
Crystal structures of the σ2 receptor template large-library docking for selective chemotypes active in vivo

By Assaf Alon, Jiankun Lyu, Joao M. Braz, Tia A. Tummino, Veronica Craik, Matthew J.O’Meara, Chase M. Webb, Dmytro S. Radchenko, Yurii S. Moroz, Xi-Ping Huang, Yongfeng Liu, Bryan L. Roth, John J. Irwin, Allan I. Basbaum, Brian K. Shoichet, and Andrew C. Kruse
Excerpt from the article published in bioRxiv 2021.04.29.441652; April 30, 2021; DOI: https://doi.org/10.1101/2021.04.29.441652
Editor’s Highlights
- The σ receptors are integral membrane proteins widely expressed in both the central nervous system and in peripheral tissues, including the liver and kidney.
- The σ2 and the σ1 receptors are promiscuous, both binding to cationic amphiphiles, and as expected, there is broad cross-reactivity between the two receptors.
- The σ2 receptor has been pharmacologically enigmatic for 30 years.
- The σ2 receptor is distantly related to emopamil-binding protein (EBP), the mammalian Δ8,7 sterol isomerase required for cholesterol synthesis, as well as to other proteins in this family, including TM6SF2, which regulates liver lipid homeostasis.
- The evolutionary connection of σ2 to EBP and the structure of the receptor bound to cholesterol strongly imply an ability to recognize sterols.
- σ2 ligands achieve peak antiallodynic effects 24 hours after dosing.
Abstract
The σ2 receptor is a poorly understood transmembrane receptor that has attracted intense interest in many areas of biology including cancer imaging, Alzheimer’s disease, schizophrenia, and neuropathic pain. However, little is known regarding the molecular details of the receptor, and few highly selective ligands are available. Here, we report the crystal structure of the σ2 receptor in complex with the clinical drug candidate roluperidone and the probe compound PB28. These structures, in turn, templated a large-scale docking screen of 490 million make-on-demand molecules. Of these, 484 compounds were synthesized and tested, prioritizing not only high-ranking docked molecules, but also those with mediocre and poor scores. Overall, 127 compounds with binding affinities superior to 1 μM were identified, all in new chemotypes, 31 of which had affinities superior to 50 nM. Intriguingly, hit rate fell smoothly and monotonically with docking score. Seeking to develop selective and biologically active probe molecules, we optimized three of the original docking hits for potency and for selectivity, achieving affinities in the 3 to 48 nM range and to up to 250-fold selectivity vs. the σ1 receptor. Crystal structures of the newly discovered ligands bound to the σ2 receptor were subsequently determined, confirming the docked poses. To investigate the contribution of the σ2 receptor in pain processing, and to distinguish it from the contribution of the σ1 receptor, two potent σ2-selective and one potent σ1/σ2 non-selective ligand were tested for efficacy in a mouse model of neuropathic pain. All three ligands demonstrated timedependent decreases in mechanical hypersensitivity in the spared nerve injury model, supporting a role for the σ2receptor in nociception, and a possible role for σ1/σ2 polypharmacology. This study illustrates the opportunities for rapid discovery of in vivo active and selective probes to study under-explored areas of biology using structurebased screens of diverse, ultra-large libraries following the elucidation of protein structures.
Introduction
The σ receptors are integral membrane proteins widely expressed in both the central nervous system and in peripheral tissues, including the liver and kidney. The σ receptors are divided into σ1 and σ2 “subtypes” based on differences in tissue distribution and in pharmacological profile1, but despite their names, the two proteins are entirely unrelated in sequence. Cloned in 1996, the σ1 receptor has no paralog within the human genome; its closest homolog of known function is the yeast Δ8,7 sterol isomerase ERG22. Pharmacological studies conducted on σ1 knockout mice3 showed that the σ2 is not a splice variant or other modified form of σ1, but rather derives from an unrelated gene. The molecular identity of the σ2receptor remained unknown until recently, when we purified it from calf liver tissue4 and showed that it is TMEM97, an ER-resident membrane protein that regulates the sterol transporter NPC15,6. TMEM97 is predicted to be a four-helix bundle protein with both amino and carboxy termini facing the cytoplasm. A member of the EXPERA family7, the σ2 receptor is distantly related to emopamil-binding protein (EBP), the mammalian Δ8,7 sterol isomerase required for cholesterol synthesis, as well as to other proteins in this family, including TM6SF2, which regulates liver lipid homeostasis8.
Despite relatively little being known about the role of σ2 in baseline physiological processes, the receptor has been implicated in multiple disease states. For example, the σ2 receptor is overexpressed in proliferating cells and in many tumors9, and labeled σ2 ligands have been proposed as tools for diagnosis and therapy for various cancers10,11. Additionally, the σ2receptor was recently identified as an interaction partner of the SARS-CoV-2 viral protein, Orf9c, during cellular infection12. Recently it was reported that a ternary complex between the σ2 receptor, PGRMC1, and the LDL receptor increases the rate of LDL internalization13. Consistent with its high expression in the nervous system, the σ2 receptor has also been proposed as a target for the treatment of multiple nervous system disorders. The σ2 receptor ligand Elayta (CT1812) is currently in clinical trials for mild to moderate Alzheimer’s disease14, and another ligand, roluperidone, (MIN-101) is in clinical development for treatment of the negative symptoms of schizophrenia15–17. When tested in animal models, σ2receptor ligands reduce alcohol-withdrawal symptoms18,19 and have a neuroprotective effect in brain injury20. Finally, recent studies have implicated σ2 in chronic pain19,21,22, with σ2ligands having anti-allodynic effects in nerve-injury induced models of neuropathic pain. As this is also thought to be true of σ1 ligands, and because most σ2 ligands cross-react with the σ1 receptor, probe ligands selective for σ2 over σ1 would help illuminate σ2 biology and could be leads for novel therapeutics. However, given the relatively recent determination of the σ2receptor’s molecular identity relatively little is known regarding its molecular architecture, ligand recognition, or amenability to methods like virtual screening for ligand identification23–30. Here, we employed a biochemical and structural approach combined with computational docking to address these issues.
Crystallization and structure determination
The human σ2 receptor was expressed in Sf9 insect cells and was extracted with lauryl maltose neopentyl glycol (LMNG) detergent and purified as described4. Size exclusion chromatography multi-angle light scattering (SEC-MALS) experiments showed that the receptor is a dimer in solution, and that the presence of ligands did not perturb this oligomeric state (Supplementary Information Fig. 1a). As the human σ2 receptor did not lend itself to structural studies, further experiments were performed with the bovine σ2 receptor, which was more tractable. Circular dichroism (CD) experiments showed that the bovine σ2 receptor has a 74% helical content (Supplementary Information Fig. 1b), in agreement with secondary structure predictions. CD thermal unfolding experiments demonstrated that the receptor is remarkably stable compared to most mammalian membrane proteins, with a midpoint of the unfolding transition (Tm) of 54 °C (Supplementary Information Fig. 1c). The Tm increased by 1-3 °C when the receptor was incubated with various ligands.
Several rounds of construct optimization and of crystallization conditions led to high-quality diffracting crystals of the bovine σ2 receptor by the lipidic cubic phase method (Supplementary Information Fig. 2a-c). Three data sets were collected, one with receptor bound to the high-affinity non-selective ligand PB2831, to a resolution of 2.94 Å, another with receptor bound to the schizophrenia drug candidate roluperidone at a resolution of 2.7 Å, and yet another with the receptor bound to a ligand tentatively modeled as cholesterol at a resolution of 2.6 Å. The σ2 receptor has no homologs with known structure, and a BLAST search against the Protein Data Bank yielded no results. A more sensitive hidden Markov model search with HHpred32 identified EBP as a distant homolog, consistent with both proteins being members of the EXPERA family7. We used the structure of EBP33 to build a homology model of the σ2 receptor and performed a molecular replacement search that generated a marginally interpretable electron density map. Manual placement of an additional copy of the σ2 receptor followed by iterative manual building and reciprocal space refinement led to high-quality final structure (Supplementary Information Table 1).
Overall structure of the σ2 receptor
The three initial σ2 receptor crystal structures are highly similar, with a backbone root mean square deviation (RMSD) of 0.71 Å. As anticipated from multi-angle light scattering, the structures showed that σ2 is an intimately associated homodimer, burying 890 Å2 of surface area in dimer interface that is mainly formed by transmembrane helix 3 (TM3; Fig. 1a). The two protomers adopt the same conformation (backbone RMSD of 0.34 Å), with each protomer showing the expected four-helix bundle fold with both the amino and carboxy termini facing the same face of the membrane, likely the cytosolic face.
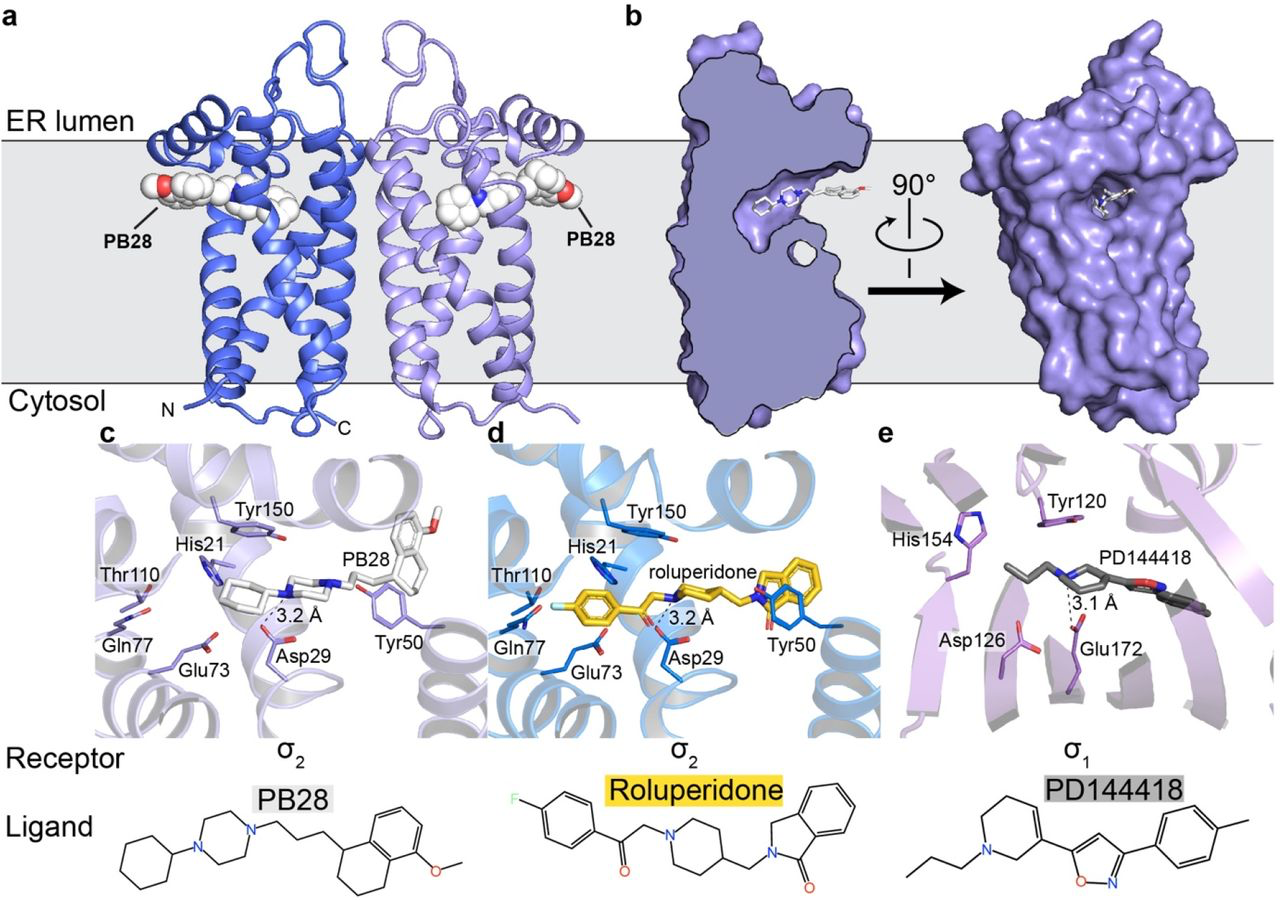
Overall structure of the σ2 receptor and binding site ligand recognition.
a, Structure of the σ2 receptor bound to PB28. Amino- and carboxy-termini are indicated. Membrane boundaries were calculated using the PPM server37. b, Cross-section of the σ2 receptor binding pocket (left) and view of the entrance to the binding pocket from the membrane. c, View of PB28 binding pose, showing charge–charge interaction with Asp29 (black dotted line) and contacts with other binding pocket residues. d, Analogous structure of the roluperidone binding pose. e, Structure of the σ1receptor bound to PD144418. Amino acids that serve similar roles and positioned in a similar orientation to amino acids in the σ2 receptor are indicated.
The four transmembrane helices of the protein are all kinked due to the presence of proline residues in each helix, creating a cavity slightly above the center of the membrane, nearer the ER-facing side of the membrane. Surprisingly, the ligand-binding cavity is entirely occluded from solvent by extracellular loops 1 and 2, which form a well-ordered cap over the luminal surface of the protein. Asp56, which was shown to be crucial for ligand binding4, is located in extracellular loop 1 and is involved in a network of hydrogen bonds likely important for proper folding (Supplementary Information Fig. 2d), hence the deleterious effect of mutating this residue on binding4. Instead of opening to the lumen of the ER, the pocket opens laterally into the lipid bilayer (Fig. 1b), reminiscent of lipid-binding G protein-coupled receptors34. The opening to the binding pocket is lined with hydrophobic and aromatic residues. Ligands may enter through this opening in their neutral, deprotonated form, and then become protonated in the binding site, allowing formation of a salt bridge with the conserved Asp29 (Fig. 1c-d). A second highly conserved acidic residue, Asp73, is located 3 Å away from Asp29, suggesting that these residues are hydrogen-bonded to each another, implying that Asp73 is likely protonated.
The two σ receptors are not homologs and do not share the same fold; the σ2 receptor is a four-helix bundle, while the σ1 receptor has a β-barrel cupin fold35. Despite this, the binding pockets of the two receptors are remarkably similar (Fig. 1c-e), placing functionally similar amino acids in cognate spatial positions, which is perhaps the result of convergent evolution and explains how two very different folds can share the same pharmacology.
It is noteworthy that both σ receptors are homologs of proteins that catalyze the same step in sterol biosynthesis. The σ1 receptor is a homolog of ERG2, the yeast Δ8,7 sterol isomerase; the σ2 receptor is a homolog of EBP, the mammalian enzyme that performs the same catalytic step in the biosynthesis of cholesterol. Both EBP and ERG2 rely on two similarly placed acidic residues in their active site for catalysis, which is thought to occur by protonation of the substrate at carbon 9 (C9) followed by proton abstraction from C7, which shifts the double bond into the C8-C7 position. All necessary components for catalysis appear to be present in σ2 receptor, in addition to the conserved polar residues His21, Gln77, and Thr110, located at the distal end of the ligand cavity, which may aid in recognizing the hydroxyl moiety of sterols. However, the σ2 receptor cannot act as a sterol isomerase. It can neither function in vivo to rescue a strain of yeast that lacks ERG2 (Supplementary Information Fig. 3a) nor can it function in vitro to convert zymostenol to lathosterol (Supplementary Information Fig. 3b). The same is true for the σ1 receptor, which also has all the residues expected to be required for catalysis and also cannot rescue yeast that lack a sterol isomerase. It was recently reported that Δ8-9 sterols serve as signaling molecules36, which may hint at a possible physiological function of the σ receptors as sensors of these molecules evolved from enzymes that would modify them.
Docking 490 million molecules against the σ2 receptor
A prospective docking screen against the σ2 receptor had two goals. The first was to discover novel chemotypes with potential σ2 selectivity. The second goal was to investigate whether docking scores predict binding likelihood38. This second goal we undertook quantitatively, with a 3-fold larger library than previously used38. Moreover, the σ2 site, with its high propensity to bind ligands, promised a higher dynamic range than the first study against the dopamine receptor. We modeled a hit rate curve as a function docking score against the σ2receptor. Guided by score supplemented by manual selection among the highest-ranking docked molecules, we chose 484 make-on-demand molecules spread among 14 scoring bins covering the highest-ranking (−65 to −57.5 kcal/mol), mid-ranking (−55 to −40 kcal/mol), and low-ranking docking scores (−37.5 to −22.5 kcal/mol). We note that these ranges are true for σ2, but they will vary among other targets. Typically, around 40 molecules per scoring bin were selected. Overall, 412 molecules were picked automatically, whereas 72 were picked by manual visual inspection. We tested compounds at 1 μM concentration and defined as “hits” those that displaced greater than 50% [3H]-DTG σ2 binding. Based on this threshold, 127 of 484 molecules qualified as hits, equal to an overall hit rate of 26% of compounds tested, and a hit rate of over 60% at the peak among the top-ranked molecules (Fig. 2a). Plotting docking score vs hit-rate resulted in a curve where, with one key exception (see below), hit-rates fell monotonically with score, with a slope of −4.2%/(kcal/mol) in the inflection region. The curve dropped from a hit rate of 61% at a docking score of about −60 kcal/mol, to 0% at −40 kcal/mol, where it essentially remained for the next 4 (worse) scoring bins (Fig. 2b).
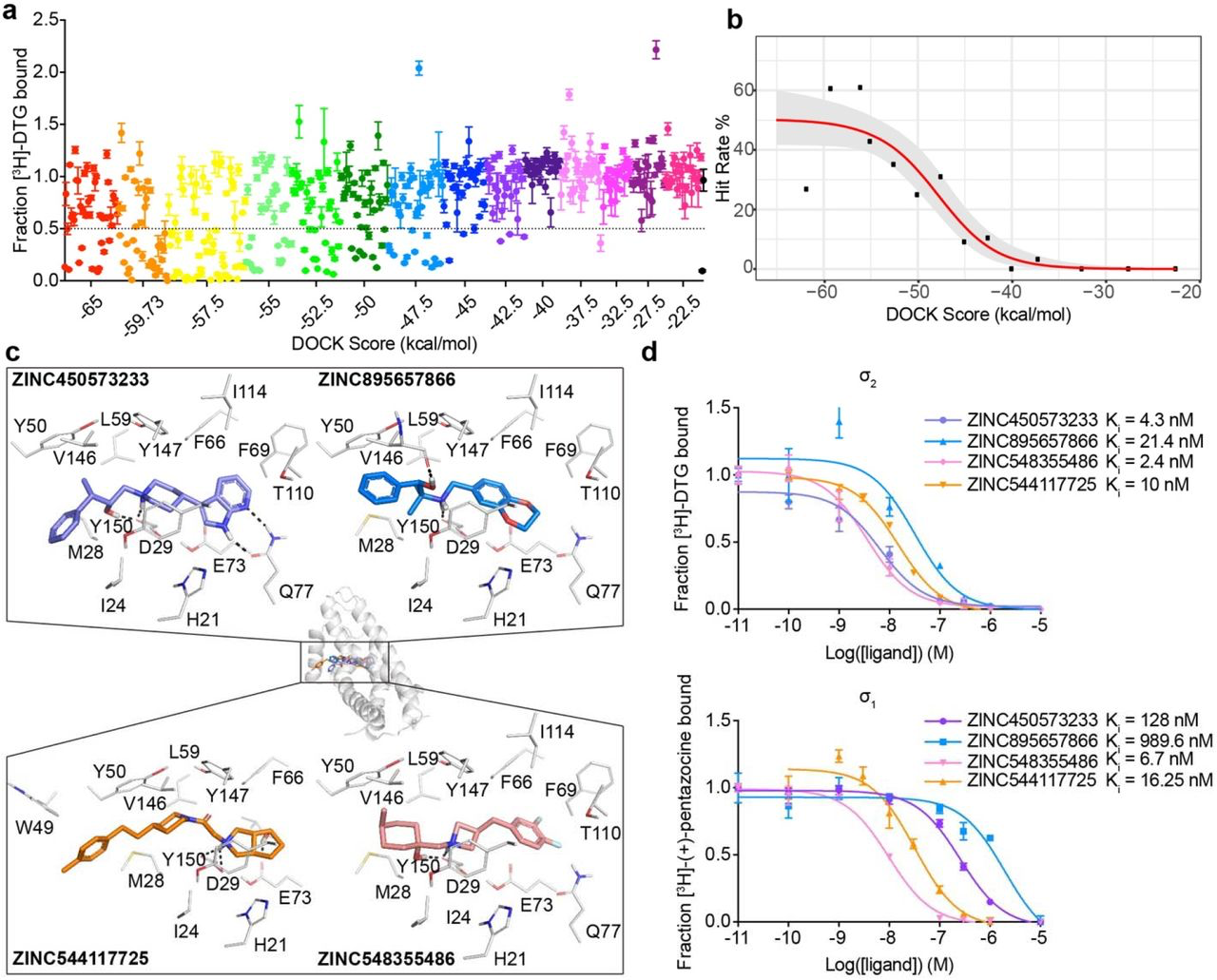
Docking 490 million molecules from ZINC libraries against the σ2 receptor.
a, Displacement of the radioligand [3H]-DTG by each of the 484 molecules tested at 1 μM (mean ± SEM of three technical replicates). The molecules are colored by the docking score at which they were selected. Dashed line indicates 50% radioligand displacement. Dot below the dashed line represent confirmed binders, which are diminished with increasing docking score. b,The hit-rate of 484 experimentally tested compounds was plotted against docking energy. The hit rate at the top plateau is 50% and at the bottom plateau is 0%, and the docking score (dock50) and slope at the maximum (slope50) are −48 kcal mol-1 and −4.2% per kcal mol-1, respectively. c, Docked poses of four representative binders, each a different scaffold. d, Doseresponse curves of radioligand displacement assays of the four molecules in c. against the σ2 receptor (upper panel) and the σ1 receptor (lower panel). The data are the mean ± SEM from three technical replicates.
Intriguingly, the hit rate among the very top scoring compounds, 27%, was meaningfully lower than those of the following four bins, and much lower than the 61% hit-rate observed in the 2nd-best scoring bin. This dip in the hit-rate curve illuminates holes in the scoring function that can be optimized in future studies. Examination of the 41 molecules (out of 490 million docked) that rank in this very top bin showed that many of these cationic molecules have unexpectedly low desolvation penalties versus electrostatic interaction energy (Supplementary Information Fig. 4a and 4b, the left column). Conversely, their van der Waals energies are undistinguishable among the first three bins (Supplementary Information Fig. 4c, the left column). These results implied that the desolvation penalties were underestimated for the molecules in the very top bin, especially for an electrostatics-driven site like the σ2 receptor site with a charged anchor residue (Asp29). A possible explanation for this drop in hit rate among the top-ranked molecules was an underestimation of ligand desolvation penalties. DOCK3.7 pre-calculates these energies based on one conformation from among hundreds of that are docked, and not necessarily the conformation that is the highest scoring against a target. Whereas different conformations of the same molecule typically have similar desolvation energies, they can differ, especially for charged molecules. Thus, a molecule can adopt a conformation that optimally complements a receptor electrostatically while only paying the desolvation cost of another, less costly conformation. Accordingly, we recalculated ligand desolvation energy using the docked conformation for all the molecules experimentally tested against σ2 and D4 receptors. After recalculation, the molecules in the first, top-scoring bin received much higher desolvation penalties than the following bins (Supplementary Information Fig. 4d). This supports the idea that the artifactually favorable scores of the very top-ranked molecules come at least partly from inappropriate desolvation penalties; this, and other holes in the scoring function, merit further investigation and optimization.
Naturally, in addition to testing docking-based prioritization, we were interested in finding new, potent, and selective σ2 receptor ligands. To supplement molecules prioritized by score alone, we also picked high-ranking molecules by human inspection39. As a comparable number of high-ranking molecules (the first 3 scoring bins) were picked manually and by docking score alone, we could investigate what human inspection added, if anything. In these top three scoring bins, covering 139 molecules (all high scoring), the hit rate by human inspection (67%) was higher than the hit rate by docking score alone (33%) (Supplementary Information Fig. 5a and 5b). Human-picked molecules were biased toward higher affinities, at least for those picked from the highest ranks by score: four had Ki values < 5 nM and twelve had Ki values < 50 nM. For those machine-picked only by score, two had Ki values < 5 nM and seven had Ki values < 50 nM (Supplementary Information Fig. 5c). While it seems clear that the human-picked molecules were more likely to be active, whether their potencies were significantly different from those picked by score alone was less clear. It’s important to emphasize that all of these molecules, human- and machine-prioritized, had highly favorable scores that put them at the very top of the rank-ordered list.
To find potent leads to selective probes for the σ2 receptor, we measured concentration-response curves for the primary screen-derived 14 docking hits with the best radioligand displacement at 1 μM. Ki values ranged from 2.4 to 68 nM. To measure selectivity, we ran competition binding assays against the σ1 receptor, observing Ki values from 1.6 nM to 1.5 μM (Fig. 2d, Supplementary Information Table 2 and 3). Several compounds showed substantial selectivity for σ2 over σ1, including ZINC450573233 and ZINC895657866, which were 30- and 46-fold selective, respectively.
We sought to improve the affinities of three potent ligands, each in a different scaffold class (Supplementary Information Fig. 6) from the docking screen. To do so, 20,000 analogues identified in SmallWorld (https://sw.docking.org/, NextMove Software, Cambridge UK) from a 28 billion make-on-demand library were docked into the σ2 site (Methods, Supplementary Table 3). Of these, 105 high-scoring analogues were sourced and tested. Encouragingly, the affinity of each scaffold was improved by 2-to 18-fold (Supplementary Information Fig. 6and Supplementary Table 3), and σ2 selectivity of two chemotypes improved to 47- and >250-fold (Z1665845742 and Z4857158944), respectively.
Crystal structures of σ2 receptor bound to optimized analogues
To validate our docking poses we determined the crystal structure of the two high affinity analogues Z1241145220 (σ2 Ki = 3.7 nM; PDB ID: 7M95) and Z4857158944 (σ2 Ki = 4 nM; PDB ID: 7M96). The electron density maps confirmed the docking predictions, with RMSD values between the crystallized and docked poses of 0.88 and 1.4 Å, respectively (Fig. 3a, Fig. 3b and Supplementary Information Table 1). Newly predicted hydrogen-bond interactions with Gln77 and the backbone carbonyl of Val146, which were not seen in the roluperidone or PB28 complexes, corresponded well between docked and crystallographic poses. The higher resolution of this structure, 2.4 Å, revealed an ordered water molecule in one of the binding site sub-pockets, coordinated by residues His21, Tyr103, and Gln77, and by an azaindole nitrogen in Z1241145220 (Fig. 3b).
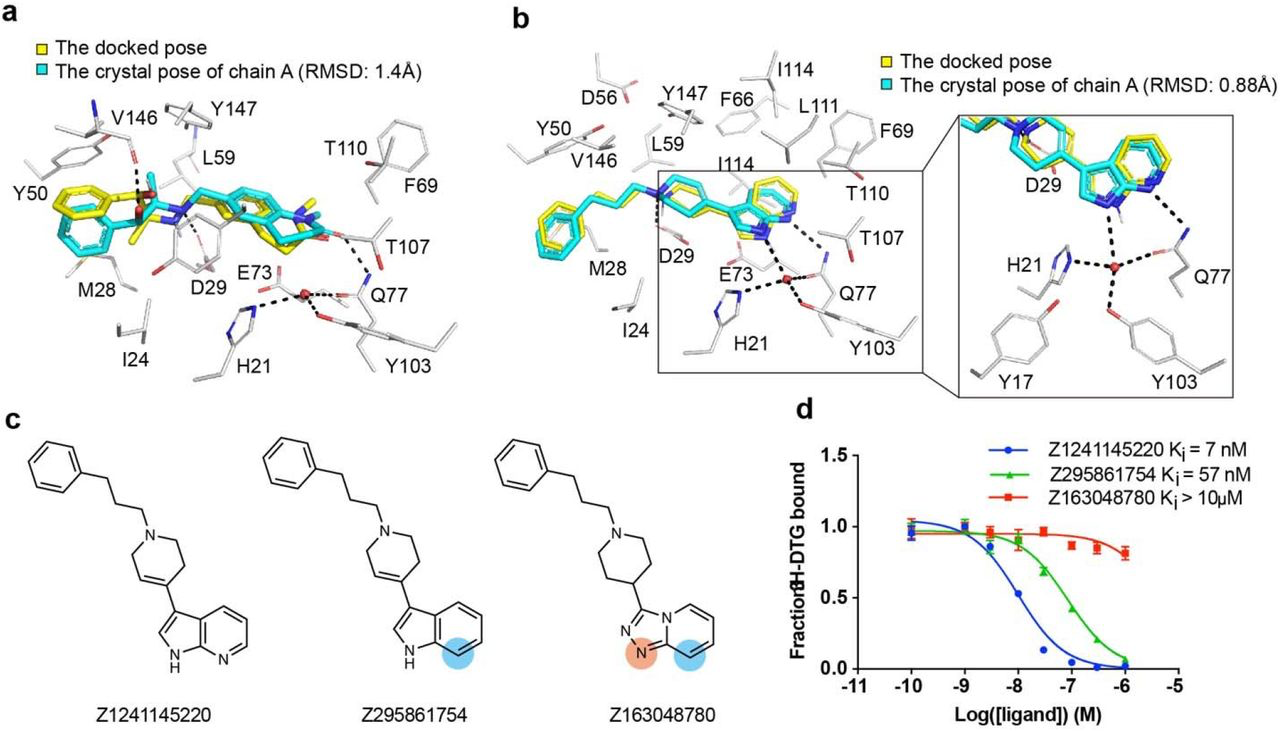
High structural fidelity between docked-predicted and crystallographically-determined poses of the new σ2 receptor ligands.
Crystal structures of the ligands (carbons in cyan) are overlaid with their respective docking predictions (yellow). σ2 receptor carbon atoms are depicted in grey, oxygens in red, nitrogens in blue, sulfurs in yellow, Hydrogen bonds are shown as black dashed lines. a, The complex with Z4857158944 (PDB code: 7M96; RMSD = 1.4 Å). b, The complex with Z1241145220 (PDB code: 7M95; RMSD = 0.88 Å). c, Two analogues that disrupt the hydrogen bonds with Gln77 and the structural water. Differences between the analogues and the parent compound Z1241145220 are depicted with light blue and apricot circles. d, Competition binding curve of the three compounds show reduced affinity at σ2.
To investigate whether this ordered water is important for ligand recognition, we tested two analogues that were designed to disrupt the hydrogen bonds between Gln77 and the water (Fig. 3c). Z295861754, which is only expected to hydrogen-bond with the water but not with Gln77, showed an ~8-fold decrease in binding affinity whereas Z163048780, which is not expected to hydrogen bond with either Gln77 or the water, had a Ki value > 10 μM (Fig. 3d), indicating a crucial role of the water in the binding of Z1241145220 to the σ2 receptor. To generalize these findings further, and to test whether this water is a structural element of the binding site, we generated a series of σ2 mutants in which the coordination of this water molecule was disrupted. We measured the affinity of the radioligand probe [3H]-DTG to these mutants and then performed competition binding assays with Z1241145220 (Supplementary Information Fig. 7). These experiments showed that mutating either His21 or Gln77 reduces the affinity of Z1241145220 by an order of magnitude. Taken together, these results demonstrate that the ordered water is an integral part of the binding pocket and is required for high-affinity binding of Z1241145220, and potentially for other ligands as well. This is reminiscent of a structural water molecule required for ligand recognition at the μ-opioid receptor40.
Novel σ2 ligands reduce mechanical hypersensitivity in a mouse model of neuropathic pain
There is strong genetic41,42 and pharmacological43–45 evidence supporting a contribution of σ1 to chronic pain46, but only recently, with the discovery of the gene encoding for σ24, has it been fully possible to understand and distinguish the roles of σ2 and σ1 in these processes21,22. However, the limited availability of highly selective σ2 probes22 has hindered disentangling the contribution of σ1 and σ2 in these processes. To investigate this, we treated mice with three high-affinity σ2 ligands with differing degrees of selectivity: Z4857158944 (4 nM; >250-fold σ2/σ1 selectivity), Z1665845742 (5 nM; 47-fold σ2/σ1 selectivity), and Z4446724338, a 3 nM non-selective ligand (Fig. 4a). As a comparator, we also treated with the well-known reagent PB28, a 5 nM σ1/σ2 non-selective ligand31. In pharmacokinetics experiments, the three docking-derived ligands had substantial brain permeability, with brain to plasma ratios ranging from 3 to 16, and brain half-lives ranging from 1.2 to 12 hours (Supplementary Information Table 4). The non-selective PB28 also had high brain permeability and a relatively long half-life, though its brain Cmax was three to eight-fold lower than that of the new docking-derived compounds. Even so, the high brain exposures of all four compounds encouraged us to examine them in a neuropathic pain model in mice.
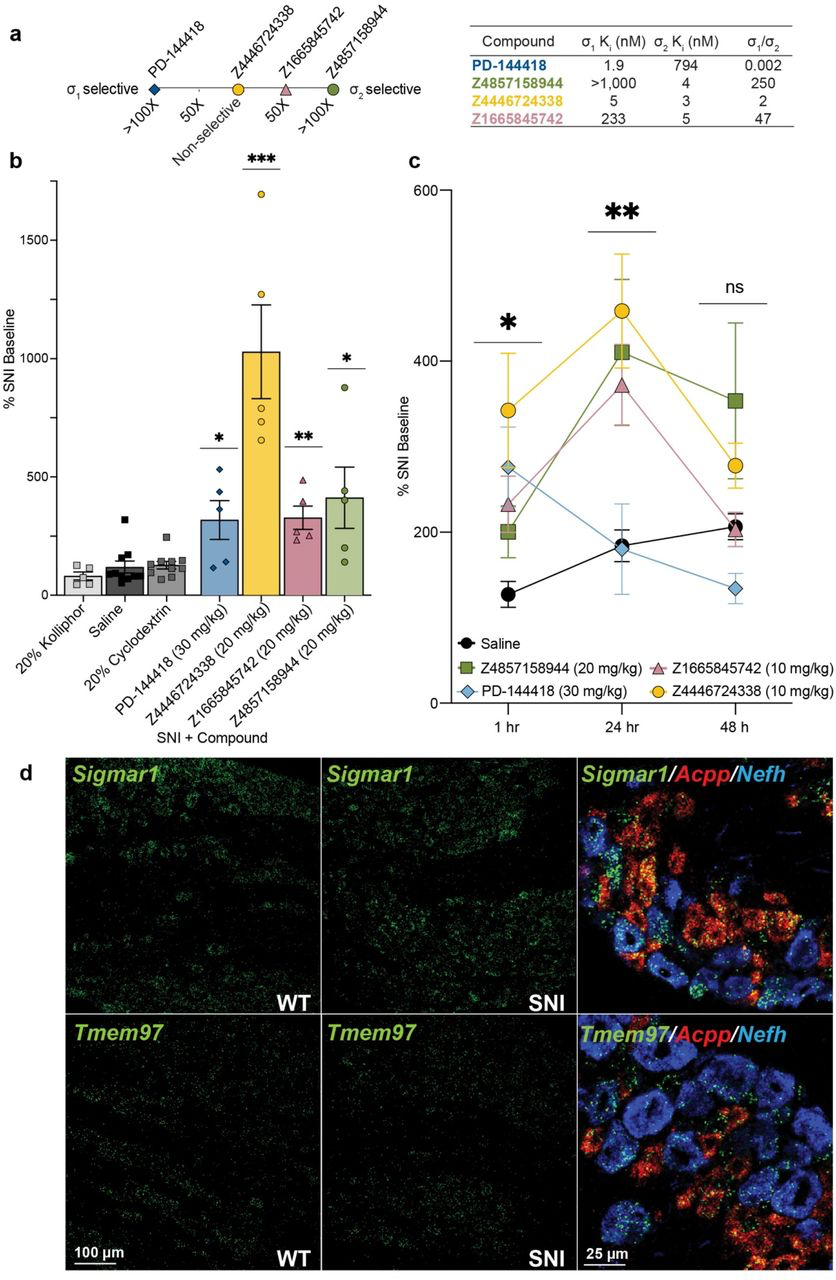
Systemic σ1/2 ligands are anti-allodynic in a model of neuropathic pain.
a, Selectivity of five ligands at σ1 and σ2, based on their experimental Ki ratios. PD-144418 values taken from the literature12. b, Response of mice to a Von Frey filament after spared nerve injury (SNI). All four ligands reduce the SNI-induced mechanical hypersensitivity compared to their vehicle (PD-144418 vs kolliphor; Z4446724338 and Z4857158944 vs cyclodextrin; Z1665845742 vs saline; one-way ANOVAs; * p < 0.05, ** p < 0.01, *** p < 0.001). Data shown are mean ± SEM. Data replotted, and statistical testing results described in Supplementary Information Fig. 8a. c, The anti-allodynic effects of σ2, but not σ1, ligands increases over time, with a peak effect at 24 hours postinjection. Significance levels determined using Dunnett’s multiple comparisons Post-hoc test reflect the difference between Z4446724338 and saline for simplicity (two-way ANOVA; time x treatment interaction: F(8,80) = 2.25, p = 0.03; time: F(2,76) = 5.09, p = 0.009; treatment: F(4,40) = 5.40, p = 0.001; four treatment groups (n = 10) except PD-144418 (n = 5); ns = not significant, * p < 0.05, ** p < 0.01). Data shown are mean ± SEM. d, in situ hybridization of mouse dorsal root ganglion (DRG) sections for Sigmar1 (σ1) and Tmem97 (σ2) genes illustrates expression in myelinated (Nefh-positive; blue) and unmyelinated (Acpp-positive; red) subsets of sensory neurons and no change after SNI.
We tested the efficacy of these σ1 and σ2 ligands in the spared nerve injury (SNI) mouse model of neuropathic pain, in which two out of three branches of the sciatic nerve are transected47, resulting in significant mechanical hypersensitivity (allodynia) transmitted by the uninjured peripheral (sural) nerve. SNI mice systemically injected with either of the σ2-selective ligands Z1665845742 or Z4857158944 were strongly anti-allodynic when dosed in SNI mice, significantly increasing mechanical thresholds versus vehicle (Fig. 4b, Supplementary Information Fig. 8a). Intriguingly, anti-allodynic effect of Z1665845742 and Z4857158944 was comparable to that attained by a systemic injection of the σ1-selective ligand PD-144418. To investigate the possible synergistic effect of targeting both σ receptors we tested the nonselective ligands PB28 and Z4446724338. Systemic injection Z4446724338 dose-dependently increased the mechanical thresholds of SNI mice, 1-hour post-injection (Fig 4b, Supplementary Information Fig. 8a), with the highest dose completely reversing the SNI-induced mechanical allodynia (i.e., thresholds returned to pre-injury levels). In contrast, systemic injections of PB28, a well-established sigma receptor ligand with high affinity for both subtypes48, produced mixed results, with anti-allodynic effects observed only in 60% of the mice (Supplementary Information Fig. 8a). The much stronger anti-allodynia of Z4446724338 versus PB28 may reflect the former’s substantially higher brain permeability as measured by their respective brain Cmax values (Supplementary Information Table 4). Importantly, none of the new σ1 and σ2 ligands were sedative on the rotarod test (Supplementary Information Fig. 8b), indicating that their anti-allodynic effect was not due to motor impairment.
On its face, substantial anti-allodynic effects of the σ2-selective ligands Z1665845742 and Z4857158944 suggest that this receptor is a potential target for managing neuropathic pain in the clinic. However, because σ2 ligands are notorious for activity at other targets, especially GPCRs49,50, we counter-screened all three docking-derived ligands against a panel of potential off-targets. In a TANGO screen51 of 320 GPCRs, the molecules did not act as agonists or inverse agonists against most targets. Some activity was observed at the 5HT1A receptor and κ-opioid receptors (Supplementary Information Fig. 9), but this activity did not replicate in subsequent concentration-response assays. Because a key pain target, the μ-opioid receptor, is often relatively inactive in TANGO assays, we further tested the molecules in G protein assays against this receptor; here too, no substantial activity was observed (Supplementary Information Fig. 9). Since the TANGO assay is limited to detecting agonism (and in some cases inverse-agonism) at GPCRs, we further screened the compounds in binding assays against a panel of 44 targets including GPCRs, ion channels, and transporters. No binding was observed for any pain-related targets (Supplemental Information Table 5). Taken together, these observations suggest that the primary mechanism of action of the selective ligands is via the σ2 receptor (naturally, other targets, not tested here, cannot be excluded). The even stronger activity of the σ1/2 ligand Z4446724338 suggests that σ1/2 polypharmacology may further increase activity.
σ2 ligands achieve peak antiallodynic effects 24 hours after dosing
In earlier studies, σ1/2 ligands showed peak antiallodynic effects up to 48 hours after dosing21. This unusual behavior was observed with ligands with mid-nanomolar potency for the σ2 receptor, and 9 to 14-fold selectivity vs. the σ1 receptor. We thought it interesting to further explore this with the low nanomolar affinity selective ligands, Z4857158944 and Z1665845742, and the joint σ1/2 ligand, Z4446724338. The three molecules were tested post SNI, at 1 hour, 24 hours, and 48 hours after dosing. Consistent with earlier reports, the anti-allodynic effects of the three novel σ ligands increased over time, reaching maximal effect 24 hours post-injection (Fig. 4c). In contrast, the anti-allodynic effect of the selective σ1 ligand PD-144418 was not sustained 24-or 48-hours post-injection. Furthermore, although the σ2-selective compounds exhibited reduced anti-allodynia efficacy at early time points compared to the mixed agonist Z4446724338, all three compounds produced similar levels of analgesia by 24 hours. We note that this long-term activity cannot be easily explained by pharmacokinetics, as the brain half-life of all three compounds suggests minimal exposure past 12 hours (Supplementary Information Table 4). Rather, this time course may reflect longer term signaling or regulatory effects, the exact nature of which remains a question for ongoing research21. Regardless of the basis, these results confirm earlier work suggesting that the antiallodynic effects of σ2 are prolonged, which may be useful in the management of chronic pain disorders.
σ1/2 receptors are expressed on primary afferent neurons
in situ hybridization of dorsal root ganglia (DRG) sections, where the cell bodies of sensory neurons that transmit the “pain” message to the spinal cord reside, revealed that both σ1 and σ2 receptors are expressed in a wide variety of DRG neurons, including myelinated and unmyelinated subsets (Fig. 4d). We additionally found that the expression level of σ1 or σ2did not change in DRG neurons 7 days after SNI. Although the sigma receptor has a broad distribution, we suggest that our new ligands exert their antinociceptive action via interaction with primary afferent neurons.
Discussion
The σ2 receptor has been pharmacologically enigmatic for 30 years. Its implication in diverse biological processes and lack of molecular data has hindered progress in understanding its biological role. Four key observations from this study begin to illuminate these issues. First, high-resolution crystal structures of the σ2 receptor complexed with roluperidone and with PB28 reveal a ligand-binding site deeply embedded in the membrane (Fig. 1a-b), suggesting the possibility of a lipid as an endogenous ligand. The evolutionary connection of σ2 to EBP and the structure of the receptor bound to cholesterol strongly imply an ability to recognize sterols. The structures explain the simple pharmacophore of σ2 ligands—a positively charged amine that ion-pairs with Asp29, while flanking hydrophobic and aromatic moieties are recognized by nearby aromatic residues, such as Phe66, Phe69, His21 and Tyr50. The structures also highlight nearby polar residues, such as Gln77, and Thr110, that seem rarely exploited by classic σ2 ligands, but which may provide new selectivity determinants for ligand discovery (Fig. 1c and 1d). Second, by testing 484 compounds across docking ranks from a library of 490 million molecules, a clear and quantitative relationship emerged between docking score and the likelihood of binding (Fig. 2). Encouragingly, crystal structures of docking-derived ligands confirmed the docking predictions with low RMSDs (Fig. 3a and 3b). Third, from among the top-ranking docking hits were 31 novel scaffolds with potent binding affinities (Ki < 100 nM) (Supplementary Information Table 2-3). Optimization of two of these led to ligands with low nanomolar affinities and 47-fold to >250-fold selectivity for the σ2 over the σ1 receptor (Supplementary Information Fig. 1). Fourth, three new σ2 chemotypes, one non-selective but potent vs. σ1/σ2, and two others selective for σ2 over σ1, were tested for efficacy in a mouse model for neuropathic pain. All three showed antiallodynic effects (Fig. 4); the expression pattern of the receptor and the activity of the σ2-selective ligands confirm a contribution of this receptor in pain processing and suggest its potential relevance in pain management.
The σ2 and the σ1 receptors are promiscuous, both binding to cationic amphiphiles, and as expected, there is broad cross-reactivity between the two receptors. Although selective σ1ligands, like PD-144418 and (+)-pentazocine, have been described, there are relatively few selective ligands22,52 for the σ2 receptor, which has hindered understanding its role in biology and in disease. We sought to optimize for selective ligands from among our potent docking actives. We adopted a chemical novelty approach described previously29,53,54 prioritizing novel scaffolds, modeled to interact with diverse groups within the σ2 structure. We reasoned that this would identify more potent molecules that would also be selective vs the σ1 receptor. This strategy has been productive previously, while the more rational approach of intentionally counter-docking vs. the off-target, here the σ1 receptor, remains a research area55. A cycle of structure-based analoging resulted in improved selectivity for two chemotypes. Increasing the linker length in ZINC450573233 by one carbon, along with two other small changes, results in Z1665845742, with selectivity improved from 30-fold to 47-fold. At the same time, replacing the 2,3-dihydro-1,4-benzodioxine group in ZINC895657866 with a 3,4-dihydro-1H-quinolin-2-one to engage with Gln77 (Fig. 3a), led to Z4857158944, with selectivity increased from 46-fold to >250-fold, making it among the most potent and selective σ2 ligands of which we are aware. We combined one of these selective molecules with a close analog that is inactive on the σ2 receptor, affording a “probe pair” (Z1665845742 and Z1665798906) (Supplementary Information Fig. 10). Such probe pairs are useful to understand the role of receptors, such as the σ2 in cellular and in vivo studies, where the activity of the inactive member controls for off-targets that any molecule inevitably has. The approach should also facilitate disentangling the role of the σ2 receptor in indications for which it has been widely mooted, including cancer9–11, schizophrenia17, and Niemann Pick disease5,6. We make this probe pair openly available via Sigma-Millipore’s probe collection (Cat. No. TBD).
Certain caveats bear airing. While our ultimate goal was to find σ2-selective ligands from the docking, ligands with a spectrum of affinities and selectivities for both σ receptors emerged, reflecting the similarities of their pockets and the historical precedence in their overlapping pharmacology (Fig. 4c-e). Human inspection of molecules revealed an unusually high 66% hit rate, as well as competitive ligand potency, likely reflecting a site that is well-suited to ligand binding, despite its solventocclusion and hydrophobicity. These high hit rates, potencies, and selectivities often do not translate to other targets—this target was unusually propitious for library docking. Additionally, while the docking-predicted pose for Z4857158944 and for Z1241145220 closely resembled the subsequent crystallographic structure, important differences remain, namely the water-bridging interaction for Z1241145220. Although modeling this water was not necessary for ligand discovery, and the docked ligands fit well without it, including this water modeling may further improve future structure-based efforts against this target. Modeling waters in docking remains a research area in molecular docking56.
The key observations of this work should not be obscured by these caveats. The high-resolution crystal structures of σ2 receptors reveal the origins of its molecular recognition, and template structure-based campaigns for novel ligand discovery. This work emphasizes the value of a structure-based approach to screen vast new libraries of chemotypes unrelated to known ligands with unique properties that illuminate the biology of the σ2 receptor. Applications to other targets should undoubtedly be considered.