
TRPV1 channels and the progesterone receptor Sig-1R interact to regulate pain

By Miguel Ortíz-Rentería, Rebeca Juárez-Contreras, Ricardo González-Ramírez, and Sara L. Morales-Lázaro
Excerpt from the article published in PNAS, January 29, 2018, 115 (7) E1657-E1666, DOI: https://doi.org/10.1073/pnas.1715972115
Significance
The TRPV1 ion channel has been widely associated with the generation of painful responses. The responses of cells expressing this ion channel and, presumably, the overall pain response of an organism may be regulated by controlling the amount of TRPV1 channels in the plasma membrane. TRPV1 levels can be regulated by its interaction with intracellular proteins, but there are no studies describing TRPV1 or any other mammalian TRP channel’s association with chaperones or how these interactions may affect the perception of pain. Here, we show that TRPV1-dependent pain is decreased through Sig-1R antagonism by progesterone and determine the presence of a physical interaction between these two proteins that may reduce pain under physiological conditions such as pregnancy.
Abstract
The Transient Receptor Potential Vanilloid 1 (TRPV1) ion channel is expressed in nociceptors where, when activated by chemical or thermal stimuli, it functions as an important transducer of painful and itch-related stimuli. Although the interaction of TRPV1 with proteins that regulate its function has been previously explored, their modulation by chaperones has not been elucidated, as is the case for other mammalian TRP channels. Here we show that TRPV1 physically interacts with the Sigma 1 Receptor (Sig-1R), a chaperone that binds progesterone, an antagonist of Sig-1R and an important neurosteroid associated to the modulation of pain. Antagonism of Sig-1R by progesterone results in the down-regulation of TRPV1 expression in the plasma membrane of sensory neurons and, consequently, a decrease in capsaicin-induced nociceptive responses. This is observed both in males treated with a synthetic antagonist of Sig-1R and in pregnant females where progesterone levels are elevated. This constitutes a previously undescribed mechanism by which TRPV1-dependent nociception and pain can be regulated.
Introduction
Nociception is a neuronal process initiated by the activation of specialized sensory receptors that enable the detection of noxious stimuli. Activation of specialized receptors in the plasma membrane of nociceptors leads to the generation of electrical and chemical signals that reach the central nervous system, providing information on potentially hazardous situations (1).
Among these molecular sensors is TRPV1 (Transient Receptor Potential Vanilloid 1), a nonselective cation channel that plays important roles in the transduction of noxious stimuli such as high temperatures (≥42 °C), extracellular acidic pH, mechanical damage, exogenous chemical stimuli such as capsaicin (2), and endogenous compounds like anandamide and lysophosphatidic acid, by-products derived from the lipoxygenase pathway and ATP (3) that are released during inflammation and tissue injury. When TRPV1 is activated by such stimuli, the resulting physiological responses are pain and/or itch (2, 4).
For these reasons, the TRPV1 channel has become an important pharmacological target for the treatment of these pathophysiological processes (5, 6). Thus, research has been geared toward identifying endogenous inhibitors of TRPV1’s activation, for example, cholesterol and oleic acid, which directly bind to TRPV1 and reduce the open probability of the channel (Po), thus rendering it refractory to stimulation by agonists (7, 8).
Another mechanism by which the activity of TRPV1 can be regulated is through its interaction with proteins that may alter its function and/or trafficking to the plasma membrane. Among such proteins are the A-kinase anchor protein (AKAP79/150) (9), GABAA receptor-associated protein (10), beta 2 subunit of voltage-gated K+ channel (Kvβ2) (11), and the ankyrin-rich membrane-spanning protein/kinase D-interacting substrate adaptor protein (ARMS) (12). Another protein with which TRPV1 interacts is Whirlin, a cytoskeletal PDZ scaffold protein that increases ion channel expression and stability (13). However, to date, the interactions of TRPV1 with proteins such as chaperones that could potentially regulate trafficking to the plasma membrane, as well as the physiological implications of this regulation, have not been described.
A human chaperone with a recently resolved crystal structure is the Sigma 1 receptor (Sig-1R) (14). This endoplasmic reticulum (ER)-resident chaperone is the only one known to be ligand-operated (15); its ligands can be either synthetic or endogenously produced and can function as agonists [e.g., N,N dimethyltryptamine and PRE-084 (16, 17)] or antagonists [e.g., BD1063 and progesterone (P4) (18)]. Although Sig-1R is an ER-resident chaperone, under certain conditions, such as cocaine exposure (19) or binding of other agonists (20), this protein may translocate to the plasma membrane, where it interacts with G-coupled protein receptors (GPCRs) (21) and some ion channels (19, 22–24).
Sig-1R is also an important mediator of pain, since in animal models the use of synthetic antagonists results in the attenuation of pain (25). However, the mechanism by which Sig-1R modulates pain attenuation is currently unresolved, and the molecular mechanisms by which this chaperone modulates pain through TRPV1 (or any other TRP channel) have not been elucidated.
P4, a natural antagonist of Sig-1R (26), is a neurosteroid with pleiotropic effects, which include neuroprotection through the inhibition of some voltage- and ligand-gated ion channels (27) and repair after several types of brain injury (28), and also promotes myelin repair by Schwann cells (29).
In some neuropathic pain animal models, P4 has also been shown to reduce pain (30). In experiments using Sig-1R–knockout mice or mice treated with Sig-1R antagonists (like BD1063), an inhibition of capsaicin-induced mechanical hypersensitivity was observed, but no link to actions of P4 on TRPV1 through Sig-1R have been proposed (31).
Here we studied the role of Sig-1R in the modulation of TRPV1 and the consequences of this modulation on pain processes associated with the functioning of this ion channel. Specifically, in mice we show that pain associated with TRPV1 activation by capsaicin is decreased by BD1063.
We also demonstrate that both endogenous and synthetic inhibitors of Sig-1R, P4 and BD1063, respectively, down-regulate TRPV1 expression in dorsal root ganglion (DRG) neurons and in a heterologous expression system through a mechanism that involves protein degradation by a proteasome-dependent pathway. Moreover, knockdown experiments using a specific siRNA for Sig-1R show that TRPV1 levels at the plasma membrane are decreased and that TRPV1 and Sig-1R form protein–protein interactions that are disrupted by Sig-1R antagonists such as P4 and BD1063. Finally, we show that capsaicin-induced pain is decreased in pregnant female mice, a condition in which P4 levels are elevated (32). In summary, disruption of the association of Sig-1R with TRPV1 by synthetic or natural antagonists leads to degradation of TRPV1 protein and to decreased levels of the ion channel protein as well as to a decrease in TRPV1-associated pain.
Results
Pain-Related Behavior in Response to Capsaicin Is Diminished by the Antagonism of Sig-1R.
Given that Sig-1R has been associated with capsaicin-induced mechanical hypersensitivity (31, 33), we first tested whether these effects, which had not been directly linked to changes in TRPV1 activity or expression, can be explained by a role of Sig-1R on TRPV1-mediated pain processes.
We began by assessing whether treatment of mice with BD1063 (32 mg/kg) for 24 h before pain-assessment assays would reduce capsaicin responses. For these experiments, mice were injected intrascapularly with BD1063 24 h before injection of capsaicin (2.8 µg) into one hind paw, and the total paw-licking time (PLT) was evaluated over a period of 10 min. The results from these experiments (Fig. 1A) show that pretreatment with BD1063 produced a significant decrease (52 ± 8%) in the capsaicin-associated pain response, demonstrating that antagonism of Sig-1R modifies the TRPV1-dependent painful response.
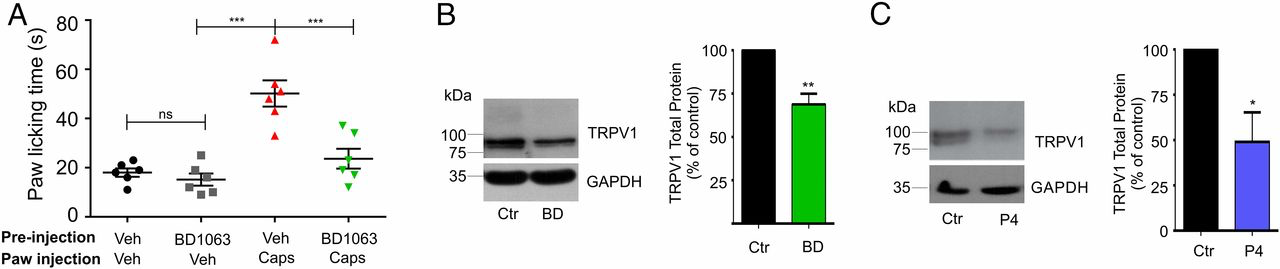
TRPV1-mediated pain is dependent upon Sig-1R activity.
(A) Response to 2.8-µg capsaicin injection into to the hind paw of male mice that had been pretreated for 24 h by intrascapularly injecting either vehicle (water) or a Sig-1R antagonist (BD1063) (32 mg/kg in water). PLT in response to capsaicin were 50 ± 5 s for vehicle-pretreated (n = 6) and 24 ± 4 s for BD1063-pretreated (n = 6) mice; ***P < 0.0001; ANOVA. PLT for mice injected only with vehicle or BD1063 were 19 ± 3 s and 16 ± 4 s, respectively; nonsignificant (ns), ANOVA. (B) Isolation of total protein from mouse DRGs injected 24 h before with vehicle (water) or with BD1063 (BD) (32 mg/kg) for TRPV1 and GAPDH (loading control) immunodetection. Normalized data are 69 ± 6% for BD1063 vs. control (Ctr), n = 3; **P < 0.001, Student’s t test. (C) Primary cultures of DRG neurons from mice treated with 25 µM P4 in 0.25% methanol for 24 h and from control cultures (0.25% methanol). Total protein was analyzed by Western blot (WB) for immunodetection of TRPV1 and GAPDH. The mean value for total TRPV1 protein in P4-treated cultures was 57 ± 14%; n = 4; *P < 0.05, Student’s t test.
Regulation of TRPV1 Protein Expression Levels by Sig-1R Antagonists.
To determine if changes in the pain responses to capsaicin after treatment with BD1063 were due to changes of TRPV1 levels in the sensory cells where it is expressed, we performed experiments where we initially confirmed the expression of Sig-1R in mice DRGs and trigeminal ganglia (TG) (Fig. S1A). Then we evaluated TRPV1 protein levels in DRGs obtained from control and BD1063-treated (32 mg/kg) mice and found that 24 h after the injection of vehicle and BD1063 there was a 31 ± 6% decrease in total TRPV1 protein levels in animals treated with BD1063 compared with control animals (Fig. 1B).
As with BD1063, we found that exposure of DRG neurons to 25 µM P4 for 24 h resulted in a 43 ± 14% decrease in TRPV1 protein levels compared with the control experiments (Fig. 1C), thus showing, in native neurons, a relevant role for this natural steroid in the regulation of TRPV1 expression.
To better elucidate the mechanisms by which Sig-1R produces changes in TRPV1 expression, we transiently transfected rat TRPV1 in HEK293 cells, which normally express Sig-1R (Fig. S1A). HEK293 cells were cultured in the presence of BD1063 (25 µM) or with P4 (25 µM) for 24 h and were evaluated for TRPV1 total and plasma membrane protein levels. The data show that in control cells the TRPV1 protein is detected as a double band (Fig. 2 A–D, arrows), a result in agreement with the previously reported core and hyperglycosylated (in residue N604) forms of rat TRPV1 (34, 35).
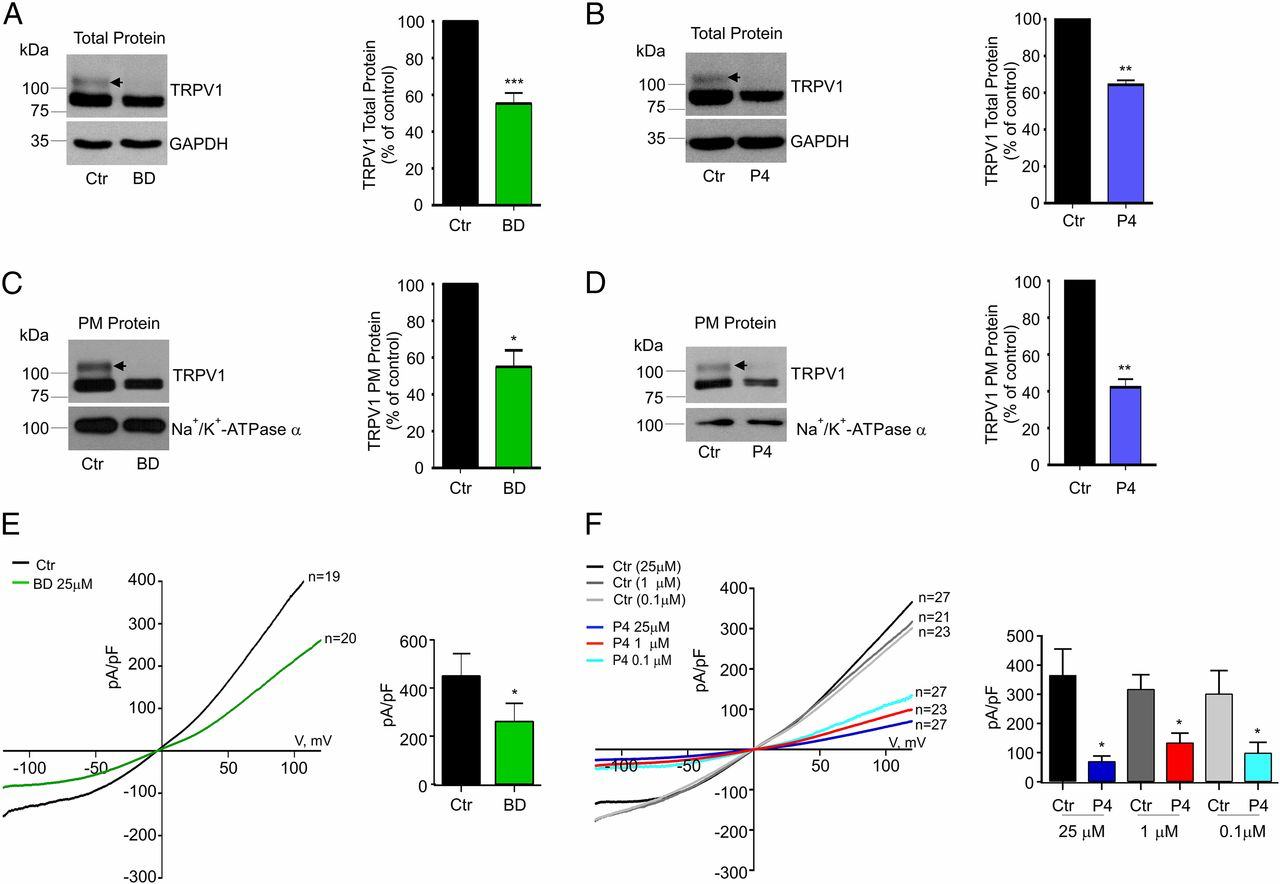
Antagonism of Sig-1R leads to changes in TRPV1 expression in HEK293 cells.
(A and B, Left) Immunodetection of TRPV1 and GAPDH total proteins from HEK293 cells expressing TRPV1 treated for 24 h with 25 µM BD1063 (BD) (A) or P4 (B) and control (Ctr) [water in A; methanol (0.25%) in B]. (A, Right) Mean values of TRPV1 for the BD group were 55 ± 6% (n = 14) with respect to control. ***P < 0.001, Student’s t test. (B, Left) Mean values of TRPV1 for P4 treatments were 65 ± 2% (n = 21) with respect to control (n = 28). **P < 0.01, Student’s t test. (C and D, Left) Plasma membrane (PM) proteins from control and 25 µM BD1063-treated cells (C) and P4-treated cells (D) were isolated for immunodetection of TRPV1 and Na+/K+-ATPase α (load control). (C, Right) PM TRPV1 mean values for BD1063 condition yielded 55 ± 9% with respect to control; (n = 3); *P < 0.05, Student’s t test. (D, Right) For P4 treatments, PM TRPV1 levels were 41 ± 5%; (n = 3); **P < 0.001, Student’s t test. Arrows point to the hyperglycosylated TRPV1 form, which decreases with BD1063 and P4 treatments. (E, Left) Average current–density plots from TRPV1-expressing HEK293 cells on the current density evoked by 4 µM capsaicin under control and BD1063-treated conditions. (Right) Averaged current density at +120 mV was 451 ± 91 pA/pF for control (water) (n = 19) and 261 ± 74 pA/pF for BD1063 treatment (n = 20). *P = 0.05; Student’s t test. (F, Left) Average current–density plots from TRPV1-expressing HEK293 cells on current density evoked by 4 µM capsaicin under control and P4-treated conditions. (Right) The averaged current density at +120 mV for control cells treated for 24 h with 0.25% methanol: 375 ± 87 pA/pF (black bar; n = 27); for control cells treated for 24 h with 0.01% methanol: 317 ± 49 pA/pF (gray bar; n = 21); for control cells treated for 24 h with 0.0001% methanol: 322 ± 85 pA/pF (light gray bar; n = 23); for cells treated for 24 h with 25 µM P4: 67 ± 17 pA/pF (dark blue bar; n = 27); for cells treated for 24 h with 134 ± 33 pA/pF (red bar; n = 23); and for cells treated for 24 h with 0.1 µM P4: 99 ± 36 pA/pF (aqua bar; n = 27). *P < 0.01 Student’s t test.
Fig. 2 A and B shows that, compared with controls, cells treated with BD1063 or P4 exhibit diminished total TRPV1 protein levels (45 ± 5% for BD1063 and 35 ± 2% for P4). With regard to TRPV1 protein in the plasma membrane, we found that, relative to control cells, BD1063 produced a 45 ± 9% decrease (Fig. 2C) while P4 decreased these levels by 69 ± 5% (Fig. 2D). Note that BD1063 and P4 produce more prominent decreases in the hyperglycosylated form (or mature protein) of TRPV1 than in the core form of TRPV1. This indicates that when Sig-1R is antagonized the core or immature form of the ion channel remains in the ER, and, consequently, it does not transit to the Golgi apparatus where it is hyperglycosylated.
To demonstrate that this effect is not due to the fact that the concentration of loaded protein was below detection limits, we performed experiments where the total protein concentration of TRPV1 was varied (20, 40, 60, and 80 μg) for channel immunodetection and found that in cells treated with P4 or BD1063 the hyperglycosylated form of TRPV1 decreased, thus suggesting that the hyperglycosylated form of TRPV1 is more affected by these treatments (Fig. S2 A and B). Additional experiments to quantify the hyperglycosylated and core forms of TRPV1 revealed that treatment with BD1063 produced 56% and 22% decreases in the hyperglycosylated and core forms, respectively (Fig. S2A). Similarly, P4 produced a 40% and a 10% decrease, respectively, in the hyperglycosylated and core forms (Fig. S2B). These experiments demonstrate that the hyperglycosylated form of TRPV1 is the one that is most affected by treatment with Sig-1R antagonists.
As a control, to demonstrate that not all membrane proteins are affected by the antagonism of Sig-1R, we performed experiments using Na+/K+-ATPase α and determined that antagonism of Sig-1R by BD1063 does not produce changes in plasma membrane protein expression (113 ± 15% for the BD-1063–treated cells with respect to control cells), indicating that protein translation, stability, or bilayer integration processes in general are not affected (Fig. S2C).
A decrease in TRPV1 levels at the plasma membrane should result in a decrease in the number of responsive channels leading to lower current-density levels of capsaicin-evoked currents. Compared with control cells, in cells treated for 24 h with 25 µM BD1063 the current density was diminished by 42 ± 16% (Fig. 2E). Likewise, compared with control cells, P4 also diminished the TRPV1 current-density levels (82 ± 4% for 25 µM, 56 ± 9% for 1 µM, and 69 ± 11% for 100 nM) (Fig. 2F). Thus, two ligands with antagonistic effects on the function of Sig-1R exhibit similar effects on TRPV1 expression.
To further substantiate the role of Sig-1R activity on TRPV1 expression, we cotransfected HEK293 cells with TRPV1 and a siRNA directed against Sig-1R to evaluate changes in TRPV1 membrane levels. Fig. S3A shows that 48 h after transfection Sig-1R protein levels were down-regulated (55.2 ± 5.3%) by the presence of the siRNA compared with those cells transfected with the scrambled siRNA. In cells cotransfected with Sig-1R siRNA and TRPV1, compared with cells transfected with scrambled siRNA and TRPV1, we observed a 46 ± 12% decrease in total TRPV1 protein (Fig. S3B). We also assessed the effects of these treatments by determining changes in TRPV1 current-density levels and, as shown in Fig. S3C, inhibition of Sig-1R expression by siRNA resulted in a 48 ± 12% decrease in current density compared with scrambled siRNA-treated cells. These data indicate that Sig-1R is necessary for TRPV1 expression.
Antagonism of Sig-1R Leads to Changes in TRPV1 Expression Through a Proteasome-Dependent Pathway.
It has been reported that antagonists of Sig-1R promote its interaction with another ER-resident protein, BiP (Binding Ig Protein) (15), resulting in the blockage of Sig-1R chaperone activity that, in turn, leads to misfolding of this chaperone’s targets (e.g., IP3R and Insig) (15, 36). These misfolded or incompletely processed polypeptides are then ejected from the ER for degradation by the 26S proteasome (37).
For this reason, we explored if a proteasomal-degradation pathway underlies the effects for the down-regulation of TRPV1 protein levels by BD1063 and P4. Fig. S4 shows that incubation of TRPV1-expressing HEK293 cells with BD1063 or P4 together with 25 µM MG132, an inhibitor of the proteasomal pathway, can almost completely rescue TRPV1 protein from degradation, compared with cells treated only with BD1063 (BD1063 = 55 ± 5% and BD1063 + MG132 = 82 ± 8%) (Fig. S4A) or P4 (P4 = 68 ± 6% and P4 + MG132 = 93 ± 6%) (Fig. S4B). These data provide additional evidence that Sig-1R plays an important role in TRPV1 protein expression. To delve further into this process, we assessed if overexpression of Sig-1R would confer resistance to degradation by prolonged exposure to capsaicin, since this agonist results in TRPV1 degradation by the lysosomal pathway (38). The data in Fig. S5 show that overexpression of Sig-1R does not rescue TRPV1 expression upon a 20-min exposure to capsaicin, indicating that the chaperone does not confer resistance to degradation by the lysosomal pathway. These data are consistent with our observation that, when cells are treated with a Sig-1R antagonist, TRPV1 protein levels are diminished through a pathway that involves the proteasome pathway (Fig. S4).
Although BD1063 is presumably specific for Sig-1R (39), P4 has been shown to bind to numerous receptors. Thus, to eliminate the possibility that down-regulation of TRPV1 protein levels by P4 was through pathways associated with its nuclear receptors, we evaluated the presence of mRNA for the nuclear progesterone receptors A and B (PR A/B) (40) in DRG and HEK293 cells. As shown in Fig. S6A, neither of these cell-types expressed PR A/B.
We also carried out experiments to exclude the possibility that low levels of PR A/B mRNA went undetected in our conventional RT-PCR measurements. To test for this, we used a PR A/B antagonist, 25 µM RU486 (RU), to determine if it would reverse the effects of P4 on TRPV1 protein levels. We hypothesized that, if the effects of P4 on TRPV1 expression levels were due to activation of the PR A/B, inhibition of these receptors should reverse the process. Unexpectedly, our findings showed that the incubation with 25 µM RU alone or together with 25 µM P4 for 24 h not only did not counteract the down-regulatory effects of P4 on TRPV1 protein expression but further decreased TRPV1 total protein levels (73.5 ± 13.5% and 54 ± 8% for P4 + RU and RU, respectively) (Fig. S6B). Nonetheless, these experiments did show that the PR A/B signaling pathway was not involved in down-regulation of TRPV1 levels, but rather another mechanism was at play. Both P4 and RU decreased TRPV1 protein levels independently of nuclear receptors, and this is consistent with reports that show that these compounds are able to induce alterations in the homeostasis of the ER (41, 42).
Still another possible explanation for the effects of P4 on TRPV1 expression could be through the activation of plasma membrane receptors of P4, such as the membrane receptors α, γ, δ, and ε or by P4 receptor membrane components 1 and 2 (PGRMC1 and PGRMC2) (40). We found that mPRα, mPRγ, PGRMC1, and PGRMC2 are expressed in DRG and HEK293 cells, whereas mPRε is expressed only in HEK293 cells (Fig. S6A). These results imply that at least four of the five membrane receptors assayed could be potentially involved in P4’s effects on TRPV1 protein expression. We tested this possibility by using a membrane-impermeable P4 conjugated with BSA to establish if any membrane-bound P4 receptor could be responsible for the effects of this steroid on TRPV1’s expression levels. Note that this compound binds to the plasma membrane P4 receptors with an affinity similar to that of the unconjugated steroid (43). Fig. S6C shows that a 24-h treatment with 25 µM of P4-BSA in HEK293 cells did not produce changes in total TRPV1 protein levels, excluding a role for membrane P4 receptors on TRPV1 protein-level down-regulation.
In summary, the data presented above indicate that the mechanism for regulation of TRPV1 expression by P4 does not depend on the activity of its classical nuclear or membrane receptors. On the other hand, the data did indicate that an internalization of P4 is required for it to exert its effects, as confirmed by the experiments using the P4-BSA conjugate.
Sig-1R and TRPV1 Association.
Another interesting question is whether regulation of TRPV1 levels is due to a protein–protein interaction with Sig-1R contributing to adequate TRPV1 expression in the plasma membrane. Consequently, we explored if there is a direct physical interaction of Sig-1R with TRPV1. We began by determining with confocal microscopy that TRPV1 and Sig-1R colocalize in HEK293 cells that overexpress Sig-1R-GFP [previously it has been shown that the GFP fusion on the C terminus yields a functional chaperone (15); Fig. S1B shows the expression of this construct as a 55-kDa protein fusion] and TRPV1-mCherry constructs (this fusion does not alter TRPV1 function; Fig. S7). The data show that these proteins do in fact colocalize in the heterologous expression system (Fig. S8). To determine what cellular compartments Sig-1R and TRPV1 interact in, we next evaluated whether they were present in the ER compartment (calnexin label). Fig. 3A shows that TRPV1-mCherry–labeled (panel 1) and Sig-1R-GFP–labeled (panel 2) proteins colocalize (panel 4) at the ER region (panel 3). We also explored whether both proteins are colocalized to the plasma membrane region. Fig. 3B shows that Sig-1R and TRPV1 do not predominantly colocalize to the plasma membrane (panel 4), although TRPV1 is highly expressed in this cellular structure, as shown by the purple signal which represents the merge between TRPV1 and the plasma membrane label (panel 4).
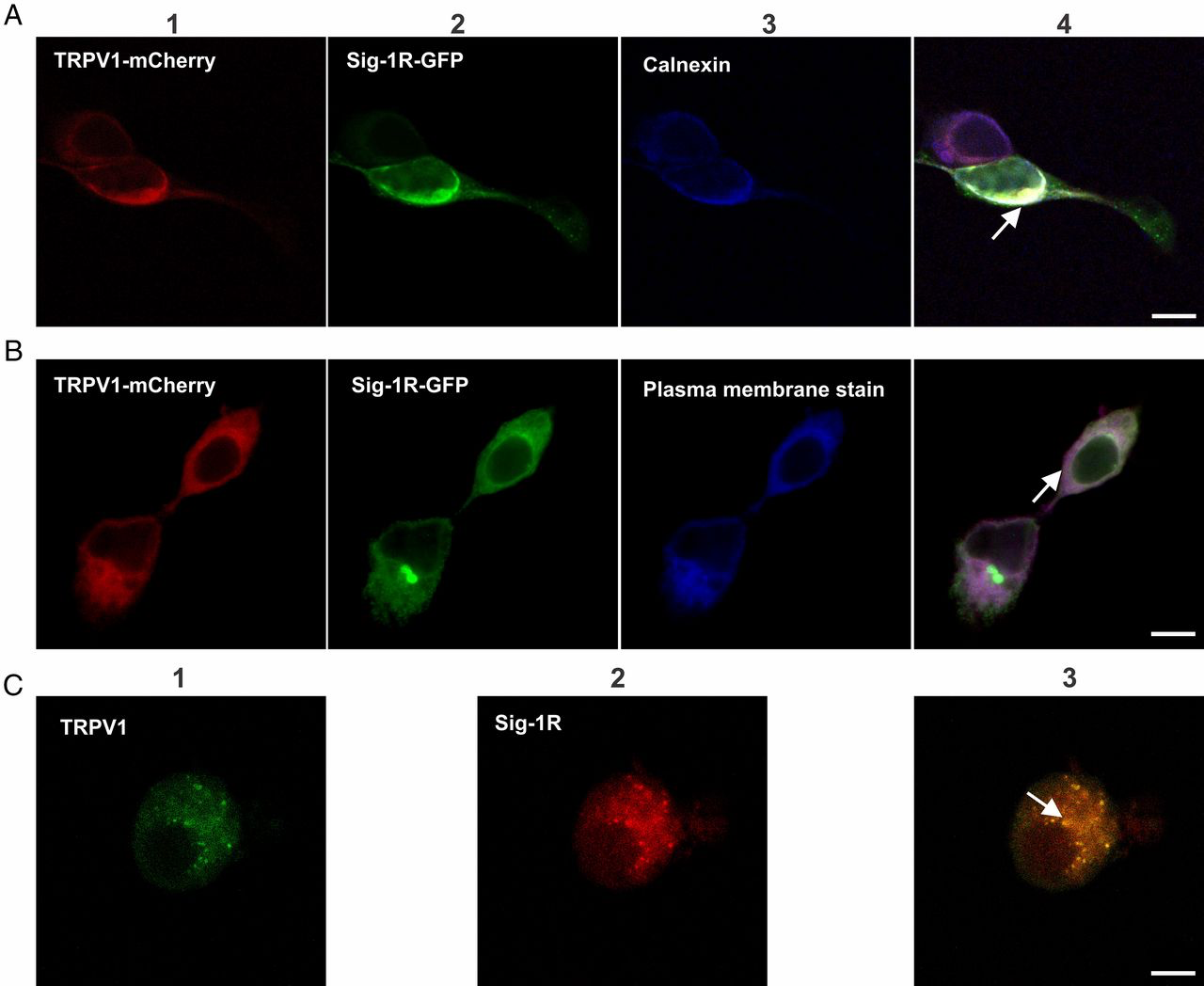
Colocalization of TRPV1 and Sig-1R in HEK293 cells and DRG neurons by confocal microscopy.
(A) Panels 1and 2 show signals detected for the TRPV1-mCherry (1, red) and Sig-1R (2, green) proteins. Panel 3 shows labeling of the ER by rabbit anti-calnexin and anti-rabbit Cy5 antibodies (blue). Colocalization of TRPV1 and Sig-1R in the ER is shown in panel 4 (arrow indicates the merge in white). (B) Panels 1 and 2 are the same as in A. Panel 3 shows the plasma membrane labelled with CellMask Deep Red (blue). Panel 4 shows the merged image (purple) and strong localization of TRPV1 (red) to the plasma membrane. (C) Colocalization of endogenous TRPV1 and Sig-1R in primary cultures of mouse DRG neurons. TRPV1 immunodetection with goat anti-TRPV1 and secondary anti-goat Alexa Fluor 488 antibodies (green, panel 1) and Sig-1R immunodetection with rabbit anti–Sig-1R and secondary anti-rabbit Alexa Fluor 594 antibodies (red, panel 2). Merge (yellow, panel 3) shows colocalization of both proteins. (Scale bars: 10 μm.) All arrows indicate colocalized signals.
We also assessed whether TRPV1 and Sig-1R colocalize in DRG neurons, a native system where the proteins are not overexpressed. The data in Fig. 3C confirm that both proteins colocalize in the endogenous expression system.
By performing biochemical assays, we also found that Sig-1R and TRPV1 from HEK293 cells coimmunoprecipitated (Fig. 4A, Left and Right, lane 2). Interestingly, Sig-1R coimmunoprecipitated with the TRPV1 core form (Fig. 4A, Left, lane 2) but not with the hyperglycosylated form of the protein. Moreover, when cells are treated with BD1063 or P4, the association of Sig-1R with TRPV1 is diminished compared with control cells without treatment (lane 5 in Fig. 4 B and C, lane 5).
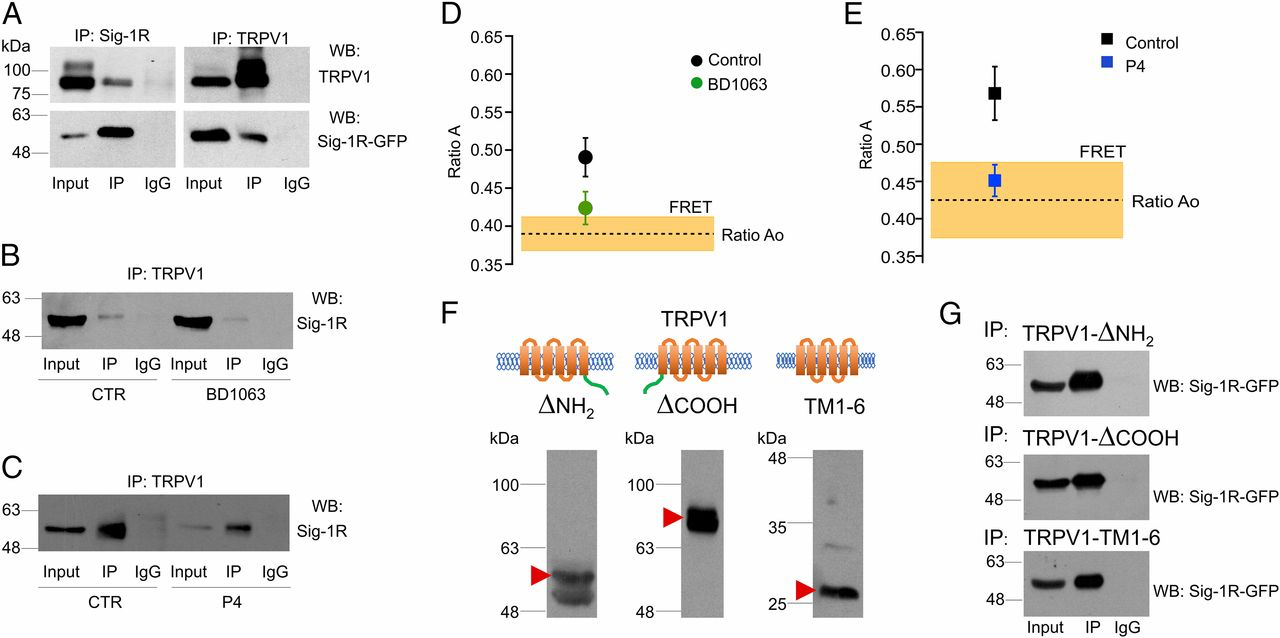
TRPV1 and Sig-1R form protein–protein complexes.
(A, Left) Coimmunoprecipitation of TRPV1 with Sig-1R antibody and immunodetection of TRPV1 (Upper) and Sig-1R (Lower). (Right) Coimmunoprecipitation of Sig-1R with TRPV1 antibody and immunodetection for TRPV1 (Upper) and Sig-1R (Lower). IgG served as negative control. The figure is representative of three independent experiments. Input is 1/10th of the total protein used in each immunoprecipitation. (B) Immunoprecipitation was carried out with TRPV1 antibody for the total protein from control cells (Ctr, Left) and BD1063-treated cells (Right). Differences between control and BD1063-treated cells were determined in the signal for immunoprecipitated Sig-1R (lane 5). The figure is representative of three independent experiments. (C) Immunoprecipitation was carried out as above with total protein from control cells (Left) and P4-treated cells (Right). Differences between control and P4-treated cells were determined in the signal for immunoprecipitated Sig-1R (lane 5). The figure is representative of three independent experiments. (D and E) FRET between TRPV1-mCherry and Sig-1R-GFP. The ratio Ao is the fractional excitation of mCherry in the absence of donor (GFP), and its average value is indicated by the dashed line. A value of ratio A, when both donor and acceptor are present, greater than ratio Ao is indicative of FRET. The coexpression of TRPV1 and Sig-1R produces an apparent FRET efficiency of 0.49 ± 0.02 (n = 17). (D) Incubation of the cells in the presence of 25 µM BD1063 reduces the apparent FRET efficiency to 0.42 ± 0.02 (n = 6), which is not different from the ratio Ao. (E) Treatment with 25 µM progesterone (P4) has an effect similar to BD1063. Control apparent FRET efficiency is 0.57 ± 0.03 (n = 10), and progesterone treatment reduces it to 0.45 ± 0.02 (n = 10). (F) Western blot analysis for deletion constructs of TRPV1 protein shows the bands corresponding to the expected sizes: TRPV1-ΔNH2 (58 kDa, Left), TRPV1-ΔCOOH (77 kDa, Center), and TRPV1-TM1-6 (28 kDa, Right). (G) Coimmunoprecipitation of different TRPV1 deletion constructs with Sig-1R. Coimmunoprecipitation of the TRPV1-ΔNH2 (Top) and TRPV1-ΔCOOH (Middle) deletion constructs and of TM1-6 transmembrane domains only (Bottom) with Sig-1R. Each deletion construct of TRPV1 was coimmunoprecipitated with Sig-1R-GFP (IP) by using a specific antibody against a specific epitope of the segment.
The biochemical assays described above suggested an interaction between Sig-1R and TRPV1. To confirm a direct interaction between both proteins, we used FRET analysis (44). We measured in vivo (live cultured cells) FRET in HEK293 cells expressing Sig-1R-GFP and TRPV1-mCherry. The results seen in Fig. 4 D and E demonstrate that there is a close interaction between Sig-1R-GFP and TRPV1-mCherry. FRET is observed as the difference between the direct excitation of the acceptor (TRPV1-mCherry) at the donor excitation wavelength (ratio Ao) and the sensitized emission of the acceptor when the donor (Sig-1R-GFP) is present and excited (ratio A). These measurements indicate that, when coexpressed, Sig-1R and TRPV1 directly interact. This interaction was significantly reduced, as expected, when the cells were treated with BD1063 or P4 for 24 h, as FRET levels diminished significantly (Fig. 4 D and E, respectively).
Taken together, these diverse lines of evidence show that Sig-1R can associate with TRPV1 in a direct protein–protein interaction and that this interaction regulates the membrane concentration of TRPV1. To determine whether Sig-1R was binding to the N or C terminus or transmembrane domains of TRPV1, we produced deletion constructs of TRPV1 (Fig. 4F) and determined that removal of the N and C termini still allowed its interaction with Sig-1R, showing that the chaperone interacts with the transmembrane region of the ion channel (Fig. 4G).
TRPV1-Dependent Pain Is Decreased in Pregnant Mice.
With respect to pain, it has been shown by several studies that males (human and other animals) exhibit higher pain thresholds in response to mechanical and thermal stimuli than their female counterparts (45). Although the difference in responses to noxious stimuli between genders is still controversial (46, 47), it has been established that rats and humans exhibit elevated thresholds to noxious stimuli during the gestation and parturition processes (48, 49). It has also been demonstrated that during pregnancy, when P4 levels are higher than in males (50), pain thresholds in females are increased and that during this physiological state agonists of Sig-1R are unable to interact with it, since P4 competes for the binding site on this receptor (51).
To show the role of Sig-1R during pregnancy, we used the paw-licking assay to elicit pain-related behavior by the injection of capsaicin in nonpregnant and pregnant female mice. The results show an increased pain-associated response when capsaicin was injected (43 ± 2 s and 24 ± 1 s, respectively) compared with saline injection (6.4 ± 2 s and 3.4 ± 1 s, respectively) (Fig. 5A). From these experiments it was also evident that the pregnant mice exhibited a less-pronounced pain-like behavior in response to capsaicin compared with their nonpregnant counterparts, and these data point to a role of elevated circulating P4 levels in this response.
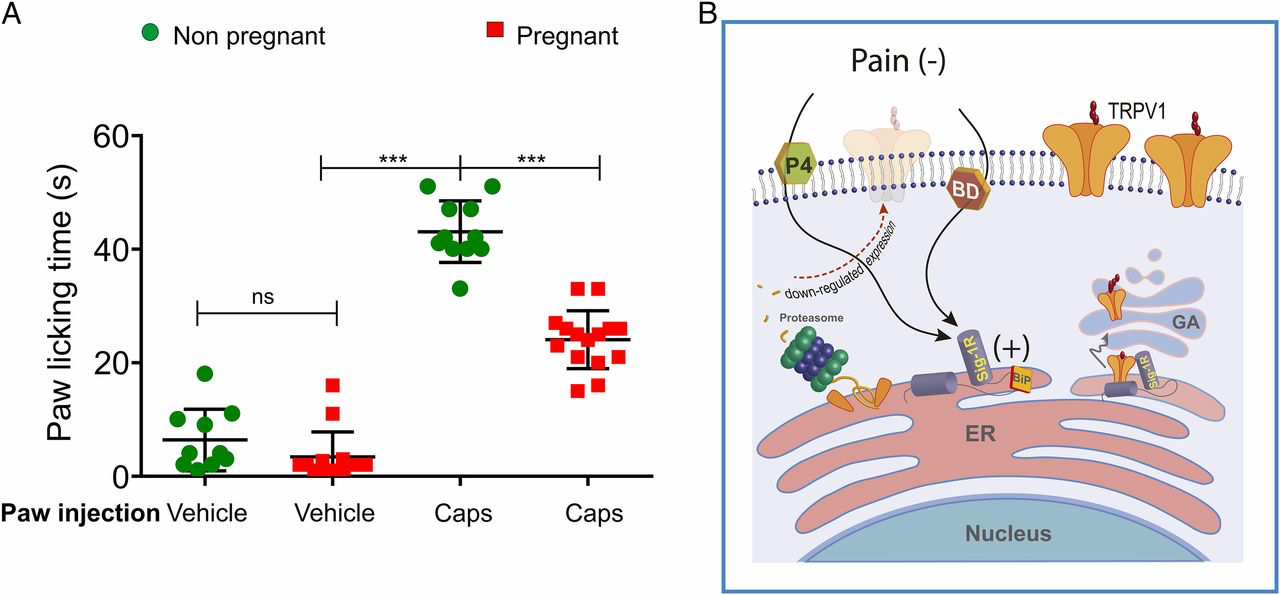
Antagonism of Sig-1R affects TRPV1 expression and its role in pain.
(A) Nonpregnant and pregnant (15 d of gestation) mice were intradermally injected into to a hind paw with vehicle- or capsaicin (2.8 µg)-containing solutions. PLTs for vehicle injections of nonpregnant (n = 18) and pregnant females (n = 12) were 5.2 ±1.2 s and 2.5 ± 0.9 s, respectively. For capsaicin injection PLTs were 43 ± 2 s in nonpregnant females and 24 ± 1 s in pregnant females. ***P < 0.0001; ns, not statistically significant; ANOVA. (B) Antagonism of Sig-1R promotes the association of Sig-1R with BiP leading to Sig-1R inactivation, protein misfolding in the ER, and, finally, to ER-associated protein degradation through the proteasome which, in turn, results in decreased TRPV1 expression levels and capsaicin-induced pain. In the absence of antagonists such as progesterone (P4) and BD1063 (BD), Sig-1R can positively regulate TRPV1 protein expression in the plasma membrane, as shown on the right side of the figure where interactions between Sig-1R and TRPV1 are not disrupted and the channel transits correctly to the Golgi apparatus (GA) where it is hyperglycosylated and then transported to the plasma membrane.
Discussion
In a series of studies of the effects on mechanical allodynia of Sig-1R agonists under control conditions and with pretreatment with capsaicin, Entrena et al. (31, 33) found that Sig-1R agonists did not alter sensitivity to mechanical stimulation under baseline conditions, and mechanical allodynia was present only when the system was pretreated with capsaicin. These effects were reversed by Sig-1R antagonists and absent in Sig-1R–knockout mice. Those observations prompted us to explore if there was a connection between Sig-1R and TRPV1, since no mechanism had been established.
Here we have investigated the interaction of two physiologically important ligand-activated proteins involved in producing pain: the ion channel TRPV1 (2) and the chaperone Sig-1R (25). Both these proteins have been extensively studied, given their important roles in pain. TRPV1 can be regulated by changing the Po and/or by controlling the available number of channels (N) in the plasma membrane of cells. Both these processes can result in a decrease of nociceptor excitability and in the reduction of pain. The Po of TRPV1 can be decreased by some natural regulators (7, 8). On the other hand, N can be reduced by removing channels from the plasma membrane by proteins that affect trafficking.
Among these latter proteins are the Sig-1R receptor, a progesterone-inhibited chaperone which has been shown to interact with and affect trafficking of some ion channels (19, 22, 23, 52) and GPCRs (21). However, there is a paucity of information on how and if chaperones interact with TRP channels in general and TRPV1 in particular. In this respect, it has only been shown that the Drosophila TRP channel and XPORT chaperones can form complexes that are important for the exit of this channel from the ER (53).
In this study, we have elucidated the molecular pathway by which the chaperone, Sig-1R, reduces pain produced by capsaicin-activated TRPV1 channels. This was accomplished by showing that the pain produced by injecting capsaicin into a mouse’s paw is reduced by an antagonist of Sig-1R, BD1063 (Fig. 1A). We also observed that BD1063 and a natural antagonist of Sig-1R, P4, reduced the levels of TRPV1 protein of nociceptors (DRGs) and transfected HEK293 cells (Figs. 1 B and C and 2 A–D). This resulted in diminished capsaicin-evoked TRPV1 currents (Fig. 2 E and F) due to a decrease in the number of functional channels (N) in the plasma membrane. While agonists of Sig-1R promote the mono- and/or dimeric conformations that allow interactions of the chaperone with other proteins, antagonists such as BD1063 and P4 promote a multimerized form of the protein that does not allow its functions (54). Such conformations of Sig-1R are a consequence of the binding of ligands to a hydrophobic region in the center of the C terminus of the chaperone (14). For example, it has been shown that BD1063 competes with the agonist (+)-pentazocine for the same binding site in the C terminus (39, 55). Thus, it is likely that BD1063 and P4 promote a conformation of Sig-1R that does not allow the interaction with TRPV1, leading to a decrease in the expression of the ion channel. Although we did not evaluate the effects of Sig-1R on TRPV1 trafficking, our results (Fig. S5) show that degradation of TRPV1 by prolonged agonist activation is not affected by Sig-1R overexpression. It had been previously shown by Sanz-Salvador et al. (38) that capsaicin-induced internalization of TRPV1 results in lysosomal degradation, a process different from the one proposed in the present study involving the proteasomal pathway.
As mentioned before, Whirlin is a protein involved in the stability and increased expression of TRPV1 (13). In this respect, the interaction between TRPV1 and Whirlin confers resistance to lysosomal degradation by prolonged exposure to capsaicin. This is also a distinct mechanism from the one proposed in the present study where Sig-1R confers resistance to proteasomal degradation.
The responses of the two tested antagonists were mimicked by the essential elimination of Sig-1R with a specific siRNA (Fig. S3). Moreover, direct proof of the interaction of TRPV1 with Sig-1R was obtained by immunoprecipitation and FRET experiments (Fig. 4 A–E). Finally, we showed that BD1063 and P4 treatments lead to degradation of TRPV1 by a proteasome-dependent pathway (Fig. S4). Taken together, our data identify an interaction of a mammalian TRP channel with a chaperone and highlight that the TRPV1-Sig-1R pathway is a fruitful target for therapeutic developments regarding the reduction of pain (Fig. 5B).
We have also shown that antagonism of Sig-1R and Sig-1R knockdown down-regulated TRPV1 expression (Fig. 2 A–D and Fig. S3B) and reduced capsaicin-activated currents (Fig. 2 E and F and Fig. S3C). Although P4 has been known to interact with other types of receptors, including the nuclear and membrane progesterone receptors (56), we show that inhibiting these receptors does not prevent the down-regulation of TRPV1 (Fig. S6). Moreover, by using a membrane-impermeable P4, we demonstrated that P4 must enter the cell to exert its effects (Fig. S6C).
Previous studies have shown that Sig-1R antagonists sequester Sig-1R in its BiP-associated form or promote its multimerized conformation, where it is rendered inactive (15, 54). This, in turn, leads to misfolded protein accumulation in the ER and the activation of the proteasome degradation pathway (Fig. 5B), a process that provides quality control (i.e., proper folding) of nascent proteins and avoids cellular stress (57). Our experiments using MG132, a selective antagonist of the 26S proteasome pathway, show that it renders BD1063 and P4 ineffective and essentially rescues TRPV1 protein expression (Fig. S4). These data show that Sig-1R in the ER is necessary for the correct folding of TRPV1 and for avoiding proteasomal degradation, thus allowing the exit of TRPV1 from the ER to the plasma membrane, constituting the mechanism by which both Sig-1R antagonists regulate TRPV1 function (Fig. 5B).
We also observed that when Sig-1R is antagonized by BD1063 or P4, the hyperglycosylated (or mature) form of the TRPV1 protein decreases (Fig. 2 A–D and Fig. S2 A and B). This result can be explained by noting that antagonism of Sig-1R results in misfolding of the TRPV1 protein, and thus there is no trafficking of the protein to the Golgi apparatus (Fig. 5B), where it is hyperglycosylated and then transported to the plasma membrane (58). Although we found that the core form of TRPV1 is not completely abolished by treatment with Sig-1R antagonists, this could be explained by having this form of the protein retained in the ER to be eventually degraded by the proteasome. In contrast, a correctly folded protein (one that has not been subjected to proteasomal degradation) is trafficked from the ER to the Golgi apparatus where it is hyperglycosylated and then transported to the plasma membrane.
We also identified an interaction between Sig-1R and TRPV1. This was accomplished by colocalization of fluorescently tagged Sig-1R and TRPV1 in the ER and confirmed the presence of these protein–protein complexes through coimmunoprecipitation and FRET experiments. Moreover, this interaction was disrupted by BD1063 and P4 (Fig. 4 B–E). To further characterize this interaction, we defined that Sig-1R binds to the transmembrane domain of TRPV1 (Fig. 4G), similar to what has been found for Kv1.3 channels (22). This constitutes evidence of such an association between a chaperone and a mammalian TRP channel.
The stoichiometry of interaction of ASIC (acid-sensing ion channels) and Sig-1R has been obtained where one Sig-1R associates with one ASIC1 subunit, meaning that the ASIC complex (a trimeric protein) will bind no more than three Sig-1R receptors (59). Although analysis of the FRET experiments shown here suggests a 1:1 interaction between Sig-1R and TRPV1, future studies directed to answering this specific question are needed.
The mechanism by which the interaction of TRPV1 with Sig-1R is disrupted is through the sequestering of Sig-1R chaperone activity by BD1063 (or P4), as they promote its association with BiP (Fig. 5B) (15). One study with P4 showed it down-regulated the expression of hERG channels by altering ER homeostasis (42), whereas another study showed that Sig-1R regulates the expression of these channels through a posttranslational mechanism (52). However, neither of these studies shows a relationship between Sig-1R and P4 in regulating hERG channel expression. In the light of our results, we suggest that the interaction of P4 with the chaperone could be responsible for disrupting the interaction of Sig-1R with hERG channels, as it has been shown that Sig-1R directly interacts with these proteins (23).
What are some of the physiological relevancies of the antagonism of Sig-1R? It has been shown that during periods of pregnancy or acute stress, when P4 is elevated, its concentration can reach 300 nM (32), a concentration well above 100 nM that decreases TRPV1 in the plasma membrane (Fig. 2F). In this regard, in our behavioral experiments we found that pregnant mice exhibited a decreased response to capsaicin-induced pain compared with nonpregnant females (Fig. 5A), thereby pointing to a role for antagonism of Sig-1R by P4 on TRPV1-dependent pain. In summary, our results elucidate a mechanism in which chaperones, specifically Sig-1R, can interact with TRPV1 in a manner that reduces pain.