
Sigma-1 receptor ablation impedes adipocyte-like differentiation of mouse embryonic fibroblasts

By Huan Yang, Hongtao Shen, Jing Li, Kristin I. Stanford, and Lian-Wang Guo
Excerpt from the article published in Cell Signal. 2020 Nov;75:109732. DOI: 10.1016/j.cellsig.2020.109732. Epub 2020 Aug 2. PMID: 32750415; PMCID: PMC7530065.
Editor’s Highlights
- Genetic ablation of sigma-1 receptor (Sig1R) impairs, and agonist activation of Sig1R enhances adipocyte-like phenotype of induced mouse embryonic fibroblasts (MEFs)
- Adipocytes play a major role in health and disease, especially with regard to their contribution to the development of obesity and diabetes
- Sig1R ablation impedes the body weight gain of male mice on high-fat diet
Abstract
The sigma-1 receptor (Sig1R) is a unique ligand-operated endoplasmic reticulum (ER) protein without any mammalian homolog. It has long been a pharmacological target for intervention of psychiatric disorders, and recently garnered refreshed interest for its neuroprotective potential. Though reported to modulate various intracellular events, its influence on cell identity is little known. We explored a role for Sig1R in adipocyte differentiation.
We induced adipogenic differentiation of mouse embryonic fibroblasts (MEFs) with a differentiation medium. MEFs were isolated from Sigmar1−/− and Sigmar1+/+ mice. The induced adipocyte-like phenotype was detected through Western blots of master transcription factors (PPARγ, CEBPA, SREBP1, SREBP2), lipogenic proteins (FABP4, ACC1, ACAT2), and Oil-Red-O staining of lipids. We found that the induced upregulation of these proteins and lipid accumulation were severely mitigated in Sigmar1−/− (vs Sigmar1+/+) MEFs. Sig1R activation with a selective agonist (PRE084) increased Sig1R protein and further enhanced the induced adipocyte-like phenotype in Sigmar1+/+ MEFs. We also determined mouse body weight gain induced by high-fat diet for 6 months, which was impeded in Sigmar1−/− (vs Sigmar1+/+) male mice.
In summary, genetic ablation of Sig1R impairs, and agonist activation of Sig1R enhances adipocyte-like phenotype of induced MEFs. In vivo, Sig1R ablation impedes the body weight gain of male mice on high-fat diet. This study warrants further investigation of a previously unrecognized role for Sig1R in adipocyte differentiation.
Introduction
The sigma-1 receptor (Sig1R) is a drug-binding site residing mostly in the endoplasmic reticulum (ER) [1,2]. Research on Sig1R has traditionally centered on its pharmacological targeting to treat psychotic disorders; some of the S1R ligands are in clinical use [3]. Recent pharmacological and genetic studies highlighted a potential in Sig1R modulation to combat neurodegenerative diseases [4]. Human genetics also pointed to a link between Sig1R mutations and familial motor neuron disease [5,6]. Efforts on mechanistic interpretation identified Sig1R-associated modulations of a wide variety of intracellular activities, ranging from ER stress, oxidative stress, inflammation, Ca2+ homeostasis, ion channel functions, GPCR signaling, to autophagy [4,[7], [8], [9], [10]]. While most of these align with Sig1R being a molecular chaperone [1], the exact context-specific Sig1R molecular actions remain largely obscure.
Earlier work implicated a potential role for Sig1R in lipid trafficking or metabolism. Sig1R contains steroid binding domain-like sequences [11]. It binds cholesterol and sphingolipids [[12], [13], [14]] and may influence ER microdomain formation and ER-mitochondria interaction [15]. Aside from the physical interaction of Sig1R with lipid molecules, Sig1R regulates the stability of UDP-galactose:ceramide galactosyltransferase (CGalT) [16]. In addition, Sig1R was also found on lipid droplets as revealed by Sig1R-GFP subcellular fluorescence [13]. Since accumulation of lipid droplets is a prominent feature of adipocytes, we became curious as to whether Sig1R is involved in adipocyte differentiation.
Adipocytes play a major role in health and disease, especially with regard to their contribution to the development of obesity and diabetes [17]. These cells originate from adipose-derived stromal cells (ASC) or their equivalents in most tissues [18]. Adipogenesis involves orchestrated gene regulations and protein changes [19]. Peroxisome proliferator-activated receptor gamma (PPARγ) and CCAAT-enhancer-binding protein alpha (CEBPA) are master transcription factors dictating late-stage adipocyte differentiation [20]. The nuclear receptor PPARγ is critical for both differentiation and phenotype maintenance of adipocytes.
Here we explored Sig1R’s influence on adipocyte phenotype using mouse embryonic fibroblasts (MEFs) that were isolated from Sigmar1−/− and Sigmar1+/+ mice. We found that the adipocyte-like phenotype induced by a differentiation medium, that is, increase of PPARγ, CEBPA and other markers and lipid storage [20], were abated in the absence of Sig1R; and, agonist activation of Sig1R had an opposite effect. In vivo, Sig1R ablation reduced high fat diet-induced body weight gain in male mice. This previously unrecognized Sig1R-specific effect on induced adipocyte-like phenotype may inspire future studies on Sig1R as a potential player in adipogenesis.
Results
Sig1R ablation abates the upregulation of lipogenesis-associated proteins in MEFs induced for adipocyte differentiation
To investigate the possible involvement of Sig1R in adipogenic cell differentiation, we first determined the effect of Sig1R ablation on adipocyte marker proteins. We opted to use primary mouse embryonic fibroblasts (MEFs) for differentiation towards adipocyte-like cells – a well-established method applied in studies of adipogenesis21. MEFs were isolated from Sigmar1−/− and littermate Sigmar1+/+ control mice, which were bred from the colonies established in our published studies22. MEF adipogenic induction was performed using the standard adipocyte differentiation medium23, and the levels of lipogenic marker proteins were determined. As seen in Figure 1, Sig1R knockout was confirmed by Western blot. Interestingly, whereas the differentiation medium induced upregulation of FABP4, ACC1, and ACAT2 in Sigmar1+/+ MEFs, this upregulation did not occur in Sigmar1−/− cells (Figure 1B). In contrast to the above three marker proteins, GOT2, a mitochondrial inner-membrane protein essential for the malate-aspartate shuttle in the glycolysis pathway, did not show appreciable changes across the four experimental conditions (Figure 1B). GOT2 thus conferred a negative control indicative of the functional specificity of Sig1R observed here. It is noted that in the basal growth medium (which does not induce adipogenic differentiation), MEFs from Sigmar1−/− and Sigmar1+/+ did not exhibit a major difference in the levels of the three lipogenic proteins. This result indicated that these proteins were not disturbed in the basal condition without Sig1R, supporting a specific role for Sig1R in the induction of adipogenic differentiation.
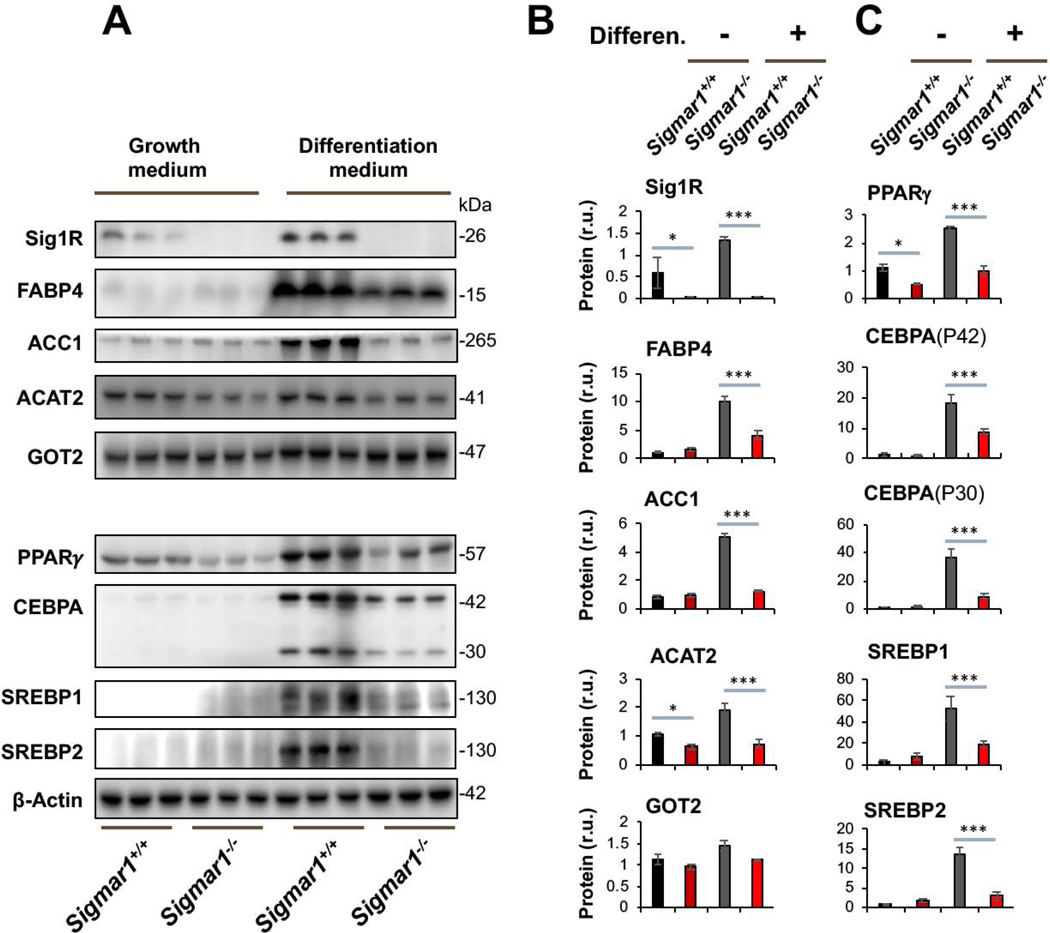
Lower marker protein levels in Sigmar1−/− (vs Sigmar1+/+) MEFs induced for adipocyte differentiationMEFs isolated from Sigmar1−/− or Sigmar1+/+ mouse embryos were cultured in the growth medium (undifferentiated control) or in the differentiation medium for adipogenic induction, as described in Methods. The cells induced for 8 days were harvested for Western blot analysis.
A. Western blots. The replicate bands of each condition represent MEF samples from 3 different embryos. Densitometry was normalized to β-actin. The value for Sig1R in Sigmar1−/− cells is the background on the blot; r.u., relative unit.
B and C. Quantification. Mean ± SD, n = 3 embryos as different sources of MEFs. Statistics: One-way ANOVA with Bonferroni test, *P<0.05, ***P<0.001.
Sig1R ablation blocks the upregulation of master transcription factors in MEFs induced for adipocyte differentiation
Western blot analysis further revealed that while PPARγ and CEBPA were dramatically upregulated in Sigmar1+/+ MEFs by adipogenic induction, neither were upregulated in Sigmar1−/− cells (Figure 1C). These two proteins are well established master transcription factors that drive adipocyte differentiation, both in development in vivo and in response to adipogenic differentiation of MEFs in vitro20. CEBPA encodes an mRNA that contains alternative translation initiation sites resulting in two major protein isoforms: the fully translated CEBPA (P42), and an N-terminally truncated protein (P30) lacking the N-terminal transactivation domain. The upregulation of both CEBPA isoforms in response to induction of adipogenesis was blunted in the absence of Sig1R (Figure 1C). We also determined protein levels of SREBP1 and SREBP2, master transcription factors governing fatty acid and cholesterol synthesis, transport, and homeostasis, as well as active participants in adipocyte differentiation24. SREBP1 and SREBP2 behaved similarly to PPARγ and CEBPA.
Taken together, these data indicate that while increased protein production of PPARγ, CEBPA, SREBPs, FABP4, ACC1, and ACAT2 were stimulated in the in vitro model of MEF adipogenic differentiation, this process was mostly averted in the absence of Sig1R. As these proteins are key factors in adipocyte differentiation and maintenance, our results suggest a novel role for Sig1R in induced adipogenesis.
Sig1R ablation prevents accumulation of Oil Red O-stained lipids in MEFs induced for adipocyte differentiation
After identifying the Sig1R influence on the marker proteins, we further characterized Sigmar1−/− and Sigmar1+/+ MEFs after adipogenic induction for their adipocyte-like phenotype at the cellular level. We performed Oil Red O staining of lipids in intact cells, a standard approach to the characterization of adipocyte differentiation. As shown in Figure 2A, Oil Red O staining was intensified in Sigmar1+/+ cells after adipogenic induction, an effect not obviously seen in the Sigmar1−/− MEF culture. This observation was corroborated with quantified data (Figure 2B and and2C).2C). The Oil Red O staining was 6-fold higher in Sigmar1+/+ cells after adipogenic induction. However, no significant difference between the two medium conditions was seen in Sigmar1−/− cells. Furthermore, the black/white images showed that there was no obvious change in cell density when compared between Sigmar1−/− and Sigmar1+/+ cells in the differentiation medium (Figure 2D); rather, there were fewer oil-rich Sigmar1−/− (vs Sigmar1+/+) cells. Together, these data provide direct evidence for the inhibitory effect of Sig1R ablation on adipogenic-induced MEF differentiation.
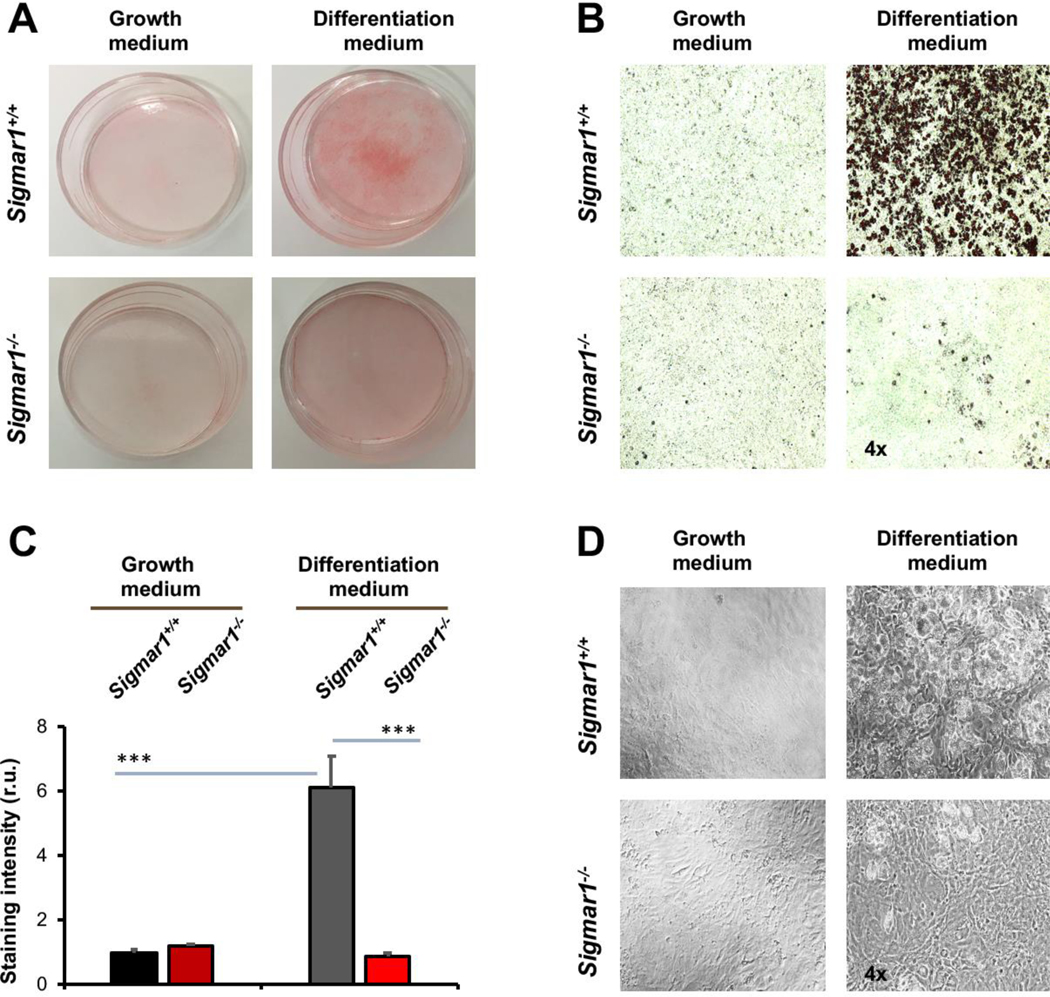
Lower lipid storage in Sigmar1−/− (vs Sigmar1+/+) MEFs induced for adipocyte differentiationMEFs isolated from Sigmar1−/− or Sigmar1+/+ mouse embryos were cultured in the growth medium (undifferentiated control) or in the differentiation medium for adipogenic induction. Cell cultures induced for 8 days were used for Oil Red O staining.
A. Overview of Oil Red O-stained cells in culture dishes.
B. Microscopic images of Oil Red O-stained cells.
C.Quantification of staining intensity in B (using ImageJ). Mean ± SD, n = 3 embryos as different sources of MEFs; r.u., relative unit. Statistics: One-way ANOVA with Bonferroni test, ***P<0.001.
D.Black and white bright-field image of cell culture in the dish.
Agonist activation of Sig1R enhances induced adipocyte-like differentiation of MEFs
Sig1R is best known as an ER-residing drug binding site, and highly Sig1R-selective agonists and antagonists have been developed and widely applied in clinical and preclinical studies10. Taking advantage of these known agonists and antagonists, we treated Sigmar1+/+ MEFs with the commonly used Sig1R-selective agonist PRE084 and antagonist BD1047 in order to further characterize the role for Sig1R in adipogenic differentiation (Figure 3). Agonist activation of Sig1R served as gain-of-function complementary to the above loss-of-function experiments using Sigmar1−/− MEFs. Consistent with a general observation in the literature with different cell types25, the treatment of MEFs with PRE084 increased Sig1R protein after adipogenic induction, thus validating the efficiency of this agonist used here. More importantly, while the protein levels of PPARγ, CEBPA, SREBPs, FABP4, and ACC1 increased in response to adipogenic induction, treatment with PRE084 (vs vehicle) further markedly elevated these proteins (Figure 3). In contrast, treatment with the Sig1R-selective antagonist BD1047 did not produce this effect, indicative of the functional specificity of Sig1R agonist activation. No significant change was observed with GOT2, a result consistent with that from the Sigmar1−/− MEF experiment (Figure 1), hence adding another layer of specificity to Sig1R’s role in adipogenic differentiation. Moreover, further determination via Oil Red O staining indicated that the Sig1R agonist and antagonist significantly increased or decreased, respectively, lipid accumulation in response to adipogenic-induced differentiation (Figure 4).
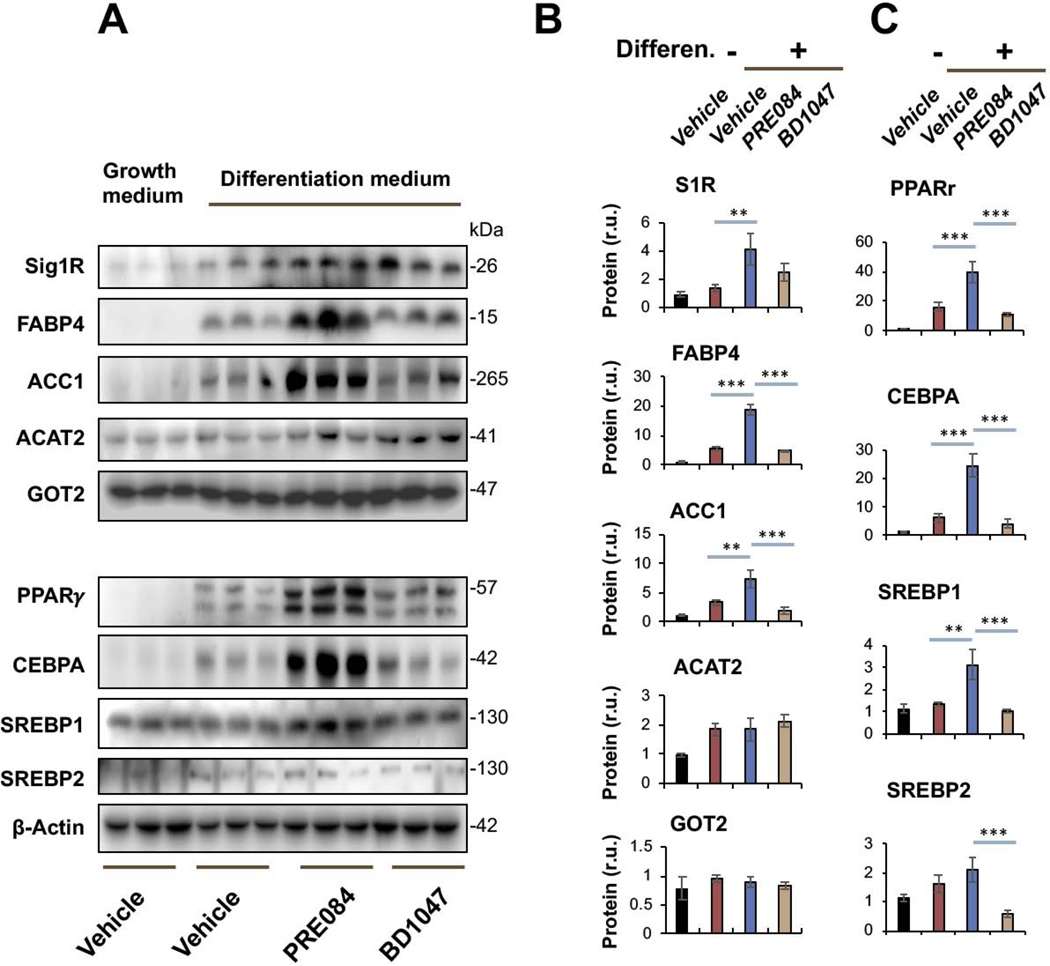
Agonist-enhanced production of adipocyte-like phenotype marker proteins in Sigmar1+/+ MEFs induced for adipocyte differentiationMEFs isolated from Sigmar1+/+ mouse embryos were cultured in the growth medium (control) or in the differentiation medium for adipogenic induction. Vehicle (DMSO) or Sig1R agonist (PRE084, final 0.5 μM) or antagonist (BD1047, final 0.2 μM) was included when changing to the differentiation medium. The cells induced for 8 days were harvested for Western blot analysis.
A. Western blots. The replicate bands of each condition represent MEF samples from 3 different embryos. Densitometry was normalized to β-actin.
B and C. Quantification. Mean ± SD, n = 3 embryos as different sources of MEFs. Statistics: One-way ANOVA with Bonferroni test, *P<0.05, ***P<0.001.
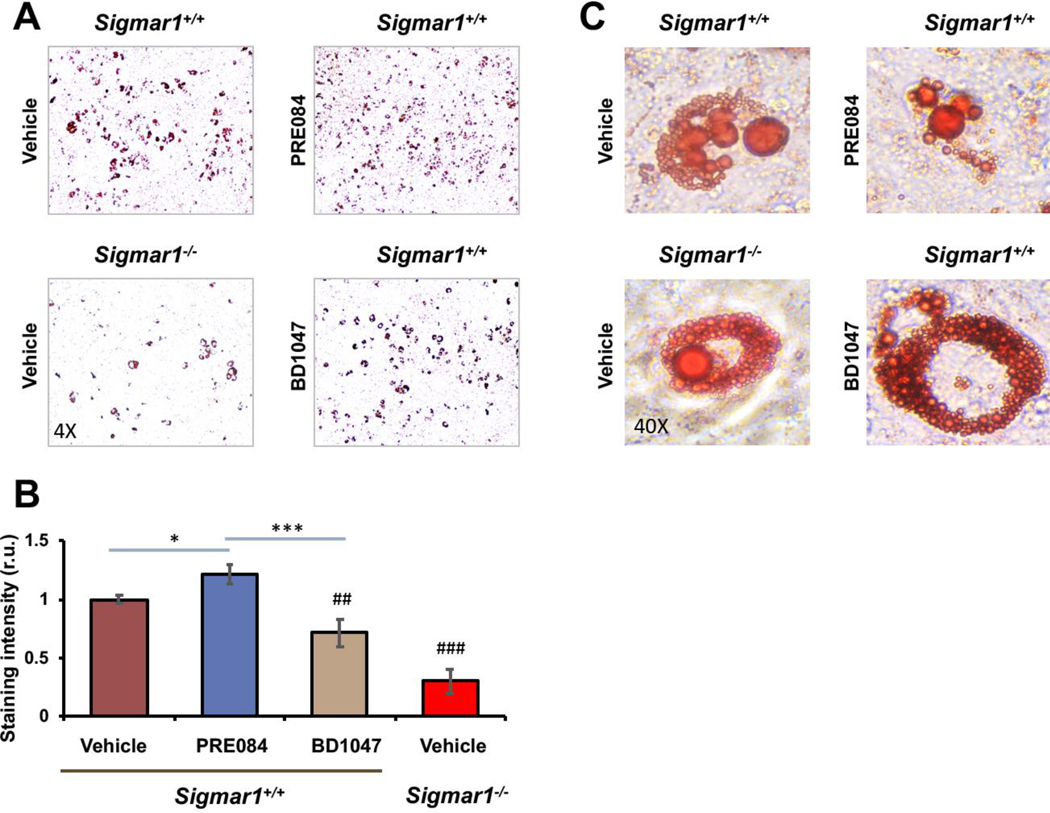
Agonist-enhanced lipid accumulation in Sigmar1+/+ MEFs induced for adipocyte differentiationMEFs isolated from Sigmar1−/− or Sigmar1+/+ mouse embryos were cultured in the differentiation medium for adipogenic induction. Cell cultures induced for 8 days were used for Oil Red O staining.
A. Microscopic images of Oil Red O-stained cells.
B. Quantification of staining intensity (ImageJ). Mean ± SD, n = 3 embryos as different sources of MEFs; r.u., relative unit. Statistics: One-way ANOVA with Bonferroni test, *P<0.05, ***P<0.001; ##P<0.01, ###P<0.001, both compared to the Sigmar1+/+ vehicle control (the first bar).
C. High-magnification images showing Oil Red O-stained lipid droplets.
Sig1R ablation impairs the body weight gain of male mice fed on high-fat diet
Up to this point, our in vitro loss- and gain-of-function data revealed a profound influence of Sig1R on the salient adipocyte-like, biochemical and cellular phenotypic parameters, including levels of master transcription factors, lipogenic proteins, and lipid accumulation (Figures 1–4). As adipocyte differentiation is a well-known contributor to body weight gain, we investigated the role of Sig1R in vivo in mice fed with a high-fat diet (HFD). When fed on a normal diet, the Sigmar1−/− animals were fertile and healthy without showing a difference in body weight compared to age-matched Sigmar1+/+ mice (Figure 5A), as previously shown26. However, Sigmar1−/− mice had a markedly lower average body weight compared to Sigmar1+/+mice after 4 weeks on a HFD. This HFD-induced phenotype continued throughout to the end of the 6-month time course, and was observed in male, but not female mice (Figure 5B). This difference in body weight was independent of food intake; rather, the food intake normalized to net body weight gain was actually higher in Sigmar1−/− mice compared to Sigmar1+/+ mice (Figure 5C).
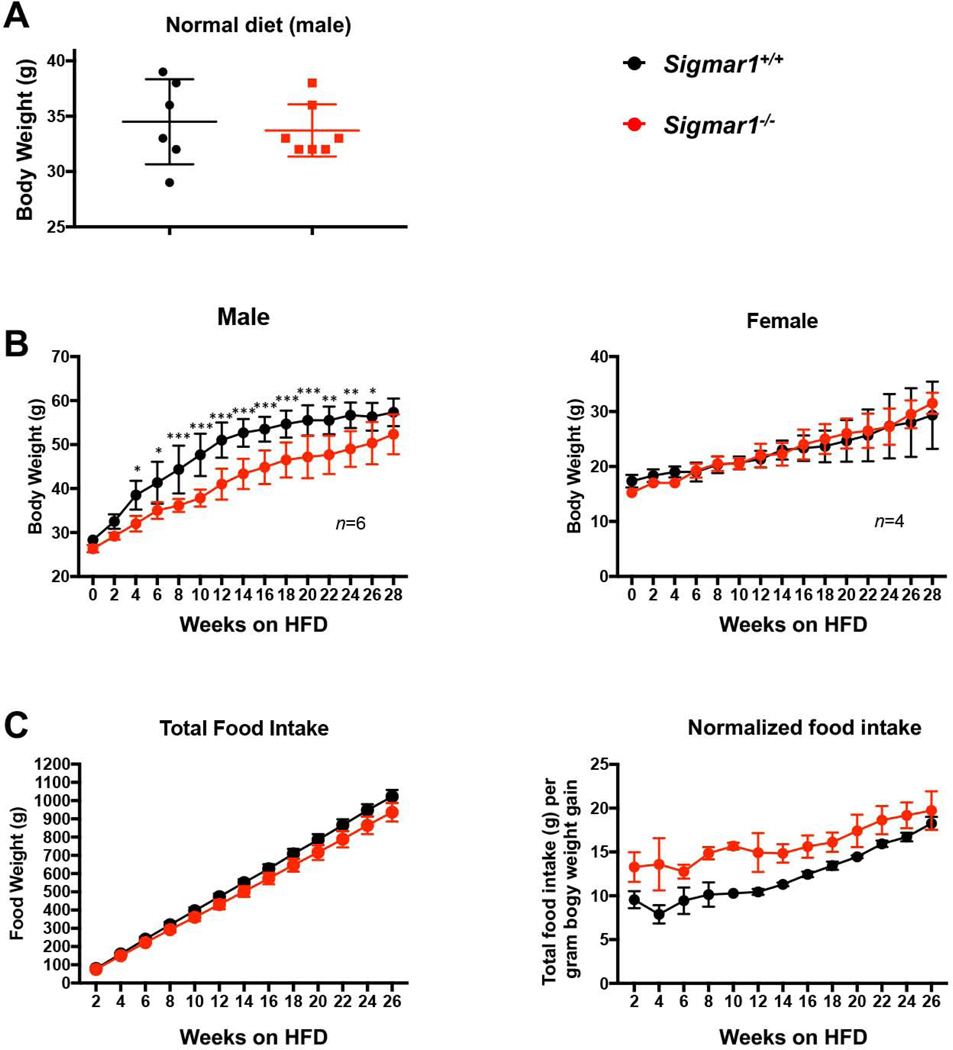
Impeded body weight gain of Sigmar1−/− (vs Sigmar1+/+) male mice on high fat dietMale and female Sigmar1−/− and Sigmar1+/+ mice at age of 8 weeks (referred to as 0 week on the plots) were put on high fat diet (HFD) for the following 6 months. Body weight and food intake were measured every two weeks.
A. Comparison of body weights between male Sigmar1−/− and Sigmar1+/+ mice on normal diet. In parallel to the HFD diet, male mice of equivalent age were kept on normal diet. The plotted data were collected at the time point corresponding to 28 weeks in B. Statistics: unpaired Student’s t-test, no significance (P>0.05); n = 6 Sigmar1+/+ mice and 7 Sigmar1−/− mice.
B.Time course of body weight recording throughout 28 weeks. Statistics: Repeated-measures 2-way ANOVA followed by multiple comparisons corrected by Sidak test; *p<0.05, **p<0.01, ***p<0.001; n= 6 males and 4 females.
C. Time course of cumulative food intake, plotted as net food intake or intake normalized to net body weight gain at each time point.
In summary, the in vitro experiments showed profound influence of Sig1R genetic ablation or pharmacological activation on the adipocyte-like phenotype of MEFs under adipogentic induction with differentiation medium (Figure 1–4). The in vivo data revealed that when fed on HFD, body weight gain was impeded in Sigmar1−/− vs Sigmar1+/+ male mice (Figure 5).
Discussion
Sig1R has been the target of intensive studies of medicinal chemistry in pursuit of Sig1R-selective compounds for intervention of psychiatric disorders9. However, the interpretation of Sig1R-specific molecular/cellular activities lagged behind27. On the one hand, a pharmaceutical potential is enticing. On the other hand, there are no mammalian Sig1R homologs for deducing its functional pattern. In particular, Sig1R’s influence on cell identity has been poorly explored14. In this regard, it is intriguing to observe the pronounced effect of Sig1R loss- or gain-of-function on adipocyte phenotype, which was not previously identified in published data.
Reports showed that a broad spectrum of intracellular events and signaling players were influenced by the expression level or ligand regulation of Sig1R; in most cases, the influence was moderate. In a general sense, Sig1R is deemed a pluripotent modulator which fits its character as a chaperone25. In accordance, Sig1R knockout mice do not show overt phenotypes in normal conditions26, 28. Likely for this sake, the biological importance of Sig1R has been overall underestimated, as partly reflected by the lack of knowledge regarding Sig1R in cell identity change or maintenance. It was therefore somewhat striking for us to see the profound effect of Sig1R ablation on induced adipocyte phenotype, as detected via the levels of lipogenic enzymes, adipogenic master transcription factors, and lipid accumulation.
Among these critical players in adipocyte differentiation, PPARγ and CEBPA are master transcription factors that govern late-stage differentiation and phenotype maintenance, and they regulate each other19. A major target gene of PPARγ is the intracellular lipid transporter FABP4 which is highly expressed upon adipocyte differentiation24 as also observed here. While participating in adipogenic differentiation, SREBPs mainly control the transcription of lipogenic enzymes including ACC1 and ACAT224. ACC1 catalyzes carboxylation of acetyl-CoA to produce malonyl-CoA, a critical step in de novo fatty acid synthesis. ACAT2 is the chief enzyme responsible for the esterification of cholesterol. Fatty acids and esterified cholesterol are the major contents of lipid droplets. This string of events, transcription factor action -> lipogenic enzyme production -> lipid storage in droplets, is well established in the literature20. As such, that this process was overall markedly altered due to the absence of Sig1R is a significant observation.
Based on the aforementioned string of adipogenic events, it is reasonable to speculate that Sig1R influenced adipogenic differentiation of MEFs by altering the protein levels of master transcription factors. However, it has been notoriously challenging to explain the Sig1R-associated molecular mechanisms. One major hurdle is that aside from the evidence for Sig1R being a chaperone1, the exact molecular function of Sig1R is largely unknown27. A pioneering report showed that Sig1R knockdown in Chinese hamster ovary cells upregulated UDP-galactose:ceramide galactosyltransferase (CGalT)16. This finding may not be directly relevant to the current study, since the Sig1R negative regulation of CGalT does not explain its positive effect on lipogenic enzymes during MEF adipogenic differentiation. Another layer of complication exists; i.e., since Sig1R is an anti-stress signaling modulator, loss of the Sig1R protein per se can be an intracellular stress22. With this in mind, it remains an open question as to whether Sig1R influences the adiopogenic proteins directly via a chaperone/client relationship or indirectly through intracellular signaling such as altered cellular stress levels. For the former, a body of work is available indicating that adipogenesis is regulated by chaperone proteins including HSP9029, HSPA12A30, and PGRMC231. For the later, reports exist that ER stress and autophagy (where Sig1R plays an important role)1, 32 influence adipocyte differentiation33, 34. In either case, it is worth further investigation especially given the magnitude of influence imposed either by Sig1R genetic depletion or its pharmacological activation.
In this study we opted to use the model of induced adipogenic differentiation with primary MEFs35 directly isolated from Sigmar1−/− and Sigmar1+/+ mouse embryos. By doing so we avoided a siRNA or CRISPR approach, which may not completely eliminate the Sig1R protein yet may result in off-target complications. Moreover, primary cells are more authentic than immortalized cell lines for interpreting endogenous cellular physiology21. In future experiments, the results from MEFs will be reevaluated using ASC isolated from Sigmar1−/− and Sigmar1+/+ mice. On the other hand, permanent Sig1R depletion is possibly accompanied by compensatory signaling. To address this concern, we also performed instant activation of Sig1R with an agonist which complemented the permanent knockout setting. Importantly, the result from this gain-of-function approach agreed with that from using loss-of-function (knockout) cells. It is noteworthy that albeit showing an overall trend of mitigated differentiation (vs vehicle control), antagonist-treated cells did not exactly phenocopy knockout cells. The reason could be twofold. First, antagonist occupancy blocks the agonist binding in Sig1R but does not necessarily reduce the protein. Thus it may have a different functional consequence than the Sig1R knockout. Whereas antagonist binding promotes Sig1R oligomerization36, the physical expulsion of the Sig1R protein may alter the ER membrane microdomain architecture15. Second, the putative endogenous agonists bind Sig1R with very low affinities (over 10 μM)26, 37. If Sig1R was not already endogenously activated before adding BD1047, an effect of the antagonist would not manifest. This may also explain the prominent effect of the exogenous high-affinity Sig1R agonist PRE084 which can readily compete with endogenous ligands for binding Sig1R. Nonetheless, by applying agonist activation of Sig1R which also raised endogenous Sig1R protein levels, we were able to avoid ectopically overexpress Sig1R which disrupts the endogenous signaling and, if not properly folded, can cause extra stress5.
Reduced weight gain in male Sigmar1−/− mice is another interesting result that has not been previously reported. It could be rationalized by impaired differentiation of Sigmar1−/− MEFs to an adipocyte-like phenotype as observed here in vitro, since adipose tissue is an important contributor to body weight gain. We did not see significantly changed adipocyte marker levels (except for adiponectin) or cell size in Sigmar1−/− (vs Sigmar1+/+) mouse adipose tissues collected at the end of 6 months of HFD feeding (Figures S1 and S2). However, there are caveats in these results. First, the markers were detected using tissues which contained different cell types. Second, body weights were nearly plateaued at the end of 6 months, and the weight of Sigmar1−/− mice appeared to “catch up” with that of Sigmar1+/+ mice. Thus, to attain conclusive comparison of adipogenesis between Sigmar1−/− and Sigmar1+/+ animals, further investigation would be performed using adipocytes isolated from fresh adipose tissues, preferably at fast-growth earlier time points.
In a recent report38, HFD-induced peripheral neuropathy was alleviated in Sigmar1−/− mice, but no body weight data was available. In another study, treating mice with a Sig1R antagonist (BD1063) reduced overconsumption of palatable (high-sucrose) food during the first hour of feeding39. This short-term effect appears to be more neuropsychiatry-related, an analogy being that Sig1R is a cocaine-binding site involved in drug addiction26. Interestingly, in a multiple-treatments meta-analysis to compare 15 antipsychotic drugs, haloperidol along with ziprasidone and lurasidone were the only ones not associated with significantly more weight gain than placebo in adult patients40. Though a known Sig1R antagonist, haloperidol is not Sig1R selective12, 26. Moreover, antipsychotic drugs often impact food intake41. Therefore, caution must be taken in the interpretation of the relationship between these drugs and body weight changes. In contrast to the pharmacological studies, in our long-term HFD feeding experiments showing lower Sigmar1−/− vs Sigmar1+/+ body weights, we did not see reduced food intake of Sigmar1−/− mice compared to the wild type animals over a 6-month period. Nevertheless, the well-documented importance of Sig1R in animal behaviors should be taken into account when thinking of Sig1R for metabolic intervention. Sex is another important variable. Sig1R is a sterol binding protein with some putative endogenous ligands being steroid hormones25. However, body weight is also subject to other factors such as housing environment, type of HFD, bedding, or even the season when measurement is made. Therefore, the difference in body weight observed between male and female mice needs to be re-examined in continued experiments. As body weight is a complex issue directly related to obesity, our findings may open a potential avenue for future research into the utility of SigR1 as an anti-obesity therapeutic target.
Conclusion
The influence of Sig1R on cell identity is an underexplored area. We found that induced adipocyte-like phenotypic parameters of MEFs were down- and up-regulated, respectively, due to Sig1R ablation and its pharmacological activation. The observed effect is deemed Sig1R-specific because primary MEFs were isolated from Sigmar1−/− and Sigmar1+/+ mouse embryos. With many Sig1R-binding drugs in cache and ligand-bound Sig1R structures recently solved27, 42, we hope this study would prod interest in further evaluating the potential of targeting Sig1R for modulation of adipogenesis.