
Ubiquilin-2 regulates pathological alpha-synuclein

By Stephanie S. Sandoval-Pistorius, Julia E. Gerson, Nyjerus Liggans, Jaimie H. Ryou, Kulin Oak, Xingli Li, Keyshla Y. Negron-Rios, Svetlana Fischer, Henry Barsh, Emily V. Crowley, Mary E. Skinner, Lisa M. Sharkey, Sami J. Barmada, and Henry L. Paulson
Excerpt from the article published in Scientific Reports 13, 293 (2023). 06 January 2023, DOI: https://doi.org/10.1038/s41598-022-26899-0
Editor’s Highlights
- Parkinson’s disease (PD) and Lewy Body Dementia (DLB) are characterized by the accumulation and misfolding of disease-specific proteins.
- Ubiquilin-2 (UBQLN2), a protein quality control protein, has been linked to PD and DLB due to its colocalization with accumulated α-syn in PD and DLB post-mortem tissue.
- Of the three brain-expressed UBQLNs, UBQLN2 has been most widely implicated in neurodegenerative diseases and a significant role for UBQLN2, but not UBQLN1, in regulating α-syn is consistent with this concept.
- UBQLN2 is intrinsically prone to form condensates and aggregates and accumulates in various neurodegenerative proteinopathies, including Lewy bodies in PD.
- UBQLN2 has been implicated in proteasomal and lysosomal degradation pathways, pathways by which α-syn is regulated. The presence of UBQLN2 restricts a key pathogenic form of α-syn, pS129, clearance to the proteasomal pathway.
- The role of UBQLN2 in regulating pS129 sheds light on one potential reason why UBQLN2 is found in Lewy bodies in synucleinopathies: to decrease pS129 levels.
Abstract
The key protein implicated in Parkinson’s disease and other synucleinopathies is α-synuclein, and a post-translationally modified form of the protein, phosphorylated at serine 129 (pS129), is a principal component in Lewy bodies, a pathological hallmark of PD. While altered proteostasis has been implicated in the etiology of Parkinson’s disease, we still have a limited understanding of how α-synuclein is regulated in the nervous system. The protein quality control protein Ubiquilin-2 (UBQLN2) is known to accumulate in synucleinopathies, but whether it directly regulates α-synuclein is unknown. Using cellular and mouse models, we find that UBQLN2 decreases levels of α-synuclein, including the pS129 phosphorylated isoform. Pharmacological inhibition of the proteasome revealed that, while α-synuclein may be cleared by parallel and redundant quality control pathways, UBQLN2 preferentially targets pS129 for proteasomal degradation. Moreover, in brain tissue from human PD and transgenic mice expressing pathogenic α-synuclein (A53T), native UBQLN2 becomes more insoluble. Collectively, our studies support a role for UBQLN2 in directly regulating pathological forms of α-synuclein and indicate that UBQLN2 dysregulation in disease may contribute to α-synuclein-mediated toxicity.
Introduction
Like many neurodegenerative diseases, Parkinson’s disease (PD) and Lewy Body Dementia (DLB) are characterized by the accumulation and misfolding of disease-specific proteins1. A key pathological feature of PD and DLB is the presence of α-synuclein (α-syn)-rich Lewy bodies (LBs) and Lewy neurites (LNs) in neurons of affected brain regions2,3. Several factors contribute to the accumulation of aggregated α-syn in synucleinopathies, including post-translational modifications (such as phosphorylation at serine 129), increased production (duplication or triplication of the SNCA gene), missense mutations (e.g., A30P and A53T)4,5and altered degradation of α-syn protein6. Perturbations in proteostasis have been implicated in the pathogenesis of synucleinopathies7. Evidence supports regulation of α-syn by multiple quality control systems, including proteasomal and lysosomal pathways6,8,9. The specific aggregation state of α-syn may also influence which pathway(s) it is handled by.
Ubiquilin-2 (UBQLN2), a protein quality control protein, has been linked to PD and DLB due to its colocalization with accumulated α-syn in PD and DLB post-mortem tissue10. UBQLN2 is one member of the family of ubiquilins that function as shuttle proteins for the ubiquitin–proteasome system (UPS)11. Ubiquilins are similar in structure, containing N terminus ubiquitin-like (UBL) and C-terminus ubiquitin-associated (UBA) domains that facilitate the shuttling of ubiquitinated substrate proteins to the proteasome for degradation12,13. UBQLNs also contain STI1 motifs that may mediate binding to chaperones14. Among the UBQLNs, UBQLN2 uniquely contains a proline-rich domain (PXX)15. The prevailing view is that UBQLNs function predominantly as UPS shuttle proteins but likely participate in other quality control pathways as well, including autophagy11,16,17,18.
The importance of UBQLNs in neurodegenerative disease was underscored by the discovery that mutations in UBQLN2 directly cause frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS)15,19,20. UBQLN2 also forms liquid condensates and can aggregate in vitro17,21 and in vivo22, with pathogenic mutations increasing UBQLN2 aggregation22,23,24,25,26. UBQLN2 also co-localizes with aggregates of disease proteins such as huntingtin in Huntington’s disease, and TDP-43 and dipeptide repeat proteins in ALS/FTD10,15. Collectively, these results support the view that UBQLN2 contributes to multiple neurodegenerative diseases. Precisely how, however, is poorly understood.
Outside of the colocalization of UBQLN2 with Lewy bodies in PD and DLB, little is known about the role of UBQLN2 in synucleinopathies. For example, it is unknown whether UBQLN2 sequestration into protein aggregates supports a role for UBQLN2 in regulating α-syn, either normally or in disease states. Here, we use human disease tissue and transgenic mouse and cellular models to show that UBQLN2 regulates a key pathogenic form of α-syn, pS129, by targeting it to the proteasome for degradation.
UBQLN2 insolubility is increased in PD
Previous studies established that UBQLN2 accumulates in proteinaceous aggregates in several neurodegenerative diseases10,15. To determine whether UBQLN2 solubility is altered in synucleinopathies, we assessed soluble and insoluble levels of UBQLN2 in protein lysates from PD and DLB patient cingulate cortex. Compared to age-matched controls, PD patient brain lysates exhibited increased insoluble UBQLN2 (p = 0.03; Fig. 1A) and DLB brain lysates exhibited a trend towards increased insoluble UBQLN2 (p = 0.07; Fig. 1B). Taken together with previous studies showing that UBQLN2 colocalizes with LBs10, this finding suggests that UBQLN2 is sequestered in insoluble material.
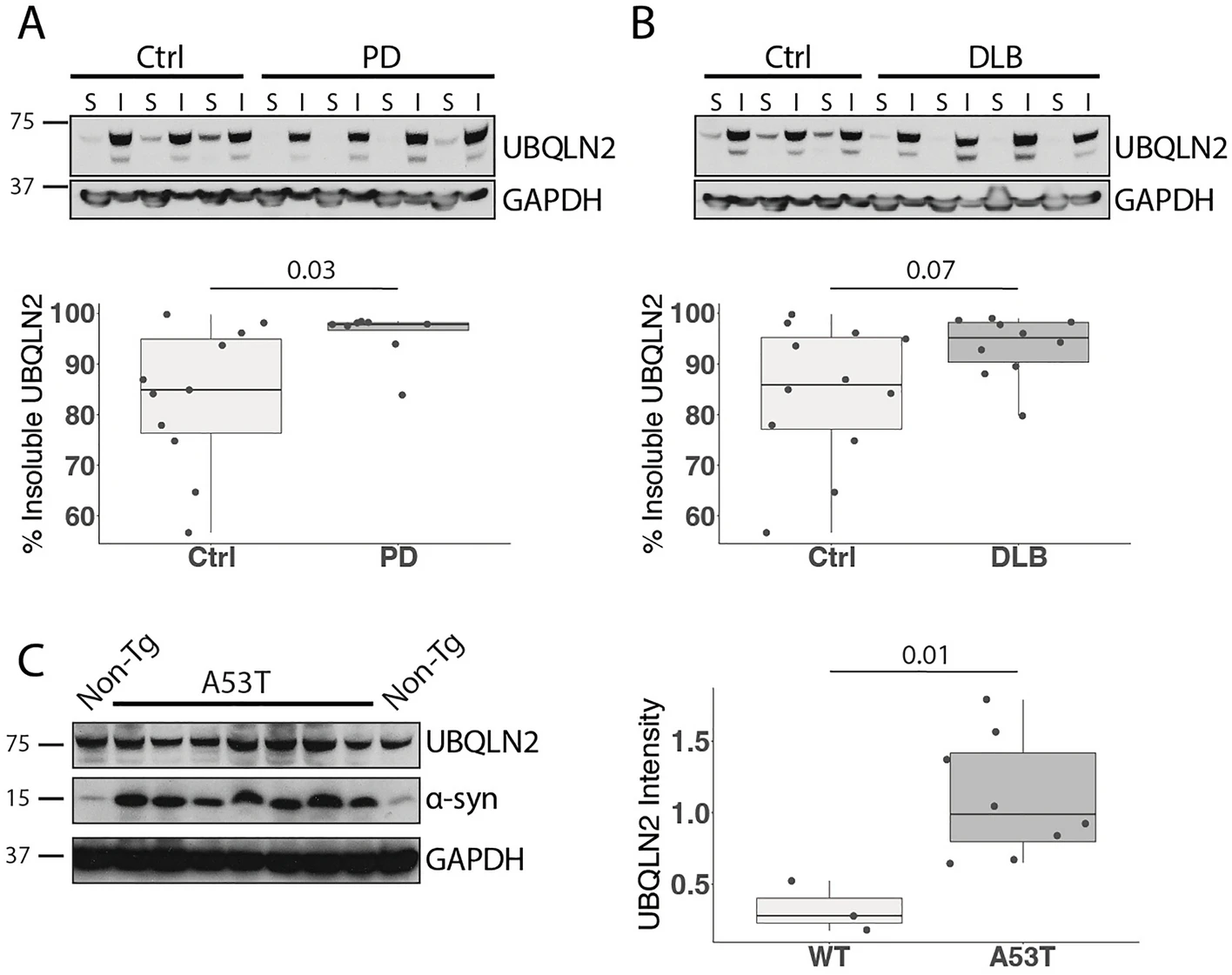
UBQLN2 insolubility is increased in Parkinson’s disease human tissue and mouse models. (A,B) Western blots of lysates from post-mortem cingulate cortex of PD and DLB patients show an increase in insoluble UBQLN2 in (A) PD (n = 8) and (B) DLB (n = 10) compared to age-matched controls. S PBS soluble, IPBS insoluble/sarkosyl soluble. (C) Representative western blot of whole brain lysate from mice overexpressing human mutant α-syn A53T solubilized in sarkosyl (n = 8) shows elevated levels of insoluble UBQLN2 compared to non-transgenic controls (n = 3). Western blots were cropped to focus on protein(s) of interest. Uncropped Western blots are shown in Supplemental Figure S1.
To determine whether a rodent model of PD recapitulates this observed increase in insoluble UBQLN2 in human disease, we analyzed levels of UBQLN2 in whole brains from 10-month-old transgenic mice expressing a pathogenic form of α-syn (A53T) implicated in familial PD27. Similar to our observations in human disease tissue, insoluble UBQLN2 was increased in brains from A53T mice compared to non-transgenic (Non-Tg) controls (p = 0.01; Fig. 1C). This result suggests not only that the A53T transgenic mouse is a relevant model for the study of UBQLN2 in synucleinopathies, but also that UBQLN2 solubility is affected by mutant α-syn.
UBQLN2, but not UBQLN1, preferentially regulates pS129 α-syn
UBQLN2’s function as a shuttle protein in protein quality control pathways and its colocalization with various disease protein aggregates10,15 suggest that it may regulate misfolded disease proteins such as α-syn by altering protein levels. LBs are enriched for pS12928,29, and based on studies showing that UBQLN2 colocalizes with LBs in disease10, we hypothesized that UBQLN2 may regulate α-syn, particularly levels of phosphorylated forms such as pS129. Preliminary studies in HEK-293 cells transiently transfected to express UBQLN2 and α-syn supported the view that UBQLN2 decreases levels of α-syn (data not shown). However, HEK-293 cells contain endogenous UBQLNs which can confound analysis. Accordingly, we used UBQLN triple knockout cells (TKO)30, which are devoid of UBQLNs 1, 2 and 4 to test the role of UBQLN2, or closely similar UBQLN1, in regulating α-syn, absent any potential compensatory effects of other UBQLNs. We transiently transfected α-syn with plasmids encoding UBQLN1, UBQLN2 or an empty vector (EV) and found that UBQLN2, but not UBQLN1, significantly decreased total α-syn (p ≤ 0.001) and pS129 levels (p = 0.03) compared to EV control (Fig. 2B). UBQLN1 expression led to an accumulation of pS129 levels (p = 0.01; Fig. 2B), but unlike UBQLN2, UBQLN1 has not been broadly implicated in neurodegeneration and does not display altered solubility in PD and DLB31. These results point to a preferential effect of UBQLN2 in altering levels of α-syn, including pS129.
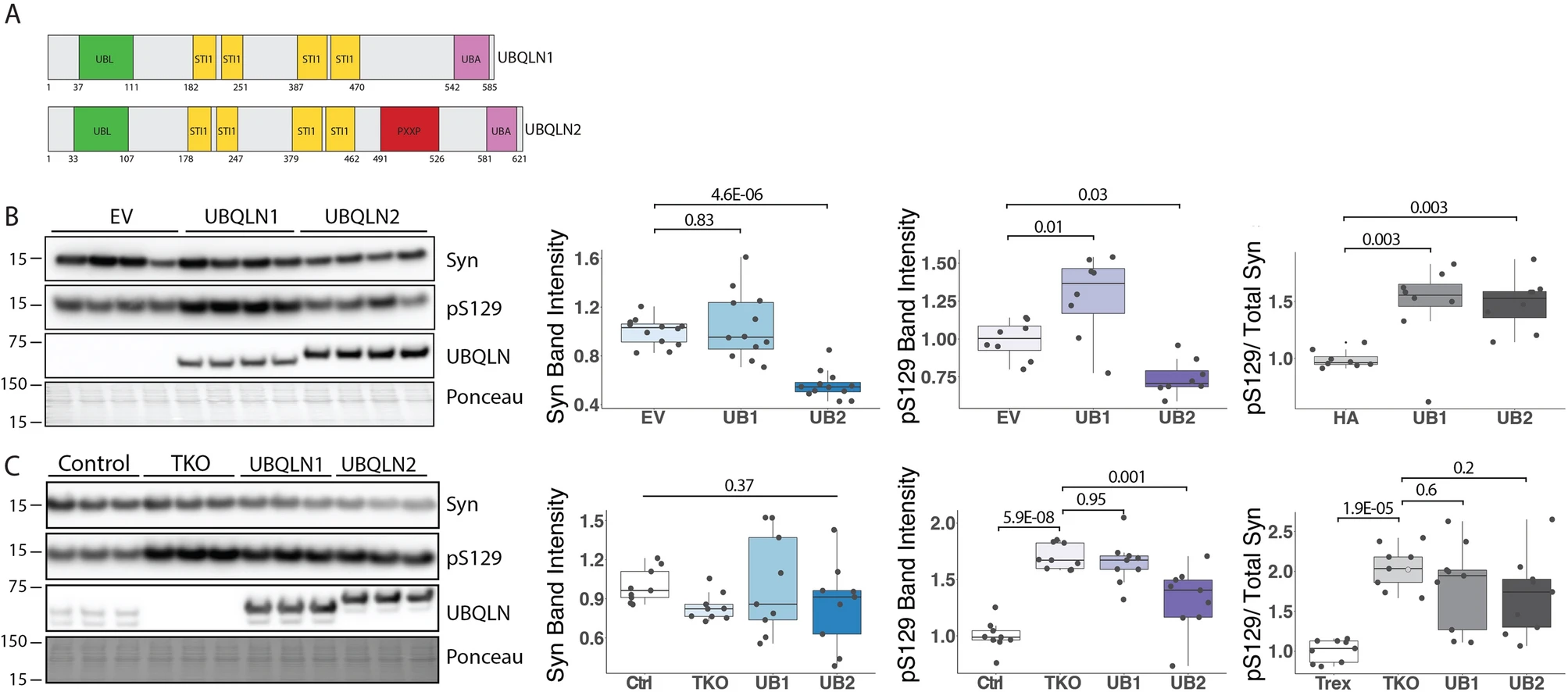
UBQLN KO leads to pS129 accumulation while UBQLN2 overexpression decreases levels of pS129 in vitro. (A) Diagram of UBQLN2 and UBQLN1 protein structure. (B) Representative western blot of UBQLN 1,2, 4 Total Knock out (TKO) cells transfected to express WT α-syn alone or together with UBQLN1 (UB1) or UBQLN2 (UB2). Total α-syn and pS129 levels are decreased by co-expression of UBQLN2 but not UBQLN1. The ratio of pS129/total Syn is increased in both UBQLN1 and UBQLN2 co-expression (n = 8). (C) Representative western blot of TKO, UBQLN1 rescue, UBQLN2 rescue, and control cells were transfected with α-syn showing elevated pS129 levels in TKO cells compared to control cells, which is partially reversed by inducible co-expression of UBQLN2 but not UBQLN1. The ratio of pS129/total Syn is significantly increased in TKO compared to control and not reversed by UBQLN1 or UBQLN2 rescue (n = 8). Western blots were cropped to focus on protein(s) of interest. Uncropped Western blots are shown in Supplemental Figure S2.
UBQLNs function as shuttle proteins for the UPS and form heterodimers that may be important for their function32. To assess the role of UBQLN 1 or 2 on α-syn and pS129 protein levels when expressed at closer to endogenous levels and independent of other UBQLNs, we transiently expressed α-syn in TKO UBQLN rescue cells that inducibly express either UBQLN1 or UBQLN2 when treated with doxycycline30. These UBQLN rescue lines overexpress UBQLN1 or UBQLN2 to a lesser extent than when transiently transfected, allowing us to assess UBQLN1 and UBQLN2 regulation of α-syn in a more biologically relevant environment. Control cells that express endogenous levels of UBQLN 1, 2 and 4 were also evaluated. In the absence of all UBQLNs, levels of pS129 were significantly increased in TKO cells, and induction of UBQLN2 significantly decreased pS129 levels, compared to TKO cells (p = 0.001; Fig. 2C). In contrast, UBQLN1 induction had no effect on pS129 levels compared to TKO cells (p = 0.95; Fig. 2C), suggesting UBQLN1 may only cause pS129 accumulation when an overabundance of UBQLN1 is expressed. Total α-syn levels were unchanged in TKO and UBQLN 1 or 2 rescue cells compared to control cells (p = 0.37; Fig. 2C). Analysis of the ratio of pS129 to total Syn revealed no significant difference between TKO and UBQLN 1 (p = 0.6; Fig. 2C) or UBQLN 2 (p = 0.2; Fig. 2C). These data further support a role for UBQLN2 in regulating α-syn, including a pathogenic isoform, pS129.
To further explore UBQLN2 regulation of α-syn, we performed immunofluorescence on HEK-293 cells in which we transiently expressed or knocked down UBQLN2 while over-expressing wild-type α-syn. Immunofluorescence analysis supported the finding that UBQLN2 overexpression decreases levels of α-syn (p = 0.004; Fig. 3A,C). However, α-syn levels were unchanged upon UBQLN2 knock-down (p = 0.7; Fig. 3A,C), perhaps due to compensatory effects of other UBQLNs and the presence of remaining UBQLN2 following partial knock-down. In contrast, levels of α-syn were not altered by UBQLN1 overexpression or knock-down (p = 0.96; Fig. 3B,D). A direct comparison of the number of α-syn-positive cells expressed as a ratio to UBQLN2- or UBQLN1-positive cells again showed far fewer α-syn-positive cells with UBQLN2 than with UBQLN1 (p = 0.005; Fig. 3E), further supporting the conclusion that UBQLN2 preferentially regulates α-syn.
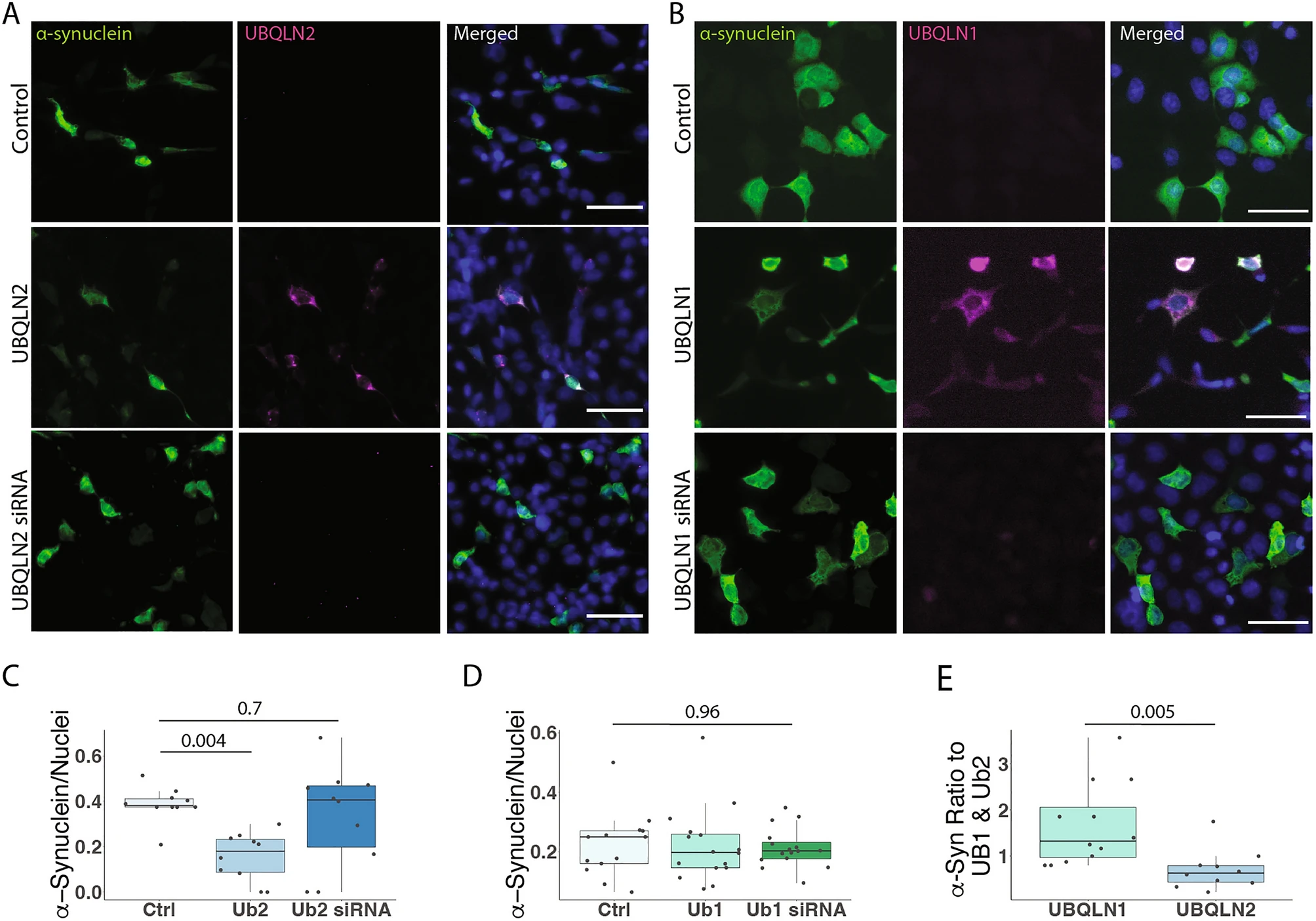
UBQLN2, but not UBQLN1, lowers α-syn levels in HEK-293 cells. (A–D) Representative immunofluorescence images show that the number of α-syn-containing cells are decreased when co-expressed with UBQLN2 (A,C; n = 10 field of cells over 3 independent experiments) but not with UBQLN1 (B,D; n = 10 field of cells over 3 independent experiments). Scale bar 100 μm (E) Expressed as a ratio of α-syn positive cells to UBQLN1 and UBQLN2, α-syn is significantly reduced in UBQLN2-expressing cells.
UBQLN2 KO robustly increases the levels of pathogenic pS129 α-syn
Our results showing UBQLN2 regulation of α-syn in cell models (Fig. 2) and increasing UBQLN2 insolubility in human and mouse brains harboring α-syn accumulation (Fig. 1), led us to test whether UBQLN2 regulates α-syn in vivo. To assess this, we measured levels of total α-syn in RIPA-soluble brain lysates from transgenic mice expressing pathogenic α-syn (A53T) crossed to either a transgenic mouse line overexpressing UBQLN2 (Ub2-Hi; Fig. 4A) or to the UBQLN2 KO mouse (Ub2-KO; Fig. 4A). Contrary to our in vitro studies (Figs. 1, 2), A53T mice devoid of UBQLN2 (A53TxUb2-KO) displayed an accumulation of total α-syn compared to A53T control mice (p = 0.0003; Fig. 4B), while A53T mice overexpressing UBQLN2 (A53TxUb2-hi) showed a trend toward decreased levels of α-syn compared to A53T (n = 7; p = 0.1). To further assess total α-syn levels in vivo, we performed immunofluorescence to measure levels of α-syn in the deep cerebellar nucleus (DCN), a brain region known to be affected in the A53T mouse model27. We did not detect a significant difference in α-syn immunofluorescence between A53T and A53TxUb2-hi (p = 0.9) or A53TxUb2-KO (p = 0.1) mice (Fig. 4C).
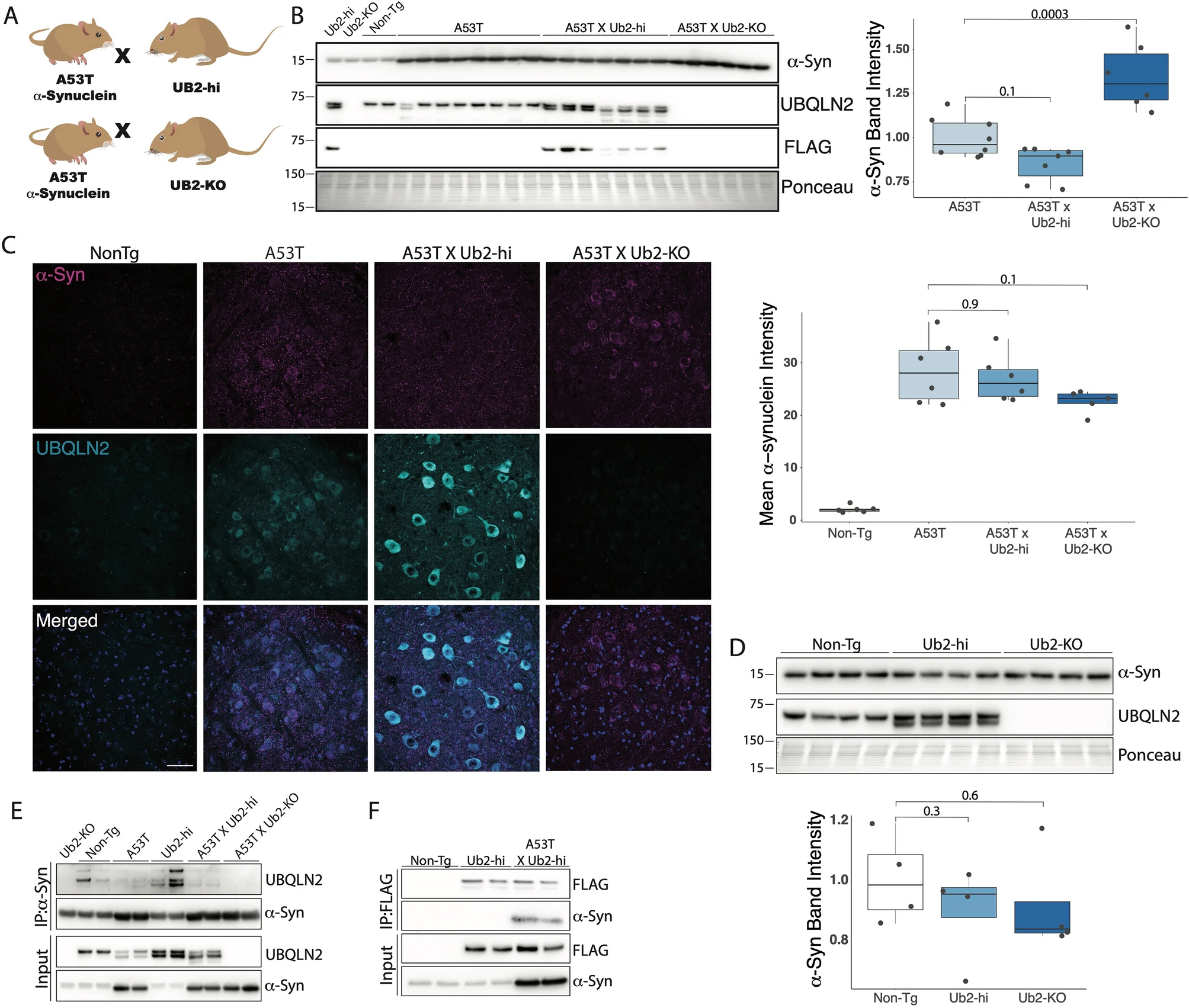
UBQLN2 interacts with and may alter α-syn levels in A53T mice. (A) Schematic depicting breeding strategy to obtain A53TxUb2-hi and A53TxUb2-KO mice. (B) Western blot of RIPA-soluble brain lysates showing increased levels of total α-syn in A53TxUb2-KO mice (n = 6), while A53TxUb2-hi (n = 7) mice show a trend toward decreased α-syn levels compared to non-transgenic (Non-Tg) controls (n = 8). (C) Representative images of total a-syn (magenta) and UBQLN2 (cyan) immunofluorescence in the deep cerebellar nucleus of 12-month-old Non-Tg (n = 6), A53T (n = 6), A53TxUb2-hi (n = 6), and A53TxUb2-KO (n = 5) mice and quantification show no significant difference in total a-syn expression between genotypes. (D) Western blot showing total endogenous α-syn levels were unchanged in both Ub2-hi (n = 4) and Ub2-KO mice (n = 4) compared to Non-Tg controls (n = 4). Western blots were cropped to focus on protein(s) of interest. Uncropped Western blots are shown in Supplemental Figure S3). (E) Immunoblot showing endogenous and transgenic UBQLN2 is pulled down when α-syn is immunoprecipitated from mouse brain lysate (n = 4). (F) Immunoblot showing α-syn coprecipitates with FLAG-tagged UBQLN2 in A53TxUb2-hi mice when FLAG is immunoprecipitated from mouse brain lysate (n = 2).
To determine if UBQLN2 plays a role in handling α-syn in non-disease states, we measured levels of total endogenous murine α-syn in Ub2-hi and Ub2-KO mice as well as in non-transgenic littermate controls by western blot. We did not detect a significant change in total endogenous α-syn in Ub2-Hi (p = 0.3) or Ub2-KO (p = 0.6) mice compared to controls (Fig. 4D). To determine whether UBQLN2 and α-syn interact, we performed immunoprecipitation assays which captured an interaction between UBQLN2 and α-syn with reciprocal pull downs using either α-syn or UBQLN2 antibodies (Fig. 4E,F). These results suggest that UBQLN2 interacts with α-syn, despite not decreasing levels of total α-syn in vivo.
The accumulation and deposition of pS129 α-syn into LBs is a hallmark of synucleinopathies29. UBQLN2 can also be found in LBs10. To determine whether UBQLN2 regulates levels of pS129 α-syn we measured pS129 levels in A53TxUb2-hi and A53TxUb2-KO mice. Western blot analysis revealed robust accumulation of pS129 in brain lysates of A53TxUb2-KO mice compared to controls (p = 0.002; Fig. 5A,B), while A53TxUb2-hi mice showed no difference in pS129 levels (p = 0.96; Fig. 5A,B). Analysis of the ratio of pS129 to total Syn levels (Fig. 4B) revealed no significant difference in pS129/total Syn between groups (p = 0.17; Fig. 5C). Further assessment of pS129 levels by immunohistochemistry revealed an accumulation of pS129 in A53TxUb2-KO mice compared to A53T controls (Fig. 5D). We were also able to detect endogenous levels of murine pS129 in Ub2-hi and Ub2-KO mice despite the generally low amount of pS129 in non-diseased brains29,33 (Fig. 5E,F). We observed a trend towards decreased murine endogenous pS129 in Ub2-hi mice (p = 0.2; Fig. 5E,F) and no change in pS129 levels in Ub2-KO mice (p = 0.8; Fig. 5E,F). The ratio of endogenous pS129 to total endogenous Syn (Fig. 4D) revealed no significant differences between Non-Tg and Ub2-hi mice (p = 0.2; Fig. 5G) or Ub2-KO (p = 1; Fig. 5G). Together these results suggest that UBQLN2 regulates pS129 in vivo, both under normal conditions and in disease states.
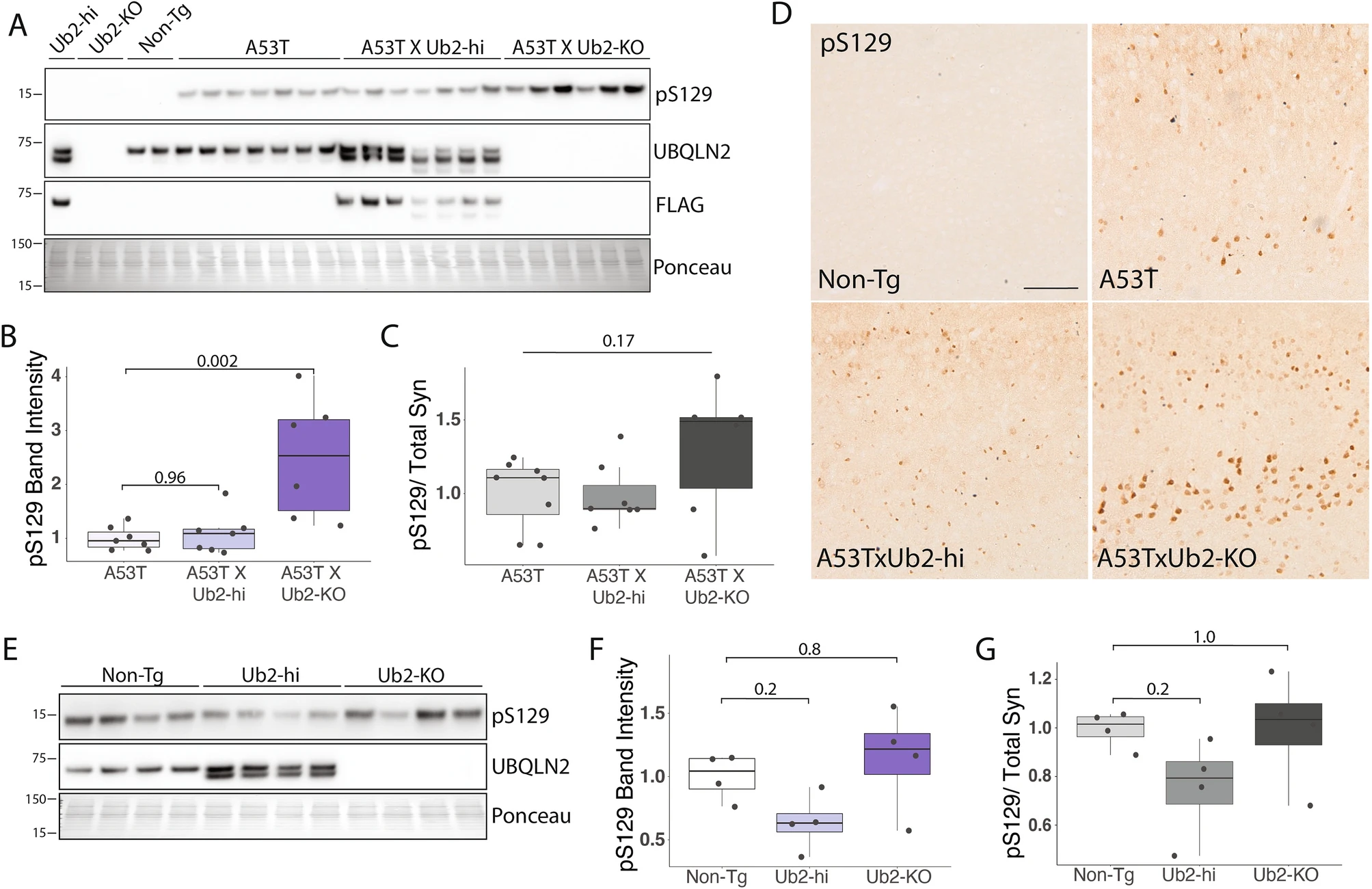
pS129 α-syn accumulates in A53T mice lacking UBQLN2. (A) Representative western blot of pS129 in RIPA-soluble brain lysates from the indicated transgenic mouse crosses. (B) Quantification from A showing accumulation of pS129 in A53TxUb2-KO mice (n = 6) and unchanged in A53TxUb2-hi mice (n = 7) compared to A53T controls (n = 8). (C) Ratio of pS129 to total Syn (see Fig. 4) does not differ significantly between groups (n = 8). (D) Representative images of pS129 expression in the cortex of A53T (n = 3), A53TxUb2-hi (n = 3) and A53TxUb2-KO (n = 3) show increased pS129 expression in A53T mice lacking UBQLN2 (12-month-old mice). (E) Representative western blots of endogenous pS129 in UBQLN2 transgenic mice. (F) Quantification of E showing a trend toward decreased endogenous murine pS129 levels in Ub2-hi mice (n = 4). (G) Analysis of the ratio of endogenous pS129 to total endogenous Syn (obtained from Fig. 4) shows no difference between groups (n = 4). Western blots were cropped to focus on protein(s) of interest. Uncropped Western blots are shown in Supplemental Figure S4.
UBQLN2 targets pS129 α-syn to the proteasome for degradation
To test whether overexpression of UBQLN2 leads to an increased rate of α-syn degradation we used optical pulse labeling to measure alpha-synuclein degradation over time34. For this experiment we used human α-syn tagged with Dendra2 (hSyn-Dendra2), a photoconvertible protein whose fluorescence changes from green to red following 405 nm light exposure (Fig. 6a). Rat primary cortical neurons were transfected with hSyn-Dendra2 and either UBQLN2-iRFP or iRFP. Following photoconversion, red fluorescence was tracked over time, allowing us to determine the rate of hSyn-Dendra2 turnover on a single-cell level. UBQLN2 overexpression had no effect on hSyn-Dendra2 decay compared to the iRFP control, suggesting that UBQLN2 does not play a significant role in nonpathogenic α-syn degradation (p = 0.6; Fig. 6b,c). Analysis of photoconverted hSyn-Dendra2 half-life further confirmed that UBQLN2 overexpression did not significantly affect normal α-syn degradation (p = 0.28; Fig. 6d,e). A limitation of optical pulse labeling, as applied here, is that we are unable to assess phosphorylated (pS129) α-syn at the same time.
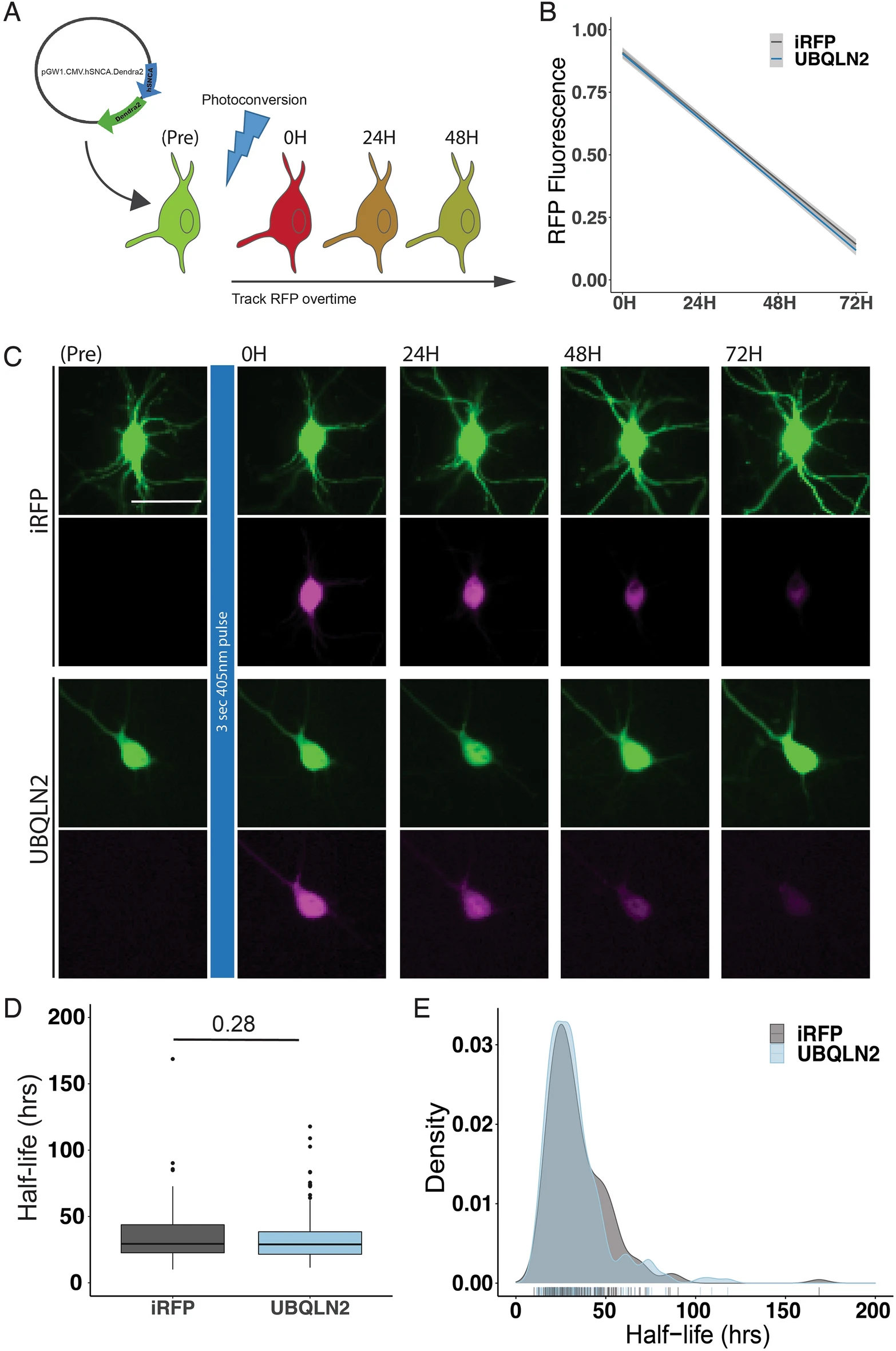
Optical Pulse Labeling reveals no effect of UBQLN2 on α-syn clearance in neurons. (A) Schematic of Optical Pulse labeling experiment. Rat primary cortical neurons were transfected with hSyn-Dendra2 and UBQLN2-iRFP or iRFP. Dendra2 was photoconverted 24 h post-transfection with 405 nm light, imaged immediately and every 24 h thereafter for 3 days. Scale bar 25 μm. (B) Optical pulse labeling reveals no change in red fluorescence intensity over time in neurons transfected with UBQLN2 (n = 213) compared to control (n = 182; p = 0.6). (C) Representative images of optical pulse labeling of hSyn-Dendra2 in rat primary cortical neurons transfected with hSyn-Dendra2 and UBQLN2-iRFP or iRFP. (D) Optical pulse labeling in primary rate neurons reveals no change in half-life of hSyn-Dendra2 upon transfection with UBQLN2 vs control (iRFP; p = 0.28). hSyn-Dendra2 half-life was determined by measuring the loss of photoconverted hSyn-Dendra2 (RFP) signal over time for each cell. (E) Density plot of hSyn-Dendra2 half-life measurements from individual neurons transfected with control (n = 182) or UBQLN2 (n = 213). Values were pooled from eight wells per condition, from each of two biological replicates and analyzed by the Kruskal–Wallis test.
α-syn and pS129 are regulated by both proteasomal and lysosomal degradation35,36,37. Though UBQLN2 may function in lysosomal protein degradation16,18, it is thought primarily to facilitate proteasomal degradation16,22. To investigate whether UBQLN2 targets α-syn to the proteasome for degradation, we treated TKO cells transfected to express α-syn and UBQLN2 or EV with either the proteasome inhibitor lactacystin38 (10 μM) or the proteasome activator Rolipram39 (50 μM). In the presence of overexpressed UBQLN2, proteasome inhibition led to a robust increase in pS129 (p ≤ 0.001) but not total α-syn (p = 1.0), whereas in the absence of UBQLN2 there was no such effect on pS129 (p = 0.98) or α-syn (p = 1.0) levels (Fig. 7A). Immunofluorescence supported the finding that overexpression of UBQLN2 leads to an accumulation of pS129 when the proteasome is inhibited (p = 0.002; Fig. 7B). These results suggest that while α-syn likely can be cleared by multiple, redundant quality control pathways, overexpressed UBQLN2 preferentially targets pS129 to the proteasome for degradation.

UBQLN2 targets pS129 α-syn to the proteasome for degradation. (A) Representative western blot of TKO cells overexpressing α-syn and transfected with either EV or UBQLN2 and treated with 10 μM lactacystin (Lac), 50 μM rolipram (Rol), or vehicle (Veh). Quantification of total α-syn and pS129 levels shows that pS129 accumulates in cells transfected with UBQLN2 and treated with Lac while total α-syn levels were not changed between groups (n = 9–10). Total α-syn and pS129 levels were each normalized to α-syn levels of EV, vehicle-treated cells. Western blots were cropped to focus on protein(s) of interest. (B) Representative images showing pS129 fluorescence in cells transfected with either EV or UBQLN2 and treated with either Veh, Lac, or Rol. Quantification of pS129 fluorescence intensity further supports that pS129 α-syn is increased when treated with Lac and in the presence of UBQLN2 (n = 11). pS129 fluorescence intensity was normalized to that of vehicle treated, EV cells. Scale bar 50 μm. (C) Representative western blot of TKO cells overexpressing α-syn and transfected with either EV or UBQLN2 and treated with 10 μM chloroquine (CLQ), 300 nM rapamycin (RAP), or vehicle (Veh). Quantification of pS129 or total α-syn levels reveals no differences between treatment groups (n = 10). Total α-syn and pS129 levels were each normalized to α-syn levels of EV-transfected, vehicle-treated cells. Uncropped Western blots are shown in Supplemental Figure S5.
To determine whether UBQLN2 also can regulate α-syn or pS129 via lysosomal degradation, we treated TKO cells transfected to express α-syn and UBQLN2 or EV with either the autophagy inhibitor chloroquine40 (CLQ; 10 μM) or the autophagy activator rapamycin41 (RAP; 300 nM). pS129 levels were not significantly altered in cells transfected with EV and treated with either CLQ or RAP, compared to vehicle controls (Fig. 7C). Similarly, overexpression of UBQLN2 and subsequent treatment with CLQ or RAP had no effect on levels of pS129 levels (Fig. 7C). Our results suggesting that total α-syn is likely handled by multiple quality control pathways (Fig. 7A) is supported by our finding that inhibition or activation of lysosomal degradation in the presence or absence of UBQLN2 has no effect on total α-syn levels (Fig. 7C). These data suggest that UBQLN2 regulates α-syn via the proteasome when autophagy is impaired and highlight UBQLN2’s preferential regulation of pS129 via the proteasome.
Discussion
Although UBQLN2 has been linked to several neurodegenerative diseases10,15,31 and is known to accumulate in Lewy bodies in disease10, whether this ubiquitin-linked quality control protein regulates α-syn under normal conditions or in disease states is unknown. Here we used human disease tissue, transgenic mouse models and cellular models to query whether UBQLN2 regulates α-syn. Our findings support a role for UBQLN2 in regulating levels of a key pathological form of α-syn, namely pS129.
UBQLN2 is intrinsically prone to form condensates and aggregates10,15,21,25 and accumulates in various neurodegenerative proteinopathies, including Lewy bodies in PD10. Our results reveal that UBQLN2 is more insoluble in PD. This is consistent with the previous finding that UBQLN2 colocalizes with aggregates of α-syn in PD10. Protein solubility may correlate with functionality, with decreased solubility correlating with decreased normal function. As UBQLN2 is sequestered in protein aggregates in disease, it may lose its normal function of regulating protein turnover. Conversely, as UBQLN2 solubility decreases, it could also undergo a gain-of-toxic-function42. In PD, UBQLN2 may be sequestered in aggregates, precipitating a loss of normal UBQLN2 function and in turn accelerating pS129 accumulation and potentially α-syn toxicity and disease progression.
Of the three brain-expressed UBQLNs, UBQLN2 has been most widely implicated in neurodegenerative diseases15,31,43 and our results supporting a significant role for UBQLN2, but not UBQLN1, in regulating α-syn are consistent with this concept. The UBQLNs are highly similar proteins except for the PXX domain that is unique to UBQLN2. Among UBQLNs, UBQLN2 and UBQLN1 are most similar in structure, sharing 79% homology44, but do not appear to share the role of regulating specific disease proteins such as α-syn and tau45. The PXX domain may be a driver of UBQLN2’s potential function as a regulator of such disease proteins. Of their many functions, proline-rich domains aid in protein binding46 so conceivably the PXX domain of UBQLN2 favors its interaction with client proteins independent of ubiquitination. Intriguingly, the PXX domain is also the site of most of the pathogenic UBQLN2 mutations that directly cause hereditary neurodegeneration15. Further investigations into the role of the PXX and other UBQLN2 domains are warranted to better understand how UBQLN2 shuttles pS129 to the proteasome for degradation.
A recent study assessing the effects of UBQLN2 on another disease-linked protein, tau, reported a greater effect of UBQLN2 on phosphorylated tau than on total tau45, reminiscent of our finding of a greater effect of UBQLN2 on pS129 α-syn than on total α-syn. Together, these studies support the view that UBQLN2 preferentially affects pathological forms of proteins, such as those that are phosphorylated or aggregated. Limitations of the current study include that fact that we have only assessed the most commonly identified site of pathological phosphorylation on α-syn, and the lack of phosphatase inhibitors in our biochemical analyses which could result in underrepresentation of the extent of UBQLN2’s effect on phosphorylated α-syn. Further mass spectrometry studies to evaluate additional phosphorylation sites47,48,49,50,51 (Ser87, Tyr136, Tyr125, etc.) will be needed to define a potentially broader role for UBQLN2 in regulating pathogenic forms of α-syn. Conceivably, UBQLN2 regulates protein phosphorylation, but there is no evidence that UBQLN2 regulates kinase or phosphatase activity45. Another possibility is that UBQLN2 preferentially targets aggregated proteins for degradation and disease aggregates are often enriched in phosphorylated proteins. Assessing this possibility in the current studies was complicated by the mild phenotype seen in hemizygous A53T mice and the lack of significant pathology until after 22 months of age27. An effect of UBQLN2 on A53T might be captured with the more robust homozygous mouse model or in vitro studies. Alternatively, the A53T mutation may alter UBQLN2’s regulation of α-syn considering that A53T α-syn is known to alter proteasome activity52, further complicating UBQLN2’s role on α-syn in PD.
UBQLN2 has been implicated in proteasomal and lysosomal degradation pathways16,18,53, pathways by which α-syn is regulated6,9,36,54,55. We sought to determine whether UBQLN2 acts on α-syn at the proteasomal or lysosomal level. In the absence of UBQLN2, pS129 appears to be cleared by other pathways when the proteasome is inhibited, but the presence of UBQLN2 restricts pS129 clearance to the proteasomal pathway. UBQLN2 may sequester pS129 at or near the proteasome, limiting accessibility to other clearance pathways. Our results further suggest that normal (nonpathogenic) α-syn can be regulated by redundant protein quality control pathways and that the presence or absence of UBQLN2 does not greatly affect normal α-syn degradation by those pathways. This may reflect a limited role for UBQLN2 in normal α-syn degradation or an ability of UBQLN2 to regulate normal α-syn via multiple clearance pathways. The former is supported by our kinetic studies showing that UBQLN2 does not significantly alter normal α-syn half-life. Further optical pulse labeling kinetic studies employing a phospho-mimetic of pS129 could help determine the basis of UBQLN2’s selective effect on this pathogenic form of α-syn.
In summary, we have established a role for UBQLN2 in regulating a pathological form of α-syn by targeting it to proteasomal degradation. This knowledge of the role of UBQLN2 in regulating pS129 sheds light on one potential reason why UBQLN2 is found in Lewy bodies in synucleinopathies: to decrease pS129 levels. Although further studies will be needed to understand the differential roles of UBQLN2 in regulating normal, post-translationally modified, and mutant forms of α-syn, our results here defining UBQLN2 as a regulator of pS129 α-synuclein point to UBQLN2 as a potentially important modifier of synucleinopathies and as a component of altered proteostasis in neurodegenerative disease.