
Small molecule modulator of sigma 2 receptor is neuroprotective and reduces cognitive deficits and neuroinflammation in experimental models of Alzheimer’s disease

By Bitna Yi, James J. Sahn, Pooneh Memar Ardestani, Andrew K. Evans, Luisa L. Scott, Jessica Z. Chan, Sangeetha Iyer, Ashley Crisp, Gabriella Zuniga, Jonathan T. Pierce, Stephen F. Martin, and Mehrdad Shamloo
Excerpt from the article published in Journal of Neurochemistry, 07 December 2016, 140: 561-575. DOI: https://doi.org/10.1111/jnc.13917
Editor’s Highlights
- Genetic inhibition of Sig2R is neuroprotective, cognitive enhancing, and antiinflammatory in experimental models of AD and excessive amyloidosis.
- Regulation of intracellular calcium concentration by Sig2R modulation could be responsible for these beneficial effects.
Abstract
Accumulating evidence suggests that modulating the sigma 2 receptor (Sig2R) can provide beneficial effects for neurodegenerative diseases. Herein, we report the identification of a novel class of Sig2R ligands and their cellular and in vivo activity in experimental models of Alzheimer’s disease (AD). We report that SAS-0132 and DKR-1051, selective ligands of Sig2R, modulate intracellular Ca2+ levels in human SK-N-SH neuroblastoma cells. The Sig2R ligands SAS-0132 and JVW-1009 are neuroprotective in a C. elegans model of amyloid precursor protein-mediated neurodegeneration. Since this neuroprotective effect is replicated by genetic knockdown and knockout of vem-1, the ortholog of progesterone receptor membrane component-1 (PGRMC1), these results suggest that Sig2R ligands modulate a PGRMC1-related pathway. Last, we demonstrate that SAS-0132 improves cognitive performance both in the Thy-1 hAPPLond/Swe+transgenic mouse model of AD and in healthy wild-type mice. These results demonstrate that Sig2R is a promising therapeutic target for neurocognitive disorders including AD.
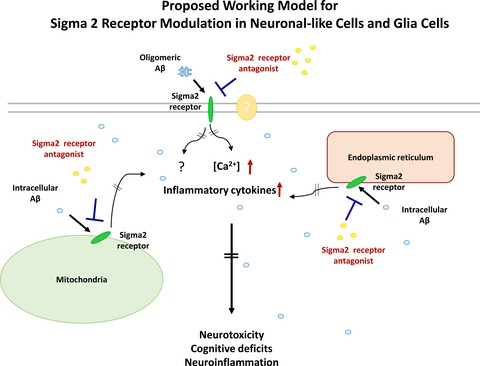
Alzheimer’s disease (AD) is currently the sixth leading cause of death in the US (James et al. 2014; Reitz and Mayeux 2014). The hallmarks of AD include neuronal loss, neuroinflammation, and the accumulation of amyloid plaques and neurofibrillary tangles (Mattson 2004; Citron 2010; Huang and Mucke 2012). The multifaceted pathology of AD makes it difficult to develop effective treatments. Current therapies for AD rely upon transiently mitigating some of its symptoms. However, these medications do not alter pathology underlying the disease (Cummings et al. 2014), and there remains an unmet need for therapeutic approaches to treat AD.
It is notable that modulating sigma receptors, for which the two known subtypes are sigma 1 (Sig1R) and sigma 2 (Sig2R) (Matsumoto et al. 2007; Nguyen et al. 2014a), has only recently been explored as a possible strategy for treating neurological disorders. Both sigma receptor subtypes are expressed in the central nervous system (CNS) and distinguished from one another based upon their affinity for different ligands and biological profiles. Sig1Rs, which have been cloned, are chaperone proteins that reside in the endoplasmic reticulum–mitochondrion interface as well as in nuclear and plasma membranes (Su et al. 2010, 2016; Hashimoto 2013); the structure of a trimer of Sig1R has been characterized by X-ray crystallography (Schmidt et al. 2016). Although Sig2Rs have not been cloned, they are widely distributed in the CNS and are implicated in cellular processes relevant to cancer and CNS disorders (Bowen 2000; Mach et al. 2013; Huang et al. 2014; Guo and Zhen 2015). Recent photoaffinity and fluorescent probe experiments suggest that the putative Sig2R binding site is in the progesterone receptor membrane component-1(PGRMC1) protein complex (Xu et al. 2011; Zeng et al. 2016). PGRMC1 is a cytochrome-like single-transmembrane protein that has been structurally characterized as a heme-bound dimer (Kabe et al. 2016) and is involved in neuroprotection and axonal migration (Runko and Kaprielian 2004; Cahill 2007; Rohe et al. 2009). Several reports conclude PGRMC1 and Sig2R are distinct molecular entities (Abate et al. 2015; Chu et al. 2015). These two observations are not necessarily mutually exclusive, and further work is needed to clarify the identity and function of the Sig2R. Notwithstanding this controversy, we originally identified the compounds described herein as Sig2R ligands and found they modulate a PGRMC1-related pathway; thus, we will refer to the biological target by the descriptor Sig2R/PGRMC1.
We became interested in Sig2R/PGRMC1 several years ago as a possible target for drug discovery in neurodegenerative diseases because of its widespread occurrence in the CNS and its putative involvement in calcium homeostasis. Within the CNS, Sig2R/PGRMC1 is highly expressed in the cortex and hippocampus, two regions known to be important for cognitive function (French and Pavlidis 2011; Izzo et al. 2014b). Moreover, Sig2R/PGRMC1 signaling has been implicated in cellular processes relevant to CNS diseases, including neuroprotection, axonal migration, and mitochondrial protection (Cahill 2007; Qin et al. 2015). For example, progesterone, which binds to both Sig2R and PGRMC1 (Chu et al. 2015), exerts neurogenic and neuroprotective effects in neural progenitor cells derived from adult rat brain (Liu et al. 2009). In the context of AD-related pathology, afobazole, which is a pan-selective Sig1R-Sig2R ligand, reduces neurotoxic microglia stimulation and apoptosis induced by fragments of amyloid beta (Aβ) (Behensky et al. 2013). Recent work conducted by Cognition Therapeutics also shows that Aβ oligomers are Sig2R/PGRMC1 ligands and that blocking the binding of these oligomers to Sig2R/PGRMC1 mitigates the synaptotoxic effects of Aβ oligomers (Izzo et al. 2014a,b). Moreover, exposure of cultured neurons to Aβ oligomers produces progressive up-regulation of Sig2R/PGRMC1 (Izzo et al. 2014b). This study also revealed that density of Sig2R/PGRMC1 remains unchanged in severely demented AD patients compared to the age-matched normal individuals despite a high degree of cell loss, suggesting that Sig2R/PGRMC1 may be up-regulated in AD patients. These findings suggest Sig2R/PGRMC1 is involved in AD pathology. Thus, we hypothesized that ligands which modulate Sig2R/PGRMC1 may hold significant promise as medicinal agents to treat AD.
As part of a broad effort to identify novel compounds with possible applications in neuroscience (Martin 2013; Sahn et al. 2014), we screened collections of substituted heterocyclic compounds against a variety of CNS proteins in the Psychoactive Drug Screening Program (PDSP) (Besnard et al. 2012). We thus identified a subclass of substituted norbenzomorphans that bind to Sig2R with high affinity (Sahn and Martin 2012; Sahn et al. 2016). This is an important discovery because norbenzomorphans have compact molecular scaffolds and may be easily modified for structure-activity relationship studies. Moreover, they are structurally distinct from all other known Sig2R ligands (Mach et al. 2013; Huang et al. 2014; Guo and Zhen 2015). Of the initial set of compounds, SAS-0132 emerged as an attractive candidate for further study because it exhibits ninefold selectivity for Sig2R over Sig1R, and has low off-target affinity for most other CNS-relevant targets. With this newly identified Sig2R/PGRMC1 ligand in our hands as a pharmacological tool, we tested our hypothesis that modulation of Sig2R/PGRMC1 might lead to beneficial effects in the treatment of AD.
Here, we report that the Sig2R/PGRMC1 antagonist SAS-0132 produces neuroprotective, cognitive enhancing, and antiinflammatory effects in animal models of neurodegeneration. These results suggest that Sig2R/PGRMC1 represents a promising target for the treatment of neurodegenerative diseases.
…
Results
Biological properties of SAS-0132 and related Sig2R/PGRMC1 ligands in vitro
Sigma receptor binding affinities of some norbenzomorphans
SAS-0132 (Fig. 1) is a member of a novel family of Sig2R ligands with a ninefold preference for Sig2R (Ki 90 nM) over Sig1R (Ki 841 nM) (Sahn et al. 2016). It also exhibits low off-target affinity (Ki ~0.5–10 μM) for most CNS-relevant targets, including opioid, adrenergic, nAChR, mAChR, and NMDA receptors, as well as the neurotransmitter transporters and ion channels (Table S2). Two structurally related compounds, DKR-1005 and DKR-1051, also show a similar degree of selectivity for Sig2R over Sig1R, whereas JVW-1009 is approximately equipotent for both SigR subtypes (Fig. 1).
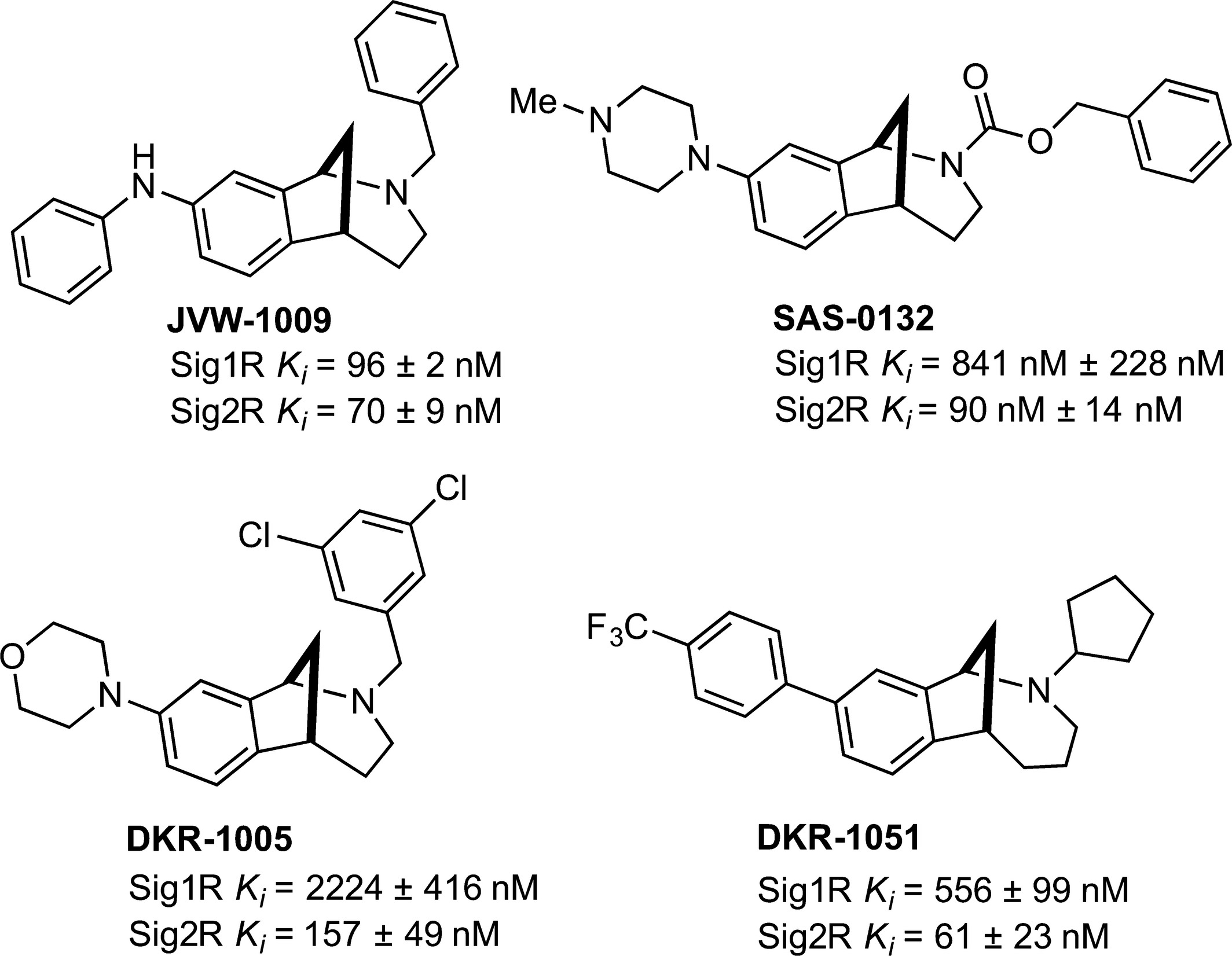
Sig2R ligands with neuromodulatory (JVW-1009, SAS-0132, DKR-1005, and DKR-1051) and cognition enhancing activity (SAS-0132).
Effects of Sig2R ligands on intracellular calcium
Sig2R agonists have been reported to modulate intracellular Ca2+ levels (Vilner and Bowen 2000; Cassano et al. 2009). Thus, we tested the effects of DKR-1051 and SAS-0132 on intracellular Ca2+ levels in human SK-N-SH neuroblastoma cells. DKR-1051 induces concentration-dependent increases in intracellular Ca2+ concentration, suggesting that it has agonist properties (Fig. 2a). SAS-0132 fails to produce a significant calcium response at concentrations up to 30 μM, but induces significant increases in intracellular Ca2+ at 100 μM (Fig. 2b). At 100 μM, the effects of SAS-0132 on intracellular Ca2+ are weaker than the effects of DKR-1051. As SAS-0132 has no effect on intracellular Ca2+ at concentrations up to 30 μM, we examined whether these lower concentrations of SAS-0132 antagonize the calcium response induced by DKR-1051. To evaluate the antagonistic effects of SAS-0132, SAS-0132 was applied prior to adding the putative Sig2R/PGRMC1 agonist DKR-1051 in order to allow SAS-0132 to first interact with the receptor in the absence of DKR-1051. To maximize the signal window for detecting antagonistic effects, we also used the high concentration of DKR-1051 (i.e., 100 μM), which produces a robust calcium response. Notably, SAS-0132 significantly attenuates the DKR-1051-induced calcium response at the lower concentrations (e.g., 0.1-30 μM) that are not per se associated with a significant calcium response (Fig. 2c).
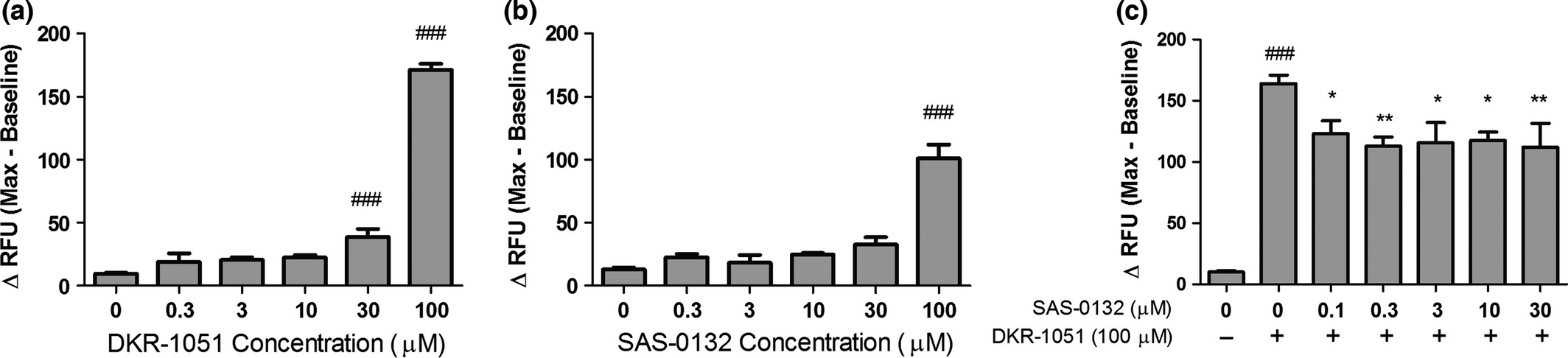
Effects of the Sig2R ligands on intracellular calcium in SK-N-SH neuroblastoma cells. (a) DKR-1051 induces concentration-dependent increases in intracellular calcium (###p < 0.001 vs. DKR-1051 (0 μM), one-way anovafollowed by Dunnett’s multiple comparison test). (b) SAS-0132 does not induce significant increases in intracellular calcium up to 30 μM, but produces a significant calcium response at 100 μM (###p < 0.001 vs. SAS-0132 (0 μM), one-way anova followed by Dunnett’s multiple comparison test). (c) SAS-0132 attenuated the calcium response induced by DRK-1051 (###p < 0.001 vs. DKR-1051 (0 μM); *p < 0.05 and **p < 0.01 vs. DKR-1051 (100 μM), one-way anova followed by Dunnett’s multiple comparison test). Data are presented as mean ± SEM from three to four independent experiments performed in duplicate or triplicate.
Sig2R/PGRMC1 antagonists are neuroprotective
Given that Sig2R/PGRMC1 appears to be involved in neuroprotection (Cahill 2007; Rohe et al. 2009; Izzo et al. 2014b), we queried whether any of our compounds might improve the survival of neurons in a C. elegans model of neurodegeneration. This novel transgenic model has a single copy insertion of human amyloid precursor protein 695 (SC_APP) under a pan-neuronal promoter in addition to the endogenous worm APP-like protein, apl-1. VC class neurons along the ventral cord are genetically manipulated to express a fluorescent label, so neurodegeneration in the transparent bodies of live worms can be easily monitored (Fig. 3a and b). Degeneration in two cholinergic neurons, VC4 and VC5, is characterized by shrinking somas visible in both fluorescence and phase-contrast images, and dimming GFP labeling (Fig. 3b). The presence of SC_APP in this worm model increases the rate of degeneration (% degeneration out of the total number of neurons scored) of these cholinergic neurons in an age-dependent manner (Fig. 3c). Notably, neuronal degradation in the SC_APP strain was significantly diminished by two different loss-of-function alleles of vem-1, a C. elegansortholog of PGRMC1 (Fig. 3d; Fisher’s exact test (n =194–230), SC_APP strain vs. each SC_APP; vem-1(null) strain, p < 0.01 and 0.001, respectively). Neurodegeneration was similarly diminished by pharmacological modulation of VEM-1 with Sig2R/PGRMC1 ligands (Fig. 3e). A 5-day treatment of SC_APP worms with SAS-0132 and the structural analog JVW-1009 (200 μM) decreased neurodegeneration on day 5 of adulthood when compared to the vehicle-treated group (Fig. 3e; Fisher’s exact test (n = 116–378), SAS-0132 or JVW-1009 vs. vehicle, p < 0.01). In contrast, neurodegeneration was exacerbated in SC_APP worms treated with DKR-1005 relative to the control group (Fig. 3e; Fisher’s exact test (n = 110–378), DKR-1005 vs. vehicle, p < 0.01). Importantly, JVW-1009 and SAS-0132 both reduced neurodegeneration in SC_APP C. elegans but provided no further neuroprotection in animals with vem-1 (PGRMC1) expression knocked down by RNAi (Fig. 3f; Fisher’s exact test (n =110–142), sham vs. RNAi, p < 0.001; RNAi vs. JVW-1009 or double treatment, n.s., Fig. 3g; Fisher’s exact test (n =130–138), sham vs. RNAi, p <0.01; RNAi vs. SAS-0132 or double treatment, n.s.). This suggests that neuroprotection occurs via inhibition of a PGRMC1-mediated pathway. Because the effects of the Sig2R/PGRMC1 null mutations are redundant to the effects of JVW-1009 and SAS-0132 and contrary to the effects of DKR-1005, we have tentatively classified JVW-1009 and SAS-0132 as Sig2R/PGRMC1 antagonists and DKR-1005 as a putative Sig2R/PGRMC1 agonist.
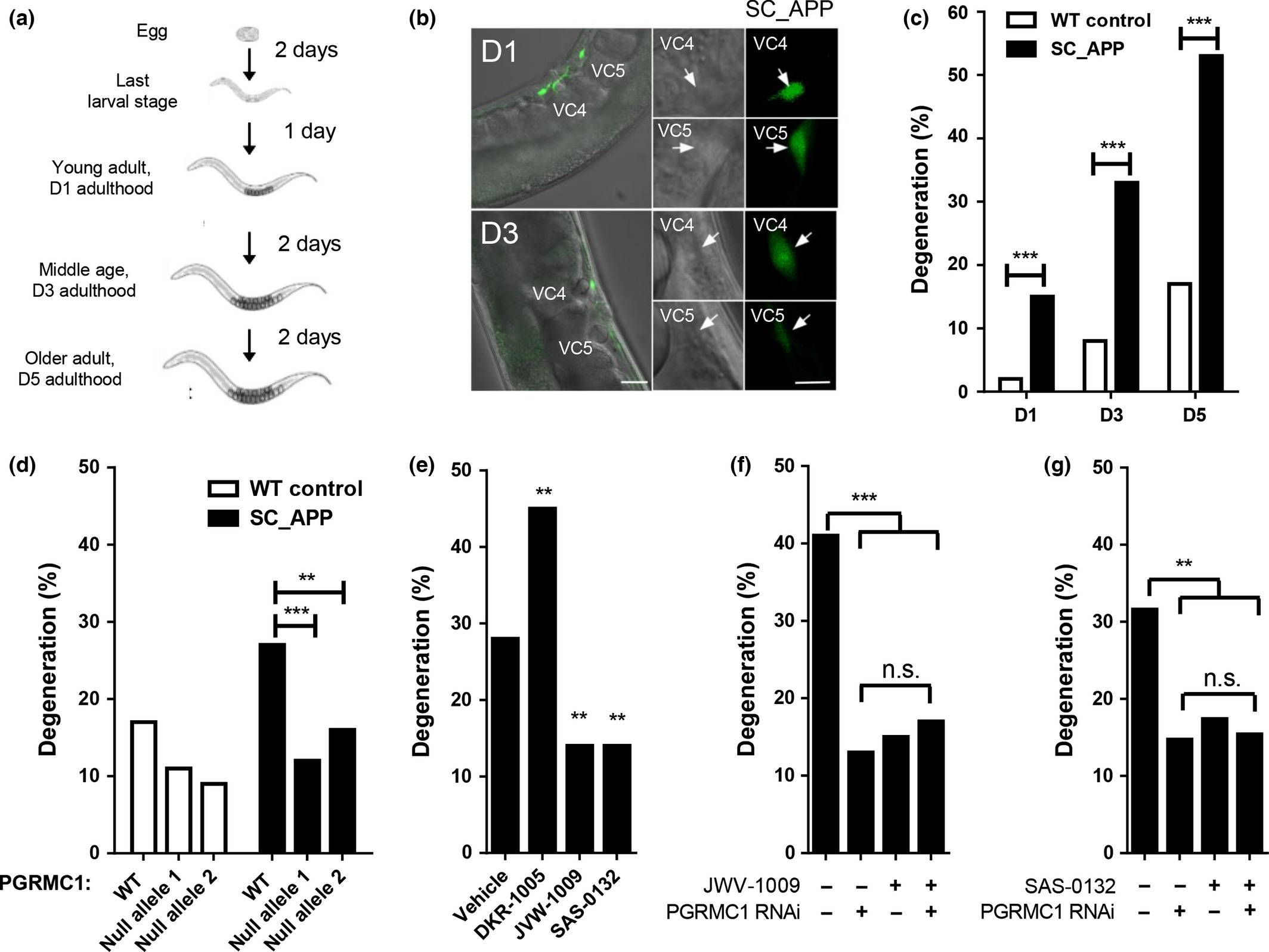
Neuroprotective effects of SAS-0132. (a) In our C. elegans model of neurodegeneration, transgenic worms carry a single copy insertion of human amyloid precursor protein (SC_APP). VC class cholinergic neurons express GFP. (b) Representative fluorescence and phase-contrast images of VC4 and 5 neurons in transgenic SC_APP C. elegans (on day 1 and 3 of adulthood). (c) Transgenic C. elegans expressing SC_APP display age-dependent degeneration of VC4 and 5 compared to wild-type (WT) controls (***p < 0.001 vs. WT control, planned comparisons with Fisher’s exact test, n = 60–128). (d) Neurodegeneration in these transgenic C. elegans is significantly diminished with two different null alleles of vem-1, an ortholog of PGRMC1 (**p < 0.01 and ***p < 0.001 vs. transgenic hAPP animals with WT PGRMC1, planned comparisons with Fisher’s exact test, n = 194–230) (e) DKR-1005 exacerbates the neurodegeneration shown in the transgenic SC_APP worm, while JVW-1009 and SAS-0132 decrease the neurodegeneration in the transgenic SC_APP worm (**p < 0.01 vs. vehicle group, planned comparisons with Fisher’s exact test, n = 110–378) (f) Knockdown of PGRMC1 or pharmacological modulation of Sig2R/PGRMC1 with JVW-1009 ameliorates the neurodegeneration shown in the transgenic C. elegans (***p < 0.001 vs. JVW-1009 (-)/PGRMC1 RNAi (-) group, planned comparisons with Fisher’s exact test, n = 110–142). JVW-1009 provides no further effects in animals with reduced PGRMC1 expression. (G) Knockdown of PGRMC1 or pharmacological modulation of Sig2R/PGRMC1 with SAS-0132 ameliorates the neurodegeneration shown in the transgenic C. elegans(**p < 0.01 vs. SAS-0132 (-)/PGRMC1 RNAi (-) group, planned comparisons with Fisher’s exact test, n = 130–138). SAS-0132 provides no further effects in animals with reduced PGRMC1 expression.
SAS-0132 is bioavailable and crosses the blood–brain barrier
The finding that the Sig2R antagonists SAS-0132 and JVW-1009 have neuroprotective effects suggested that they might mitigate neuronal damage in neurodegenerative diseases such as AD. Toward evaluating this intriguing possibility, we determined the PK properties of SAS-0132. Detectable levels of SAS-0132 were measured in plasma up to 3 h post-administration of 10 mg/kg through intravenous, subcutaneous, and oral administration (Fig. 4a). The bioavailability of SAS-0132 was 45% and 7% after subcutaneous and oral administration, respectively (Fig. 4b). Consistent with its favorable ClogD (3.5; pH = 7.4), SAS-0132 readily crosses the blood–brain barrier and achieves a brain concentration of 3.8 μM (subcutaneous administration) and brain/plasma ratio above 7 through all routes of administration at 3 h post-administration (Fig. 4c and d). The finding that only a trace (1.6 ng/g) of SAS-0132 could be detected in brain homogenates 24 h after a single injection of SAS-0132 (10 mg/kg, subcutaneous administration) suggests that it does not accumulate in the brain.
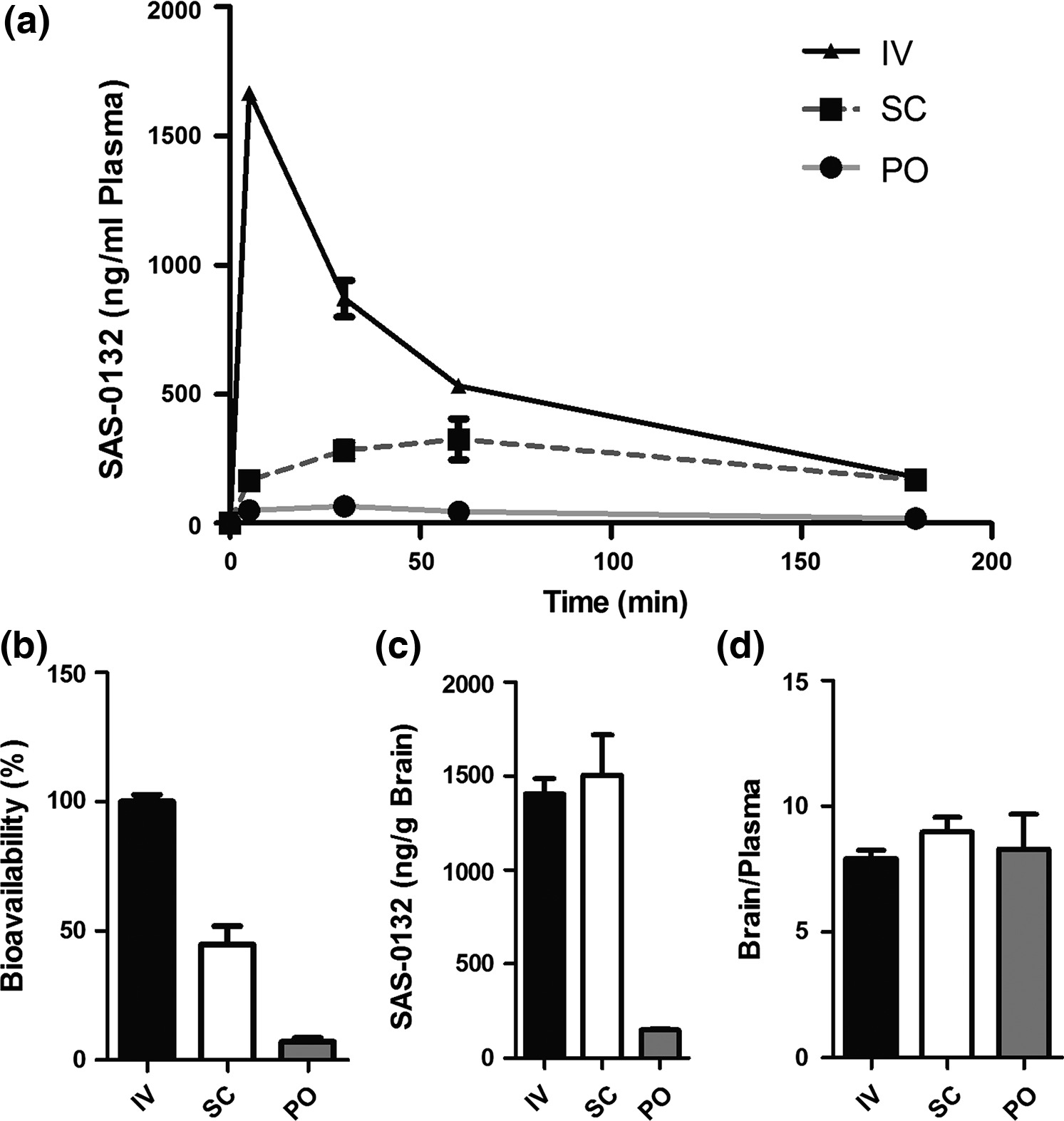
Pharmacokinetics of SAS-0132. (a) Plasma concentration of SAS-0132 in mouse plasma collected over 3 h after a single injection of SAS-0132 (10 mg/kg) via intravenous (IV), subcutaneous (SC), and oral (PO) administration (n =3 per route). (b) Bioavailability of SAS-0132 by SC and PO dosing (n =3 per route). (c and d) Brain concentration and brain/plasma ratio of SAS-0132 3 h after the administration of SAS-0132 (n = 3 per route). Data are represented as mean ±SEM.
SAS-0132 improves cognitive performance in APPLond/Swe+ mice
Having confirmed the bioavailability and brain permeability of SAS-0132, we assessed whether chronic treatment with SAS-0132 might alleviate the cognitive deficits associated with AD in an animal model of the disease. We selected the APPLond/Swe+ transgenic mouse model, which expresses human APP751 cDNA containing the London (V717I) and Swedish (K670M/N671L) mutations under the regulatory control of the murine Thy1 gene (Rockenstein et al. 2001, 2007); these animals exhibit cognitive deficits in several behavioral tests (Faizi et al. 2012; Coutellier et al. 2014; Nguyen et al. 2014b). Two separate cohorts of mice were used to assess chronic dosing and chronic dose–response effects of SAS-0132. Importantly, all behavioral tests were conducted prior to administration of the daily dose of SAS-0132 when SAS-0132 from the previous day’s dosing is expected to have been cleared from the brain.
In the chronic dosing study, daily dosing of APPLond/Swe+ mice and their WT littermates with SAS-0132 (10 mg/kg/day) was initiated at 4.5 months of age and continued for 64 days. In the sociability test performed after 35 days of dosing, vehicle-treated WT animals showed normal sociability by showing preference for the cup containing a mouse over the empty cup; this preference was not significantly altered by SAS-0132 treatment (Fig. 5a, t-test, mouse vs. empty cup, *p < 0.05). In contrast, vehicle-treated APPLond/Swe+ mice exhibited impaired sociability and showed no preference between the mouse and empty cup (Fig. 5a, t-test, mouse vs. empty cup, n.s.). The SAS-0132-treated APPLond/Swe+ mice showed a distinct preference for the cup containing a mouse over the empty cup (Fig. 5a, t-test, mouse vs. empty cup, *p < 0.05). In the Y-maze test performed after 50 days of dosing, vehicle-treated APPLond/Swe+ mice were impaired as assessed by low spontaneous alternation behavior in contrast to the WT mice (Fig. 5b, one-sample t-test vs. 50% theoretical mean, **p < 0.01, ***p < 0.001). APPLond/Swe+ mice treated with SAS-0132 showed an alternation level comparable to vehicle-treated WT mice (Fig. 5b, one-sample t-test vs. 50% theoretical mean, *p < 0.05). Total arm entries were not different among the four groups (Fig. 5b).
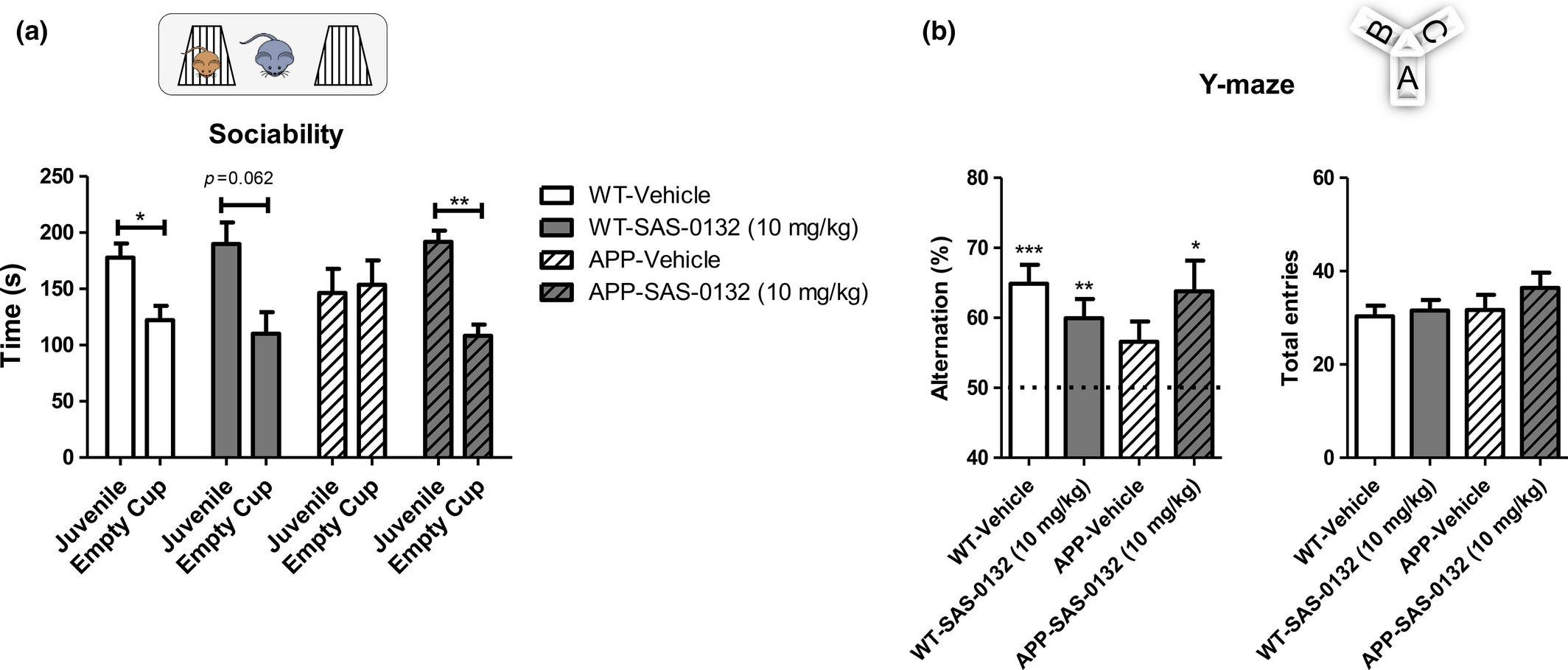
Effects of SAS-0132 on cognitive performance in the chronic dosing study. (a) In the sociability test, vehicle-treated wild-type (WT) mice explored a cup containing a C57Bl/6 mouse more than an empty cup, showing normal sociability. APPLond/Swe+ mice treated with vehicle showed lack of sociability, while APPLond/Swe+ mice treated with SAS-0132 (10 mg/kg) showed normal sociability (t-test, mouse vs. empty cup, *p < 0.05, **p <0.01, n = 9–12 per group). (b) In the Y-maze, WT mice showed spontaneous alternation behavior, indicating normal spatial memory. The vehicle-treated APPLond/Swe+ mice showed impaired spontaneous alternation performance. The APPLond/Swe+ mice treated with SAS-0132 (10 mg/kg) showed spontaneous alternation behavior to a level comparable to the WT mice (One-sample t-test vs. 50% theoretical mean, *p < 0.05, **p <0.01, ***p <0.001, n = 9–12 per group). The number of total entries was not significantly different among groups (one-way anova). Data represent mean ± SEM. Wild-type, WT; APPLond/Swe+, APP.
In the chronic dose–response study, APPLond/Swe+ mice and their WT littermates were aged to 6.7 months prior to initiating the administration of SAS-0132 at doses of 3, 10, and 30 mg/kg/d for a total of 60 days. In a social discrimination task performed after 30 days of dosing, all groups showed normal sociability except for WT and APPLond/Swe+ mice treated with 10 mg/kg/d of SAS-0132 (data not shown). We attribute this observation to animal variations because the higher dose of 30 mg/kg did not impair sociability. The social discrimination test was performed 1 day after the sociability test, and only APPLond/Swe+ mice treated with 3 mg/kg of SAS-0132 showed a significant preference for a novel mouse over a familiar one (Fig. 6a, t-test, familiar vs. novel). In the Y-maze test, which was performed after 45 days of dosing, vehicle-treated APPLond/Swe+ mice showed a deficit in spontaneous alternation behavior (Fig. 6b, one-sample t-test vs. 50% theoretical mean, n.s.), whereas those treated with 3 and 10 mg/kg of SAS-0132 showed normal spontaneous alternation behavior (Fig. 6b, one-sample t-test vs. 50% theoretical mean, n.s.). Administration of 3 and 10 mg/kg of SAS-0132 to WT mice did not affect their performance, but those dosed with 30 mg/kg/d showed impaired alternation behavior, suggesting that higher doses of SAS-0132 might have adverse effects on memory as measured in this test (Fig. 6b, one-sample t-test vs. 50% theoretical mean, n.s.). The number of total arm entries was not different among groups (Fig. 6b). The MWM test was initiated after 35 days of dosing. During the hidden platform learning period, WT mice treated with 3 and 30 mg/kg of SAS-0132 found the escape platform faster than the vehicle-treated WT mice (Fig. 6c, repeated measures of anova, *p < 0.05, Dunnett’s post hoc analysis, *p < 0.05). When the platform was removed, vehicle-treated WT mice did not show target quadrant preference, whereas WT mice treated with 3 mg/kg/d of SAS-0132 showed a clear preference for the target quadrant (Fig. 6c. t-test, target vs. non-target quadrant, *p < 0.01). Administration of SAS-0132 had no effect on the cognitive functions of APPLond/Swe+ animals during the hidden platform learning phase (Fig. 6d). After the spatial learning phase, the reversal learning hidden platform trial was performed. On day 2 of this training, WT mice treated with 10 and 30 mg/kg of SAS-0132 found the escape platform faster than WT mice treated with vehicle (Fig. 6e, one-way anova, *p < 0.05, Fisher’s LSD, *p ≤0.05). With the exception of the group treated with 3 mg/kg of SAS-0132, all WT groups demonstrated recall and showed target quadrant preference during the reversal learning probe trial (Fig. 6e, paired t-test, target vs. non-target quadrant, *p < 0.05, ***p <0.001). There were no significant effects of SAS-0132 treatment in APPLond/Swe+ mice during the reversal learning trials (Fig. 6f). During the reversal learning probe trial, vehicle-treated APPLond/Swe+ mice did not show a preference for the target quadrant over the non-target quadrants, whereas APPLond/Swe+ mice treated with 10 mg/kg of SAS-0132 showed target quadrant preference, indicating that SAS-0132 treatment reversed the reversal training probe deficits in the APPLond/Swe+ group (Fig. 6f, paired t-test, target vs. non-target quadrant, *p <0.05).
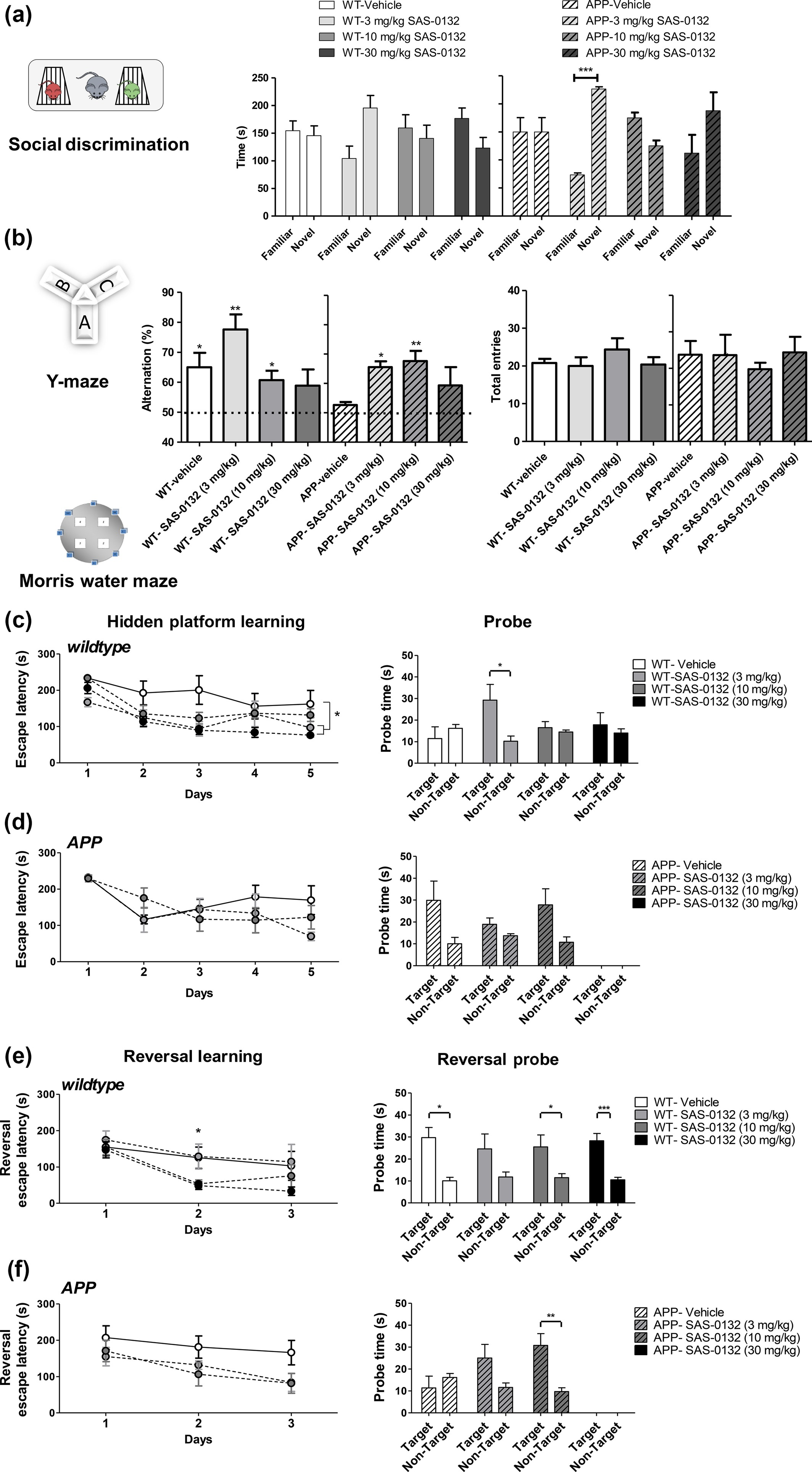
Effects of SAS-0132 on cognitive performance in the chronic dose–response study. (a) In the social recognition test, only the APPLond/Swe+ mice group treated with 3 mg/kg/d of SAS-0132 distinguished between a novel and familiar mouse, preferring a novel mouse (t-test, familiar vs. novel, ***p < 0.001, n = 3–5 per group). (b) In the Y-maze test, the APPLond/Swe+ mice showed impaired spontaneous alternation behavior, which was restored by chronic administration of SAS-0132. Both wild-type (WT) and APPLond/Swe+ mice dosed with 30 mg/kg/d showed impaired spontaneous alternation behavior (one-sample t-test vs. 50% theoretical mean, *p < 0.05, **p <0.01, n = 3–5 per group). The number of total entries was not significantly different among groups (one-way anova). (c) WT mice groups treated with 3 and 30 mg/kg/d SAS-0132 showed faster escape latency compared to vehicle-treated WT mice during the hidden platform training (repeated measures of anova, *p <0.01 vs. vehicle-treated WT, n = 3–5 per group). During the probe trial, WT mice treated with 3 mg/kg/d of SAS-0132 showed target quadrant preference (t-test, target vs. non-target quadrant, *p <0.01), while the other three groups did not show this preference (n = 3–5 per group). (d) APPLond/Swe+ mice showed signs of learning as indicated by preference for the target over the non-target quadrant during the probe trial, although not significant (n = 3–5 per group). Three of the five mice in the 30 mg/kg/d APPLond/Swe+ group did not perform the task. Thus, this group was excluded from the analyses. (e) On the second day of the reversal learning training, WT mice treated with 10 and 30 mg/kg/d SAS-0132 found the escape platform significantly faster relative to vehicle-treated WT mice (one-way anova, *p <0.05 post hoc,n = 3–5 per group). During the reversal probe trial, all WT groups showed target quadrant preference with the exception of the 3 mg/kg dose group (paired t-test, *p <0.05, ***p <0.001, n = 3–5 per group). (f) The reversal learning training revealed no statistically significant effect of drug on learning in APPLond/Swe+ mice (repeated measures of anova), although a non-significant decreased escape latency was observed for APPLond/Swe+ mice treated with 3 and 10 mg/kg/d SAS-0132 compared to the vehicle-treated APPLond/Swe+mice (n = 3–5 per group). During the reversal probe trial, APPLond/Swe+ vehicle-treated mice did not show target quadrant preference. APPLond/Swe+ mice treated with 10 mg/kg/d SAS-0132 showed preference for the target over the non-target quadrant (paired t-test, **p < 0.01, n = 3–5 per group). Three of five mice in the 30 mg/kg/d APPLond/Swe+ group did not pass the swimming performance criteria. Thus, this group was excluded from the analyses. Three additional mice in three other groups (WT-vehicle, APPLond/Swe+–3 and –10 mg/kg/d), were also excluded for the same reason. Data represent mean ± SEM. Wild-type, WT; APPLond/Swe+, APP.
In both chronic dosing and chronic dose–response studies, animals treated with SAS-0132 remained healthy and exhibited no detectable abnormalities. Locomotor activity measured in the activity chamber test after 24 days of treatment was not significantly different between vehicle- and SAS-0132-treated mice, indicating that general locomotor activity is not affected by SAS-0132 (data not shown). Comprehensive blood chemistry and histopathology studies conducted using plasma and organs obtained from WT mice treated with vehicle and SAS-0132 (10 mg/kg/d for 64 days) revealed no signs of SAS-0132-induced toxicity (Tables S3–4).
Effects of SAS-0132 treatment upon Aβ pathology, synaptic loss, and neuroinflammation
Upon completion of the behavioral tests, we queried whether the cognitive enhancing effects of SAS-0132 might be attributable to its effects on AD-related pathological features. In vehicle-treated APPLond/Swe+ mice from the chronic dosing study (6.5 months old at tissue collection), increased intracellular Aβ immunoreactivity was observed in the cortex, hippocampus, and amygdala, relative to vehicle-treated WT group (Fig. S1a, ***p <0.001 vs. WT groups, independent samples t-test). APPLond/Swe+ animals treated with SAS-0132 displayed Aβ immunoreactivity similar to that seen in the vehicle-treated APPLond/Swe+groups, indicating that chronic treatment with SAS-0132 does not affect levels of intracellular Aβ (Fig. S1a). Similarly, ELISA analysis using brains of these animals revealed no significant difference in soluble and insoluble Aβ40 and Aβ42 between vehicle- and SAS-0132-treated APPLond/Swe+ mice (Fig. 1b, independent samples t-test). Collectively, these data indicate that SAS-0132 does not affect Aβ burden in the APPLond/Swe+ mouse model of AD.
Because Aβ is known to be toxic to synapses and to induce a neuroinflammatory response (Shankar et al. 2007; Halle et al. 2008; Meyer-Luehmann et al. 2008; Koffie et al. 2009), we determined whether SAS-0132 affects synaptic pathology and the neuroinflammatory response. Quantifying the immunoreactivity of the pre-synaptic marker synaptophysin revealed a significant reduction in synaptophysin levels in the CA3 region of APPLond/Swe+mice compared to WT mice (chronic dosing study cohort, 6.5 months old at tissue collection), but chronic treatment with SAS-0132 (10 mg/kg/d) did not alter the extent of synapse loss (Fig. S2). With respect to neuroinflammation, quantitative immunohistochemistry with iba1 did not reveal any differences in microglia/monocyte-derived macrophages in vehicle-treated APPLond/Swe+ mice relative to vehicle-treated WT mice (chronic dosing study cohort, 6.5 months old at tissue collection). There were no drug-related effects on immunoreactivity for iba1 in either WT or APPLond/Swe+ mice (Fig. S2). On the other hand, qRT-PCR analysis revealed that APPLond/Swe+ animals had elevated inflammatory markers TNFα, CD14, and IL1β at 8.75 months (dose–response cohort), although elevation of the IL1β was not significant at 8.75 months (dose–response cohort) (Fig. 7). Notably, treatment with SAS-0132 decreased the expression of the inflammatory cytokine IL1β (Fig. 7).
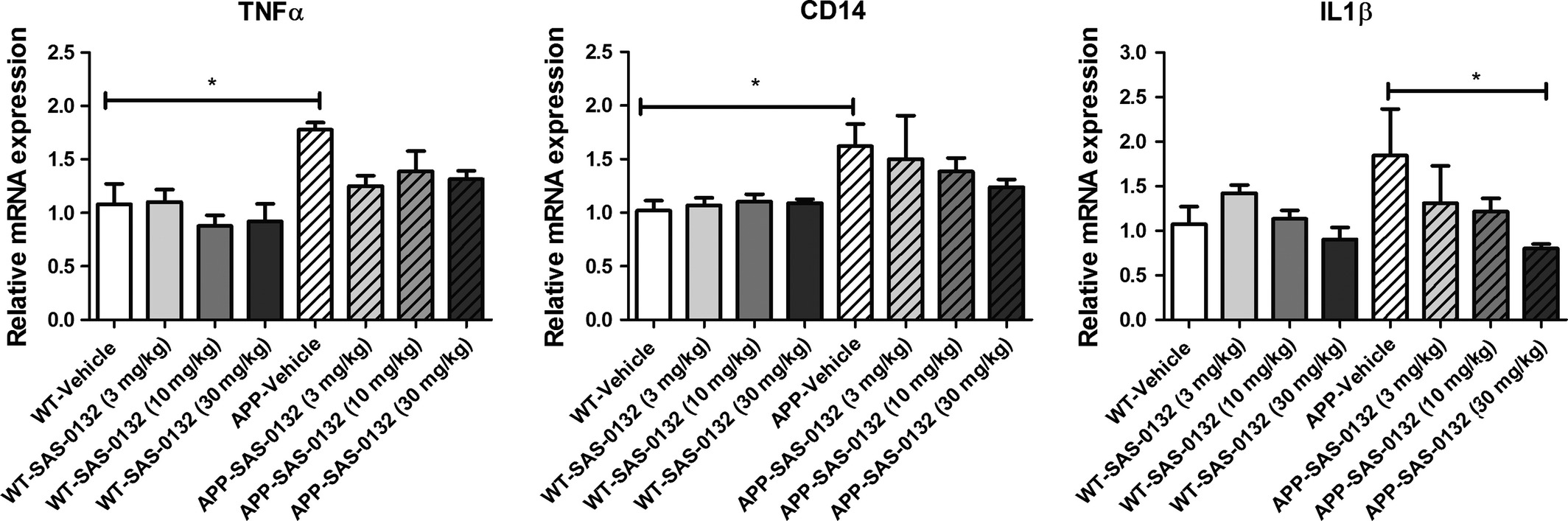
Effects of SAS-0132 on neuroinflammation in the chronic dose–response study. TNFα, CD14, and IL1β expression was elevated in the vehicle-treated APPLond/Swe+ mice compared to wild-type (WT) counterparts, although not significantly. SAS-0132 treatment attenuated increases in IL1β expression. Bar graphs depict mRNA expression in cortical tissue from WT and APPLond/Swe+ mice. mRNA expression was normalized relative to vehicle-treated WT mice for each gene. Data represent mean ± SEM. n = 3–5 per group. Wild-type, WT; APPLond/Swe+, APP.
Discussion
In this study, we report the identification of a novel norbenzomorphan class of Sig2R/PGRMC1 ligands and their cellular and in vivo activity. The Sig2R/PGRMC1 ligands reported here have Ki values in the low nanomolar range, which are comparable to the Kivalues of other known Sig2R/PGRMC1 ligands (Vilner and Bowen 2000; Huang et al. 2014; Guo and Zhen 2015). The three ligands (i.e., SAS-0132, DKR-1005, and DKR-1051) display selectivity for Sig2R/PGRMC1 over the Sig1R, with selectivity ratios > 9. The discovery of this novel class of Sig2R/PGRMC1 ligands is of great importance, because these norbenzomorphans have favorable pharmacokinetic attributes required for CNS indications. In addition, they have compact molecular scaffolds and are amenable to structure-activity relationship studies. Thus, norbenzomorphan analogs may be promising candidates for further development into drug leads.
Regulation of intracellular Ca2+ is one important cellular process mediated by Sig2R/PGRMC1, and structurally diverse Sig2R agonists are known to promote the release of calcium from the endoplasmic reticulum and mitochondria (Brent et al. 1996; Vilner and Bowen 2000; Cassano et al. 2009). We have shown that DKR-1051 produces concentration-dependent increases in intracellular Ca2+ levels, suggesting that DKR-1051 has agonist properties (Fig. 2a). The other Sig2R ligand SAS-0132 failed to produce a significant calcium response at up to 30 μM. At 100 μM, however, SAS-0132 increased intracellular Ca2+ levels, but its effects on intracellular Ca2+ levels were weaker than the effects of DKR-1051 (Fig. 2a and b). Interestingly, at the lower concentrations that have no effect on intracellular calcium levels, SAS-0132 attenuates the calcium response induced by DKR-1051 (Fig. 2c). Collectively, these observations suggest that SAS-0132 may have partial agonist activity. At the molecular level, Sig2R/PGRMC1 could potentially modulate intracellular Ca2+ by indirectly influencing the activity of ion channels expressed in the endoplasmic reticulum, mitochondria, and cell membrane (Vilner and Bowen 2000). As Sig2R/PGRMC1 is localized in high density to subcellular fractions known to store calcium (Basile et al. 1992), another possibility is that Sig2R/PGRMC1 directly gate the release of calcium. Further studies will be needed to determine the exact mechanisms by which Sig2R agonists increase intracellular calcium concentrations. Sig2R/PGRMC1-mediated calcium mobilization has important clinical implications because intracellular Ca2+ plays a critical role in learning and memory, and is strongly implicated in neuronal health (Bezprozvanny 2009). Perturbed Ca2+ homeostasis also appears to be involved in the dysfunction and death of neurons (Mattson 1994; Demuro et al. 2010), and its probable role in AD pathogenesis is supported by studies of animal models of AD as well as of patients (Bezprozvanny and Mattson 2008; Chakroborty et al. 2012; Zhang et al. 2015). Because Ca2+ dyshomeostasis is a pathological feature of AD and other neurodegenerative diseases (Berridge 2010), restoring Ca2+ homeostasis via modulation of Sig2R/PGRMC1 may be a promising new approach to treat neurodegenerative disorders.
C. elegans is well suited as a model organism for human diseases because it has clear orthologs for two-thirds of all human genes (Sonnhammer and Durbin 1997; Consortium 1998; Lai et al. 2000; Kuwabara and O’Neil 2001; Hulme and Whitesides 2011). Relevant to our study, C. elegans expresses vem-1, an ortholog of PGRMC1, as well as other genes involved in AD-related pathways (Runko and Kaprielian 2004; Link 2006; Haynes and Ron 2010; Simonsen et al. 2012; Safra et al. 2013). Our results demonstrate that single copy insertion of a single copy of the human APP gene into the C. elegans genome leads to age-dependent degeneration of cholinergic neurons. While previous studies have not examined age-dependent neurodegeneration, multi-copy over-expression of the C. elegans ortholog of APP (apl-1) or human APP has been shown to cause behavioral dysfunction and partial lethality in C. elegans (Hornsten et al. 2007; Ewald et al. 2012). Importantly, the neurodegeneration we observe in transgenic APP worms is diminished either by genetic manipulation of vem-1, the worm ortholog of PGRMC1, or by pharmacological inhibition with SAS-0132 and JVW-1009. Conversely, neurodegeneration is exacerbated by treatment with the agonist DKR-1005 (Fig. 3). These findings provide in vivo genetic evidence that PGRMC1 is critically involved in APP-mediated neurodegeneration in this animal model, and the Sig2R antagonists SAS-0132 and JVW-1009 exert neuroprotection via a PGRMC1-related pathway.
In chronic dosing and chronic dose–response studies, significant cognitive enhancing effects of SAS-0132 were observed in both transgenic and WT animals. In the chronic dosing study, SAS-0132 rescues AD-related sociability deficits and spatial memory deficits (Y-maze-test). In the chronic dose–response study, SAS-0132 rescues deficits in the social discrimination test (3 mg/kg) as well as deficits in spatial memory in the Y-maze test (3 mg/kg and 10 mg/kg). SAS-0132 also leads to improvements in spatial and long-term memory in both WT (3, 10, and 30 mg/kg) and APPLond/Swe+ mice (10 mg/kg) in the MWM test. The cognitive enhancing effects of SAS-0132 shown in the chronic dose–response study should be interpreted cautiously because of the small sample size; however, it is noteworthy that the effects of SAS-0132 are sufficiently large to be detected despite the low statistical power. Importantly, cognitive performance of the animals was evaluated prior to administration of the daily dose of SAS-0132. Because SAS-0132 is largely cleared from the brain within 24 h, the observed improvements in cognitive performance likely arose from sustained modulation of Sig2R/PGRMC1 rather than from an acute effect from treatment with SAS-0132.
There are several mechanisms that might explain the cognition enhancing effects of SAS-0132. First, SAS-0132 could produce pro-cognitive effects by being neuroprotective against APP-mediated neuronal toxicity. For example, the Sig2R ligands CT01344 and CT01346 were found to block binding of Aβ oligomers to Sig2R/PGRMC1 and restore AD-related behavioral deficits in the APPLond/Swe+ mouse model of AD (Izzo et al. 2014a). Second, by regulating intracellular calcium levels, SAS-0132 treatment may counteract Aβ-related dysregulation of calcium homeostasis. For example, application of Aβ oligomers to cultured neurons induces an unregulated flux of Ca2+ through the plasma membrane via various mechanisms that include activating endogenous receptors coupled to Ca2+ influx, disrupting membrane lipid integrity, and forming Ca2+-permeable channels (Arispe et al. 1993; Rovira et al. 2002; Kayed et al. 2004; Demuro et al. 2010). Dysregulated Ca2+ homeostasis can trigger detrimental processes such as apoptosis and free radical formation, and these adverse events can eventually lead to cognitive deficits. Thus, with its ability to modulate intracellular Ca2+concentration, SAS-0132 may lead to cognitive enhancing effects. Last, the benefits of SAS-0132 might not be exclusively related to its effects on APP-related pathways, but may result from more generalized mechanisms that are not specific to APP. For example, in our dose–response study, we discovered that chronic treatment of WT mice with SAS-0132 improved spatial and long-term memory in the MWM test. Although further studies are required to confirm this finding, the cognitive enhancing effects of SAS-0132 in healthy animals certainly warrant further investigation.
In contrast to its significant effect on cognition, SAS-0132 has no discernible effects upon several markers of AD pathology, including Aβ burden and synaptic loss. As neuroinflammation is one of the main pathological features of AD, we also sought to determine the effects of SAS-0132 on neuroinflammation. In the chronic dosing study, we didn’t evaluate the effects of SAS-0132 on AD-related neuroinflammation, as APPLond/Swe+mice showed little evidence of neuroinflammation. This is consistent with a previous study that reported the absence of signs of neuroinflammation in this mouse model at 8 months of age (Lull et al. 2011). Multiple lines of transgenic models of AD have been shown to develop AD-related neuropathology in an age-dependent manner (Rockenstein et al. 2001; Sly et al. 2001; Abbas et al. 2002; Oakley et al. 2006). Thus, it is possible that the APPLond/Swe+mouse model may show evidence of neuroinflammation at older age. Indeed, we observed significant increases in expression of neuroinflammatory genes in APPLond/Swe+ mice compared to WT mice in the chronic dose–response study (8.7 months of age at tissue collection). Chronic treatment with SAS-0132 was shown to decrease AD-related neuroinflammatory responses as measured by reduced brain levels of the inflammatory cytokine IL1β. Because chronic inflammation in response to neuronal damage is thought to accelerate and potentially underlie disease progression in AD (Heneka et al. 2015), the antiinflammatory effects of SAS-0132 might be partly responsible for its cognitive enhancing effects.
In this study, we report that pharmacological and genetic inhibition of Sig2R is neuroprotective, cognitive enhancing, and antiinflammatory in experimental models of AD and excessive amyloidosis. Regulation of intracellular calcium concentration by Sig2R modulation could be responsible for these beneficial effects. Further mechanistic studies will be needed to investigate the effects of modulating Sig2R/PGRMC1 on neuroinflammation, neuroprotection, cellular signaling, and intracellular calcium homeostasis. The fact that the Sig2R/PGRMC1 antagonist SAS-0132 has significant benefits when it is chronically administered to symptomatic animals inspires the intriguing question of whether treatment of pre-symptomatic animals could prevent the development of AD and/or cognitive deficits. Our findings clearly demonstrate the significant potential of modulating pathways mediated by Sig2R/PGRMC1 as a novel mechanism to treat AD. This hypothesis is supported by recent work conducted by Izzo and coworkers, who have shown that CT01344 and CT01346: (i) block binding of Aβ oligomers to Sig2R/PGRMC1 on neurons (Izzo et al. 2014a,b); (ii) prevent and reverse the effects of Aβ oligomers on membrane trafficking and synapse loss in neuronal culture (Izzo et al. 2014a); and (iii) reverse cognitive deficits in a mouse model of AD (Izzo et al. 2014a). These collective discoveries suggest that modulating pathways mediated by Sig2R/PGRMC1 is meritorious as a new approach to study neurodegenerative conditions and, if shown to be safe and well tolerated, should be further evaluated.