
Sigma 2 Receptor/Tmem97 Agonists Produce Long Lasting Antineuropathic Pain Effects in Mice

By James J Sahn, Galo L Mejia, Pradipta R Ray, Stephen F Martin, and Theodore J Price
Excerpt from the article published in ACS chemical neuroscience, 2017 Aug 16;8(8):1801-1811. DOI: https://doi.org/10.1021/acschemneuro.7b00200 Free PMC article
Editor’s Highlights
- The sigma 1 receptor (σ1R) and the sigma 2 receptor (σ2R) proteins are involved in intracellular ion regulation and neuron survival.
- The σ2R/Tmem97 gene, is clearly expressed in structures outside of the intrathecal space and has high expression in the DRG and in non-neuronal cells in the DRG and CNS.
- σ2R/Tmem97 agonists are a novel strategy for the pharmacological management of neuropathic pain, bringing forth the possibility for patients to experience enhanced pain relief and reduced side-effects by modulating a receptor not targeted by currently approved FDA drugs.
Abstract
Neuropathic pain is an important medical problem with few effective treatments. The sigma 1 receptor (σ1R) is known to be a potential target for neuropathic pain therapeutics, and antagonists for this receptor are effective in preclinical models and are currently in phase II clinical trials. Conversely, relatively little is known about σ2R, which has recently been identified as transmembrane protein 97 (Tmem97). We generated a series of σ1R and σ2R/Tmem97 agonists and antagonists and tested them for efficacy in the mouse spared nerve injury (SNI) model. In agreement with previous reports, we find that σ1R ligands given intrathecally (IT) produce relief of SNI-induced mechanical hypersensitivity. We also find that the putative σ2R/Tmem97 agonists DKR-1005, DKR-1051, and UKH-1114 (Ki ~ 46 nM) lead to relief of SNI-induced mechanical hypersensitivity, peaking at 48 h after dosing when given IT. This effect is blocked by the putative σ2R/Tmem97 antagonist SAS-0132. Systemic administration of UKH-1114 (10 mg/kg) relieves SNI-induced mechanical hypersensitivity for 48 h with a peak magnitude of effect equivalent to 100 mg/kg gabapentin and without producing any motor impairment. Finally, we find that the TMEM97 gene is expressed in mouse and human dorsal root ganglion (DRG) including populations of neurons that are involved in pain; however, the gene is also likely expressed in non-neuronal cells that may contribute to the observed behavioral effects. Our results show robust antineuropathic pain effects of σ1R and σ2R/Tmem97 ligands, demonstrate that σ2R/Tmem97 is a novel neuropathic pain target, and identify UKH-1114 as a lead molecule for further development.
INTRODUCTION
Neuropathic pain is a common medical problem that is very poorly treated by current therapeutics. Recent meta-analyses show that the most widely prescribed
drugs for neuropathic pain only achieve 50% pain relief in 1 of 4 patients for the antidepressants (e.g., monoamine oxidase inhibitors) and are effective in as few as 1 in 7 patients for the gabapentinoids.1 Better therapeutics are clearly needed for neuropathic pain. Sigma receptors (σRs) are unique transmembrane proteins expressed throughout the CNS (central nervous system) and in certain peripheral tissues. Consisting of two subtypes, the sigma 1 receptor (σ1R) and the sigma 2 receptor (σ2R), these proteins are involved in intracellular ion regulation and neuron survival.2 σ1R has been cloned and a crystal structure of the receptor obtained.3 The involvement of this receptor in pain signaling has been studied extensively, and a variety of compounds that bind to σ1R demonstrate antinociceptive effects.4 E-52862,5a σ1R antagonist, is currently in phase II clinical trials for the treatment of neuropathic pain as both a monotherapy and in a multidrug cocktail.
Although strong evidence supports a role for σ1R in neuropathic pain, σ2R has not been explored as a pain target, and the molecular identity of this receptor has been challenging to pinpoint. Molecular cloning recently confirmed the identify of σ2R as transmembrane protein 97 (Tmem97),6 and this receptor will accordingly be referred to as σ2R/Tmem97 herein unless we are referring to the Tmem97 mouse or TMEM97 human genes. σ2R/Tmem97 is a gene product that appears to be involved in cholesterol trafficking and homeostasis and interacts with NPC-1, a protein responsible for shuttling lipids to postlysosomal locations.7–9σ2R/Tmem97 is involved in regulating intracellular Ca2+ concentration, and certain σ2R/Tmem97 ligands can induce a transient rise in intracellular Ca2+levels,2 while other compounds that bind to σ2R/Tmem97 suppress Ca2+ influx in the presence of an inducer.10 Some evidence suggests that compounds that bind to σ2R/Tmem97 modulate a signaling pathway involving progesterone receptor membrane component one (PGRMC111),10 a heme binding protein12 involved in cell survival2 and apoptosis.13 With respect to medical relevance, it is noteworthy that TMEM97 has been implicated in the metabolic disorder, Niemann-Pick disease,9 as well as in multiple neurodegenerative and neurological conditions.14 Indeed, small molecules that bind to σ2R/Tmem97 are emerging as potential therapeutics for a range of CNS disorders, including Alzheimer’s disease (AD)15 and schizophrenia.16
We have discovered that the chlorinated norbenzomorphan and methanobenzazocine scaffolds 1 and 2 (Figure 1) are excellent templates for preparing both selective and mixed affinity σ1R and σ2R/Tmem97 ligands.17 In the context of CNS lead development, these scaffolds are of particular interest because of their drug-like properties18,19 and the ease with which they cross the blood brain barrier, as demonstrated by representative compounds SAS-013210 and DKR-1051 (Figure 1). In vivo testing has revealed that some compounds derived from 1 and 2have promising medicinal properties that may warrant further preclinical development. For example, certain σ2R/Tmem97 ligands are neuroprotective10 and also restore cognitive function in transgenic AD mice10,20 as well as in aged, nondiseased animals,10 the latter of which suggests σ2R/Tmem97 may be a potential target for treating mild cognitive impairment (MCI). Despite this increasing knowledge base and the emergence of tool ligands for probing σ2R/Tmem97, the involvement of σ2R/Tmem97 in neuropathic pain has not yet been reported.
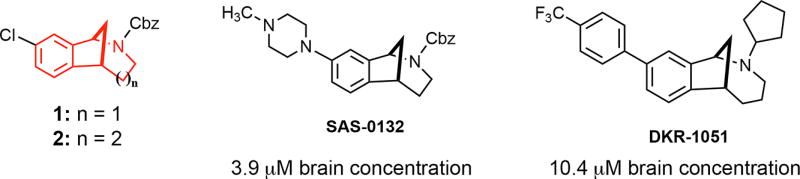
Norbenzomorphan and methanobenzazocine scaffolds 1 and 2 and the CNS penetrant σ2R/Tmem97 ligands SAS-0132 and DKR-1051.
Herein we describe the antineuropathic pain properties of a series of σ1R and σ2R/Tmem97 ligands in mice. We report that molecules that bind to σ2R/Tmem97 as putative agonists exert a profound effect on mechanical hypersensitivity in the spared nerve injury (SNI) model with a duration of action and potency that is superior to that of gabapentin. Small molecule modulation of σ2R/Tmem97 may thus represent a new approach for managing pain by a previously unexplored mechanism of action.
RESULTS
Screening of σ1R and σ2R/Tmem97 Ligands for Activity in the Mouse SNI Model
Several previous studies have demonstrated that σ1R antagonists show efficacy in rodent neuropathic pain models via a spinal mechanism of action,21–23 but the possible effect of σ2R/Tmem97 ligands in this model has not been explored. We screened a series of σ1R and σ2R/Tmem97 ligands (Figure 2) for activity in the SNI model via a single IT injection. We chose this route of administration because σ1R antagonists have been shown to be active in rodent pain models with IT injection,21,24 and because we had incomplete knowledge of compound disposition in vivo with systemic dosing.
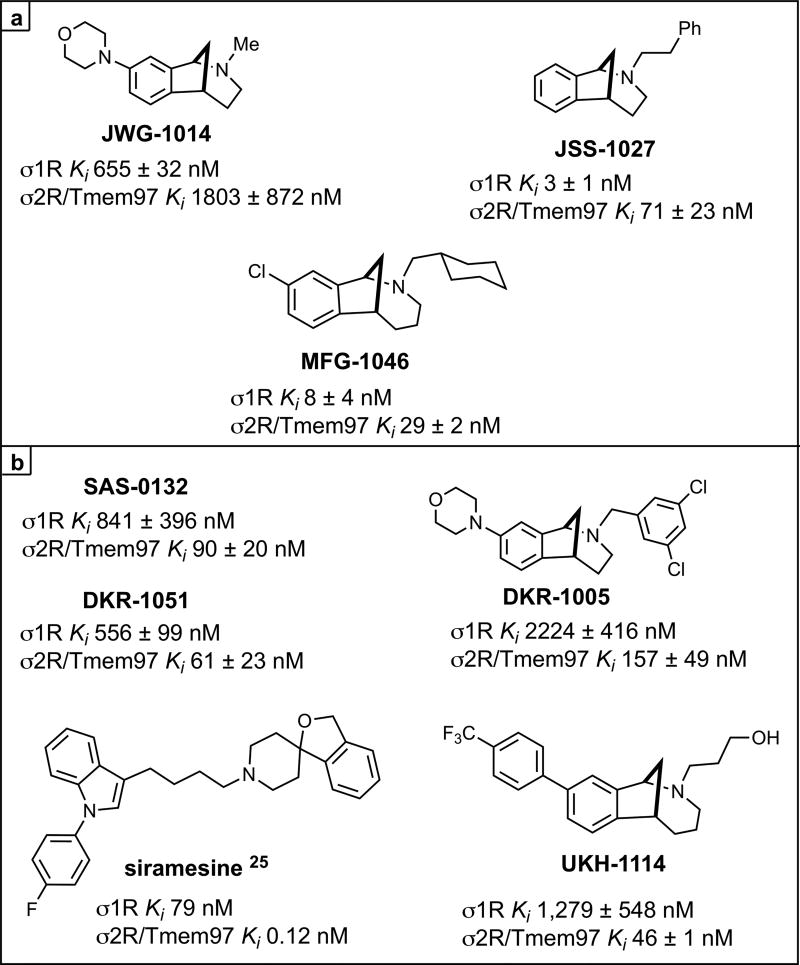
σ Receptor binding ligands.aKi are shown for each ligand at σ1R or σ2R/Tmem97 as the mean of at least two independent experiments ± standard deviation. Siramesine data is from ref 25. aFor all compounds, σ2R/Tmem97 was sourced from rat PC12 cells, with the exception of siramesine, which utilized rat brain homogentate. See Methods Section for more details.
We first assessed the σ1R preferring ligands, JWG-1014, JSS-1027, and MFG-1046, all given at 10 μg doses, and observed antipain effects for all three compounds that were significantly different from vehicle. JWG-1014 and JSS-1027 (Figure 8) both demonstrated efficacy 48 h post IT injection, and this effect persisted through 120 h after ligand administration (Figure 3). Faster onset of action was observed with MFG-1046 (Figure 9), which elicited an antipain effect at 24 h that continued through 72 h after IT injection (Figure 3).
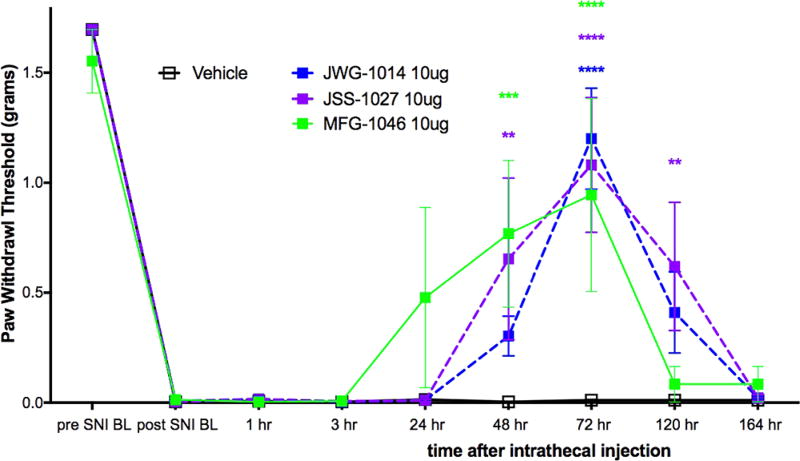
Effect of IT injection of σ1R ligands in the mouse SNI model. Compounds acting on σ1R were tested in the mouse SNI model. Mechanical sensitivity was measured at the indicated time points after IT injection of 10 μg compound. All vehicle groups, n = 6; JWG-1014, n = 5; JSS-1027, n = 5; MFG-1046, n = 4. **p < 0.01, ***p < 0.001, and ****p < 0.0001.
We then tested four σ2R/Tmem97 preferring ligands of the norbenzomorphan and methanobenzazocine structural class, as well as the known σ2R/Tmem97 agonist, siramesine.25–27 Siramesine induced a small inhibitory effect on mechanical hypersensitivity that was significant at 24 and 48 h (Figure 4A). On the other hand, DKR-1005, DKR-1051, and UKH-1114 all produced significant antimechanical hypersensitivity effects at either 24 or 48 h after IT injection (Figure 4B).
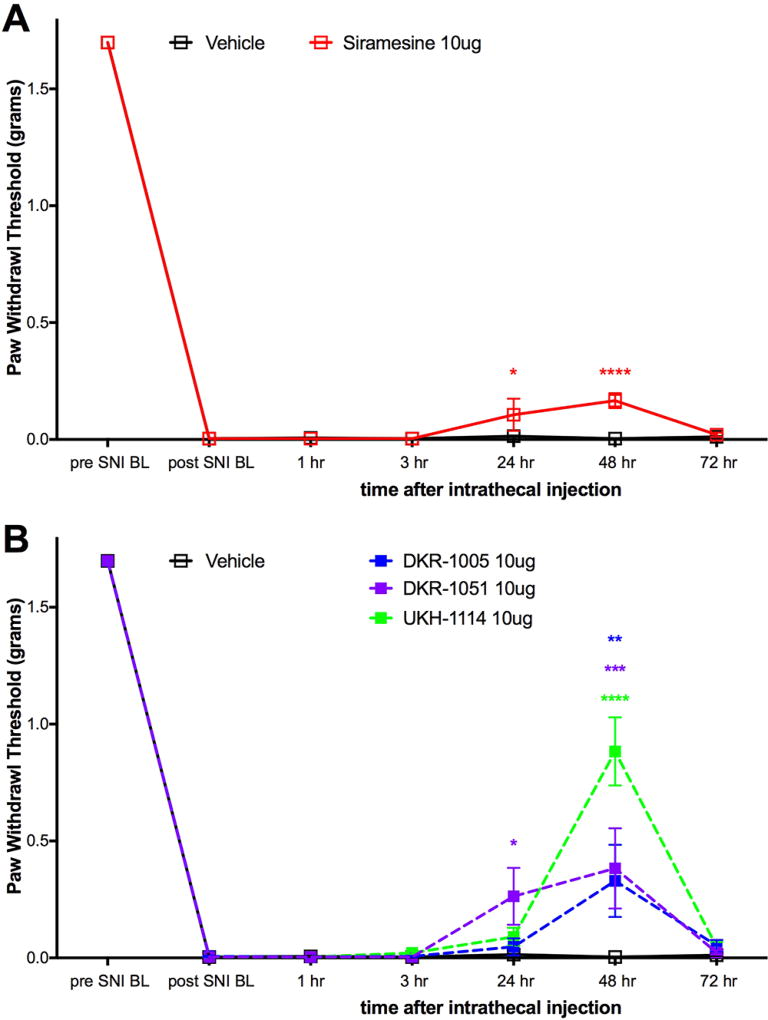
Effect of IT injection of σ2R/Tmem97 ligands in the SNI model. Mechanical sensitivity was measured at the indicated time points after IT injection of 10 μg compound. (A) Effect of siramesine (n = 4) compared to vehicle (n = 6) is shown. (B) Effect of DKR-1005 (n = 6), DKR-1051 (n = 5) and UKH-1114 (n = 11) compared to vehicle (n = 6) is shown. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Based on the observed in vivo effects of the four σ2R/Tmem97 preferring ligands examined, tentative functional activity assignments were made. Compounds DKR-1005, DKR-1051, and UKH-1114elicited pronounced and sustained antinociceptive effects. However, when UKH-1114 was administered with SAS-0132, the antinociceptive action of UKH-1114 was abolished (Figure 5). Because UKH-1114elicits a significant antipain effect that is completely blocked by SAS-0132, we surmise that UKH-1114 is a σ2R/Tmem97 agonist (or partial agonist) and that σ2R/Tmem97 agonism is responsible for the antimechanical hypersensitivity effects observed with these compounds in SNI mice. On the other hand, SAS-0132 appears to function as a σ2R/Tmem97 antagonist by suppressing the action of UKH-1114. Indeed, these findings are consistent with our previous work that showed that SAS-0132behaves as a σ2R/Tmem97 antagonist, whereas DKR-1051 acts as a σ2R agonist10 based upon their opposing effects upon Ca2+ in SK-N-SH neuroblastoma cells. Namely, treatment of SK-N-SH neuroblastoma cells with DKR-1051 induces a rapid Ca2+ transient, and this effect is attenuated when cells are pretreated with SAS-0132.
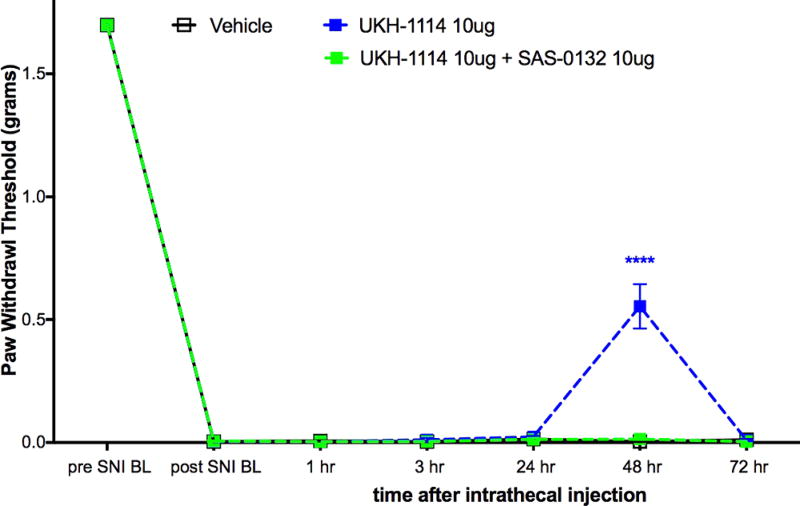
Effect of UKH-1114 is blocked by the σ2R/Tmem97 antagonist SAS-0132. σ2R/Tmem antagonist SAS-0132 at 10 μg dose given at the same time as UKH-1114, also at 10 μg dose, completely blocked the effect seen with UKH-1114 given alone. n = 6 per group. ****p < 0.0001.
Systemic Administration of σ2R/Tmem97 Agonist Alleviates Neuropathic Pain
Of the putative σ2R/Tmem97 agonists we tested, UKH-1114 had the largest behavioral effect, and its chemical properties and high selectivity for σ2R/Tmem97 versus >50 other proteins (See Tables 1 and and2)2) make it an eligible candidate for systemic dosing.
| molecular weight | 375.4 |
| ClogD (7.4) | 4.3 |
| total polar surface area | 23.5 |
| hydrogen bond donors | 1 |
| hydrogen bond acceptors | 2 |
| rotatable bonds | 5 |
Chemical Properties of UKH-1114a
aCalculated with ACD/I-Laboratories (https://ilab.acdlabs.com/iLab2/).
| target | Ki (nM) | target | Ki (nM) |
|---|---|---|---|
| 5HT1A | a | Beta2 | a |
| 5HT1B | a | Beta3 | a |
| 5HT1D | a | BZP rat brain | a |
| 5HT1e | a | calcium channel | >10 000 |
| 5HT2A | a | D1 | a |
| 5HT2B | a | D2 | a |
| 5HT2C | a | D3 | a |
| 5HT3 | a | D4 | a |
| 5HT5a | a | D5 | a |
| 5HT6 | a | DOR | a |
| 5HT7 | a | GabaA | a |
| A2B2 | a | H1 | a |
| A2B4 | a | H3 | a |
| A3B2 | a | KOR | 1383 |
| A3B4 | a | M1 | a |
| A4B2 | a | M2 | a |
| A4B2b | a | M3 | a |
| A4B4 | a | M4 | a |
| A7 | a | M5 | a |
| A7b | a | MOR | a |
| Alpha1a | a | NET | 1046 |
| Alpha1b | a | NMDA | 6724 |
| Alpha1d | a | PBR | a |
| Alpha2a | a | SERT | a |
| Alpha2b | a | V1A | >10 000 |
| Alpha1c | a | V1B | >10 000 |
| AMPA | >10 000 | V2 | >10 000 |
| Beta1 | a |
UKH-1114 Binding Profile at Non-Sigma Receptor Sites
a<50% inhibition of radioligand binding at 10 μM.
bSourced from rodent brain.
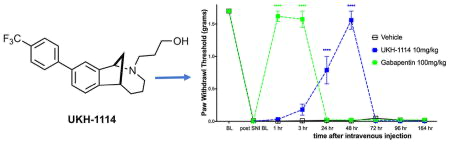
Therefore, we injected UKH-1114 IV in mice at 10 mg/kg and compared the effect of the compound to the gold-standard antineuropathic pain treatment, gabapentin (100 mg/kg). Gabapentin completely reversed mechanical hypersensitivity at 1 and 3 h after injection, but animals were fully mechanically hypersensitive again 24 h after IV injection. UKH-1114 also produced a complete reversal of mechanical hypersensitivity but with a different time course (Figure 6A). A significant effect was observed at both 24 and 48 h after injection indicating that pain relief from this mechanism lasts longer than that produced with a 10-fold larger dose of gabapentin.
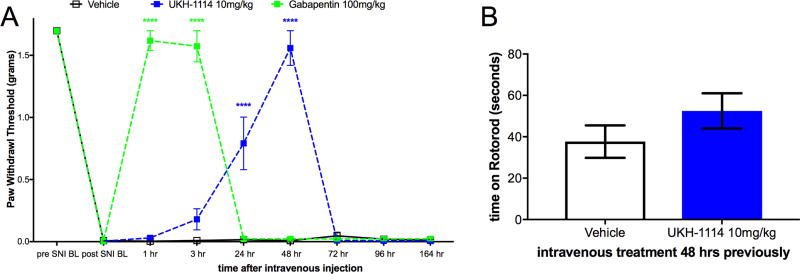
Systemic dosing with UKH-1114 leads to alleviation of neuropathic pain without motor effects. (A) Vehicle (n = 6), gabapentin (100 mg/kg, n = 6), or UKH-1114 (10 mg/kg, n = 6) were given IV and mechanical testing was done at the indicated time points. (B) Rotorod testing was done 48 h after IV injection on an accelerating rotorod reaching a maximum of 40 rotations per minute over 200 s. Latency to fall is shown on the third trial (n = 6 per group). ****p < 0.0001.
Because the mechanism of action of σ2R/Tmem97 ligands for neuropathic pain relief is not known, we were concerned that this effect could be produced by motor impairment. To test this possibility we used the rotorod test. SNI mice were given IV vehicle or UKH-1114 and tested at the peak time point for alleviation of neuropathic pain, 48 h after injection. There was no effect of UKH-1114 on motor performance (Figure 6B) ruling out the possibility of motor impairment.
Tmem97 Gene Expression Analysis in Mouse and Human Tissues
To gain insight into where σ2R/Tmem97 is expressed, we quantified Tmem97 mRNA relative abundance in mouse DRG as well as additional tissues, and their orthologous tissues in humans, based on publicly available sequencing data (human data sets: GTex project,28 ENCODE project,29 Uhlén et al.,30 Duff et al.,31 and the UTD DRG project;32mouse data sets: mouse ENCODE project,33 Rakic et al.,34 Gerhold et al.,35 Eipper-Mains et al.,36 and Huan et al.37). We find that Tmem97 gene expression and its human orthologue (TMEM97) are ubiquitously expressed in a wide range of tissues, with higher expression in the human and mouse gastrointestinal (GI) tract commensurate with its role in cholesterol trafficking7 (Figure 7A). While expression levels in the human DRG is high, both single cell38 and bulk35 RNA-seq for mouse DRGs identify gene expression, but at lower levels than in human (Figure 7A). TMEM97/Tmem97 is also relatively highly expressed in human and mouse spinal cord. While the detection rate for Tmem97across mouse DRG neuronal subpopulations is relatively low, it is clearly expressed in subpopulations of peptidergic and nonpeptidergic nociceptors (Figure 7B). Given the high TPM levels in mouse and human DRG, Tmem97 may also be expressed in non-neuronal cells in DRG (e.g., satellite glial cells or Schwann cells). Unfortunately, mouse DRG glial transcriptomes have not been characterized, so we turned to a CNS tissue where these cell populations have available transcriptomes. We find that in adult cerebral cortex,39 Tmem97 expression in cortical glial cells can be enriched 2-fold or more over neuronal expression levels (Figure 7C), lending credence to the hypothesis of glial expression of Tmem97 in the DRG and/or spinal cord.
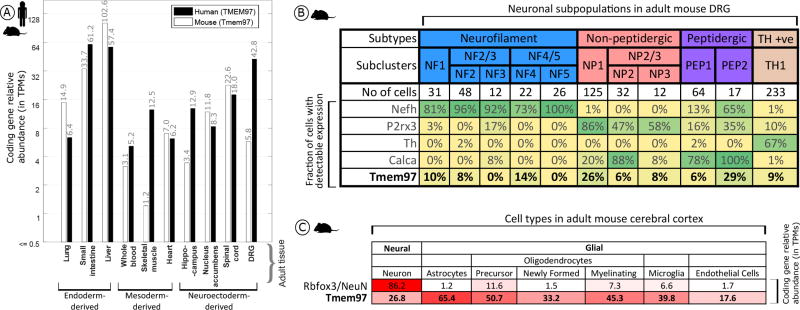
Expression analysis for Tmem97 (A), TMEM97 (human), and Tmem97 (mouse) gene expression across orthologous tissues, with greater expression in the mouse and human GI tract and the human DRG. (B) Analysis of mouse single cell data reveals a maximum detection rate of 29% for Tmem97 across all sensory neuron subpopulations as contrasted with 67% or more for known subpopulation marker genes. (C) Cortical expression of Tmem97 as contrasted with the neuronal marker NeuN. Tmem97 expression spans both neuronal and non-neuronal cells, with ~1.5–2.5-fold higher expression in non-neuronal cells.
DISCUSSION
Several primary conclusions may be reached based upon the work described herein. First, our results using distinct σ1R binding ligands are consistent with previous demonstrations that σ1R antagonists reduce nerve injury-induced mechanical hypersensitivity.21,40 This observation suggests that the σ1R binding ligands described herein might be antagonists. Second, we find that σ2R/Tmem97 ligands DKR-1005, DKR-1051, and UKH-1114 bind σ2R/Tmem97 with high affinity and produce antinociceptive effects when administered IT to SNI mice. UKH-1114 is the most efficacious of these compounds in vivo, producing a strong antinociceptive effect when administered IV that was longer lasting and equally efficacious at 1/10 the dose of gabapentin. UKH-1114 is highly selective for σ2R/Tmem97 binding with an affinity 28-fold greater at σ2R/Tmem97 than at σ1R, and with negligible affinity for 55 other targets (Table 2). While TMEM97/Tmem97 is expressed in DRG and spinal cord of humans and mice, the gene is likely expressed in a mix of neuronal and non-neuronal cells that may include key glial and/or immune cells that are thought to play an important role in the pathogenesis of neuropathic pain.41 Therefore, we conclude the σ2R/Tmem97 is a promising target for the generation of neuropathic pain drugs.
An interesting aspect of our behavioral findings is the relatively long onset of action of the compounds that we tested compared to rapid onset actions in previous studies with distinct ligands.21,40,42,43 There could be several reasons for this observation. One is that most previous studies have looked at short time points after administration of σ1R antagonists (30 or 45 min) or at repeated dosing effects21,40,42,43 so it is possible that effects with a slower onset are missed by these acute dosing schedules but are part of the sustained efficacy observed with repeated dosing. Another possibility is that while the mechanism of action of σ1R antagonists in pain was first thought to involve a spinal mechanism of action,21–23 more recent studies suggest an action of DRG neurons where σ1R is also expressed.44 It is therefore probable that the time to onset of effects is due to the time that is needed for the drug to diffuse to sites of action that are more distant from the injection site in the intrathecal space. Because σ1Rs are found on the nuclear envelop of DRG neurons,44 yet another possibility is that the effect of these compounds is transcriptional. If this were the case, this would also potentially take longer to manifest as a behavioral change. These considerations aside, our results are consistent with previous studies where σ1R antagonists produced long lasting relief of neuropathic mechanical hypersensitivity when given IT.21 Another important consideration here is that all of the compounds we examined as σ1R binding ligands also have affinity at σ2R/Tmem97 that could alter their behavioral effects, and some of them have affinity in the low nM range (JSS-1027 and MFG-1046).
To our knowledge, σ2R/Tmem97 ligands have not been previously tested for antinociceptive effects. The σ2R/Tmem97 ligands described herein have high affinity for σ2R/Tmem97, have good specificity relative to σ1R binding, and have little affinity for other targets in a small specificity screen. Indeed, UKH-1114 displays exceptional selectivity for σ2R/Tmem97 over >50 other receptors and channels. Although the previously described σ2R/Tmem97 agonist, siramesine, produced only a small effect in SNI mice, several other putative σ2R/Tmem97 agonists produced strong effects when given IT in the SNI model. This effect was blocked in the case of UKH-1114 by the known σ2R/Tmem97 antagonist,SAS-0132, strongly implicating σ2R/Tmem97 agonism as the mode of action responsible for antinociception. UKH-1114 was chosen to test via systemic administration in the SNI model. This compound produced a strong antimechanical hypersensitivity effect that was long lasting and devoid of motor impairment. The peak magnitude of effect was equivalent to the standard of care neuropathic pain drug, gabapentin, but it was much longer lasting at 10-fold lower dose. We observed a relatively long onset to antinociceptive action with putative σ2R/Tmem97 agonists, like σ1R ligands, although this onset to action was shorter when UKH-1114 was given IV. This may be explained by the expression of the Tmem97 gene, which is clearly expressed in structures outside of the intrathecal space and has high expression in the DRG and in non-neuronal cells in the DRG and CNS, suggesting the possibility of immune cell expression. We mined publicly available data sets and found that Tmem97mRNA is expressed in many mouse and human tissues, but in the mouse CNS, expression levels are apparently higher in many non-neuronal cells types, including astrocytes and microglia. This is consistent with the known expression of TMEM97 in human glioma cells.45 While beyond the scope of the present experiments, the discovery of tool ligands to manipulate σ2R/Tmem97 function will allow for testing the role of this receptor in a variety of cell types in the pain pathway.
More work is clearly needed to understand the role of σ2R/Tmem97 in pain, but these unprecedented results provide compelling support for developing σ2R/Tmem97 agonists as a novel strategy for the pharmacological management of neuropathic pain, bringing forth the possibility for patients to experience enhanced pain relief and reduced side-effects by modulating a receptor not targeted by currently approved FDA drugs. We are in the process of identifying other σ2R/Tmem97 agonists to further explore their utility as potential therapeutics for neuropathic pain.