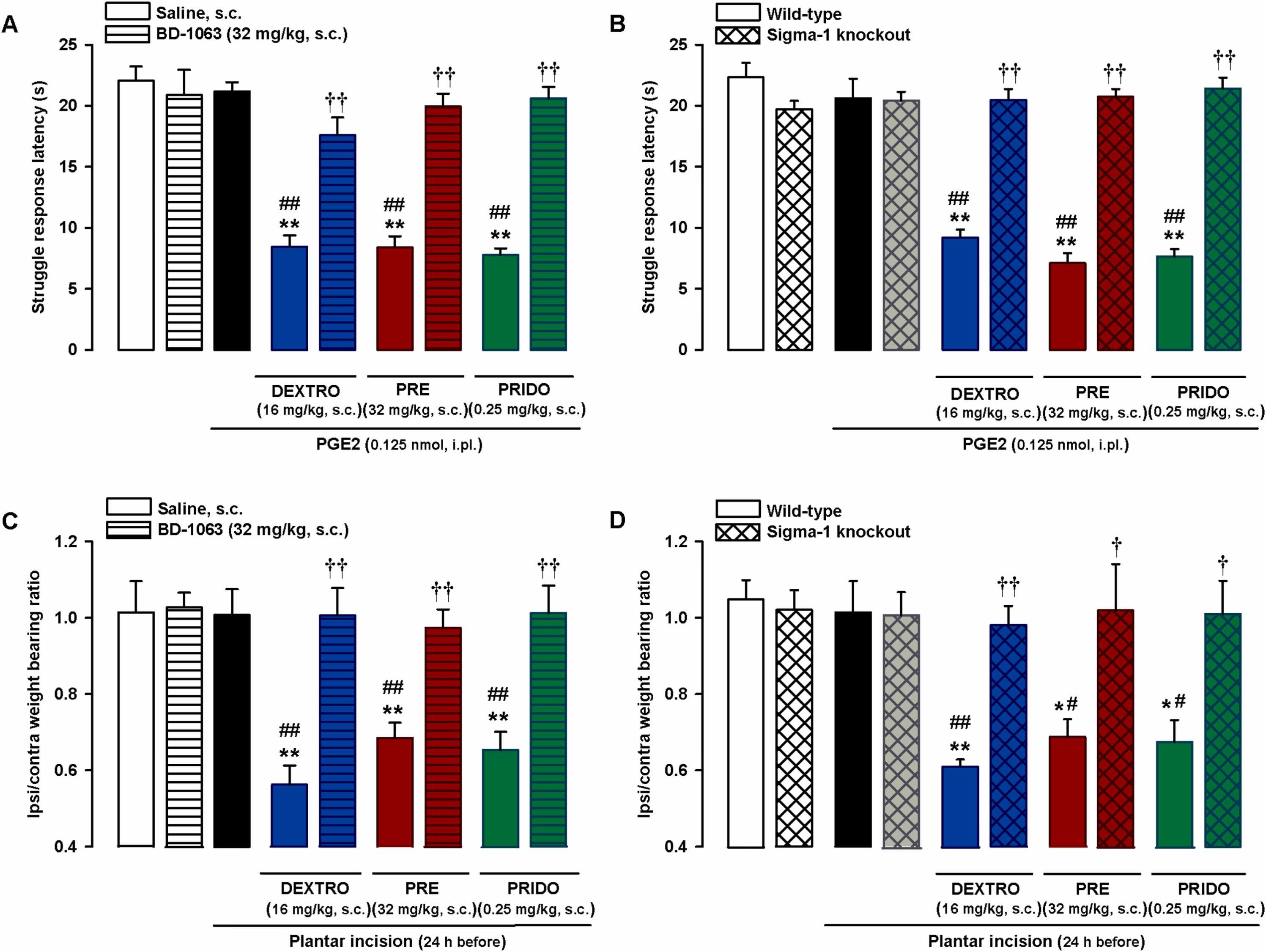
Selectivity of effects induced by systemic administration of sigma-1 agonists on PGE2-induced mechanical hyperalgesia and on hindpaw weight bearing asymmetry after plantar incision.
(A and C) The sigma-1 antagonist BD-1063 (BD) reversed the effects of sigma-1 agonists on (A) mechanical hyperalgesia induced by intraplantar (i.pl.) injection of PGE2 and on (C) hindpaw weight bearing asymmetry 24 h after plantar incision in wild-type mice. The mice were subcutaneously (s.c.) treated with BD or its solvent alone or in combination with s.c. dextromethorphan (DEXTRO), PRE-084 (PRE), pridopidine (PRIDO), or their solvents. (B and D) Comparison of pronociceptive effects of sigma-1 agonists in wild-type and sigma-1 knockout mice on (B) PGE2-induced mechanical hyperalgesia and on (D) hindpaw weight bearing asymmetry 24 h after plantar incision. (A-D) Each bar and vertical line represents the mean ± SEM of the values obtained in 6–8 mice. Statistically significant differences between the values obtained in non-sensitized control wild-type mice (left white bars) and the other experimental groups (*P < 0.05, **P < 0.01). (A and C) Statistically significant differences between the values obtained in sensitized wild-type mice injected with PGE2 or subjected to paw incision (black bars) administered sigma-1 agonists or their solvent (##P < 0.01) and between the values obtained in sensitized wild-type mice administered the sigma-1 agonist in combination with BD or its solvent (††P < 0.01). (B and D) Statistically significant differences between the values obtained in wild-type animals injected with PGE2 or subjected to paw incision (black bars) administered the sigma-1 agonists or their solvent (#P < 0.05, ##P < 0.01) and between the effects of sigma-1 agonists administered to wild-type and sigma-1 knockout mice (†P < 0.05; ††P < 0.01) (one-way ANOVA followed by Student-Newman-Keuls test).
Mice lacking the sigma-1 receptor, the purported target of the three sigma-1 agonists, were used to test the selectivity of the effects of these drugs on PGE2-induced hyperalgesia. As previously shown, wild-type and sigma-1 knockout mice exhibited similar responses to mechanical stimulation in paws injected with PGE2 0.125 nmol or its solvent (Fig. 5B). However, although s.c. dextromethorphan (16 mg/kg), PRE-084 (32 mg/kg), and pridopidine (0.25 mg/kg) markedly enhanced PGE2-induced hyperalgesia and significantly decreased struggle latency in wild-type mice, none of these agonists altered behavioral responses in PGE2-injected sigma-1 knockout mice (Fig. 5B). This absence of effect in mice lacking sigma-1 receptors suggests that off-target effects do not contribute to the potentiation of PGE2-induced hyperalgesia by these drugs.
We then tested the selectivity of the effects induced by the sigma-1 agonists on weight bearing asymmetry after plantar incision using the same strategies as above. The experiments were performed 24 h after superficial plantar incision, when as described in the section above, mice had recovered a weight bearing ratio of close to 1; subsequent s.c. administered dextromethorphan (16 mg/kg), PRE-084 (32 mg/kg), and pridopidine (0.25 mg/kg) induced a relapse in weight bearing asymmetry. Subcutaneous BD-1063 did not modify the weight bearing ratio in the control (non-injured) mice, but it fully reversed the weight bearing asymmetry induced by the sigma-1 agonists (Fig. 5C). When we compared wild-type and sigma-1 knockout mice, we found no significant differences in the weight bearing ratios of sham mice and those subjected to plantar incision 24 h before evaluation in mice of either genotype. Finally, although the sigma-1 agonists induced weight bearing asymmetry in wild-type mice, they were unable to alter the weight bearing ratio in sigma-1 knockout mice (Fig. 5D).
The above results support the selectivity of the effects induced by the sigma-1 agonists on both PGE2-induced hyperalgesia and hindpaw weight bearing asymmetry during inflammation-associated tissue damage.
3.5. Expression of sigma-1 receptors in mouse and human DRG
We analyzed expression of sigma-1 receptors in DRG by immunohistochemical staining. DRG neurons were first identified using the pan-neuronal marker PGP9.5. Sigma-1 receptor immunoreactivity was detected in most (if not all) PGP9.5 + DRG cells, indicating that both markers label an overlapping cell population (DRG neurons). The staining patterns in the neuronal bodies, however, were different: most neurons contained a central round area that was completely devoid of sigma-1 receptor staining but showed intense PGP9.5 expression (Fig. 6A, top panels). Higher-magnification photomicrographs showed that this area clearly overlapped with the area expressing Hoechst 33342 (Fig. 6A, middle panels). Because Hoechst 33342 labels the cell nuclei, these findings indicate that sigma-1 receptors are not present in this location. Sigma-1 receptor immunostainingwas not observed in DRG sections when the sigma-1 receptor primary antibody was omitted (Fig. 6A, bottom panels), supporting the specificity of the antibody used.
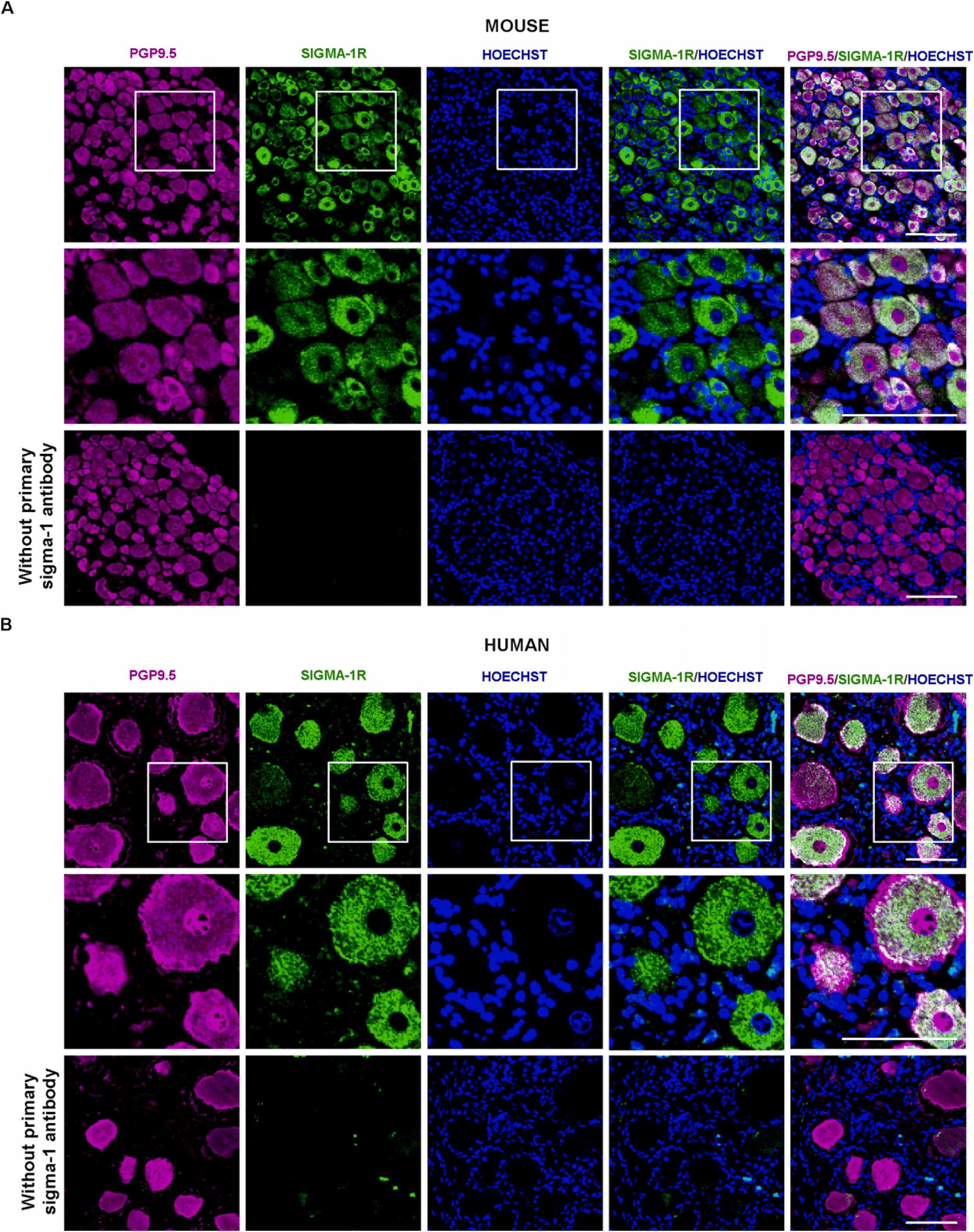
The sigma-1 receptor is selectively present in mouse and human DRG neurons.
Representative images showing labeling with the pan-neuronal marker PGP9.5 (magenta), the sigma-1 receptor (Sigma-1R, green), and Hoechst 33342 (Hoechst) (blue) in samples from (A) an L4 DRG from intact mice and (B) a human lumbar DRG. The top and bottom panels show low-magnification images of experiments performed in the presence (top) or absence (bottom) of the sigma-1 receptor primary antibody. The middle panels show a higher-magnification view of the areas squared in the top panels. Scale bar is 100 µm.
Staining for PGP9.5 and the sigma-1 receptor in human DRG samples yielded similar results, with sigma-1 staining visible in virtually all the PGP9.5 + cells although not present in the neuronal nuclei (Fig. 6B, top and middle panels). The human DRG neurons were notably larger than the mouse neurons (compare Fig. 6A and B). In contrast to findings for the mouse samples, the sigma-1 receptor antibody labeled some small extraneuronal particles in the human samples (Fig. 6B top and middle panels). Omission of the primary sigma-1 receptor antibody in the staining procedure resulted in a complete loss of sigma-1-like staining in PGP9.5 + cells, but preserved extraneuronal staining (Fig. 6B bottom panels). These results support the specificity of sigma-1 staining in human sensory neurons and also indicate that the extraneuronal labeling detected is due to nonspecific staining during the procedure.
Altogether, our results show that sigma-1 receptors are markedly present in both mouse and human peripheral sensory neurons.
3.6. Involvement of TRPV1 + nociceptors in the pronociceptive effects of sigma-1 agonism
We also tested whether the pronociceptive effects of sigma-1 agonism were mediated by TRPV1 + peripheral sensory neurons. Staining for TRPV1 and IB4 showed minimal or no overlap in DRG neurons from intact mice (see top panels of Fig. 7A for representative images). Treatment with the molecular scalpel RTX abolished TRPV1 but not IB4 expression (Fig. 7A, bottom panels), confirming the specificity of the ablation procedure.
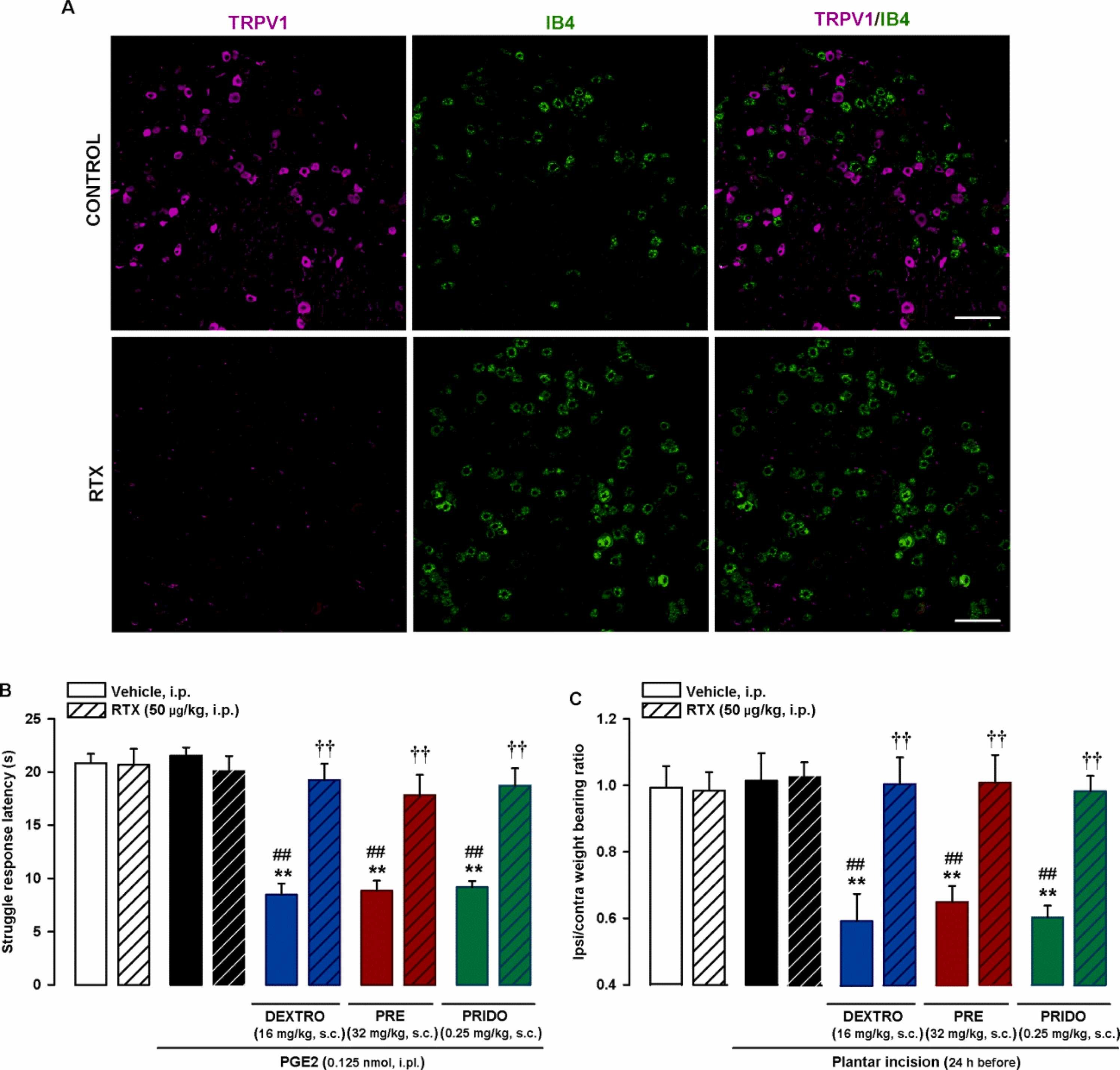
Effects of in vivo ablation of TRPV1 + neurons on the effects induced by sigma-1 agonists on PGE2-induced mechanical hyperalgesia and hindpaw weight bearing asymmetry after plantar incision.
Wild-type mice were injected intraperitoneally (i.p.) with resiniferatoxin (RTX, 25 μg/kg) or its vehicle on 2 consecutive days 5 days before sample collection or behavioral experiments. (A) Double labeling of TRPV1 (magenta) and IB4 (green) in L4 DRG. Top panels: samples from vehicle-treated mice (control). Bottom panels: samples from mice treated with RTX. Scale bar is 100 µm. (B) The results represent latency to struggle in response to a mechanical stimulus of 100 g in mice administered subcutaneously (s.c.) with the sigma-1 agonists dextromethorphan (DEXTRO), PRE-084 (PRE), pridopidine (PRIDO), or their solvent, and injected intraplantarly (i.pl.) with PGE2 (0.125 nmol) or its solvent. (C) The results represent the ratio between the weight borne by the ipsilateral (ipsi) and the contralateral (contra) hindpaw to the incision in wild-type mice administered the s.c. sigma-1 agonists or their solvent. Behavioral responses were evaluated 24 h after plantar incision. (B and C) Each bar and vertical line represents the mean ± SEM of the values obtained in 6–8 mice. Statistically significant differences between the values obtained in non-sensitized control animals (left white bars) and the other experimental groups (**P < 0.01), between the values obtained in sensitized mice injected with PGE2 or subjected to paw incision (black bars) and administered the sigma-1 agonists or their solvents (##P < 0.01), and between sensitized animals administered the sigma-1 agonists and injected with RTX or its vehicle (††P < 0.01 (one-way ANOVA followed by Student-Newman-Keuls test).
We next studied the effects of in vivo RTX ablation of TRPV1 + neurons on the effects of sigma-1 agonists. Ablation did not affect struggle latencies in mice injected with the low dose of i.pl. PGE2 (0.125 nmol) or its solvent. Of note, s.c. administration of sigma-1 agonists (dextromethorphan 16 mg/kg, PRE-084 32 mg/kg, or pridopidine 0.25 mg/kg) did not enhance PGE2-induced sensitization to the mechanical stimulus in mice treated with RTX; this result contrasts with the marked decrease in struggle response latency observed in PGE2-sensitized mice treated with solvent of this molecular scalpel (Fig. 7B).
We also explored whether TRPV1 + neurons were responsible for the effects of sigma-1 agonists on weight bearing asymmetry after plantar incision. As above, the experiments were performed 24 h after plantar incision, which is when the mice appeared to recover normal weight distribution on the injured and non-injured hindpaws. Subcutaneous treatment with the sigma-1 agonists dextromethorphan (16 mg/kg), PRE-084 (32 mg/kg), and pridopidine (0.25 mg/kg) caused weight bearing asymmetry to reappear. Ablation of TRPV1 + neurons with RTX, however, did not induce any changes in weight bearing ratios in mice that underwent plantar incision or control mice (sham procedure and solvents). It did, however, fully prevent the sensitizing effect of all three sigma-1 agonists on weight bearing asymmetry (Fig. 7C).
These results suggest that sigma-1 agonists need TRPV1 + afferents to potentiate both PGE2-induced hyperalgesia and hindpaw weight bearing asymmetry after surgical injury.
3.7. The effect of sigma-1 agonism is exerted at sensitized sites
As the pronociceptive effect of the systemically administered sigma-1 agonists depended on the presence of TRPV1 + neurons, we investigated whether local (intraplantar) administration of the TRP antagonist RR at the sensitized site would be sufficient to reverse the PGE2-induced mechanical hyperalgesia potentiated by sigma-1 agonism. An RR dose of just 32 µg administered at the site injected with PGE2 (0.125 nmol) fully reversed this hyperalgesia in mice administered s.c. PRE-084 (32 mg/kg). The effect was exerted locally, at the injection site, as no effects were observed when RR was injected into the paw contralateral to PGE2 injection (Fig. 8A). These results suggest that TRP activation at the PGE2-sensitized site is needed for systemically administered PRE-084 to exert its prohyperalgesic effect. We then explored the participation of sigma-1 receptors at the PGE2-sensitized site in the prohyperalgesic effects induced by the systemic administration of PRE-084. Injection of the sigma-antagonists BD-1063 (150 µg) and S1RA (200 µg) into the PGE2-injected paw showed that local sigma-1 antagonism at the sensitized site was able to abolish the prohyperalgesic effect of systemic sigma-1 agonism. Again, this effect was produced locally since sigma-1 antagonists were devoid of effect when administered in the paw contralateral to PGE2 injection (Fig. 8A). Since these results pointed to a relevant role for sigma-1 receptors at the sensitized site, we tested whether the administration of PRE-084 at the PGE2-injected site would be sufficient to potentiate mechanical hyperalgesia. We found that i.pl. administration of PRE-084 (50–75 µg) at the PGE2-injected site dose-dependently decreased struggle latency during mechanical stimulation. These effects were mediated locally and were not observed when PRE-084 (75 µg) was injected into the paw contralateral to PGE2 injection (Fig. 8B). Altogether, these results suggest that peripheral sigma-1 receptors at the sensitized site play a relevant role in the potentiation of PGE-induced mechanical hyperalgesia, which also depends on local TRP activation.
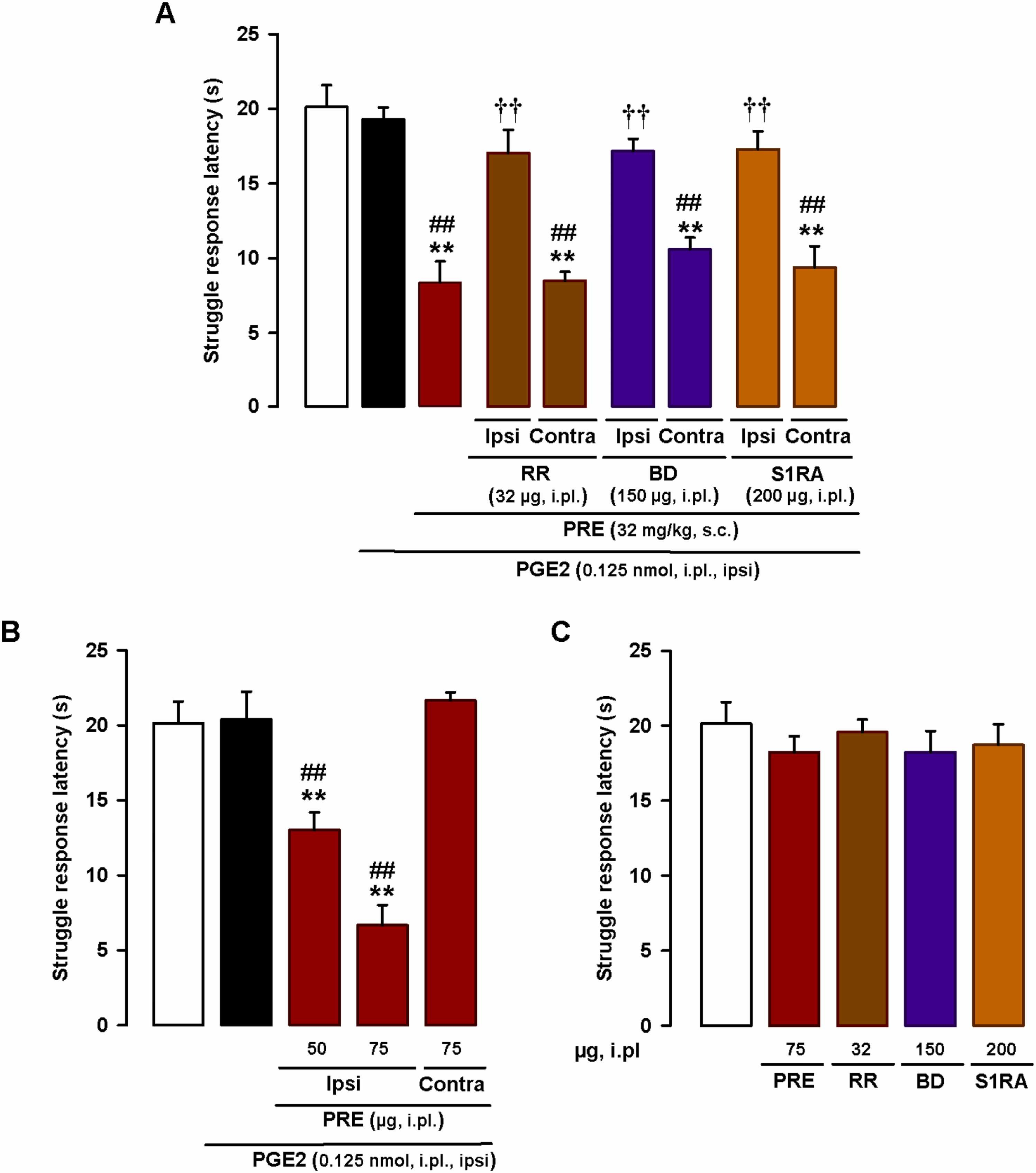
Effects of the local administration of sigma-1 drugs and the TRP antagonist ruthenium red on PGE2-induced mechanical hyperalgesia.
Struggle latencies evoked by a mechanical stimulus of 100 g in wild-type mice treated intraplantarly (i.pl.) with PGE2 (0.125 nmol) or its solvent. (A) Mice were injected subcutaneously (s.c.) with the sigma-1 agonist PRE-084 (PRE) and i.pl. with the sigma-1 antagonists BD-1063 (BD) or S1RA, or the TRP antagonist RR, or their solvents, in the paw ipsilateral (ipsi) and contralateral (contra) to the PGE2 injection. Mechanical stimulation was performed in the ipsi paw. (B) Mice were i.pl. injected with PRE in the paw ipsilateral or contralateral to the PGE2 injection. Mechanical stimulation was performed in the ipsi paw. (C) Absence of effect of i.pl. RR, BD, S1RA, and PRE in non-sensitized mice. (A-C) Each bar and vertical line represents the mean ± SEM of the values obtained in 6–8 mice. (A and B) Statistically significant differences between the values obtained in non-sensitized control animals (white bars) and the other experimental groups (**P < 0.01) and between the values obtained in sensitized wild-type mice injected with PGE2 (black bars) and administered PRE or its solvent (##P < 0.01). (A) Statistically significant differences between PGE2-sensitized animals administered PRE alone or in combination with BD, S1RA, or RR (††P < 0.01). (C) There were no significant differences between the values obtained in non-sensitized mice treated with any of the i.pl. drugs or their solvents (one-way ANOVA followed by Student-Newman-Keuls test).
Finally, we tested the effects of i.pl. injection of RR (32 µg), BD-1063 (150 µg), S1RA (200 µg), and PRE-084 (75 µg) in non-sensitized mice, and found that none of these treatments modified struggle latency (Fig. 8C). Our results, therefore, suggest that peripheral TRPs and sigma-1 receptors only influence response to mechanical stimulation during sensitizing conditions.
4. Discussion
In this study, we have shown that sigma-1 agonism enhances PGE2-induced hyperalgesia and post-incisional pain.
Systemic administration of dextromethorphan does not per se induce mechanical hypersensitivity, but it does increase mechanical hyperalgesia induced by a low, otherwise inactive, dose of PGE2. The dextromethorphan doses used in the present study are lower than those used to investigate antitussive effects in rodents (e.g. [34]). Sigma-1 agonism is thought to contribute to the antitussive effects of dextromethorphan [5], [6], but this compound has several pharmacological properties [6] that could potentially contribute to its antitussive and prohyperalgesic effects. Importantly, the effects of dextromethorphan were replicated by the selective sigma-1 agonists PRE-084 and pridopidine [8], [9], [10].
Changes to hindpaw weight distribution were observed in the immediate postoperative period (3.5 h after plantar incision), with a reduction in the weight borne by the injured limb and an increase in that borne by the non-injured contralateral limb. The incision was sufficient to induce robust behavioral effects without causing severe deep tissue injury. The weight bearing asymmetry was no longer evident at 24 h, supporting previous reports which also used a superficial incision [31]. Despite this, the incision had not fully healed, as inflammation and prominent neutrophil recruitment were still evident at the incision site. Systemic administration of the three sigma-1 agonists at this time induced similar pain-like behaviors to those seen in the immediate postoperative period (shift of body weight toward the non-injured limb). This pronociceptive effect of sigma-1 agonism was shown to be fully dependent on the presence of neutrophils at the injured site, suggesting that factors released by these immune cells (and not by other cells such as epithelia and fibroblasts) were responsible for the weight bearing asymmetry induced by sigma-1 agonism. PGE2 is one of the major algogenic chemicals produced by neutrophils [35]. Considering the enhancement of the PGE2-induced hyperalgesia observed, it could be speculated that the effects of sigma-1 agonists on weight bearing asymmetry are (at least partially) due to enhancement of the pronociceptive effects of neutrophil-derived PGE2. Neutrophils, however, are able to produce other proalgesic substances [36] that might also participate in the proalgesic effects of sigma-1 agonism. Further studies are needed to fully unveil the effects of sigma-1 agonism on a more complete repertoire of immune cell–derived peripheral sensitizers.
While the decrease in struggle latency observed in the paw pressure test during the experiments with PGE2 is clearly linked to hyperalgesia, the significance of weight bearing asymmetry is not so clear. While weight bearing changes may reflect spontaneous or pressure-evoked pain (in this case contact between the injured limb and the floor), they might also reflect pain avoidance behavior, since pain anticipation may lead to motor control changes and avoidance of activities that could induce or aggravate existing pain (such as placing weight on an injured limb) [37], [38]. There are also important methodological differences between the paw pressure and weight bearing tests. Paw pressure tests are short (typically 10–20 s) and require the mice to be held by the experimenter, an additional stressor. Hindpaw weight bearing tests, by contrast, take place over several minutes, assessing therefore sustained postural changes, and are performed in freely moving mice. Another obvious difference between the tests is that in the paw pressure test, the mice were sensitized with an intraplantar injection of a single inflammatory mediator (PGE2), whereas in the weight bearing test, they underwent plantar incision leading to subsequent incision site inflammation. Despite the differences in the pain stimuli and methodologies used, our findings show that dextromethorphan, PRE-084, and pridopidine all exerted proalgesic effects in both experimental conditions, supporting the robustness of the effects induced by these sigma-1 agonists.
Two of our findings indicate that the pronociceptive effects induced by the sigma-1 agonists tested—both the selective agonists PRE-084 and pridopidine and the non-selective agonist dextromethorphan—are mediated by sigma-1 receptors. On the one hand the effects of the agonists were reversed by the known sigma-1 antagonist BD-1063 and on the other, none of the agonists were able to induce pronociceptive effects in mice lacking sigma-1 receptors, their purported pharmacological target.
There are some discrepancies between the effects of pharmacological and genetic inhibition of sigma-1 receptors. In a previous study, we reported that sigma-1 antagonism abolished PGE2-induced hyperalgesia [22], but in this study we show that sigma-1 knockout mice exhibited the same PGE2-induced hyperalgesia as wild-type mice. This is not the first report of conflicting results of this type in sigma-1 receptor research. While some studies have shown that sigma-1 antagonists can abolish inflammatory and neuropathic heat hyperalgesia [27], [39], [40] and potentiate opioid-induced antinociception to heat stimulus (e.g. [41], [42]), others have shown that the effects of sigma-1 antagonists are not replicated in sigma-1 knockout mice and that these exhibit similar behavioral responses to wild-type mice in these situations [39], [40], [42], [43]. One proposed explanation is the development of compensatory mechanisms in the heat pain pathways of sigma-1 knockout mice [39], [40]. Although we tested PGE2-induced hyperalgesia to mechanical stimulus in this study, it should be noted that this form of sensory hypersensitivity is fully dependent on the sensitization of TRPV1 + peripheral sensory neurons [22], which while needed for mechanical hyperalgesia in this context, normally code for heat stimulus [44]. It could thus be hypothesized that this compensatory mechanism might modulate the effects of sigma-1 receptors on TRPV1 + sensory neurons.
Our findings also show strikingly similar sigma-1 receptor immunostaining patterns in mouse and human DRG samples, with reactivity detected in most (if not all) peripheral sensory neurons. TRPV1 is expressed in DRGs by peptidergic C neurons, which constitute a distinct cellular population in mice, with virtually no overlap with non-peptidergic C neurons, which can be labeled with IB4, as reported here and elsewhere [22], [45]. We showed that in vivo ablation of TRPV1 + neurons abolished the effect of sigma-1 agonists on the potentiation PGE2-induced mechanical hyperalgesia and in the relapse of weight bearing asymmetry 24 h after plantar incision. These sensory neurons are thus necessary for the pronociceptive effects of sigma-1 agonism in both circumstances. The enhancement of PGE2-induced hyperalgesia produced by the systemic administration of PRE-084 was abolished not only by administration of the standard TRP antagonist RR in the sensitized paw but also by the local administration of two different sigma-1 antagonists: BD-1063 and S1RA. Hyperalgesia was thus due to simultaneous TRP and sigma-1 activation at the sensitized site. In fact, locally administered PRE-084 at the PGE2-injected site was sufficient to significantly enhance hyperalgesia. Our results suggest that peripheral sigma-1 receptors play a prominent role in the pronociceptive actions of sigma-1 agonists. Just one previous report investigating the pronociceptive role of peripheral sigma-1 agonists showed that locally (intraplantarly) injected PRE-084 enhanced allodynia induced by activation of acid-sensing ion channels and purinergic P2X receptors [46]. Interestingly, both these targets are minimally present or even absent in mouse TRPV1 + neurons [45], [47]. We and others have shown that sigma-1 receptors can bind to [22], [48] and modulate TRPV1 activity [22], possibly explaining why the effects induced by sigma-1 agonism in this study were dependent on TRPV1.
Dextromethorphan is a widely used antitussive agent found in most over-the-counter cough-suppressing drugs [6]. Considering that more than 300 million people worldwide undergo surgery each year [49], it would not be unusual for some patients to be taking this drug at the time of surgery. Clinical studies investigating the influence of dextromethorphan on immediate post-operative pain have reported conflicting results, with some finding no apparent effect (e.g. [50], [51]) and others reporting analgesic effects, purportedly attributable to NMDA antagonism (e.g. [52], [53]). To our knowledge, no studies have examined the effects of dextromethorphan in the late postoperative recovery phase, when (according to our findings) it might enhance pain due to residual inflammation secondary to tissue injury and repair. It would be interesting to carry out a retrospective study examining associations between dextromethorphan treatment and postoperative pain duration. We also tested the selective sigma-1 agonists PRE-084 and pridopidine. PRE-084 is a prototypic sigma-1 agonist used in most preclinical sigma-1 receptor studies (e.g. [8]). It is far from being used in clinical settings. Pridopidine, by contrast, is currently being investigated for the treatment of Huntington’s disease [9] and ALS [10] in clinical trials. Pain is very common in both diseases [54], [55]. While they are eminently central nervous system disorders, blood from patients with Huntington’s disease shows increased inflammatory markers (such as C-reactive protein and IL-6) indicating peripheral inflammation [56]. A peripheral inflammatory component has also been described in ALS, and it might affect sensory neurons [57]. It could, therefore, be worth monitoring pain levels in patients with these diseases being treated with pridopidine in clinical trial settings. A similar recommendation could be made for other sigma-1 agonists, such as blarcamesine (ANAVEX2–73), which is currently being investigated in clinical trials for its potential to treat other neurodegenerative diseases, such as Alzheimer’s disease, Rett syndrome and Parkinson’s disease [58]. All these diseases cause pain [59], [60], [61] and, according to recent findings, involve a peripheral inflammatory response [62], [63], [64].
5. Conclusions
This study shows that the sigma-1 receptor is present in human and mouse DRG neurons and that sigma-1 agonism exacerbates pain-like responses in mice with mild inflammatory changes. The mechanism underlying the pronociceptive effects of sigma-1 agonism involves enhancement of the sensitizing actions of algogenic chemicals such as PGE2 that are released by immune cells and able to sensitize TRPV1 + nociceptors (Fig. 9A and B). Whether or not this potentiation of immune-driven pronociceptive effects by sigma-1 agonism also occurs in humans, either in medical practice or clinical trials, is unknown, but may merit further clinical investigation.
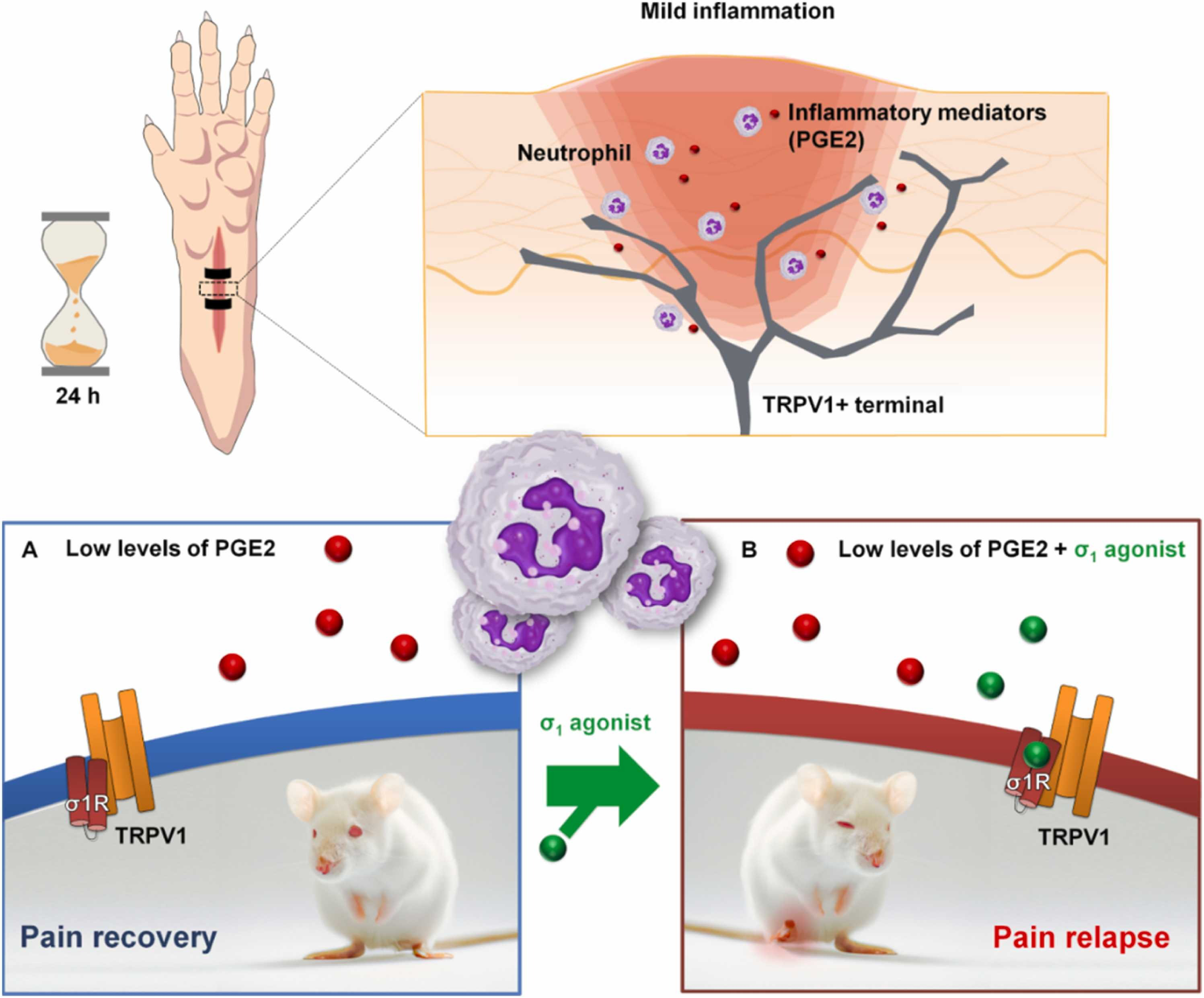
Proposed mechanism for the proalgesic actions of sigma-1 agonism during a mild inflammatory reaction.
(Upper panels) Neutrophilic inflammation is present 24 h after plantar incision. (A) Under normal conditions, the mice appear to recover from the pain induced by the surgical procedure performed 24 h earlier. (B) However, in the presence of a sigma-1 agonist, the mice once again exhibit pain-like behavior due to the enhancement of the effects of proalgesic mediators such as PGE2 released by immune cells, which recruit the actions of TRPV1 + peripheral sensory neurons.