
σ2R/TMEM97 in retinal ganglion cell degeneration

By Hua Wang, Zhiyou Peng, Yiwen Li, James J. Sahn, Timothy R. Hodges, Tsung-Han Chou, Qiong Liu, Xuezhi Zhou, Shuliang Jiao, Vittorio Porciatti, Daniel J. Liebl, Stephen F. Martin, and Rong Wen
Excerpt from the article published in Scientific Reports 12, 20753, 01 December 2022, DOI: https://doi.org/10.1038/s41598-022-24537-3
Editor’s Highlights
- The molecular nature of sigma-2 receptor (σ2R) remained elusive until 2017 when a membrane protein previously known as transmembrane protein 97 (TMEM97) was purified using a resin coupled with a high-affinity σ2R-binding ligand.
- σ2R/TMEM97 is known to be involved in regulating cellular Ca2+ levels.
- σ2R/TMEM97 is also known to regulate cellular cholesterol by controlling cholesterol trafficking from lysosomes to the ER and cholesterol uptake.
- Small molecules like synthetic σ2R/TMEM97 ligands have the advantage over biological agents of being able to penetrate the blood-retinal barrier.
- σ2R/TMEM97 ligands are excellent candidates for the development of neuroprotective agents for treating glaucoma and other degenerative retinal diseases.
- inhibiting σ2R/TMEM97 protects retinal ganglion cells (RGCs)
- σ2R/TMEM97 facilitates RGC degeneration and that inhibiting σ2R/TMEM97 function is neuroprotective.
Abstract
The sigma 2 receptor (σ2R) was recently identified as an endoplasmic reticulum (ER) membrane protein known as transmembrane protein 97 (TMEM97). Studies have shown that σ2R/TMEM97 binding compounds are neuroprotective, suggesting a role of σ2R/TMEM97 in neurodegenerative processes. To understand the function of σ2R/TMEM97 in neurodegeneration pathways, we characterized ischemia-induced retinal ganglion cell (RGC) degeneration in TMEM97−/− mice and found that RGCs in TMEM97−/− mice are resistant to degeneration. In addition, intravitreal injection of a selective σ2R/TMEM97 ligand DKR-1677 significantly protects RGCs from ischemia-induced degeneration in wildtype mice. Our results provide conclusive evidence that σ2R/TMEM97 plays a role to facilitate RGC death following ischemic injury and that inhibiting the function of σ2R/TMEM97 is neuroprotective. This work is a breakthrough toward elucidating the biology and function of σ2R/TMEM97 in RGCs and likely in other σ2R/TMEM97 expressing neurons. Moreover, these findings support future studies to develop new neuroprotective approaches for RGC degenerative diseases by inhibiting σ2R/TMEM97.
Introduction
Sigma receptors were originally identified by radioactive ligand-binding assays as a subclass of opioid receptors1, but subsequent studies indicated that they were a distinct group of receptors with two members: the sigma-1 receptor (σ1R) and the sigma-2 receptor (σ2R)2,3,4,5. σ1R was cloned in 1996, and the protein sequence showed no homology to any other mammalian protein6. On the other hand, the molecular nature of σ2R remained elusive until 2017 when a membrane protein previously known as transmembrane protein 97 (TMEM97) was purified using a resin coupled with a high-affinity σ2R-binding ligand7. The ligand-binding properties of TMEM97 match those of the enigmatic σ2R, so σ2R was finally identified as TMEM977. The structure of σ2R/TMEM97 and its complexes with several ligands have been recently reported8, and 20(S)-hydroxycholesterol (20(S)-OHC) has been identified as an endogenous σ2R/TMEM97 ligand9.
The biological function of σ2R/TMEM97 is not well understood, but its wide distribution in the body, including the brain, the liver, pancreas, testis, and other organs10,11,12, implies that it may have distinct functions in different cells. σ2R/TMEM97 is known to regulate intracellular Ca2+ levels13 and to play a role in cholesterol trafficking and uptake14,15,16. The high expression of σ2R/TMEM97 in proliferating cancer cells makes it a biomarker and target for cancer diagnosis and therapy17,18,19,20,21,22,23. σ2R/TMEM97 is also involved in various neurological disorders24. Recent studies in animal models of Alzheimer’s disease and traumatic brain injury demonstrate that small molecules binding selectively to σ2R/TMEM97 are neuroprotective25,26, suggesting that σ2R/TMEM97 is involved in pathways relevant to neurodegeneration.
Toward understanding the role of σ2R/TMEM97 in neurodegenerative processes, we characterized ischemia-induced retinal ganglion cell (RGC) degeneration in mice with or without σ2R/TMEM97 (i.e., TMEM97+/+or TMEM97−/− mice). Herein we report that in TMEM97−/− mice, the absence of σ2R/TMEM97 renders RGCs resistant to ischemia-induced cell death. Moreover, intravitreal injection of a selective, high-affinity σ2R/TMEM97 ligand DKR-1677 protects RGCs from ischemia damage in wildtype (TMEM97+/+) mice. These findings demonstrate that σ2R/TMEM97 facilitates RGC degeneration. Our present work is a breakthrough in understanding the biological function of σ2R/TMEM97 in neurodegenerative processes in RGCs and likely in other TMEM97 expressing neurons. Importantly, these results support the development of novel therapeutic approaches to prevent or slow the progression of RGC degeneration by inhibiting the function of σ2R/TMEM97.
Results
Retinas of TMEM97 −/− mice
TMEM97−/− mice are viable and fertile with no visible gross abnormalities. The retinal structure of a TMEM97−/− mouse (Fig. 1b) is indistinguishable from the retina of a wildtype (WT) animal (Fig. 1a). Scotopic and photopic ERGs (electroretinograms) from the TMEM97−/−mice (TM−/−, Fig. 1c,d, respectively) are similar to those from the wildtype mice (WT, Fig. 1e,f, respectively). In both TMEM97−/− mice and wildtype mice, scotopic b-waves were reliably recorded with flash intensity as low as − 4.4 log cd s/m2. The visual function of the TMEM97−/− mice was further assessed by measuring the visual acuity (VA) in an optomotor system27. The average VA of TMEM97−/− mice is 0.45 ± 0.024 cpd (cycle per degree, n = 4, Fig. S1), which is within the reported range of wildtype mice28.
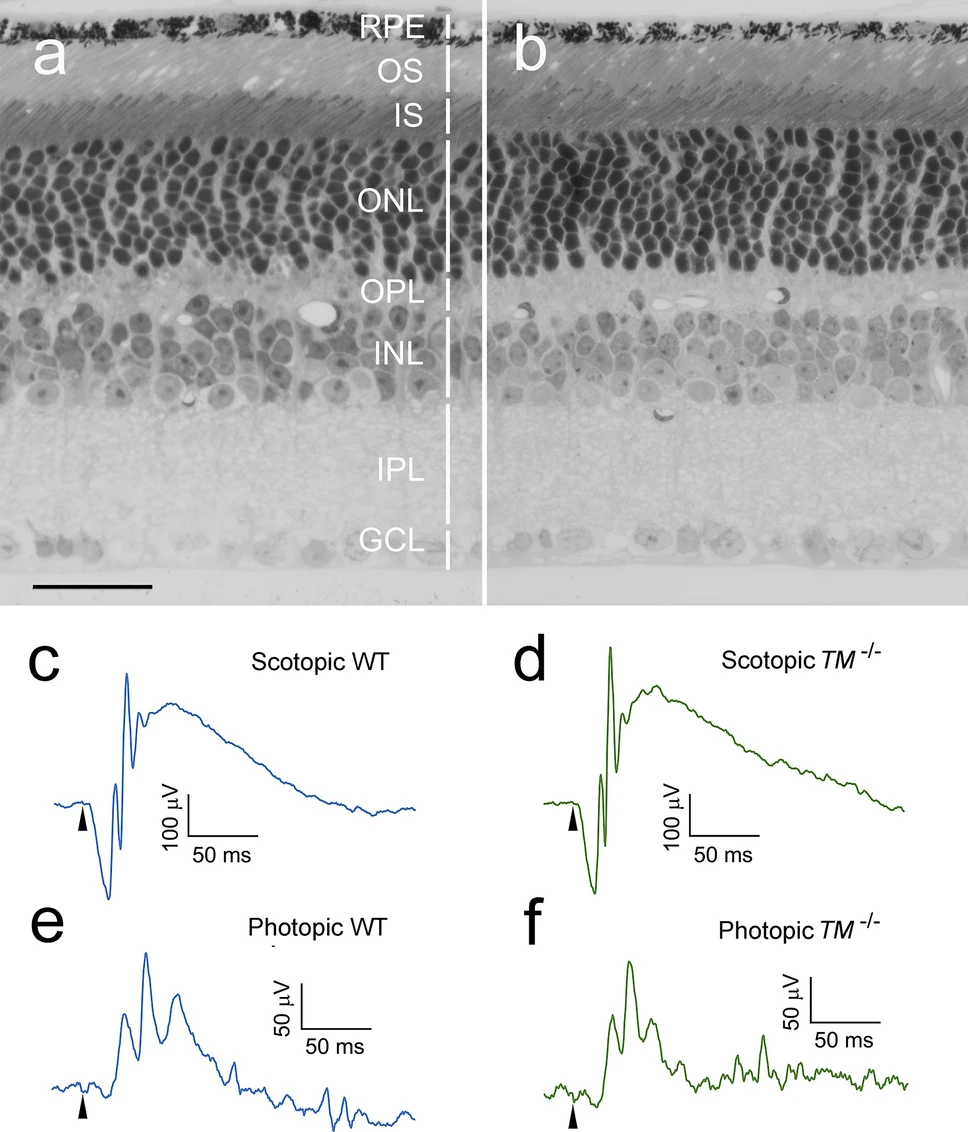
Retinal morphology and ERGs of TMEM97−/− mice. A semi-thin retinal section of a TMEM97−/− mouse (b) shows well-organized retinal layers indistinguishable from the structure of the wildtype mouse (a) (retinal layers are indicated by white vertical bars). Scotopic ERGs (evoked by flashes of 0.60 log cd s/m2) and photopic b-waves (elicited by flashes of 1.20 log cd s/m2) from the TMEM97−/− (TM−/−) mice (d,f, respectively) are very similar to scotopic and photopic ERGs from wildtype (WT) mice (c,e, respectively). RPE retinal pigment epithelium, OS photoreceptor outer segments, IS photoreceptor inner segments, ONL outer nuclear layer, OPL outer plexiform layer, INL inner nuclear layer, IPL inner plexiform layer, GCL ganglion cell layer. Scale bar: (a,b) 30 µm. The onset of the flash is indicated by arrowheads in (c–f).
In the retina of a wildtype mouse, σ2R/TMEM97 immunoreactivity was found in RGCs, the photoreceptor inner segments (IS), the RPE (retinal pigment epithelium), cells in the inner nuclear layer (INL), and sparsely in the outer nuclear layer (ONL) (Fig. 2a,b). No σ2R/TMEM97 immunoreactivity was detected in the retina of the TMEM97−/− mouse (Fig. S2).
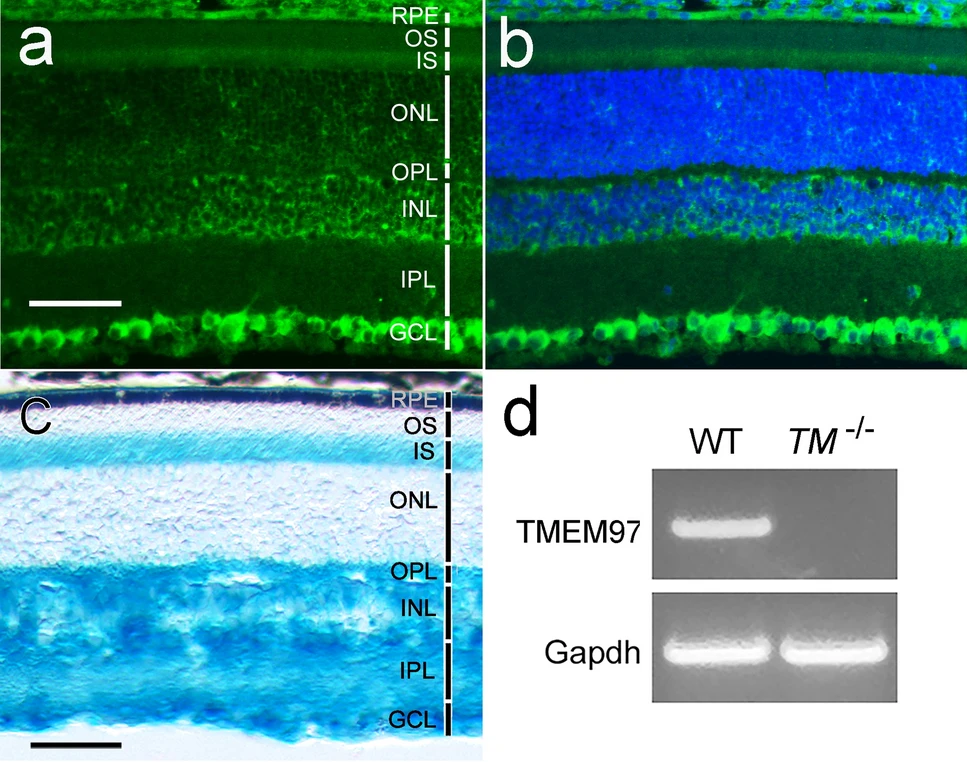
σ2R/TMEM97 expression in RGCs in the retina. σ2R/TMEM97 immunoreactivity in the retina of a wildtype mouse was detected in RGCs as well as in the RPE; the IS, sparsely in the ONL; cells in the INL (a). The same retinal section in (a) is shown in (b) with DAPI staining for cell nuclei. In the retina of a TMEM97−/−mouse (c), X-gal blue staining is present in the RGCs, and in the IS, the ONL (sparsely), cells in INL, the IPL, and the OPL. The blue stain in the RPE is mostly masked by melanin pigment (c). Retinal layers are indicated by white (a) or black (c) vertical bars. The RNA expression of TMEM97 in the retina was examined by RT-PCR (d). Robust TMEM97 expression (TMEM97) was detected in the retinas of the wildtype animals (d, WT) but absent in the retinas of TMEM97−/− mice (d, TM−/−). Expression of Gapdh served as a reference (d). Scale bars (a–c): 50 µm. The full gel image in (d) is displayed in Fig. S3.
The expression of TMEM97 in RGCs was also confirmed by X-gal staining in the TMEM97−/−mice. The TMEM97 gene in TMEM97−/− mice was replaced with a Velocigene cassette ZEN-Ub1 that has a lacZ reporter gene under the control of the TMEM97 promoter29, so the expression of the lacZ reporter correlates with TMEM97 expression. Blue X-gal staining is present in the RGCs as well as in the IS, the INL, the outer plexiform layer (OPL), the inner plexiform layer (IPL), the RPE, and sparsely in the ONL (Fig. 2c). In the RPE cells, the blue staining is mostly masked by the heavy melanin pigment in the 20 µm thick section (Fig. 2c).
The σ2R/TMEM97 distribution pattern in the wildtype mouse (Fig. 2a) and the X-gal staining pattern in the TMEM97−/− mouse overlap with each other, except in the IPL and OPL where σ2R/TMEM97 immunoreactivity is negative but blue X-gal stain is present. This discrepancy could be caused by differences in the subcellular localization of σ2R/TMEM97 and β-galactosidase because σ2R/TMEM97 is an ER membrane protein, but β-galactosidase is not.
The absence of TMEM97 expression in the retinas of TMEM97−/−mice was confirmed by reverse transcription PCR (RT-PCR). Robust TMEM97 mRNA expression was found in the wildtype retinas (WT, Fig. 2d, Fig. S3), but no TMEM97 mRNA was detected in the retinas of TMEM97−/− mice (TM−/−, Fig. 2d, Fig. S3).
Ischemia-induced RGC degeneration in TMEM97 −/− mice
Retinal ischemia was induced by elevating the intraocular pressure (IOP) to 120 mm Hg for 45 min in the left eyes of both wildtype and TMEM97−/− mice. The right eyes were untouched and served as naïve controls. Eyes were collected seven days after ischemia, and RGCs were identified by immunostaining of the RGC marker RBPMS (RNA binding protein with multiple splicing)30. A significant loss of RGCs was observed in the left eyes of wildtype mice as compared to RGCs in the untouched right eye (Fig. 3a,b). In contrast, many RGCs are present in the left eyes of TMEM97−/− mice after retinal ischemia (Fig. 3d). For comparison, the retina of the naïve fellow eye from the same animal in Fig. 3d is shown in Fig. 3c. The RGC survival rate was calculated as the ratio of RBPMS-positive cells in the left eye vs those in the right eye in the same animal. In the TMEM97−/− mice, the average RGC survival rate is 0.59 ± 0.02 (n = 9), which is significantly higher than the average rate of 0.18 ± 0.01 (n = 8, P < 0.001) in the wildtype animals (Fig. 3e). These results demonstrate that RGCs in adult animals lacking σ2R/TMEM97 are resistant to ischemia-induced cell death.
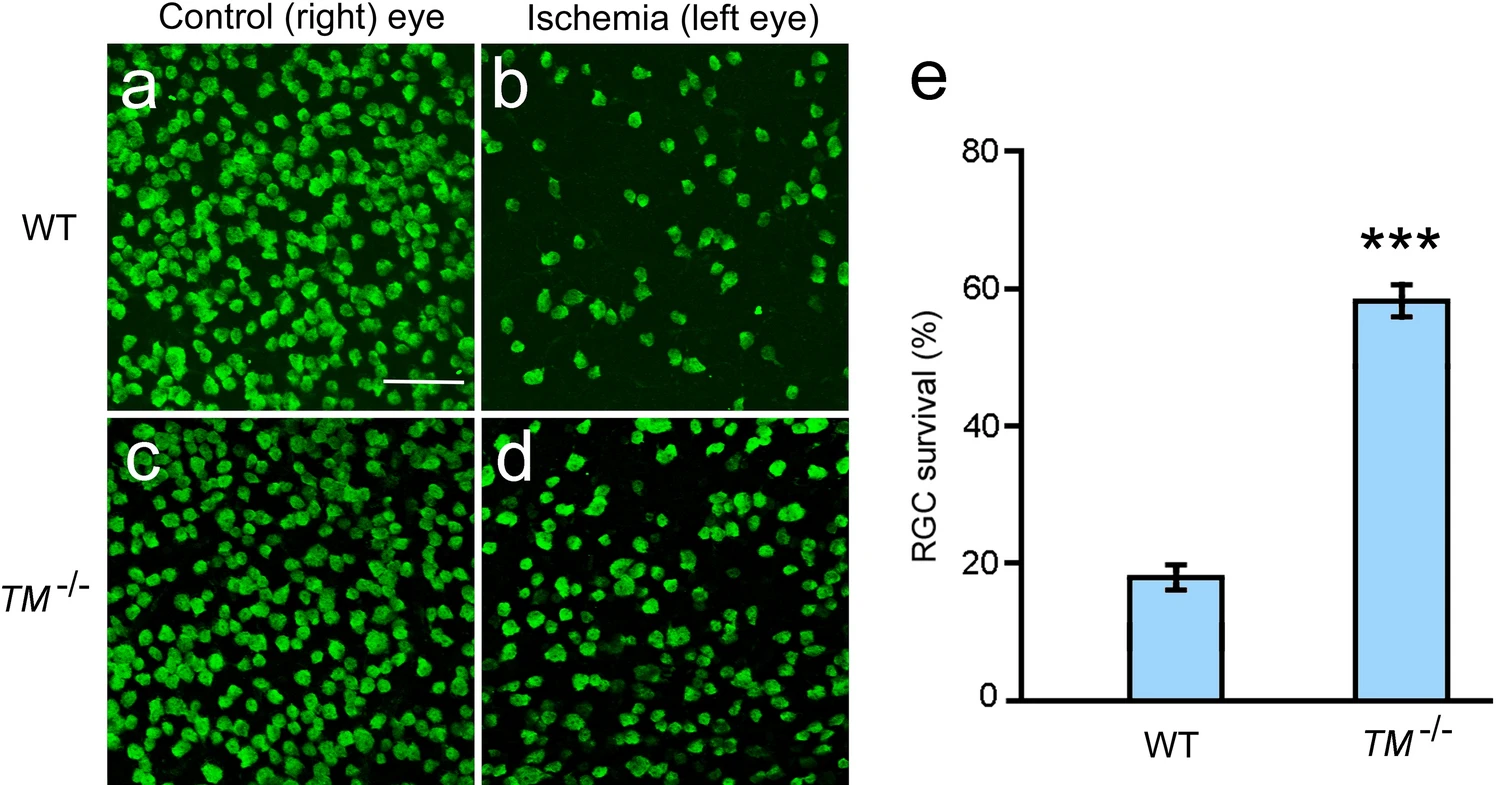
RGC survival in TMEM97−/− mice after retinal ischemia. RGCs were identified as RBPMS-positive cells in the flat-mounted retinas (a–d). A significant loss of RGCs is apparent in the left eye of a wildtype (WT) mouse (b) after ischemia as compared with the untouched fellow (right) eye of the same animal (a). In the retina of a TMEM97−/− mouse (TM−/−), many RGCs are present after ischemia (d). The retina of the untouched fellow (right) control eyes from the same animal in (d) is shown in (c) for comparison. RGC survival rates were calculated and presented in (e). The survival rate of TMEM97−/− mice is significantly higher than that of the wildtype control mice (e). Scale bar: 100 µm. Triple asterisks indicate P < 0.001.
The mean RGC density in the naïve eyes of the TMEM97−/− mice (271 ± 7/0.1 mm2, n = 9) is significantly lower than the average RGC density in the wildtype mice (366 ± 15/0.1 mm2, n = 8, P < 0.001). In contrast, the mean RGC density in eyes of TMEM97−/− mice after ischemia (157 ± 7/0.1 mm2, n = 9) is much higher than that in the eyes of wildtype animals after ischemia (65 ± 4/0.1 mm2, n = 8, P < 0.001). The lower RGC density in the naïve eyes of the TMEM97−/− mice is consistent with the lower baseline mean amplitude of PERG (pattern electroretinogram) from the naïve eyes in the TMEM97−/− mice (see below).
Pattern ERG measurement
PERG has been used as a sensitive measurement to assess the electrophysiological function of RGCs31. The PERG amplitude was measured peak-to-trough from the peak of the major positive wave at about 80 ms to the trough of the major negative component at about 300 ms. PERG latency was the time-to-peak of the major positive wave at about 80 ms. To monitor changes in RGC electrophysiological activities, we first recorded baseline PERG (Pre-IS) in wildtype and TMEM97−/− mice. The left eyes in each of these groups of animals were then subjected to ischemia, and the PERG was measured again seven days after ischemia (Post-IS).
In the retina of the wildtype mouse, ischemia markedly reduced PERG amplitude (Fig. 4a,Post-IS) as compared to the baseline (Fig. 4a, Pre-IS). No such change was observed in PERG amplitude following ischemia in the retina of the TMEM97−/− mouse (Fig. 4b, Pre-IS and Post-IS). A significant ischemia-induced decrease in mean PERG amplitude is seen in wildtype animals (Fig. 4c, WT ~ 60%, P < 0.001, n = 5), but not in the TMEM97−/− mice (Fig. 4c, TM−/−, n = 5). Ischemia-induced change in PERG amplitude in each animal was calculated as ∆Amp (Post-IS minus Pre-IS) to show that the average ∆Amp is significantly different from zero in the wildtype mice (P < 0.01, n = 5) (Fig. 4d, WT, n = 5). In contrast, the average ∆Amp in TMEM97−/− mice is not significantly different from zero (Fig. 4d, TM−/−, n = 5).
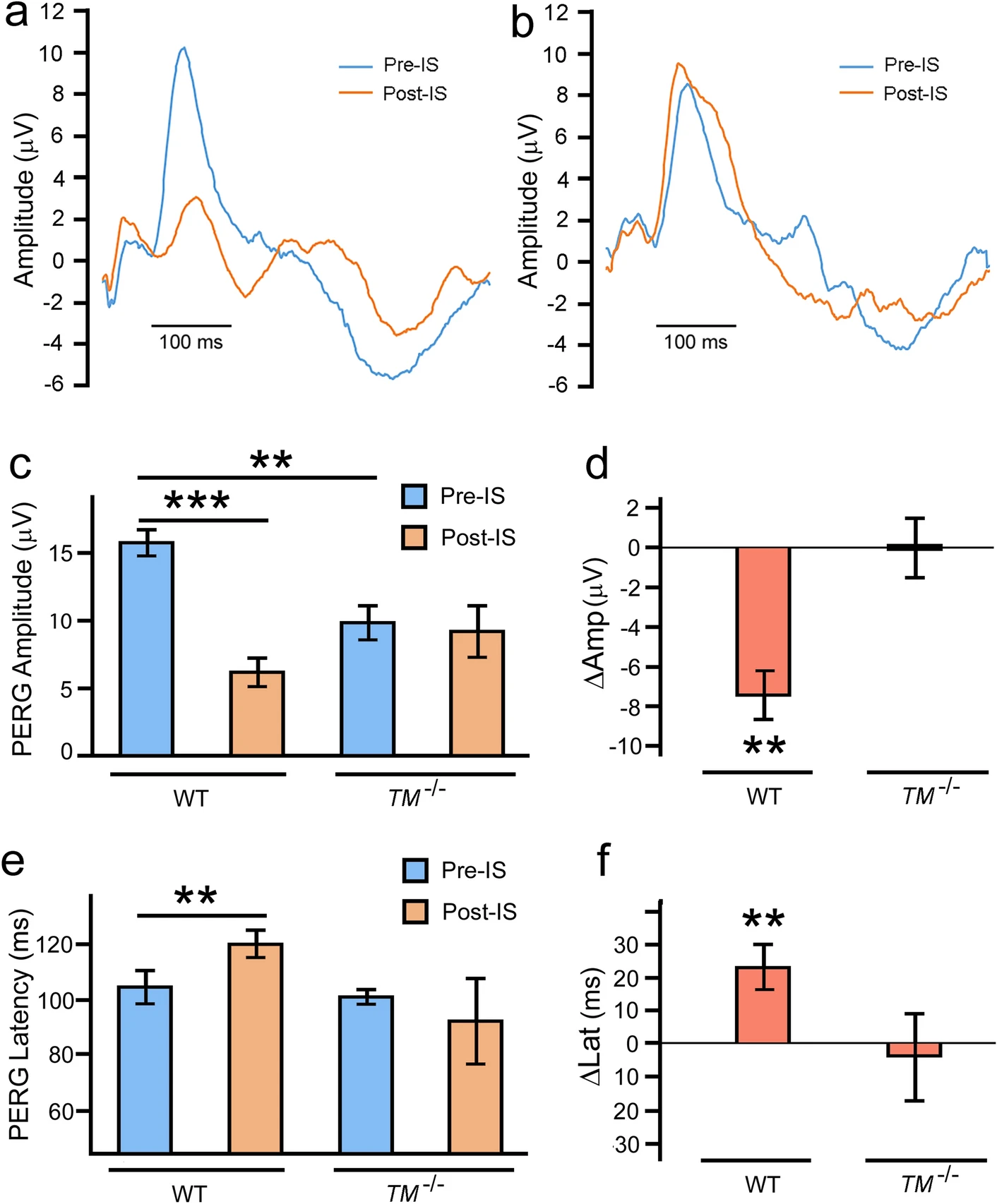
PERG measurements. Representative PERG recordings from a wildtype mouse show a significant decrease in PERG amplitude and an increase in PERG latency after retinal ischemia (a, Post-IS) as compared to the baseline amplitude of the same eye (a, Pre-IS). No significant change in PERG amplitude or latency was found in PERG recordings from the TMEM97−/− mouse (b, Pre-IS and Post-IS). The mean PERG amplitude showed a dramatic decrease in the wildtype mice after ischemia (c, WT, Post-IS) as compared to the baseline (c, WT, Pre-IS). No significant change in mean PERG amplitude was seen in TMEM97−/− mice (c, TM−/−, Pre-IS and Post-IS). The change in PERG amplitude in each animal (∆Amp, Post-IS minus Pre-IS) was calculated and the average ∆Amp is significantly different from zero in WT mice (d, WT) but not in TMEM97−/− mice (d, TM−/−). There was also an increase in mean PERG latency in wildtype mice after ischemia (e, WT), but not in TMEM97−/− mice (e, TM−/−). The change in latency in each animal (∆Lat, Post-IS minus Per-IS) was calculated and the average ∆Lat is significantly different from zero in wildtype mice (f, WT), but not in TMEM97−/− mice (f, TM−/−). The baseline mean PERG amplitude in the TMEM97−/− mice (c, TM−/−, Pre-IS) is lower than that in the wildtype mice (c, WT, Pre-IS). Double asterisks indicate P < 0.01. Triple asterisks indicate P < 0.001.
Retinal ischemia also induced an increase in PERG latency in the retina of the wildtype mouse (Fig. 4a), but not in the retina of the TMEM97−/− mouse (Fig. 4b). A significant increase in mean PERG latency (~ 20 ms, P < 0.01, n = 5) (Fig. 4e, WT, Post-IS) is observed in the wildtype mice as compared with the baseline (Fig. 4e WT, Per-IS), but there is no significant increase in mean PERG latency in TMEM97−/− mice (Fig. 4e, TM−/−, Per-IS and Post-IS). Ischemia-induced change in PERG latency in each animal was calculated as ∆Lat (Post-IS minus Per-IS) to show that the average ∆Lat in wildtype mice is significantly different from zero (P < 0.01, n = 5) (Fig. 4f), whereas the average ∆Lat in TMEM97−/− mice is not significantly different from zero (Fig. 4f, TM−/−, n = 5).
The baseline mean PERG amplitude in the TMEM97−/− mice (Fig. 4c, TM−/−, Pre-IS) is significantly lower (~ 30%, P < 0.01, n = 5) than that in wildtype mice (Fig. 4c, WT, Pre-IS), which is consistent with the lower RGC density in the TMEM97−/− mice.
Protection of RGCs by DKR-1677
The neuroprotective effect of a σ2R/TMEM97 ligand DKR-1677 on RGCs was examined in wildtype mice. DKR-1677 is a selective and high-affinity σ2R/TMEM97 ligand (Fig. 5h) that has been shown to be neuroprotective in two models of traumatic brain injury26. For intravitreal injections, DKR-1677 was dissolved in DMSO (dimethylsulfoxide) at concentrations of 2 µg/µL and 20 µg/µL. The left eyes were injected with 2 µL of either the low-dose (2 µg/µL) or the high-dose (20 µg/µL) formulation of DKR-1677. The vehicle control animals were injected with 2 µL DMSO into the left eyes. The right eyes of animals in the DKR-1677 and DMSO groups were not injected. Retinal ischemia was performed on the left (injected) eyes five days after injection, and the right eyes were untouched. RGC survival was assessed seven days after ischemia.
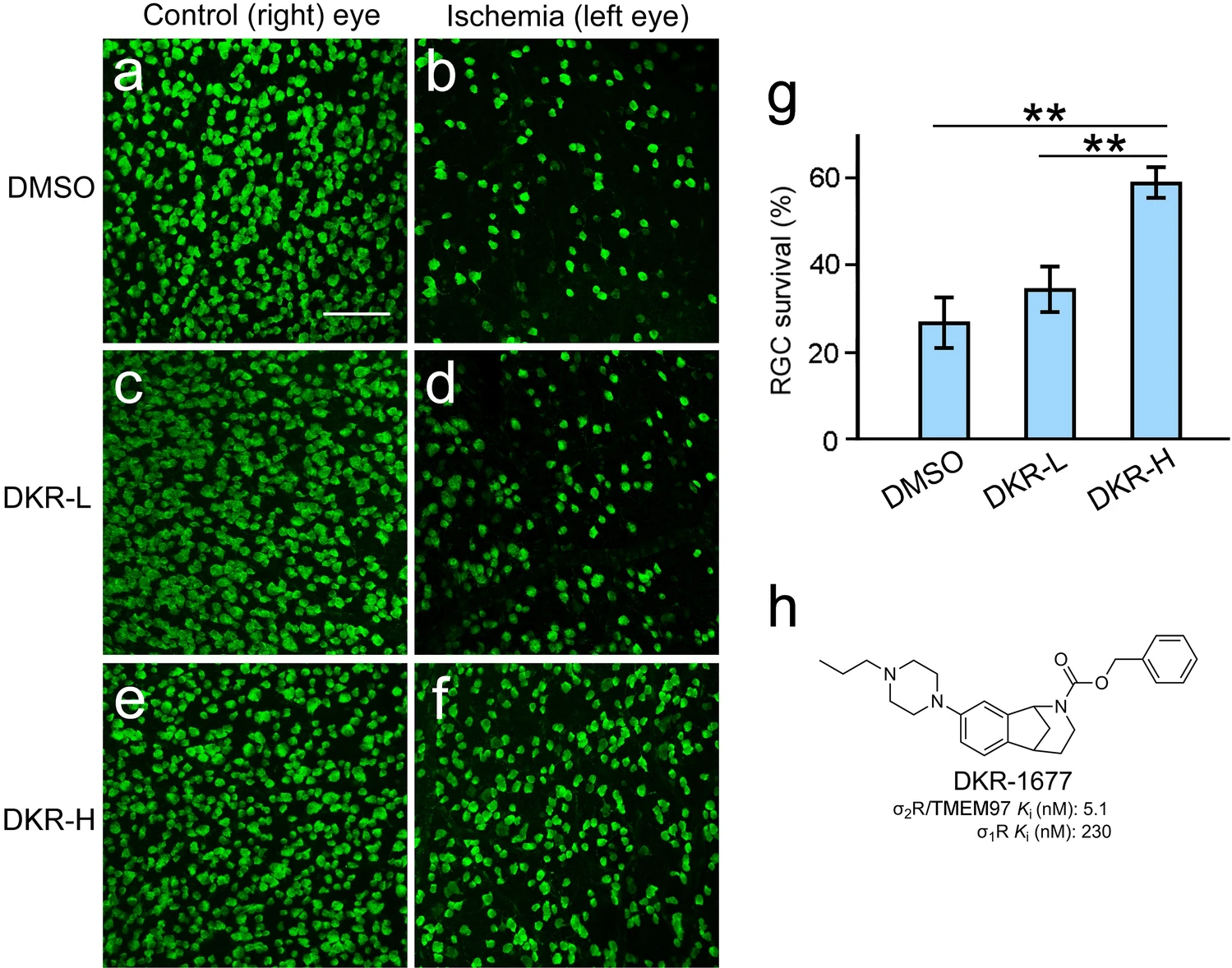
RGC protection by DKR-1677. Eyes of wildtype mice were intravitreally injected with DKR-1677 or DMSO and then subjected to ischemia. There was a significant loss of RGCs in the DMSO treated left eye after ischemia (b) as compared with the untouched fellow (right) eye of the same animal (a). Similar RGC loss was found in the eyes treated with a low-dose of DKR-1677 (DRK-L) (d), as compared to the right control eye of the same animal (c). In the eye treated with a high-dose of DKR-1677 (DRK-H), many RGCs remained after ischemia (f). The retina of the untouched fellow (right) eye of the same animal in (f) is shown in (e) for comparison. The RGC survival rate in the DKR-H group (g) is significantly higher than that in the DKR-L and DMSO group, respectively (g). The structure of DKR-1677 and its binding affinity for σ1R (guinea pig) and σ2R/TMEM97 (rat) are presented in (h). Scale bar (a–f): 100 µm. Double asterisks (g) indicate P < 0.01.
A significant loss of RGCs was seen in the left eyes in the vehicle group after ischemia, as compared with the untouched right eyes (Fig. 5a,b). Similar cell loss was found in the retina treated with low-dose DKR-1677 (DKR-L) as compared with the control right eye (Fig. 5c,d). In eyes injected with high-dose DKR-1677 (DKR-H), many surviving RGCs were present (Fig. 5e). For comparison, the retina of the naïve fellow eye from the same animal in Fig. 5f is shown in Fig. 5e. The average RGC survival rate in eyes treated with high-dose DKR-1677 (DKR-H, 0.59 ± 0.08, n = 5) is significantly higher than that in vehicle controls (DMSO, 0.27 ± 0.02, n = 5, P < 0.01) and eyes treated with low-dose DKR-1677 (DKR-L, 0.34 ± 0.03, n = 4, P < 0.01), respectively (Fig. 5g). The difference in RGC survival rate between the DKR-L and DMSO groups is not statistically significant (Fig. 5g).
The mean RGC densities in the naïve eyes of three groups (DMSO, 377 ± 15, n = 5; DKR-L, 360 ± 13, n = 4; DKR-H, 351 ± 55/0.1 mm2 n = 5) are not significantly different. On the other hand, the mean RGC density in eyes treated with high-dose of DKR-1677 and received ischemia (209 ± 18/0.1 mm2, n = 5) is significantly higher than that in the eyes subjected to ischemia in DMSO control animals (102 ± 18/0.1 mm2, n = 5, P < 0.01), or in the eyes treated with low-dose of DKR-1677 and received ischemia (123 ± 20/0.1 mm2, n = 4, P < 0.05). The difference in RGC densities between the DKR-L and DMSO groups is not statistically significant. The retinal overall structure showed no visible changes after injection of high-dose DKR-1677 (Fig. S4). These results demonstrate that the σ2R/TMEM97 ligand DKR-1677 protects RGCs against ischemia-induced cell death in a dose-dependent manner.
Discussion
A significant discovery of the present work is the loss-of-function phenotype in RGCs in the TMEM97−/− mice. When challenged by retinal ischemia, the RGCs in TMEM97−/− mice had significantly higher survival rates than the RGCs having fully functional σ2R/TMEM97 in wildtype mice. Moreover, electrophysiological activities of RGCs were much better maintained after ischemia in TMEM97−/− mice than in wildtype mice. These findings unequivocally demonstrate a role of σ2R/TMEM97 in the degenerative processes to facilitate ischemia-induced RGC degeneration. Another important finding is the neuroprotective effect of DKR-1677 against ischemia related RGC death in wildtype mice. The effect of DKR-1677 resembles the loss-of-function phenotype in RGCs in TMEM97−/− mice, indicating that the binding of DKR-1677 to σ2R/TMEM97 inhibits its function. Thus, the mechanism of action of DKR-1677, and perhaps other neuroprotective σ2R/TMEM97 ligands, is inhibiting the function of σ2R/TMEM97.
Selective σ2R/TMEM97-binding compounds have been reported to be neuroprotective in other studies. For example, DKR-1677, the σ2R/TMEM97 ligand used in the present work, has been shown to be neuroprotective in two models of traumatic brain injury26. Moreover, SAS-0132, a compound structurally similar to DKR-1677, was found to significantly improve cognitive performance in a mouse model of Alzheimer’s disease25. Furthermore, a σ2R/TMEM97-binding compound CT1812 is in clinical trials for treating Alzheimer’s disease32.
Intriguingly, the mean RGC density in the TMEM97−/− mice is lower than that in the wildtype mice. It is known that in early development of the retina, excessive RGCs are generated, and many undergo programmed cell death before the final number of RGCs is reached33,34. The lower RGC density in the TMEM97−/− mice is evidence that σ2R/TMEM97 is perhaps involved in the mechanism to determine the final RGC numbers during retinal development. This finding has important implications, and future studies will elucidate the role of σ2R/TMEM97 in regulating RGC numbers during early development.
RGCs express both σ1R and σ2R/TMEM97. σ1R has been shown to promote RGC survival35, and genetic ablation of σ1R in mice leads to an age-related inner retinal dysfunction and loss of RGCs36. σ1R agonists have also been shown to protect RGC35,37. We show herein that σ2R/TMEM97 facilitates RGC degeneration. It seems that the two receptors play opposite roles in RGC degeneration. The question remains as to whether σ1R and σ2R/TMEM97 interact with each other to regulate RGC degeneration.
The biological functions of σ2R/TMEM97 are not well understood, although σ2R/TMEM97 is known to be involved in regulating cellular Ca2+ levels13 and controlling cellular cholesterol levels14,15,16. It has long been recognized that treating cells with σ2R/TMEM97 ligands induces a biphasic increase in cytosolic Ca2+ levels13, indicating that σ2R/TMEM97 signaling could be mediated by intracellular Ca2+ levels. σ2R/TMEM97 is also known to regulate cellular cholesterol by controlling cholesterol trafficking from lysosomes to the ER and cholesterol uptake14,15,16. For example, a trimeric complex of σ2R/TMEM97, the progesterone membrane component 1 (PGRMC1), and the low-density lipoprotein (LDL) receptor has been shown to regulate cholesterol levels in cells by internalizing LDL16. A recent study identified 20(S)-OHC as an endogenous σ2R/TMEM97 ligand9. 20(S)-OHC binding to σ2R/TMEM97 significantly enhances the interaction between σ2R/TMEM97 and NPC1 (NPC intracellular cholesterol transporter 1), providing evidence that 20(S)-OHC regulate cholesterol trafficking from lysosomes to the ER9.
σ2R/TMEM97 is shown to be associated with several neurological disorders, including anxiety, depression, and addiction24. In animal models, σ2R/TMEM97 binding compounds are known to mitigate alcohol withdrawal symptoms38,39 and the effects of cocaine40, as well as to induce long-lasting relief of neuropathic pain41. A potential role of σ2R/TMEM97 in oxidative damage to RPE cells has been reported in two papers, but the results are contradictory42,43. Further studies could clarify the potential role of σ2R/TMEM97 in oxidative damage in RPE cells.
Our finding that inhibiting σ2R/TMEM97 protects RGCs could have specific clinical relevance. RGCs are output neurons that process and convey visual information from the retina to the visual cortex44. Several clinical conditions, including glaucoma, ischemic optic neuropathies, hereditary optic neuropathies, and demyelinating disease, lead to RGC death, optic nerve damage, and blindness45,46,47. Glaucoma is a heterogeneous group of optic neuropathies characterized by RGC degeneration and a leading cause of irreversible vision loss48,49,50,51. A promising therapeutic strategy for treating glaucoma is to protect RGCs46,52. The present work provides strong experimental data supporting a new neuroprotective approach for RGCs by inhibiting σ2R/TMEM97. Small molecules like synthetic σ2R/TMEM97 ligands have the advantage over biological agents of being able to penetrate the blood-retinal barrier. Accordingly, they are excellent candidates for the development of neuroprotective agents for treating glaucoma and other degenerative retinal diseases.
In summary, the present work is a breakthrough toward elucidating the function of σ2R/TMEM97 in RGCs and other σ2R/TMEM97 expressing neurons. We show that σ2R/TMEM97 facilitates RGC degeneration and that inhibiting σ2R/TMEM97 function is neuroprotective. This work also provides persuasive experimental support for a new therapeutic strategy to protect RGCs and other neurons in neurodegenerative diseases by inhibiting σ2R/TMEM97.