
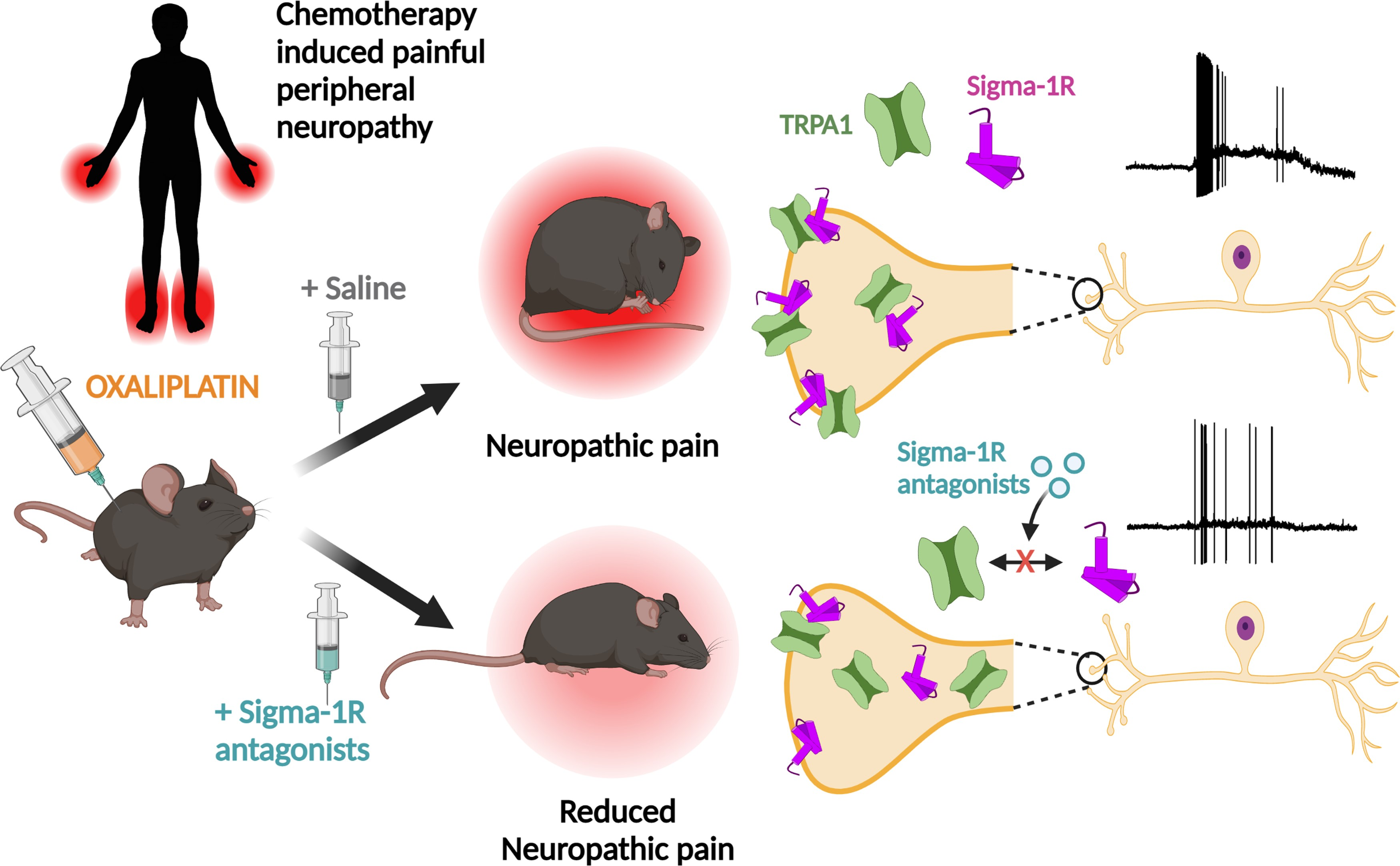
TRPA1 modulation by Sigma-1 receptor prevents oxaliplatin-induced painful peripheral neuropathy

By Aida Marcotti, Jorge Fernández-Trillo, Alejandro González, Marta Vizcaíno-Escoto, Pablo Ros-Arlanzón, Luz Romero, José Miguel Vela, Ana Gomis, Félix Viana, and Elvira de la Peña
Excerpt from the article published in Brain, Volume 146, Issue 2, February 2023, Pages 475–491, DOI: https://doi.org/10.1093/brain/awac273
Editor’s Highlights
- Transient receptor potential ankyrin 1 (TRPA1) is a polymodal, non-selective cation-permeable channel expressed in nociceptors, linked to the establishment of chemotherapy-induced peripheral neuropathy and other painful neuropathic conditions.
- Chemotherapy-induced peripheral neuropathy (CIPN) is a frequent, disabling side effect of different anticancer treatments, affecting millions of patients worldwide.
- Oxaliplatin is a platinum compound frequently used in the treatment of colorectal cancer.
- TRPA1 is downmodulated by antagonism of Sigma-1R, a multifunctional chaperone, and this inhibition reduces the painful side effects of oxaliplatin.
- Sigma-1R antagonists reduce the plasma membrane expression of functional TRPA1 channels.
- The incubation with S1RA increases the proportion of Sigma-1R multimers, possibly altering the interaction with TRPA1. This may lead to a misfolded TRPA1, interfering with the forward trafficking and reducing its functional expression at the plasma membrane without alteration in total TRPA1 protein levels.
Abstract
Chemotherapy-induced peripheral neuropathy is a frequent, disabling side effect of anticancer drugs. Oxaliplatin, a platinum compound used in the treatment of advanced colorectal cancer, often leads to a form of chemotherapy-induced peripheral neuropathy characterized by mechanical and cold hypersensitivity. Current therapies for chemotherapy-induced peripheral neuropathy are ineffective, often leading to the cessation of treatment. Transient receptor potential ankyrin 1 (TRPA1) is a polymodal, non-selective cation-permeable channel expressed in nociceptors, activated by physical stimuli and cellular stress products. TRPA1 has been linked to the establishment of chemotherapy-induced peripheral neuropathy and other painful neuropathic conditions. Sigma-1 receptor is an endoplasmic reticulum chaperone known to modulate the function of many ion channels and receptors. Sigma-1 receptor antagonist, a highly selective antagonist of Sigma-1 receptor, has shown effectiveness in a phase II clinical trial for oxaliplatin chemotherapy-induced peripheral neuropathy. However, the mechanisms involved in the beneficial effects of Sigma-1 receptor antagonist are little understood. We combined biochemical and biophysical (i.e. intermolecular Förster resonance energy transfer) techniques to demonstrate the interaction between Sigma-1 receptor and human TRPA1. Pharmacological antagonism of Sigma-1R impaired the formation of this molecular complex and the trafficking of functional TRPA1 to the plasma membrane. Using patch-clamp electrophysiological recordings we found that antagonists of Sigma-1 receptor, including Sigma-1 receptor antagonist, exert a marked inhibition on plasma membrane expression and function of human TRPA1 channels. In TRPA1-expressing mouse sensory neurons, Sigma-1 receptor antagonists reduced inward currents and the firing of actions potentials in response to TRPA1 agonists. Finally, in a mouse experimental model of oxaliplatin neuropathy, systemic treatment with a Sigma-1 receptor antagonists prevented the development of painful symptoms by a mechanism involving TRPA1. In summary, the modulation of TRPA1 channels by Sigma-1 receptor antagonists suggests a new strategy for the prevention and treatment of chemotherapy-induced peripheral neuropathy and could inform the development of novel therapeutics for neuropathic pain.
Introduction
Chemotherapy-induced peripheral neuropathy (CIPN) is a frequent, disabling side effect of different anticancer treatments, including taxanes, vinca alkaloids, bortezomid and platinum analogues, affecting millions of patients worldwide (reviewed by Staff et al.,1 Gordon-Williams and Farquhar-Smith2 and Seretny et al.3). In the case of oxaliplatin, a platinum compound frequently used in the treatment of colorectal cancer, the neuropathy affects over 70% of treated patients3 and manifests itself with paraesthesias, dysesthesias and pain, preferentially in hands and feet.1,4 The severity of CIPN painful symptoms, including mechanical and cold hypersensitivity often lead to dose reduction or early cessation of treatment, compromising therapeutic effectiveness and survival.5 The chronicity of many cases has a long-term negative impact on patient’s quality of life. There is no effective therapy to treat or prevent CIPN. The results of many clinical interventions have been negative or inconclusive6; current therapies are primarily geared towards the relief of pain symptoms.7
The mechanisms underlying painful CIPN are complex and poorly understood (reviewed by Starobova and Vetter,8 Ma et al.9 and Colvin10). Proposed mechanisms include mitochondrial dysfunction leading to oxidative stress in peripheral tissues,11–13 neuroimmune alterations14,15 and ion channel dysregulation in sensory afferents.16–20 In particular, several studies implicated the ion channel transient receptor potential ankyrin 1 (TRPA1) in the painful symptomatology observed in several animal models of CIPN,21 including those caused by oxaliplatin administration.22–25 TRPA1 is a polymodal, non-selective cation channel expressed in a fraction of nociceptors,26 where it senses noxious temperatures, mechanical forces and a broad array of exogenous and endogenous irritants, including reactive oxygen species (reviewed by Viana27 and Talavera et al.28). The role of TRPA1 as a molecular detector of mechanical and thermal stimuli is particularly evident in pathological conditions causing neuroinflammation or neuropathy.29
Sigma-1 receptor (Sigma-1R) is a ligand-regulated chaperone residing at mitochondria-associated endoplasmic reticulum membranes, expressed in many tissues, including peripheral sensory neurons.30–32 Sigma-1R is involved in multiple cellular functions, such as cellular bioenergetics, calcium homeostasis, reactive oxygen species generation, chromatin remodelling, inflammation and cell proliferation.32–36 Sigma-1R can translocate to other cellular compartments, such as the plasma membrane, regulating the expression and function of many ion channels.37–39 Previous studies linked Sigma-1R with pain modulation in animal models.40,41 Moreover, a recent phase II clinical trial established the efficacy of sigma 1 receptor antagonist (S1RA) (also known as E-52862), a highly selective Sigma-1R antagonist,42 in ameliorating oxaliplatin-induced peripheral neuropathy in patients treated for colorectal cancer.43 Treatment with S1RA significantly reduced cold pain threshold temperature and cold-evoked pain intensity. However, the cellular and molecular mechanism involved in pain relief by Sigma-1R ligands are currently unknown.
We found that antagonism of Sigma-1R inhibits TRPA1 activity by a mechanism involving alterations in TRPA1-Sigma-1R complex formation, channel membrane trafficking and reduced expression at the plasma membrane of nociceptors. In an experimental model of oxaliplatin neuropathy, in vivo Sigma-1R antagonists reduced TRPA1 activity and prevented the painful mechanical and cold symptoms caused by the drug. This finding paves the way for innovative treatments of painful CIPN by targeting Sigma-1R.
…
Results
Sigma-1R antagonists reduce calcium responses to TRPA1 agonists
To explore the modulatory action of Sigma-1R on TRPA1 channels, cultured HEK293 cells transiently transfected with human TRPA1 (hTRPA1-tGFP) were incubated with CS, 0.2% DMSO) or with different concentrations (25, 50 and 100 µM) of the highly selective Sigma-1R antagonist S1RA.42 Intracellular calcium responses to AITC, an electrophilic TRPA1 agonist, were monitored with the fluorescent calcium indicator Fura-2 (Fig. 1A and B). A 4-h incubation with S1RA caused a dose-dependent reduction in the fraction of responding cells and in the peak amplitude [half-maximum inhibitory concentration (IC50) = 47 ± 3 µM] (Fig. 1B–D). At the maximum concentration tested (100 µM) the amplitude reduction was (83%, and the number of cells responding to AITC decreased to 44%, compared to 95% responses in cells incubated in CS: Fig. 1E and F). Incubations, as short as 1 h with S1RA (100 µM), produced a clear inhibition. Longer incubations (24 h) at lower concentrations (25 µM) also produced marked reductions in the response to AITC (Supplementary Fig. 1).
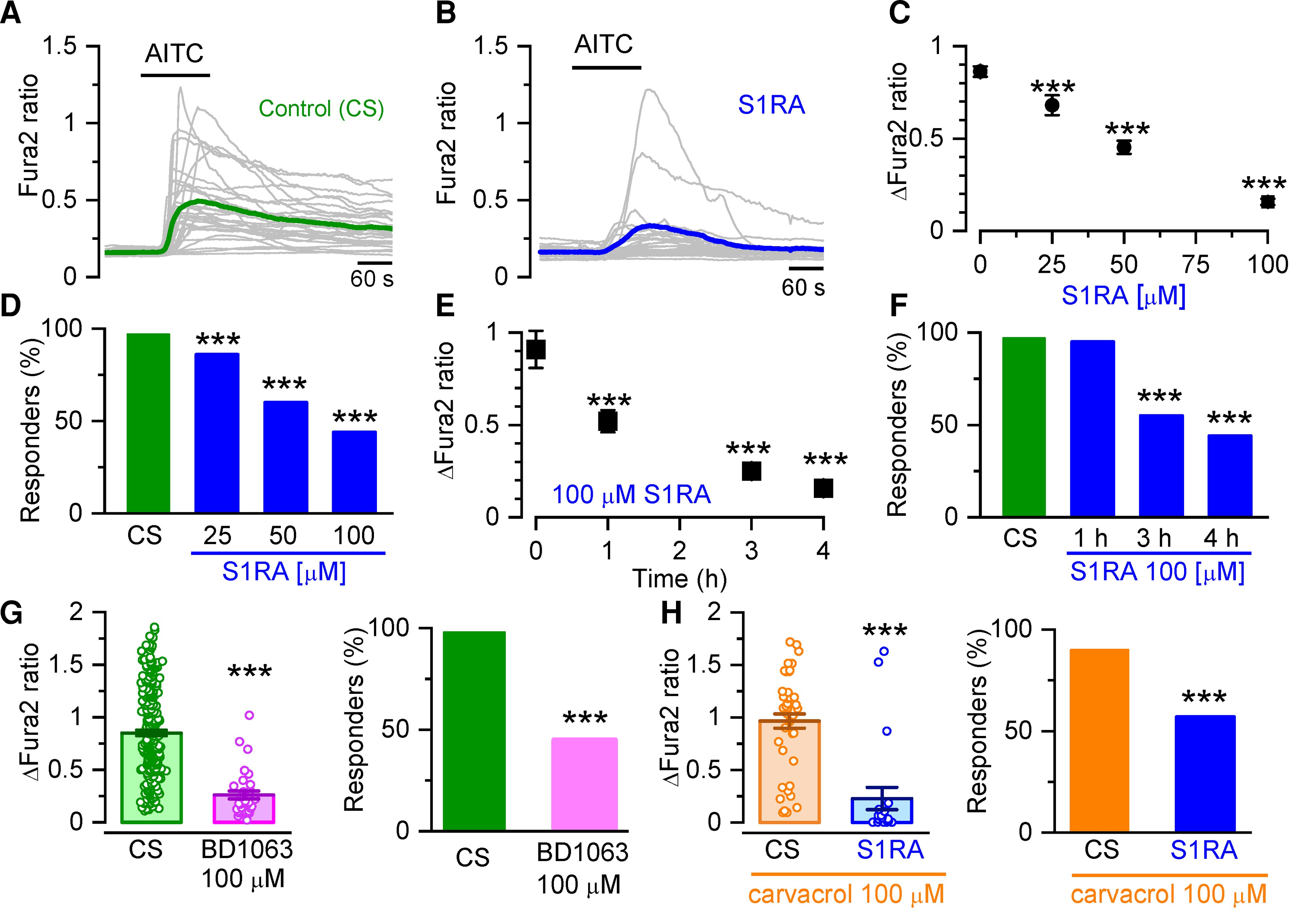
Sigma-1R antagonists inhibit intracellular calcium responses in TRPA1 expressing cells.
(A) Representative intracellular calcium responses monitored with Fura-2 in response to AITC (50 µM) after incubation in CS, 4 h). Green trace is the average of 32 cells (grey traces) expressing hTRPA1-tGFP. (B) Same as in A but after incubation with S1RA (100 µM, 4 h). The blue trace is the average of 30 cells (grey traces) expressing hTRPA1-tGFP. (C) Dose-response curve of S1RA effects. ***P < 0.001 (one-way ANOVA in combination with Dunnett’s post hoc test) CS n = 37, S1RA 25 µM n = 68, 50 µM n = 90, 100 µM n = 64. (D) Percentage of responding cells to AITC after pre-incubation (4 h) with S1RA at different concentrations. ***P < 0.001 (Fisher’s exact test) versus CS. (E) Time-dependent inhibition of TRPA1 responses to AITC (50 µM) by pre-incubation with S1RA (100 µM, 4 h). ***P < 0.001 (ANOVA in combination with Dunnett’s post hoc test). CS n = 37, 1 h n = 21, 3 h n = 77, 4 h n = 64. (F) Effect of incubation time with S1RA (100 µM, 4 h) on AITC responses. ***P < 0.001 (Fisher’s exact test). (G) Graph summarizing the effect of pre-incubation with BD1063 (100 µM, 4 h, n = 255) versus CS n = 255 ***P < 0.001 (unpaired t-test). (H) Graph summarizing the effect of pre-incubation with S1RA (100 µM, 4 h) to responses when the cells were stimulated with carvacrol (100 µM, n = 21) versus CS n = 45. ***P< 0.001 (unpaired t-test).
A similar inhibition in TRPA1 activity was observed after 4 h of incubation with BD1063 (100 µM), a structurally unrelated Sigma-1R antagonist (Fig. 1G). The amplitude was reduced to 36% of control and the percentage of responders was reduced to 45%. The neurosteroid progesterone has been shown to act as an endogenous Sigma-1R antagonist.52,53 Incubation of hTRPA1-expressing cells with progesterone also inhibited AITC-evoked responses (Supplementary Fig. 2). We also examined responses to carvacrol (100 µM), a non-electrophilic agonist of TRPA1 found in oregano. After incubating hTRPA1-tGFP-HEK293 cells with S1RA (100 µM, 4 h), the fraction of responding cells was reduced from 90 to 57% and their mean amplitude was reduced to 26% of control (Fig. 1H).
In summary, three different Sigma-1R antagonists markedly reduced human TRPA1 responses to agonists with different chemical properties and activation mechanisms, showing that the inhibitory actions of Sigma-1R antagonists on TRPA1 activity are independent of their structural properties or the TRPA1 mode of activation.
TRPA1 inhibition by Sigma-1R antagonists depends on the presence of Sigma-1R
Sigma-1R is broadly expressed in most tissues, including human kidney cells.30 To confirm that Sigma-1R is the target of S1RA effect, we transfected hTRPA1-tGFP transiently in Sigma-1R KO HEK293 cells. As expected, these cells did not express Sigma-1R protein (Fig. 2C). Cells were pre-incubated for 4 h with S1RA (100 µM) or CS and the calcium response to AITC (50 μM) was tested. In cells lacking Sigma-1R, incubation with S1RA had no effect on the amplitude of AITC-evoked responses (Fig. 2A, top). In contrast, the transient expression of hSigma-1R-mCherry in silenced cells restored the strong inhibitory action of S1RA incubation on TRPA1 activity (Fig. 2A, bottom). In these cells, the peak amplitude of AITC-evoked responses was reduced 32% by S1RA compared to hSigma-1R-mCherry transfected cells incubated with CS (Fig. 2B).
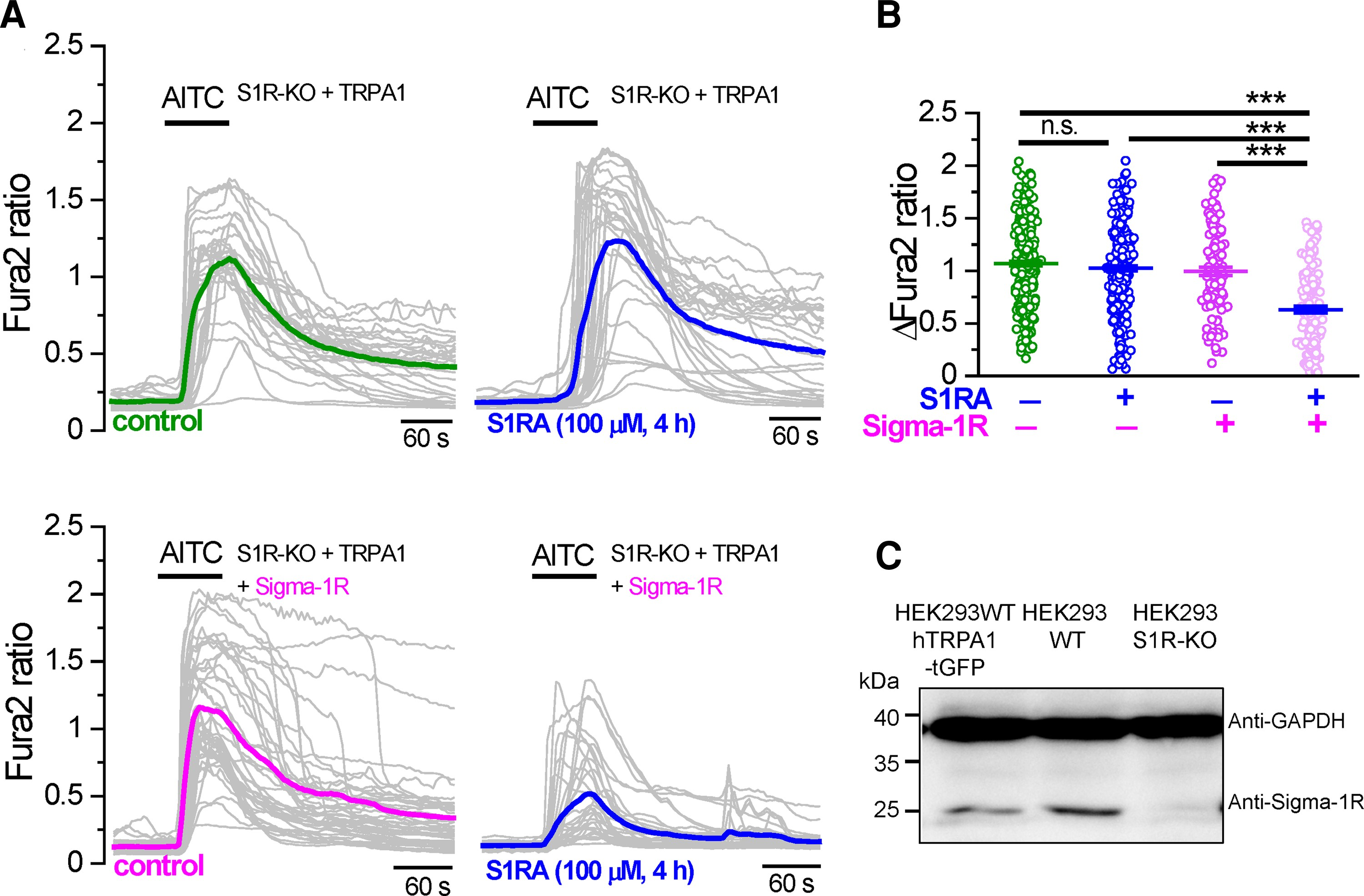
TRPA1 inhibition by Sigma-1R antagonists depends on Sigma-1R expression.
(A) Top: Representative intracellular calcium responses to AITC (50 µM) in Sigma-1R silenced HEK293 cells (S1R-KO) transfected with hTRPA1-tGFP. Cells were incubated with CS (4 h) or in S1RA (100 µM, 4 h). Green and blue traces represent the averages of individual cells in grey. Bottom: The same experiment as in A but in hTRPA1-tGFP cells cotransfected with hSigma-1R-mCherry. (B) Summary histograms of mean ± SE calcium responses to AITC in S1R-KO cells + hTRPA1-tGFP in control (green, n = 312), S1R-KO cells + hTRPA1-tGFP in S1RA (100 µM, 4 h) (blue, n = 276), S1R-KO cells + hTRPA1-tGFP+ hSigma-1R-mCherry in control (magenta, n = 118) and S1R-KO cells + hTRPA1-tGFP+ hSigma-1R-mCherry in S1RA (100 µM, 4 h) (pink, n = 144). ***P < 0.001 (ANOVA in combination with Bonferroni’s post hoc test). (C) Western blot of HEK293 cells transiently transfected with hTRPA1-tGFP, WT HEK293 cells and S1R-KO HEK293 cells, demonstrating the expression of endogenous Sigma-1R in WT cells and the absence in S1R-KO cells.
These results indicate that Sigma-1R expression is necessary and sufficient for TRPA1 inhibition after S1RA incubation. They also suggest that these inhibitory effects are not exerted directly on TRPA1 channels.
The reduction of TRPA1 currents by S1RA depends on Sigma-1R
To investigate the influence of Sigma-1R on TRPA1 channels directly, whole-cell voltage-clamp recordings were performed from HEK293 cells stably expressing hTRPA1-tGFP. Cells were pre-incubated 4 h with CS or with different concentrations of S1RA. AITC-evoked inward currents were inhibited in a dose-dependent manner (IC50 = 37 ± 11 µM) when cells were pre-incubated with S1RA (Fig. 3A and B). Incubation with 100 µM S1RA, the highest dose tested, reduced AITC-evoked currents by 81% at −60 mV, compared to cells incubated in CS (Fig. 3C). AITC-activated inward currents at −60 mV (Fig. 3A) had a mean density of 52 ± 11 pA/pF (n= 10) in CS compared to 10 ± 3 pA/pF (n = 10) after S1RA 100 µM. Of note, S1RA pre-incubation also modulated the kinetics of AITC-evoked inward currents (Fig. 3A). The time to peak increased to 65 ± 10 s (n = 18) after pre-incubation with 100 µM S1RA, compared to a time to peak of 25 ± 2 s (n = 20) in cells incubated in CS, suggesting that S1RA slowed the macroscopic activation rate.
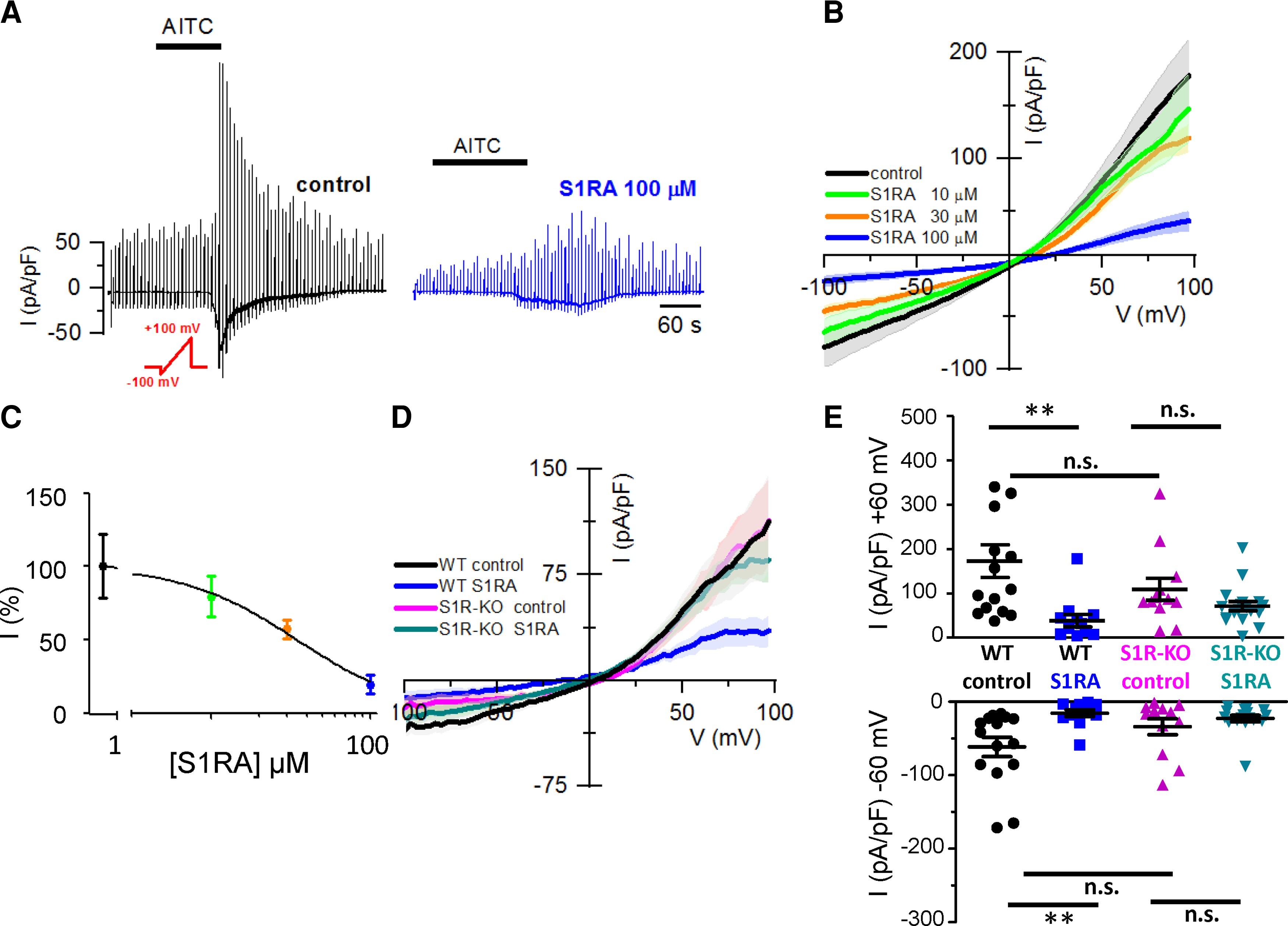
Sigma-1R expression is necessary for the reduction of TRPA1 whole-cell currents by S1RA.
(A) Representative time course of whole-cell currents, from the HEK293 hTRPA1-tGFP stable cell line, in response to the indicated repeated voltage ramp protocol (red) and AITC (50 µM), HP = −60 mV, in CS and after S1RA pre-incubation (100 µM, 4 h). Note the slow development of current and the reduced peak amplitude after S1RA incubation. (B) Average IV relationships from HEK293-hTRPA1-tGFP cells using the protocol shown in A recorded in control (n = 10) and S1RA at different concentrations: 10 µM (n = 10), 30 µM (n = 10) and 100 µM (n = 10). Average of maximal response to AITC in each trace. (C) Dose-response curve of S1RA effects on HEK293 hTRPA1-tGFP cells in response to AITC (50 µM). Normalized current density at −60 mV with respect to CS. ***P < 0.001 (ANOVA in combination with Dunnett’s post hoc test). (D) Average IV relationships from WT HEK293 cells transiently transfected with hTRPA1-tGFP in control (n = 15), S1RA pre-incubation (100 µM, 4 h, n = 12) and in Sigma-1R silenced cells transiently transfected with hTRPA1-tGFP (S1R-KO) recorded after pre-incubation in CS for 4 h (n = 12) or S1RA (100 µM, 4 h, n = 18). Average of maximal response to AITC in each case. (E) Summary of individual and mean ± SE current density at −60 and +60 mV for the different experimental conditions shown in D. **P < 0.01 (ANOVA in combination with Bonferroni’s post hoctest). Results from transient transfections were obtained in seven independent experiments.
Next, to demonstrate the specificity of S1RA actions, we examined the effect of S1RA pre-incubation on TRPA1 currents after silencing Sigma-1R (i.e. S1R-KO). In this case, incubation with S1RA had no effect on the amplitude of AITC-evoked currents (Fig. 3D and E).
These results confirm that Sigma-1R expression is necessary for the reduction in TRPA1 channel activity observed after S1RA pre-incubation.
S1RA impairs the formation of TRPA1–Sigma-1R complexes
Sigma-1R is a molecular chaperone known to interact directly with various ion channels, affecting their gating and their expression at the plasma membrane, reviewed by.54 To probe the molecular interaction between Sigma-1R and TRPA1, we performed co-immunoprecipitation experiments, using HEK293 transiently transfected with hTRPA1-tGFP and mSigma-1R-EYFP fusion proteins. Cell extracts containing mSigma-1R-EYFP were purified with GFP beads and, as shown in Fig. 4A, TRPA1 was readily pulled down only in the presence of Sigma-1R (Fig. 4A, lane 1).
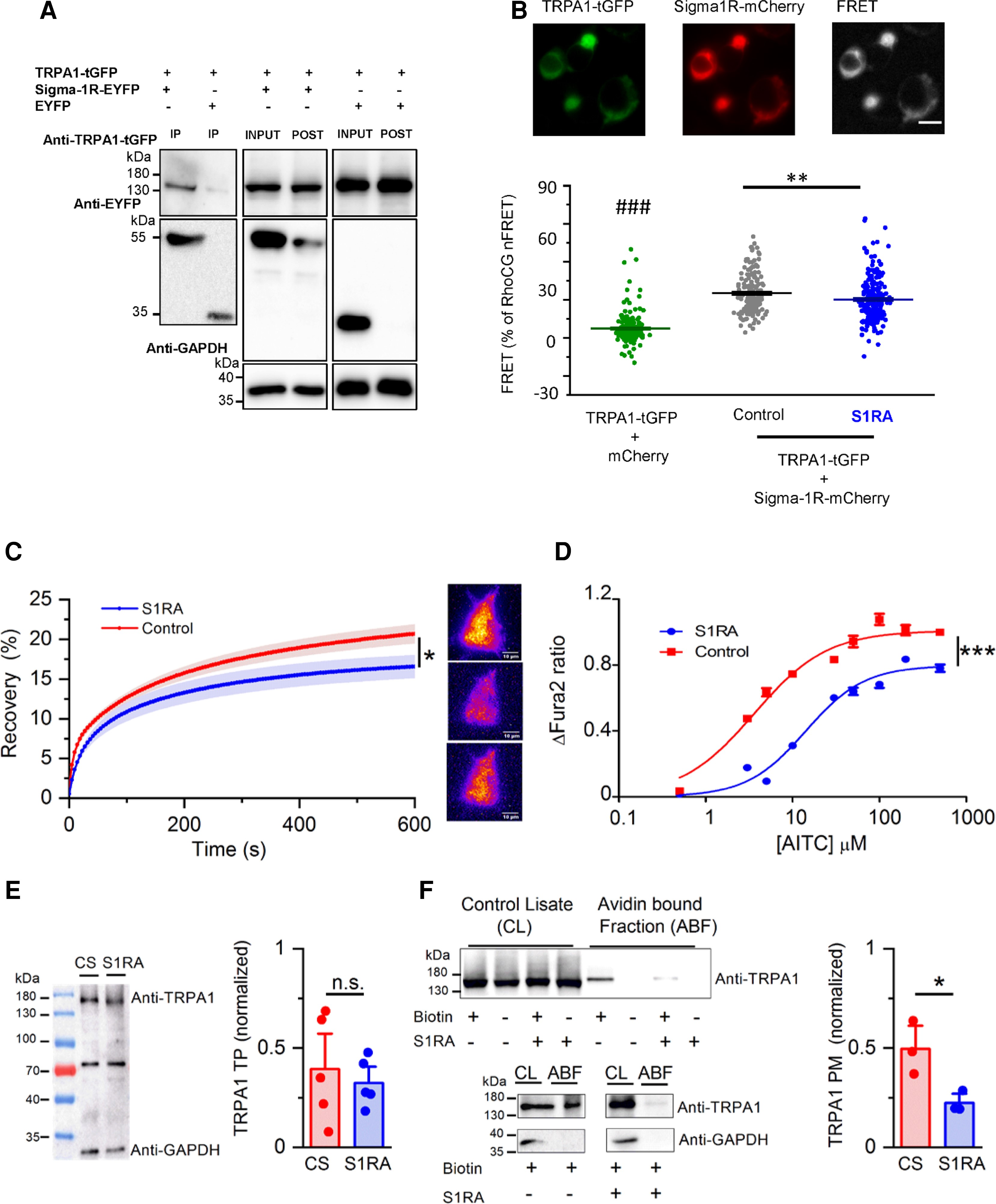
S1RA impairs the formation of TRPA1–Sigma-1R complexes and reduces TRPA1 expression at the plasma membrane.
(A) Co-immunoprecipitation of TRPA1 and Sigma-1R-EYFP in cotransfected HEK293 cells. Input is one-tenth of the total protein used in each immunoprecipitation. TRPA1 was pulled down by Sigma-1R-EYFP (lane 1) using GFP-Trap (Chromotek), while it was not when Sigma-1R-EYFP was not present (lane 2). The figure is representative of four independent protein extractions. GFP-Trap agarose beads do not recognize tGFP. (B) Detection of net sensitized emission (FRET) levels between hTRPA1-GFP and hSigma-1R-mCherry in transfected HEK293 cells. Top: Representative image of the fluorescence signal of hTRPA1-tGFP+Sigma-1R-mCherry as well as the signal in the FRET channel (scale bar = 20 µM). Bottom: Summary histogram of FRET levels (as a percentage of RhoCG FRET) of TRPA1-tGFP+mCherry (negative control, green bar) and TRPA1-tGFP+Sigma1-mCherry in control conditions (black bar) and after S1RA incubation (100 µM, 4 h) (blue bar). ###P < 0.001 (ANOVA in combination with Bonferroni’s post hoc test) test TRPA1-tGFP+soluble mCherry versus all groups, **P < 0.01 (unpaired t-test), S1RA untreated versus treated hTRPA1-tGFP+Sigma-1R-mCherry transfected cells. (C) Left: The time course of hTRPA1-tGFP TIRF fluorescence recovery after TIRF photobleaching, in control treated cells (red trace) and cells incubated with S1RA (100 µM, 4 h, blue trace). *P < 0.05 (unpaired t-test). Error bars represent SE. Representative TIRF images of HEK293 transfected with hTRPA1-GFP before (top), immediately after photobleaching (middle) and after 10 min recovery (bottom), scale bar = 10 µM. (D) Dose-response curves of AITC-evoked calcium responses in stable HEK293-hTRPA1-tGFP cells in CS (red) and pre-incubated with S1RA (100 µM, 4 h, blue). Values have been normalized to 500 µM AITC in CS. The lines represent fits to Hill functions (Y = Ymax/(1 + (EC50/X)nH)) with parameters EC50 = 14.12 ± 0.11 µM and 3.60 ± 0.03 µM, Ymax = 1.009 ± 0.002 and 0.797 ± 0.002 and nH = 1.03 ± 0.01 an 1.29 ± 0.01 in control and S1RA (100 µM, 4 h), respectively. (E) Western blot of total protein (TP) extracts from HEK293-hTRPA1-tGFP stable line incubated in CS for 24 h) or S1RA (25 µM, 24 h). A non-specific band at ∼70 kDa is exposed with anti-GAPDH (1:5000 G9545, Sigma-Aldrich). Immunodetection values in S1RA and CS, normalized with respect to GAPDH, are similar (n = 5 independent extractions, P > 0.05, unpaired t-test). (F) Representative western blot from biotinylation assays of hTRPA1- tGFP cells. Total expression of TRPA1 was assessed by directly immunoblotting 15 µg of each cell lysate. TRPA1 cell surface (Plasma Membrane protein, PM) expression was calculated by expressing the intensities of the TRPA1 bands in the avidin-bound fraction as a fraction of those in the corresponding cell lysates (n = 3, *P < 0.05, unpaired t-test). GAPDH was used as a negative control blot for the presence of a soluble protein in the avidin bound fraction (ABF).
FRET experiments were performed to define the possible formation of a complex between Sigma-1R and TRPA1, and to study the effect of S1RA on this interaction. HEK293 cells cotransfected with hTRPA1-tGFP and hSigma-1R-mCherry were excited at 488 nm to measure sensitized emission FRET between both fluorophores (see the ‘Materials and methods’ section). As shown in Fig. 4B, cotransfection of both proteins produced a level of FRET that was 35.3 ± 1.1% of RhoCG FRET (n = 153) (the FRET positive control, see the ‘Materials and methods’ section). This value was significantly higher than that observed when hTRPA1-tGFP was cotransfected with free soluble mCherry and no specific interaction was expected (10.4 ± 3.6% of RhoCG FRET, n = 199). These results indicate that the physical distance between TRPA1 and Sigma-1R is <10 nm, sufficiently close for making a protein–protein complex between them. Interestingly, when cotransfected cells were pre-incubated with S1RA (100 µM, 4 h), the levels of FRET decreased significantly (28.6 ± 1.1% of RhoCG FRET, n = 221), compared to the level obtained in cells treated with CS (P < 0.01, unpaired t-test), suggesting a reduced interaction.
This result suggests that the antagonistic effect of S1RA on TRPA1 activity could be due to an impairment in the assembly of TRPA1–Sigma-1R complexes or an alteration in the complex conformation.
S1RA incubation diminishes TRPA1 expression at the plasma membrane
So far, the results indicate that Sigma-1R interacts with TRPA1 channels and this interaction is affected by S1RA, leading to reduced TRPA1 activity. Next, we asked whether trafficking of TRPA1 channels to the plasma membrane was altered by S1RA incubation. To this end, we performed fluorescence recovery after photobleaching experiments in HEK293 cells transiently transfected with hTRPA1-tGFP, using TIRF excitation to restrict the photobleaching and recovery visualization to a thin layer of ∼200 nm containing the plasma membrane. Sustained, high-power TIRF illumination for 120 s photobleached ∼80% of fluorescence excited near the plasma membrane. Thereafter, fluorescence recovery due to hTRPA1-tGFP channel trafficking from non-bleached regions was monitored for 10 min. hTRPA1-tGFP was observed in the entire footprint of the cell as a dense mesh of punctate structures (Fig. 4C and Supplementary Fig. 3). After photobleaching, recovery of hTRPA1-tGFP takes place homogenously across the entire surface of the cell in contact with the coverslip, suggesting trafficking of the channel from the cytosol towards the plasma membrane and not by membrane lateral diffusion (Supplementary Fig. 3).51 As shown in Fig. 4C, the observed final level of fluorescence recovery after photobleaching after 600 s was significantly lower (blue trace, 16.6 ± 1.5% n = 22) in cells pretreated with S1RA (100 µM, 4 h) compared to cells incubated in CS (red trace, 20.7 ± 1.2% n = 31). Comparing the time constants of fluorescence recovery, no differences were found in recovery kinetics (data not shown). These results suggest that Sigma-1R facilitates TRPA1 trafficking to the plasma membrane, a process impaired by S1RA.
Additional evidence for the reduced expression of TRPA1 channels at the plasma membrane after S1RA incubation (100 µM, 4 h) came from dose-response curves to the agonist AITC. As shown in Fig. 4D, S1RA pre-incubation produced a rightward shift in the dose-response curve [half-maximum effective concentration (EC50) 3.61 ± 0.03 µM in control versus EC50 14.12 ± 0.11 µM after S1RA] and a significant reduction (∼20%) in the maximal calcium response.
Finally, to establish more directly an effect of S1RA on plasma membrane expression of TRPA1, we used cell surface biotinylation techniques (see the ‘Materials and methods’ section). We evaluated TRPA1 plasma membrane and total protein levels in CS and after S1RA incubation (25 µM–24 h) (Fig. 4E). Notably, total TRPA1 expression level was unaffected by S1RA treatment (Fig. 4E). In contrast, treatment produced a significant reduction in the avidin-bound fraction (i.e. biotinylated) of TRPA1. The level of TRPA1 at the surface, normalized to TRPA1 in the total protein extract, was reduced to about half in cells pretreated with S1RA (Fig. 4F).
Collectively, these results indicate that the observed decrease in TRPA1 channel activity after S1RA incubation is caused by a reduction in TRPA1 membrane expression without alteration in total TRPA1 levels.
S1RA incubation reduces endogenous TRPA1 activity in sensory neurons
To investigate the effects of Sigma-1R antagonists on TRPA1-mediated responses in nociceptors, DRGs from adult C57BL/6J mice were cultured and incubated with S1RA (100 µM, 4 h) or CS. Representative traces of neurons responding to AITC in control and pre-incubated with S1RA are shown in Supplementary Fig. 4A. The amplitude of intracellular calcium responses to AITC (50 µM) were significantly reduced from 0.46 ± 0.01 in CS (n = 314) to 0.25 ± 0.02 after S1RA treatment (n = 195). Treatment also reduced the percentage of AITC responders from 47 to 32% (Supplementary Fig. 4A and B). In TRPA1-expressing DRG neurons, obtained from Trpa1 Cre-tdTomato mice and identified by their red fluorescence, incubation with S1RA (100 µM, 4 h) produced a similar reduction in AITC-evoked responses (Supplementary Fig. 4C and D). Similar to our findings in TRPA1-expressing HEK293 cells, longer incubations (24 h) of DRG neurons with S1RA or BD1063 reduced the concentration of antagonists necessary to observe a significant reduction in AITC-evoked responses (Supplementary Fig. 5). In contrast to the effects of S1RA incubation, the acute application of a high concentration of the drug had no effect on the time course of AITC-evoked response in mouse DRG neurons (Supplementary Fig. 6).
The effect of S1RA incubation on the excitability of TRPA1(+) DRG neurons was examined with cell-attached recordings, a condition where the intracellular milieu is preserved. In CS, all tdTomato(+) neurons tested (8/8) responded to 100 µM AITC, reaching a maximum average firing of 1.4 Hz (Fig. 5A and B). In contrast, after S1RA treatment about a half (5/11) failed to respond to AITC, or decreased their firing rate: their average frequency did not exceed 0.6 Hz.
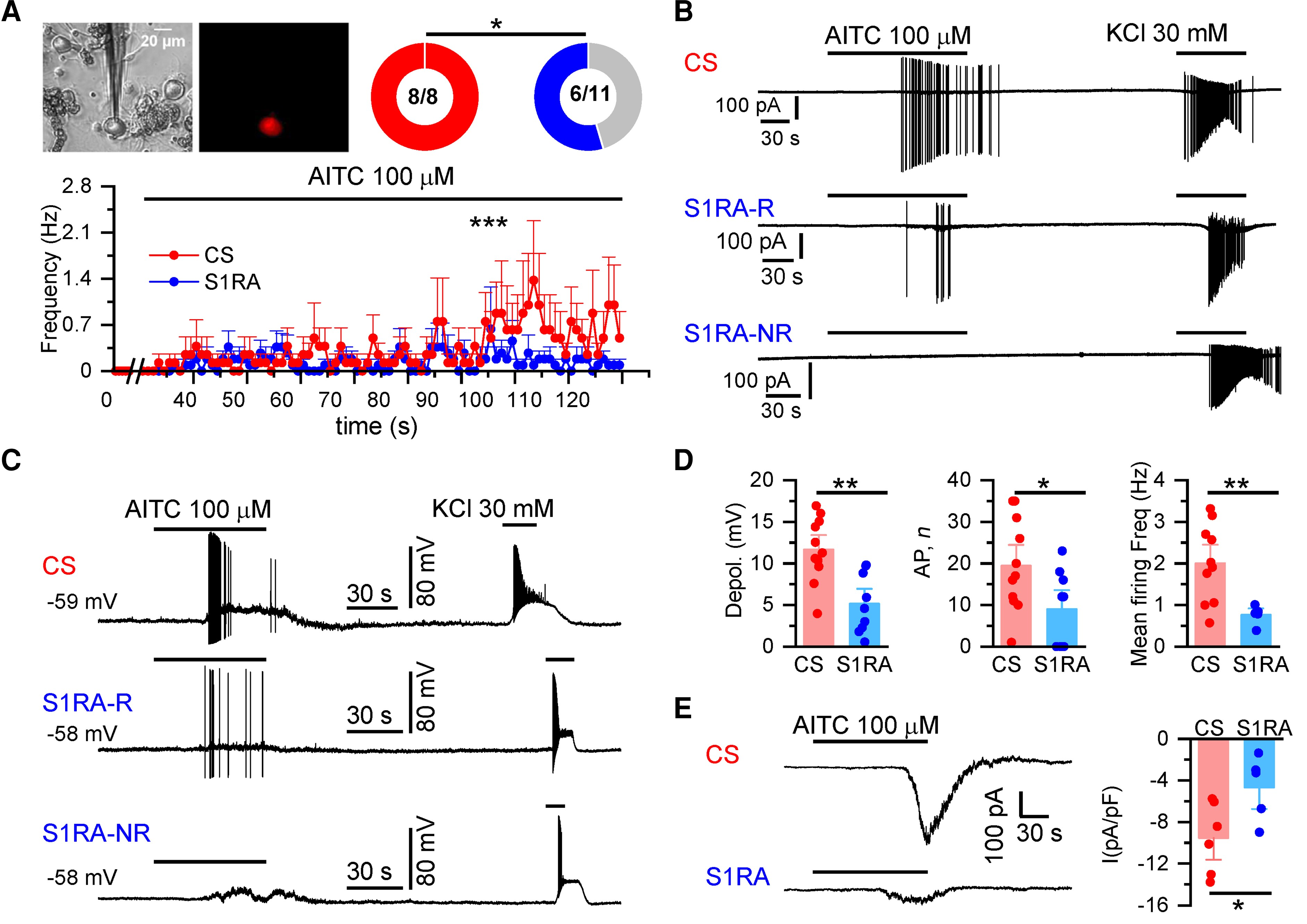
S1RA reduces TRPA1 activity in mouse DRG nociceptors.
(A) TRPA1(+) neurons in cultured DRGs from Trpa1 Cre-tdTomato mice were identified by their red fluorescence. AITC (100 µM) evoked action currents in 100% of neurons in CS (n = 8) and in 55% after S1RA incubation (6/11) (*P < 0.05, Fisher’s exact test). Firing frequency during the 2 min of AITC application. ***P < 0.001 (unpaired t-test). (B) Representative cell-attached recordings of TRPA1-expressing DRG neurons (n = 5 mice). Responses to AITC (100 µM) and high KCl (30 mM) after incubation with CS, 4 h) or S1RA (100 µM, 4 h). Note the robust firing in CS. After S1RA incubation (100 µM, 4 h), six neurons showed a weak response (S1R1-R, responder) while five did not fire action potentials (S1RA-NR, non-responder). Note that in neurons in which no response to AITC was observed, an impulse discharge could be evoked with 30 mM KCl. (C) Representative whole-cell current-clamp recordings of TRPA1(+) DRG neurons (n = 8 mice) showing the response to AITC (100 µM) and KCl (30mM) after incubation with CS-4 h or S1RA (100 µM, 4 h). After S1RA, some neurons fired AP (S1RA-R) while others showed a subthreshold depolarization but no AP firing (S1RA-NR). Both fired AP in response to high KCl. (D) Histograms summarizing the amplitude of AITC-evoked depolarization, number of action potentials and mean firing frequency of neurons: in CS (n = 11 out of 11) and after S1RA incubation (n = 5 of 9) responded to AITC. Mean ± SE **P < 0.001, *P < 0.05 (unpaired t-test). (E) Left: Representative whole-cell voltage-clamp recordings (n= 2 mice) of TRPA1(+) neurons (Vhold = −73 mV). AITC-evoked inward current in CS for 4 h incubated neurons (−9.6 ± 1.4 pA/pF n = 6) that was reduced in S1RA (100 µM, 4 h) incubated neurons (−4.8 ± 1.3 pA/pF n = 5), *P < 0.05 (unpaired t-test). Electrophysiological recordings were performed on 15 independent DRG cultures.
Activation of nociceptor endings by TRPA1 agonists produce depolarizing generator potentials and spike firing.55 We simulated these conditions in current-clamp recordings obtained from cultured TRPA1(+) DRG neurons (Fig. 5C). Only minor differences were found in the electrophysiological properties of TRPA1(+) neurons after S1RA incubation when compared to CS (Supplementary Table 2). In contrast, the maximum depolarization evoked by AITC was strongly reduced after S1RA incubation, 5 ± 1 mV (n = 9) versus 12 ± 1 mV (n = 11) after incubation in CS (Fig. 5D). In CS, all (11/11) TRPA1(+) neurons fired AP after AITC application. In contrast, this number was reduced to 56% (5/9) after S1RA incubation (P < 0.05, Fisher’s exact test). Moreover, the number of evoked action potentials were also reduced by S1RA incubation and their frequency during AITC application did not exceed 0.76 ± 0.10 Hz (average of n = 5) in the presence of S1RA while in control it reached 2.0 ± 0.3 Hz (average of n = 11) (Fig. 5D). Finally, electrophysiological recordings in whole-cell voltage-clamp mode showed a large reduction in the amplitude of AITC-evoked inward currents after S1RA incubation (Fig. 5E).
These results confirm that S1RA causes a substantial decrease in the activation of TRPA1-expressing nociceptors to a TRPA1 agonist, without significant alteration in their intrinsic excitability.
Systemic S1RA injections reduce pain-related behaviours to intradermal TRPA1 agonists
Considering that Sigma-1R antagonists mitigate nociceptive responses in different experimental pain models,41,42,56 we examined the possible role of TRPA1 as an antinociceptive drug target of Sigma-1R. To this end, we quantified responses of WT and TRPA1 KO mice to intradermal injections of AITC (10 µl at 10 mM) in their hind paws.46 One group of animals received vehicle (saline i.p.) and the other S1RA (40 mg/kg i.p.), 24-h before blind evaluation of pain-related behaviour (i.e. duration of paw licking after AITC injection). The results show that systemic S1RA injection produces a substantial reduction in pain-related responses to intradermal AITC in WT mice (Supplementary Fig. 7B). Responses to AITC were strongly diminished in TRPA1 KO mice compared to WT, and no further reduction was observed in these animals when pretreated with S1RA (Supplementary Fig. 7B). These findings demonstrate that the modulation of pain-related behaviours produced by S1RA after AITC application was dependent on TRPA1 activation.
TRPA1 channels participate in painful responses to oxaliplatin
Systemic oxaliplatin administration causes a painful peripheral neuropathy in patients and rodent models, characterized by mechanical and cold hypersensitivity in their extremities.1,8 To induce the neuropathy, mice received three injections of oxaliplatin (6 mg/kg i.p.) on alternate days (Fig. 6A), while the control group received its vehicle (glucose 5% in dH2O). Mechanical and cold sensitivity were monitored in mice before and after oxaliplatin treatment (see the ‘Materials and methods’ section).
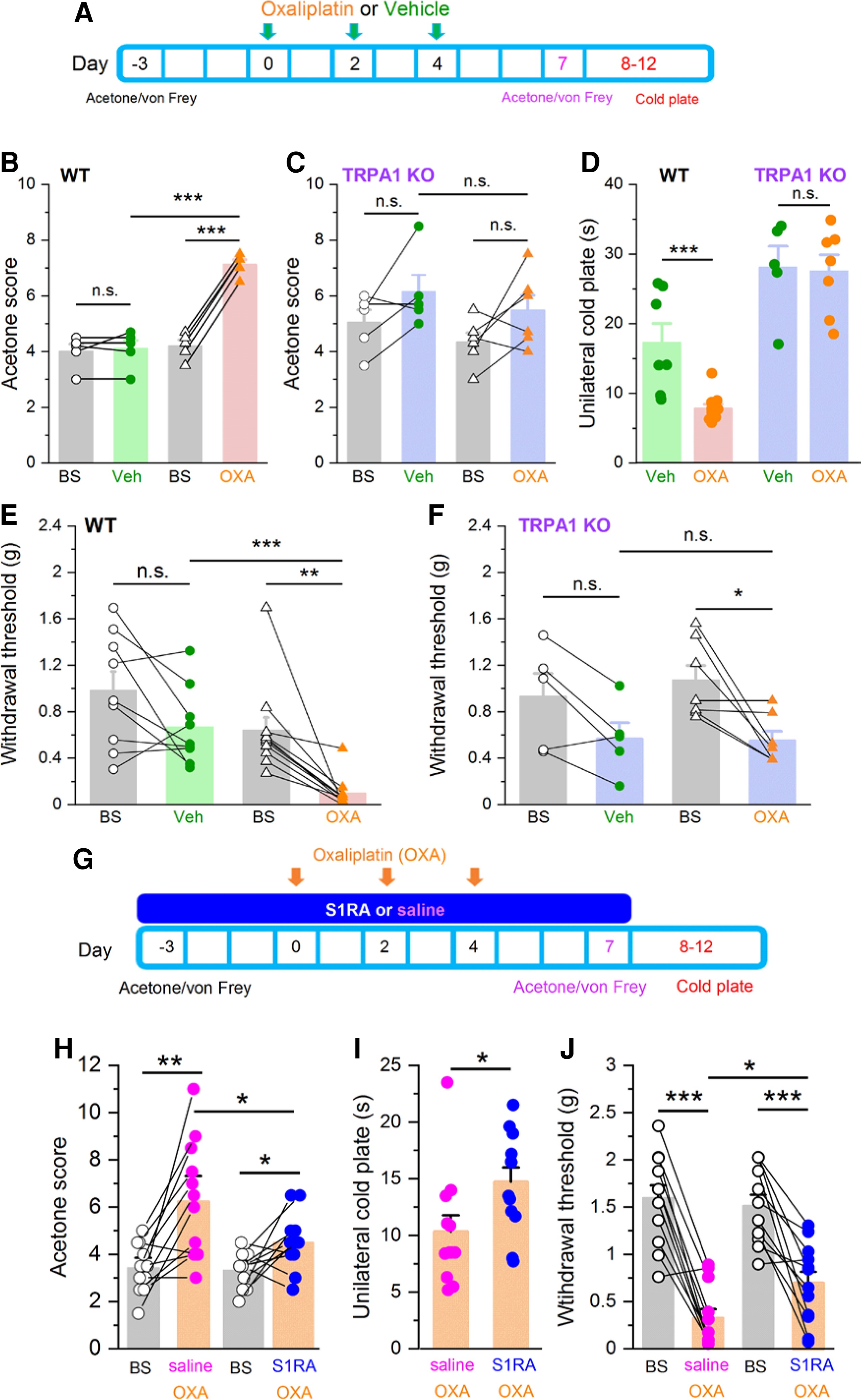
Systemic S1RA administration reduces TRPA1-dependent cold and mechanical hypersensitivity in oxaliplatin-treated mice.
(A) Time line of experimental protocol. Oxaliplatin was injected three times on alternate days. Oxaliplatin effects to mild cold (acetone) or mechanical stimulation (von Frey) were evaluated 3 days before and 7 days after first injection. Noxious cold was evaluated 8–12 days after the first oxaliplatin injection. (B) Responses to acetone in WT mice before (baseline, BS) and after vehicle (green, n = 5), and before and after oxaliplatin (orange, n = 5) ***P< 0.001, n.s. P > 0.05 (unpaired t-test). Paired t-test when responses of the same mice were compared (lines connecting symbols). (C) Cold (acetone) sensitivity in TRPA1 KO mice before (BS) and after vehicle (purple-green, n = 5), and before (BS) and after oxaliplatin (purple-orange, n = 7) injection. n.s. P > 0.05 (unpaired t-test). Paired t-test when the same mice were compared. Oxaliplatin did not induce cold allodynia in TRPA1 KO mice. (D) Evaluation of noxious cold sensitivity by unilateral cold plate test after vehicle (WT green, n = 7, TRPA1 KO purple-green, n = 5) or oxaliplatin injection (WT orange n= 11, TRPA1 KO purple-orange n = 7) ***P < 0.001, n.s. P > 0.05 (one-way ANOVA in combination with Bonferroni’s post hoc test). Oxaliplatin-induced cold hyperalgesia in WT but not in TRPA1 KO mice. (E) Mechanical withdrawal thresholds, in WT mice before and after vehicle injection (green, n = 9), before and after oxaliplatin injection (orange, n = 11). ***P < 0.001, n.s. P > 0.05 (unpaired t-test). Paired t-test when the same mice were compared. Oxaliplatin induced a clear mechanical allodynia. (F) Mechanical withdrawal thresholds in TRPA1 KO mice before and after vehicle injection (purple-green, n = 5), and before and after oxaliplatin injection (purple-orange, n = 7), *P < 0.05, n.s. P > 0.05 (unpaired t-test). Paired t-test when the same mice were compared. Oxaliplatin induces less mechanical allodynia in TRPA1 KO than in WT mice. (G) Timeline of experimental protocol to evaluate preventive effects of S1RA. Mice were evaluated 3 days before and 7–12 after first oxaliplatin (OXA) injection. (H) Mild cold (acetone) sensitivity before and after or oxaliplatin (OXA) injections. One group received saline combined with OXA (orange-pink, n = 12), and the other S1RA combined with OXA (orange-blue, n = 12) **P < 0.01, *P < 0.05 (paired t-test). (I) Unilateral cold plate test in saline + OXA injected mice (orange-pink, n = 12), and S1RA + OXA injected mice (orange-blue, n = 12). *P < 0.05 (unpaired t-test). (J) Mechanical sensitivity before (black) and after saline + OXA injections (orange-pink, n = 12) or S1RA + OXA (orange-blue, n = 12) ***P < 0.001, *P < 0.05 (paired t-test).
Systemic oxaliplatin treatment led to a marked hypersensitivity to mild (acetone application) (Fig. 6B) and noxious (10°C) cold stimuli (Fig. 6D).24,57 Notably, this thermal hypersensitivity was abolished in TRPA1 KO mice (Fig. 6C). Interestingly, baseline responses to mild cold were not altered in TRPA1 KO mice compared with WT, suggesting that other mechanisms, e.g. TRPM8, mediate innocuous cold sensitivity (Fig. 6B and C). In contrast, TRPA1 KO mice showed longer latencies in the noxious cold test, suggesting a role of TRPA1 in physiological noxious cold responses (Fig. 6D).44,58
Mechanical sensitivity was evaluated with calibrated von Frey hairs at baseline and 3 days after the end of oxaliplatin or vehicle treatment. We noticed that vehicle injections tended to reduce mechanical thresholds (Fig. 6E). This should be noted because it introduces a confounding factor when evaluating the effects of oxaliplatin. This reduction may reflect a higher stress level caused by the repeated injections required in this protocol. Oxaliplatin caused a marked mechanical hypersensitivity in WT mice (n = 11): their mean threshold was reduced from 0.64 ± 0.11 g at baseline to 0.10 ± 0.04 g after oxaliplatin (Fig. 6E). In contrast, the injection of vehicle resulted in a moderate, not significant, decrease in mechanical threshold compared to baseline (in average 0.98 ± 0.16 g versus 0.67 ± 0.11 g, n = 9). The effects of oxaliplatin on mechanical sensitivity in TRPA1 KO mice were clearly attenuated when compared to WT mice. As shown in Fig. 6F, TRPA1 KO animals treated with oxaliplatin showed a milder mechanical hypersensitivity when compared to WT animals, and this hypersensitivity was nearly identical to the one observed after vehicle injection, suggesting that it is mainly caused by the handling or repeated injections.
These results indicate that activation of TRPA1-expressing nociceptors plays a central role in the cold and mechanical hypersensitivity caused by systemic oxaliplatin treatment.
Systemic S1RA treatment prevents mechanical and cold hypersensitivity in oxaliplatin-treated mice
Since administration of oxaliplatin and other chemotherapeutics follows a planned regime,59 we asked whether previous administration of a Sigma-1R antagonist could prevent or attenuate the painful symptoms produced by systemic oxaliplatin. The treatment protocol consisted of daily dosing of systemic S1RA (40 mg/kg i.p., see the ‘Materials and methods’ section), starting 3 days before the first oxaliplatin injection (6 mg/kg), which was injected in three alternate days for 3 days (Fig. 6G). In the control group, the animals received vehicle (saline, i.p.) instead of S1RA and identical oxaliplatin treatment. Behavioural evaluation was performed before the start (baseline, BS) and after the last oxaliplatin injection, the same day or 1–5 days after the last S1RA injection (Fig. 6G), by a researcher blind to treatment. Consistent with our previous results, 7 days after the start of oxaliplatin treatment, control mice (i.e. treated with saline) developed a marked hypersensitivity to mild cold (i.e. acetone) (Fig. 6H). This hypersensitivity was greatly reduced in animals treated with S1RA (Fig. 6H). Similarly, responses to noxious cold were also substantially reduced in animals receiving S1RA instead of saline (Fig. 6I). Notably, this antiallodynic effect persisted even 3 days after the last S1RA injection (Fig. 6G), well passed the plasma clearance of the drug in mice.42,45 The effects of S1RA on attenuating mechanical allodynia following oxaliplatin treatment were also substantial (Fig. 6J). In this case, saline-treated animals showed a mechanical threshold of 0.32 ± 0.1 g (n = 12), compared to 0.69 ± 0.12 g (n = 12) after S1RA treatment (P < 0.05, unpaired t-test).
Collectively, these findings suggest that TRPA1 channels play a major role in the development of cold and mechanical hypersensitivity produced by oxaliplatin and these painful symptoms can be attenuated by a Sigma-1R antagonist applied systemically.
S1RA treatment normalizes TRPA1 responses in cultured DRG neurons from oxaliplatin-treated mice
Sigma-1R shows a very broad distribution throughout the nervous system.31 The cellular locus of S1RA antiallodynic effects is still unclear, with evidence for central and peripheral sites of action.42 Having established the antiallodynic effect that S1RA plays in oxaliplatin-induced neuropathic symptoms, and the dependence of these symptoms on TRPA1, we tested the modulatory role of Sigma-1R on sensitized peripheral neurons.
First, we investigated responses to AITC in short-term cultured DRG neurons prepared from oxaliplatin or vehicle-treated WT mice, and evaluated the effects of incubation with S1RA (100 µM, 4 h) or CS on these responses. We selected animals that showed a marked behavioural phenotype (i.e. mechanical and cold allodynia) to oxaliplatin treatment. Cold hypersensitivity was confirmed by the unilateral cold plate test (10°C) the same day of neuronal isolation. Two hours after harvesting and plating, neurons were incubated 4 h with S1RA or CS and loaded with Fura-2, during the last 45 min of incubation (see the ‘Materials and methods’ section). In sensory neurons obtained from oxaliplatin-treated mice, the average amplitude of AITC-evoked calcium responses was larger (0.55 ± 0.04, n = 118) than in neurons obtained from vehicle-injected mice (0.38 ± 0.25, n = 149) (Fig. 7A and B). Furthermore, incubation of neurons obtained from oxaliplatin-injected mice with S1RA (100 μM, 4 h), restored AITC-evoked responses to values similar to those obtained from mice treated with vehicle (0.35 ± 0.02, n = 172) (Fig. 7A and B). These results show that acute S1RA treatment restores sensitized TRPA1 responses in DRG neurons obtained from oxaliplatin-treated animals to normal values.
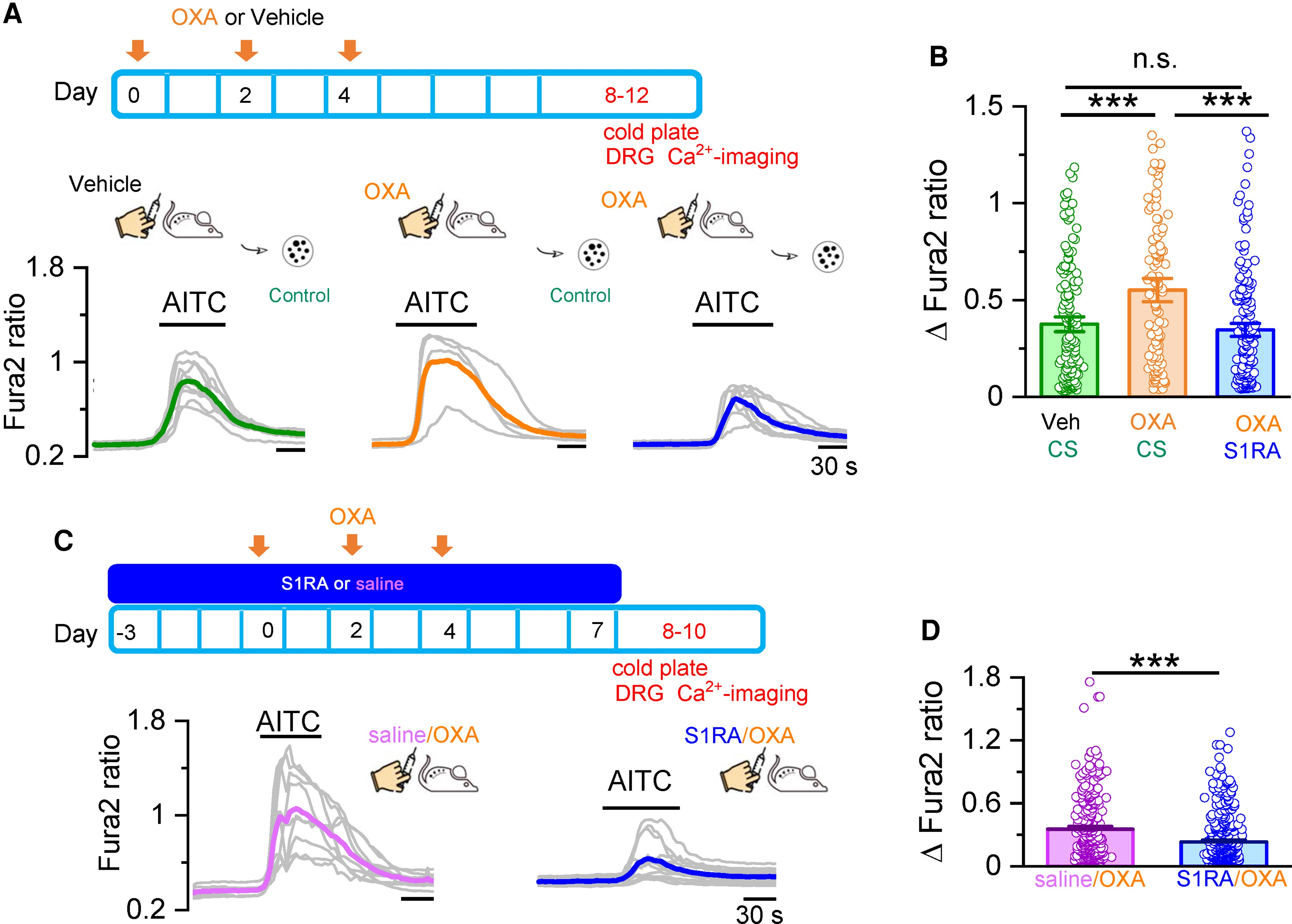
In vivo and in vitro S1RA treatment normalizes TRPA1 responses in DRG neurons from oxaliplatin-treated mice.
(A) Oxaliplatin (OXA) was injected three times on alternate days. DRG neurons were cultured and studied by Ca2+ imaging at Days 8–12, shortly after mice were evaluated by unilateral cold plate test. Calcium responses in DRG neurons from mice that received vehicle (left) and OXA injections (middle). Right: Responses from mice that received OXA injections and were incubated in S1RA (100 µM, 4 h). Individual traces of calcium elevations evoked by AITC (50 µM) for each condition are shown in grey. Coloured traces represent averages of the respective individual traces. (B) Histograms summarizing the sensitization of the AITC response by OXA and the inhibition by S1RA. Vehicle/CS n = 149, OXA/CS n = 118, OXA/S1RA n = 172, ***P < 0.001, n.s. P > 0.05 (one-way ANOVA in combination with Bonferroni’s post hoc test). (C) Mice received saline via S1RA injections starting 3 days before the first OXA injection until Day 7. DRG neurons were cultured and studied by Ca2+ imaging at Days 8–10, right after mice were evaluated by unilateral cold plate test. Representative traces of calcium elevations evoked by AITC (50 µM). DRG neurons from mice injected with Saline+OXA (left, average in pink) and S1RA + OXA (right, average in blue). (D) Histograms summarizing the preventive effect of S1RA in the sensitization of AITC responses by OXA. Saline/OXA (n = 797), S1RA/OXA (n = 1208), ***P < 0.001 (unpaired t-test).
In vivo S1RA treatment prevents TRPA1 sensitization in cultured DRG neurons from oxaliplatin-treated mice
Finally, to evaluate the preventive effect of systemic (i.p.) S1RA on oxaliplatin-mediated sensitization of TRPA1, we used the protocol shown in Fig. 7C. The effects of in vivo S1RA administration were compared to the injection of saline. Response to TRPA1 agonists were examined in short-term cultures 1–3 days after the last S1RA injection. Consistent with the behavioural results shown previously, the amplitude of AITC-mediated responses in cultured DRG neurons was reduced in animals treated in vivo with S1RA (Fig. 7C and D). These effects persisted for 72 h after systemic S1RA treatment.
These results suggest that the preventive effect of systemic S1RA in the development of the oxaliplatin-induced neuropathy involves the modulation of TRPA1 channels in nociceptors.
Discussion
CIPN is a frequent, insidious side effect of anticancer therapies. Without a full mechanistic understanding of its causes, treatments have been generally unsuccessful.2,5 The salient contribution of this study is the demonstration that TRPA1 is downmodulated by antagonism of Sigma-1R, a multifunctional chaperone, and this inhibition reduces the painful side effects of oxaliplatin in an experimental model of CIPN neuropathy. We established that antagonism of Sigma-1R impairs the formation of a molecular complex between TRPA1 and Sigma-1R, and the trafficking of functional TRPA1 channels to the plasma membrane, leading to a reduction in the excitability of nociceptor neurons sensitized by oxaliplatin.
Role of TRPA1 in CIPN
Multiple mechanisms are thought to be involved in CIPN. In particular, dysregulation of different ion channels, including sodium, potassium and TRP channels have been implicated in the painful symptoms16–19,60,61 Our behavioural studies in TRPA1 KO mice defined a key role of this ion channel in the development of mechanical and cold hypersensitivity in oxaliplatin CIPN. Our findings are in line with previous pharmacological and genetic studies in mice and rats.22,24,62 Recent in vivo imaging studies of mouse DRG neurons found that oxaliplatin treatment unmasked a population of silent peptidergic nociceptors with altered excitability to cold and noxious pinch.20 The molecular identity of the cold sensor in this subpopulation of sensory neurons was not established in the study and further work is needed.
A recurrent theme in various postulated mechanisms for CIPN is the alteration in the redox-status of peripheral sensory neurons, linked to excessive generation of reactive oxygen species by damaged mitochondria63–65. This is very relevant to our findings because TRPA1 is activated by cellular stress, including reactive oxygen species (reviewed by Sakaguchi and Mori66). In particularoxaliplatin activates TRPA1 by a glutathione-sensitive mechanism,21,23 and cytosol acidification of DRG sensory neurons.67 Other in vitro studies showed that oxaliplatin can enhance cold sensitivity of human TRPA1 by a mechanism implicating inhibition of prolyl hydroxylase and oxidation of intracellular cysteines.25,68
Current clinical guidelines recommend the use of duloxetine in the treatment of CIPN.6 This antidepressant drug showed modest analgesic efficacy in a randomized, double-blind, placebo-controlled trial for CIPN.69 It is worth mentioning that a recent study found that duloxetine inhibits TRPA1 activity, and this inhibition had neuroprotective effects on delayed neuropathy caused by exposure to organophosphates, reactive compounds present in many insecticides, herbicides and nerve agents.70 Thus, a deeper understanding of the mechanisms involved in the beneficial effects of duloxetine in CIPN, including the possible role of TRPA1 channels, may open new avenues for treatment.
Sigma-1R is necessary for S1RA effects on TRPA1
Using a KO cell line for Sigma-1R, we demonstrated that the receptor is essential for the inhibitory effect of S1RA on TRPA1. Moreover, reintroduction of Sigma-1R rescued the effect. In addition, acute application of S1RA did not reduce AITC-evoked responses. Altogether, this indicates that the actions of S1RA are not mediated by direct inhibition or block of the channel by the antagonist. This demonstration is important because some previous studies found that Sigma-1R ligands can inhibit other TRP channels (e.g. TRPC5 and TRPM3) by mechanisms independent of Sigma-1R.71 These findings also suggest that the actions of S1RA are not mediated by Sigma-2R. Despite their names, these two proteins show no sequence homology and derive from unrelated genes.72 Consistent with this view, S1RA is highly selective for Sigma-1R over Sigma-2R.42 The inhibitory effects of S1RA on TRPA1 required much higher concentrations than the estimates suggested from binding affinities of S1RA to Sigma-1R receptors.42 This discordance between affinities obtained in radioligand binding assays and IC50s in functional studies has been noted previously for S1RA and other potent Sigma-1R antagonists like BD1063,73 that normally require micromolar concentrations to be effective.37,74,75 Multiple reasons could contribute to this discrepancy, including the fact that functional studies are performed on cells with intact membranes, and the complex, multistep process, taking place between drug–receptor interaction and biological effect. The crystal structure of the human Sigma-1 receptor revealed that the ligand-binding cavity is very hydrophobic and completely occluded from solvent.76 These structural constraints account for the very slow ligand-binding kinetics characteristic of Sigma-1 receptors and help explain the discrepancies noted previously.
In calcium imaging and electrophysiological recordings, we established that Sigma-1R reduces TRPA1 activity in nociceptive neurons. In a recent study, Ortiz-Rentería and collaborators described the inhibition of TRPV1, another nociceptive channel, by Sigma-1R antagonists.37 In addition, the authors found a reduced inhibition in cells treated with siRNA, suggestive of a role of Sigma-1R in the inhibitory action. Since TRPV1 and TRPA1 colocalize in a fraction of nociceptors,26 the inhibitory effects of Sigma-1R antagonists may reflect shared signalling pathways downstream of both receptors.
Molecular interaction between Sigma-1R and TRPA1
We found that Sigma-1R antagonists reduce the plasma membrane expression of functional TRPA1 channels. The FRET experiments demonstrated that Sigma-1R and TRPA1 are close enough to form a molecular complex at the plasma membrane, a result corroborated by immunoprecipitation. We also show that FRET efficiency decreases after incubation with S1RA, indicating structural rearrangements in the TRPA1–Sigma-1R complex. Previous studies, using intramolecular FRET, showed that TRPA1 tetramers undergo notable conformational changes in the presence of agonists (e.g. calcium).77 We also show that S1RA incubation reduces biotinylated (i.e. plasma membrane TRPA1 levels) without affecting total TRPA1 expression, and impairs the repopulation of TRPA1 channels at the plasma membrane. We interpret this result as further evidence that the main effect of S1RA is on subcellular redistribution (i.e. assembly, folding or trafficking) of the channel, rather than direct interaction with the channel pore. Previous studies have shown that Sigma-1R can affect cellular excitability by modulating the traffic of various ion channels (reviewed by Soriani and Kourrich35). Indeed, the same interpretation to ours was suggested for similar results obtained in a previous study examining the interaction between Sigma-1R and Kv1.2 channels in the nucleus accumbens.38
We have not elucidated the subcellular location of the interaction between Sigma-1R and TRPA1. An obvious candidate is the endoplasmic reticulum-associated membrane, rich in Sigma-1R.34 Another possible locus of modulation is the alteration in the subcellular localization of TRPA1. Both, Sigma-1R78–80 and TRPA181reside in membrane lipid rafts, possibly facilitating their interaction. Moreover, Sigma-1R silencing shifted these channels from lipid nanodomains to non-raft fractions.80 In the case of TRPA1, disruption of lipid rafts reduced their activity.81Future studies should address the interesting possibility that cholesterol or other lipids participate in the interaction between Sigma1-R and TRPA1.
Ankyrin repeats are structural motifs found in many proteins, including TRP channels.82 These repeats are associated with protein–protein interactions and have been postulated to act as mechanosensitive springs.82–84 TRPA1 function is modulated through its long ankyrin repeat-rich amino-terminal domain.85 Previous work also demonstrated that Sigma-1R can form a complex with ankyrin B and inositol 14,5-trisphosphate receptors at the endoplasmic reticulum membrane, and this trimer was regulated by Sigma-1R agonists.86 Future mutagenesis studies can address the relevance of TRPA1 ankyrin repeats in the interaction between both proteins.
Sigma-1R can exist in various oligomeric forms, with antagonists favouring higher order oligomerizations and agonists having the opposite effect.87 The incubation with S1RA increases the proportion of Sigma-1R multimers,74 possibly altering the interaction with TRPA1, as suggested by the reduced FRET signal that we observed between both proteins. This may lead to a misfolded TRPA1, interfering with the forward trafficking and reducing its functional expression at the plasma membrane without alteration in total TRPA1 protein levels.
Sigma-1R and CIPN
Chemotherapy administration to cancer patients is a planned procedure, allowing for the design of preventive strategies for minimizing neurotoxic side effects. In a clinical trial of patients treated with oxaliplatin for colorectal cancer, S1RA showed a protective effect on neuropathic symptoms.43 Moreover, in rats, co-administration of S1RA and oxaliplatin prevented the development of cold allodynia.45 We demonstrated similar effects with the anticipated systemic injection of S1RA in oxaliplatin-treated mice. The reduction in mechanical and cold hypersensitivity extended 3 days past the last S1RA administration, suggesting a long-lasting effect on TRPA1 activity, well beyond the plasma half-life of the drug.42 This interpretation is reinforced by the demonstration of in vitro effects on TRPA1 activity, several days after the in vivo application of S1RA. However, ligand association and dissociation at the Sigma-1R is very slow,88 possibly explaining the long-lasting effects we observed. Monitoring the reduction in neuropathic symptoms over longer times after ending the oxaliplatin treatment will be useful in defining the potential use of S1RA or other Sigma-1R antagonists in preventing CIPN in cancer patients. Future studies, examining the effects of Sigma-1R antagonists on human nociceptors derived from induced pluripotent stem cells (iPSCs) should provide further insight into the mechanisms of CIPN.
TRPA1 is expressed in many barrier tissues, including the skin, gut and airways (reviewed by Viana27). Because Sigma-1R are also widely expressed in the same tissues,30 the potential for functional interactions between TRPA1 and Sigma-1R beyond the one described here is a distinct possibility worth exploring, and potentially relevant for treatment of other diseases like chronic itch, asthma and inflammatory bowel disease.