
The synergy of β amyloid 1-42 and oxidative stress in the development of Alzheimer’s disease-like neurodegeneration of hippocampal cells
By Gohar Karapetyan, Katarine Fereshetyan, Hayk Harutyunyan, and Konstantin Yenkoyan
Excerpt from the article published in Scientific Reports 12, 17883, 25 October 2022. DOI: https://doi.org/10.1038/s41598-022-22761-5
Editor’s Highlights
- Alzheimer’s disease (AD) is a neurodegenerative disease that occurs in elderly individuals and which incidence has increased year by year, and there is still no effective drug for treating it.
- Oxidative stress and mitochondrial dysfunction are the main pathological features of AD.
- Oxidative stress occurs within the bilayer, in the Aβ-induced oxidative stress hypothesis in which Aβ inserts as oligomers into the bilayer and serves as a source of reactive oxygen species (ROS).
- Aggregated Aβ (Aβ1-42) damages neurons, and glial cells proliferate to remove Aβ from the hippocampus.
- The combination of Aβ, external oxidative damage and inhibition of internal antioxidant defense is even more toxic, impairs cellular defense systems, and may represent the late phase of AD-associated cell damage.
- There is a critical role in the combination effect of Aβ and oxidative stress in the development of neurodegeneration.
Abstract
Alzheimer’s disease (AD) is a type of dementia that affects memory, thinking and behavior. Symptoms eventually become severe enough to interfere with daily tasks. Understanding the etiology and pathogenesis of AD is necessary for the development of strategies for AD prevention and/or treatment, and modeling of this pathology is an important step in achieving this goal. β-amyloid peptide (Aβ) injection is a widely used approach for modeling AD. Nevertheless, it has been reported that the model constructed by injection of Aβ in combination with a prooxidant cocktail (ferrous sulfate, Aβ, and buthionine sulfoximine (BSO) (FAB)) best reflects the natural development of this disease. The relationship between oxidative stress and Aβ deposition and their respective roles in Aβ-induced pathology in different animal models of AD have been thoroughly investigated. In the current paper, we compared the effects of Aβ 1-42 alone with that of Aβ-associated oxidative stress induced by the FAB cocktail on the neurodegeneration of hippocampal cells in vitro. We constructed a FAB-induced AD model using rat primary hippocampal cells and analyzed the contribution of each compound. The study mainly focused on the prooxidant aspects of AD pathogenesis. Moreover, cellular bioenergetics was assessed and routine metabolic tests were performed to determine the usefulness of this model. The data clearly show that aggregated Aβ1-42 alone is significantly less toxic to hippocampal cells. Aggregated Aβ damages neurons, and glial cells proliferate to remove Aβ from the hippocampus. External prooxidant agents (Fe2+) or inhibition of internal antioxidant defense by BSO has more toxic effects on hippocampal cells than aggregated Aβ alone. Moreover, hippocampal cells fight against Aβ-induced damage more effectively than against oxidative damage. However, the combination of Aβ with external oxidative damage and inhibition of internal antioxidant defense is even more toxic, impairs cellular defense systems, and may mimic the late phase of AD-associated cell damage. Our findings strongly indicate a critical role for the combination of Aβ and oxidative stress in the development of neurodegeneration in vitro.
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease that occurs in elderly individuals. Most patients show early loss of memory and progressively worsening of cognitive function, including language and mental impairments and loss of emotion and directionality1,2. With the development of society and changes in the human environment, the incidence of AD has increased year by year3. There is still no effective drug for treating AD.
Animal models are critical tools for better understanding the pathogenesis of AD and for investigating new therapeutic strategies. To date, the majority of transgenic AD mouse models exhibit the accumulation of β-amyloid (Aβ) plaques and neurofibrillary tangles4,5. However, alternative nontransgenic models are urgently needed, as indicated by the high failure rate of clinical trials on AD treatments4. This may be due to the complexity and multifactorial nature of AD pathogenesis. Oxidative stress and mitochondrial dysfunction are the main pathological features of AD6,7. Oxidative stress that occurs within the bilayer, hypothesized in the Aβ-induced oxidative stress hypothesis in which Aβ inserts as oligomers into the bilayer and serves as a source of reactive oxygen species (ROS)8. Oxidative stress in AD brain is manifested by decreased levels of antioxidant enzymes and also by increased protein oxidation (including protein carbonyls formation), lipid peroxidation, DNA oxidation, advanced glycation end products9. Mitochondria are the main consumers of oxygen and the main organelles responsible for the formation of ROS. ROS generation involves the spontaneous leakage of electrons from respiratory chain complexes and direct interaction of these electrons with oxygen molecules10. ROS attack proteins, lipids and nucleic acids, increasing the levels of end-products, such as protein carbonyls, thiobarbituric acid-reactive substances (TBARS), and malondialdehyde11. Additionally, mitochondria are unique organelles that provide ATP to the host cell by oxidative phosphorylation. Neurons are highly differentiated cells that require a high level of ATP to maintain an ionic gradient and generate potentials, as well as for neurotransmission12. Unlike other organs, the brain does not store excess ATP,thus, normal mitochondrial function is extremely important for the continuous generation and delivery of energy13.
In addition, neuronal cells are more vulnerable to ROS due to the high rate of oxygen consumption and the enrichment of polyunsaturated fatty acids in membranes14,15. Moreover, the level of redox metal ions is higher and the activity of the antioxidant system is lower in the brain than in other tissues16. There is a growing body of evidence showing the presence of markers of oxidative stress in neurofibrillary tangles and senile plaques (SPs) in AD brain. Several studies indicated significant elevation of membrane PUFA peroxidation by-product 4-hydroxynonenal (HNE) in hippocampus, amygdala, temporal cortex and cerebrospinal fluid of AD cases (W.R17. HNE neurotoxic mechanisms may be mediated by modifying neuron membrane proteins/enzymes, altered glucose and glutamate transport system, calcium metabolism, etc.18. This indicates that, in line with the primary damaging effect of reactive oxygen species on neuron membrane, a secondary, reaction by-products are capable of increasing neuron degeneration. Additionally, the level of antioxidant enzyme glutathione S-transferases (GST) is significantly decreased in AD brain19, consistent with the notion that a loss of protection against HNE might be correlated with subsequent protein modifications that lead to neuronal death. In fact, it is known that Aβ decreases activity of mitochondrial superoxide dismutase, which detoxifies the anion superoxide and protects from peroxidative damage20. Aβ is also capable of binding and inhibiting mitochondrial alcohol dehydrogenase known as ABAD (Aβ binding alcoholdehydrogenase)21. ABAD has a protective role, being responsible for the detoxification from aldehydes such as 4-hydroxynonenal.In addition, several studies have reported that Aβ affects mitochondrial DNA and proteins, leading to impairment of the electron transport chain and ultimately mitochondrial dysfunction. In turn, it should be noted that mitochondrial dysfunction causes alterations in amyloid precursor protein (APP) metabolism, enhances intraneuronal accumulation of Aβ and makes neuronal cells vulnerable22. Undoubtedly, the generation of ROS and accumulation of Aβ peptides are interconnected. It is important to note that alterations in the AD brain, such as the presence of redox metal ions, mitochondrial dysfunction, and an imbalance of antioxidant/oxidant enzymes, also induce ROS production23. However, the importance of Aβ-associated oxidative stress for neurodegeneration relative to that of other forms of AD-associated oxidative stress still needs to be investigated.
Thus, in the current study, the combination of ferrous sulfate, Aβ1-42, and buthionine sulfoximine (BSO) (FAB) was chosen to recapitulate AD pathophysiology as closely as possible. Aβ1-42 is more amyloidogenic than other Aβ peptides due to its hydrophobic characteristics. Ferrous sulfate was used as a prooxidant factor to induce oxidative stress under conditions of glutathione synthase inhibition. Glutathione synthase is an important enzyme in the antioxidant system, and inhibition of this enzyme by BSO likely contributes significantly to AD pathogenesis. Several studies have shown that administration of the FAB cocktail into the ventricular system by a pump over 4 weeks can be used to successfully model AD in rats. In the current study, the FAB cocktail was administered to primary rat hippocampal cells. We cultivated mixed cells isolated from the rat hippocampus, which included both glial and neuronal cells. This allowed us to recapitulate the complex environment of the brain, including neuroglial interactions, which play a very important role in AD pathogenesis. This cellular model of AD allowed us to monitor dynamic metabolic changes over several days. This study mainly focused on the prooxidant aspects of AD pathogenesis. Furthermore, cellular bioenergetics was assessed and routine metabolic tests were performed to evaluated cell survival and viability.
…
Results
Cell number and morphology
Hippocampal cells growth in primary culture (control cells) was characterized by a short lag phase during the first 7 days followed by a moderate increase in cell number (by 27.2%) until the 14th day of cultivation (exponential phase). The last 7 days of culture (14–21 days) was a stationary phase, during which we did not observe cell growth (Fig. 1A). The changes in neuronal morphology were evaluated using phase-contrast microscopy. Until the 7th day of culture, hippocampal cells appeared as round cells (Fig. 1B). After 7 days of culture, the hippocampal neurons extended moderate-sized apical dendrites, and early branches began to grow (Fig. 1C). On the 21st day of cultivation, although no proliferation was observed, extended axons and dendrites were observed (Fig. 1D).
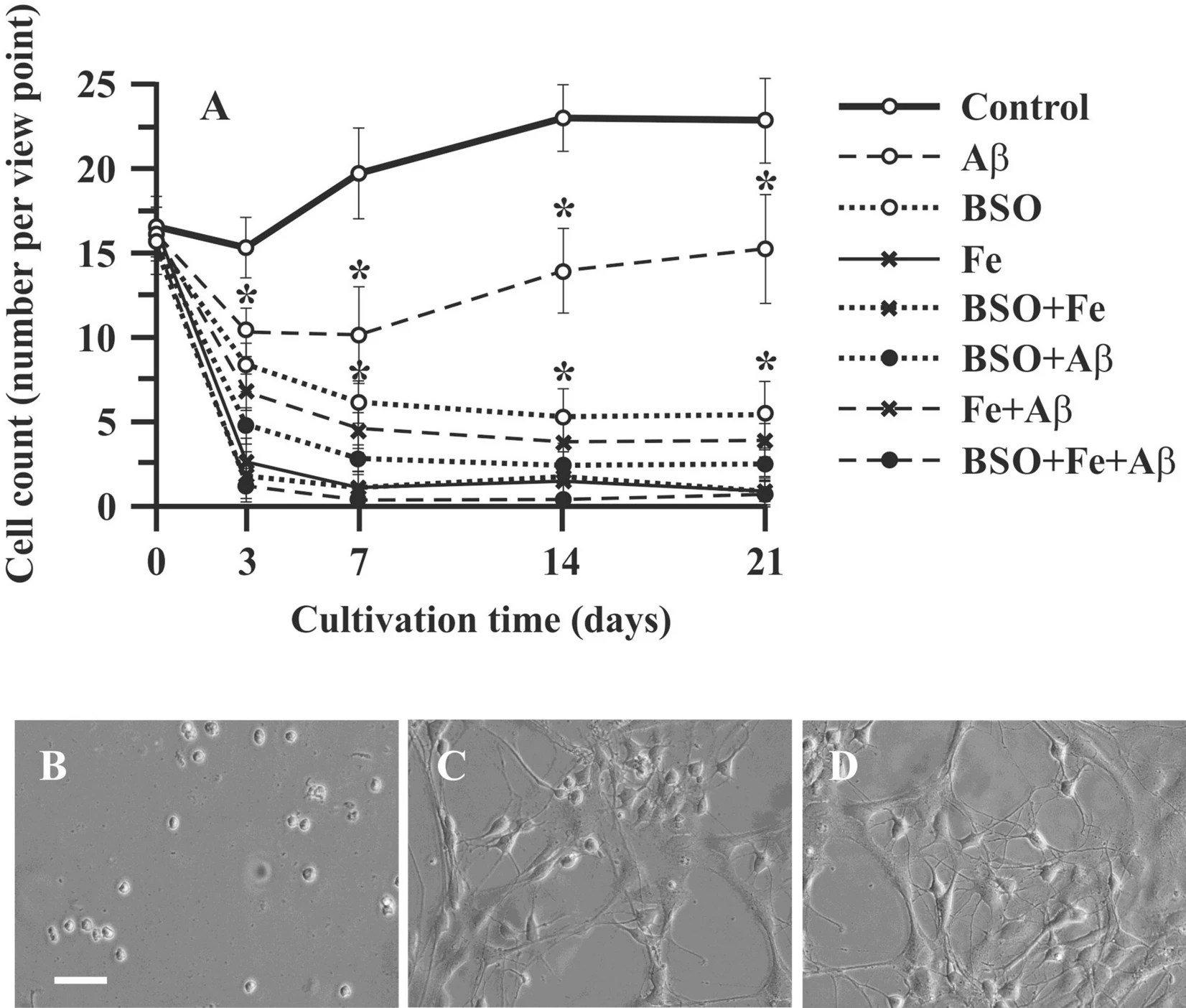
Effect of Aβ, BSO, and Fe2 + on neuronal cell count and morphology. (A) Changes in the number of primary rat hippocampal cells. (B) Neuronal cell morphology on the 7th day of culture. (C) Neuronal cell morphology on the 14th day of culture. (D) Neuronal cell morphology on the 21st day of culture. Scale bar, 100 μm. n = 10. * P < 0.05. The error bars indicate the mean ± SD. Hippocampal cells were isolated from the brains of two-month-old rats. The cells were cultured in NeuroCult medium supplemented with 10% NeuroCult proliferation supplement, 10% fetal bovine serum and 1% penicillin/streptomycin.
Aggregated Aβ (1-42) had a marked effect on neuronal cell growth in the exponential phase (7–14 days) of growth in culture; on the 7th day of culture, the cell count in the Aβ group was 51% lower (P < 0.05) than that in the control group. After this point, cells in the Aβ group retained moderate proliferative capability, and the increase in cell number in this group in the exponential phase (from the 7th to 14th days of culture) was 21% (Fig. 1A). This growth rate was not significantly different from that in the control group (P > 0.05). The other experimental groups exhibited more pronounced cell loss and no proliferation. The cell count in these groups in the lag phase was markedly decreased, and no exponential growth phase was observed. The combination of BSO or Fe and Aβ decreased the cell count by 82% and 84%, respectively (P < 0.05). The combination of all compounds (Aβ, BSO, Fe2+) in one plate (the FAB model) had in much more detrimental effects. The cell number in the FAB group was only 6.5% of that in the control group (P < 0.05).
Immunocytochemistry
In the current study, we did not purify neuronal cells but analyzed the effect of FAB on mixed hippocampal cells in culture. Therefore, in the first immunocytochemistry study, we mainly focused on hippocampal astroglia. To this end, we stained cells with an anti-GFAP antibody and DAPI. Immunocytochemistry was performed on the 14th day of cultivation in the exponential growth phase. We calculated the GFAP+/(DAPI+-GFAP+) ratio, which was 1.67 ± 0.34 in the control group (Fig. 2A, Q). This ratio was markedly increased in the cells treated with Aβ (Fig. 2B, F, Q); the GFAP+/(DAPI+-GFAP+) ratio was 4.57 ± 0.68 in the Aβ group and 2.50 ± 0.73 in the BSO + Aβ group (P < 0.05). In these groups, the cells retained the ability to form axons and dendrites. The number of GFAP+ cells in the Fe2+-treated groups was decreased, and the GFAP+/DAPI+-GFAP+ ratio was 0.78 ± 0.26 in the Fe group and 0.56 ± 0.22 in the BSO + Fe group (P < 0.05) (Fig. 2D, E, Q). Although the GFAP+/(DAPI+-GFAP+) ratio was normal in the BSO group (1.39 ± 0.41; P > 0.05), cells in this group exhibited diminished axon and dendrite formation (Fig. 2C, Q). GFAP+/DAPI+-GFAP+ ratio in the remaining groups was not significantly differ from the control samples (Fig. 2 G, H, Q).
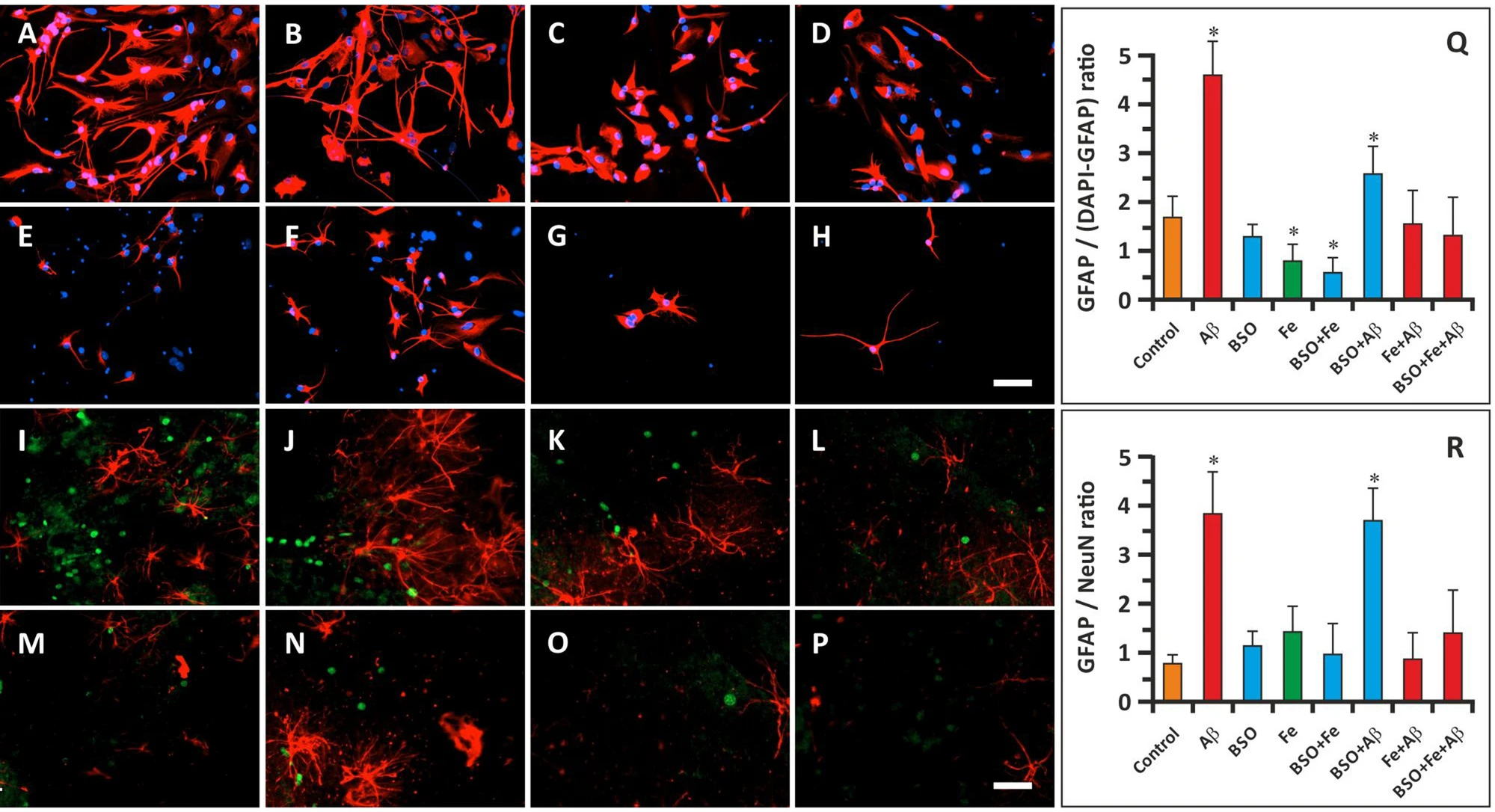
Effect of Aβ, BSO, and Fe2+ on the ratio of glial cells to neuronal cells and glial and neuronal morphology. (A–H) Immunostaining of GFAP (red) and DAPI staining (blue) in hippocampal cells on the 14th day of cultivation. Scale bar, 100 μm. (I–P) Immunostaining of GFAP (red) and NeuN staining (green) in hippocampal cells on the 14th day of cultivation. Scale bar, 200 μm. (Q) GFAP+/(DAPI+-GFAP+) cell ratio. (R) GFAP+/NeuN+ cell ratio. n = 10. * P < 0.05. The error bars indicate the mean ± SD.
In the next immunocytochemistry study, hippocampal cells were stained for GFAP and NeuN. The GFAP+/NeuN+ ratio in control samples was 0.69 ± 0.17 on the 14th day of cultivation (Fig. 2I, R). Similar to the data reported above, the GFAP/NeuN ratio was significantly increased in Aβ and BSO + Aβ samples (3.83 ± 0.87 and 3.75 ± 0.76, respectively) (P < 0.05) (Fig. 2J, N, R). The GFAP/NeuN ratio in the other groups did not differ significantly from that in the control group (Fig. 2K–M, O–R).
Analysis of neuronal metabolism and survival via determination of glucose, lactate, and LDH levels
Glucose and lactate concentrations and LDH activity in the culture medium were measured to evaluate neuronal metabolism and damage, respectively (Fig. 3). We found that in the control samples, the glucose concentration gradually decreased until the 14th day of cultivation (Fig. 3A). The glucose content in the culture medium gradually decreased from 24.91 ± 0.52 to 21.03 ± 0.37 mmol/l in the control group. During the last week of cultivation (14–21 days of culture), the glucose concentration in the culture medium did not change significantly. The change in the glucose concentration was strongly correlated with the change in the cell count presented above and reflects cell metabolism, which gradually decreased. Fe application almost completely abolished glucose utilization, likely by causing oxidative stress and cell death (Fig. 3A). In the Aβ-treated groups (the Aβ, Fe + Aβ, and BSO + Fe + Aβ groups), intermediate toxicity was observed. Glucose utilization was most markedly affected in the BSO + Fe + Aβ group but less affected in Aβ group (Fig. 3A). The change in glucose content in the culture medium was similar between the Aβ group and the control group. Interestingly, wave-like fluctuations in glucose concentrations were observed in the BSO-treated groups (the BSO, BSO + Fe, and BSO + Aβ groups). Generally, the change in glucose concentrations in these groups was similar to that in the Aβ-treated groups, but there were two periods in which glucose utilization was abolished, i.e., 3–7 day of culture and 14–21 days of culture (Fig. 3A).
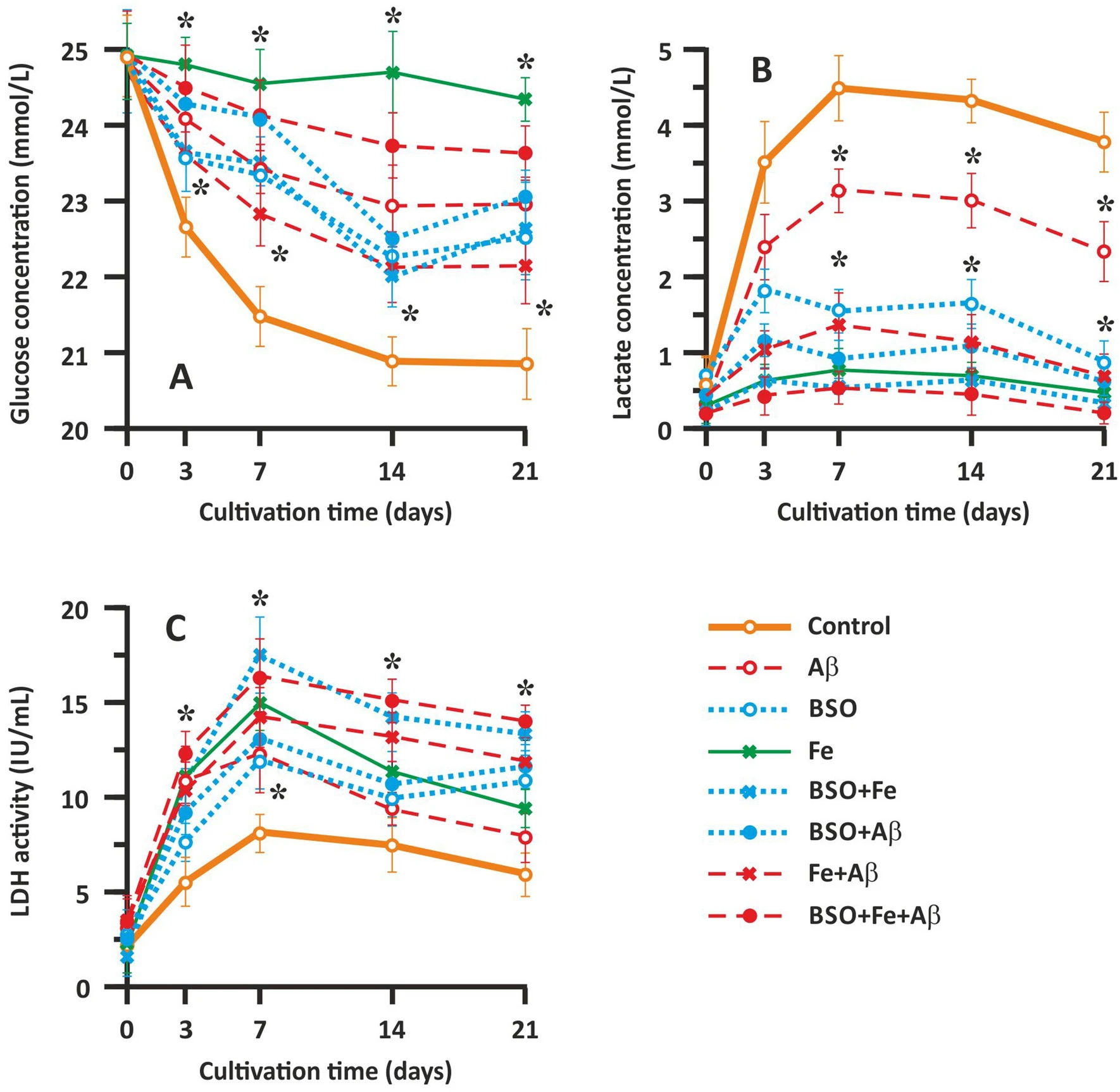
Effect of Aβ, BSO, and Fe2+ on hippocampal cell metabolism and survival. (A) Change in the glucose concentration in the culture medium. (B) Change in the lactate concentration in the culture medium. (C) Change in LDH activity in the culture medium. Biochemical parameters reflecting cellular metabolism (glucose and lactate levels) and cell damage (LDH activity) in the extracellular medium were measured using Roche Cobas C311 and Cobas E411 automatic biochemical analyzers (Roche Cobas, Switzerland) and appropriate test kits. n = 10. * P < 0.05. The error bars indicate the mean ± SD.
The change in lactate accumulation in the neuronal cell medium was opposite of the change in glucose concentrations, which is expected and normal for metabolically active cells. In the control group, we observed lactate accumulation until the 7th day of culture, when the analyte concentration reached 4.51 ± 0.49 mmol/l (Fig. 3B). The lactate concentration in the control group gradually decreased after the 7th day and until the end of cultivation and finally reached a value of 3.84 ± 0.42 mmol/l. The change in lactate accumulation in the Aβ-treated groups (the Aβ, Fe + Aβ, and BSO + Fe + Aβ groups) resembled the change in glucose concentrations. The curves representing the change in the lactate concentration in these groups showed the same shape as the curve of the control group, but the concentrations were lower. Cells treated with Aβ alone showed less marked changes, with lactate concentrations reaching the maximal value (3.12 ± 0.27 mmol/l) on the 7th day of cultivation and gradually decreasing to 2.44 ± 0.35 mmol/l (Fig. 3B). In the BSO-treated groups (the BSO, BSO + Fe, and BSO + Aβ groups), the lactate concentration reached the maximal value on the 3rd day of culture and then exhibited wave-like fluctuations until the 21st day of culture (Fig. 3B).
Intracellular LDH release indicates a change in cell membrane permeability and reflects the extent of damage. In our experiments, a gradual increase in LDH activity was observed in the control group until the 7th day of cultivation, with LDH activity peaking at 7.86 ± 0.48 IU/ml. Afterward, LDH activity gradually decreased, reaching a value of 6.03 ± 0.76 IU/ml at the end of cultivation (Fig. 3C). Overall, the change in LDH activity resembled the change in lactate levels and reflected cellular metabolism. The change in LDH activity in the Aβ-treated groups (the Aβ, Fe + Aβ, and BSO + Fe + Aβ groups) resembled the change in LDH activity in the control group, but LDH activity was much higher in the control group than in the other groups. The greatest degree of cellular damage was observed in the BSO + Fe + Aβ group, with LDH activity reaching 16.07 ± 1.93 IU/mL (P < 0.05) on the 7th day of cultivation (Fig. 3C). In the BSO-treated groups (the BSO, BSO + Fe, and BSO + Aβ groups), LDH activity was decreased on the 14th day of cultivation, which is consistent with the results of previous metabolic tests.
Oxidative modification of cellular proteins and lipids
To address the prooxidative capacity of Aβ and FAB, we measured several relevant parameters. On the 3rd day of culture, we measured protein carbonylation, lipid peroxidation (TBARS), and AOPP levels in the culture medium. The findings showed that Aβ significantly enhanced protein oxidative modification (Fig. 4A, C). In the Aβ group, the protein carbonylation level was 6.43 ± 0.94 nmol/ml and the AOPP concentration was 7.40 ± 0.89 nmol/ml, whereas in the control group, the protein carbonylation level and AOPP level were 4.02 ± 0.71 nmol/ml and 4.80 ± 1.07 nmol/ml, respectively (P < 0.05 versus the Aβ group). Further addition of BSO and BSO + Fe significantly increased in oxidative damage to cellular proteins. The protein carbonylation level was 8.49 ± 1.21 nmol/ml and 9.98 ± 1.54 nmol/ml in the BSO + Aβ and BSO + Fe + Aβ groups, respectively (P < 0.05). The AOPP level was also significantly increased. In the BSO + Aβ and BSO + Fe + Aβ groups, the AOPP level was 10.00 ± 1.38 nmol/ml and 9.98 ± 0.79 nmol/ml, respectively (P < 0.05 versus the Aβ group). Interestingly, Fe + Aβ resulted in less protein oxidation than Fe alone (Fig. 4A, C). BSO induced less protein oxidation in hippocampal neurons than Aβ. Additionally, Fe and Aβ enhanced the prooxidant effects of BSO. Similar to Aβ + BSO, Fe + BSO resulted in less oxidation than Fe alone (Fig. 4A, C).
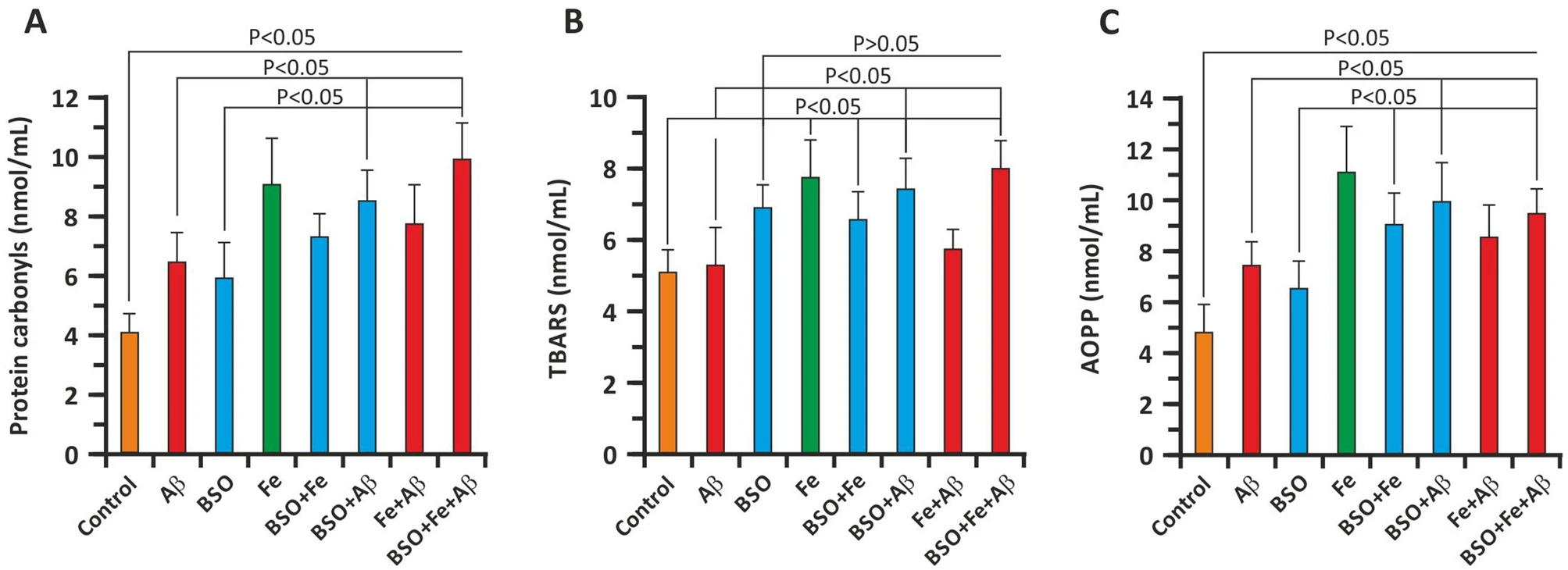
Effect of Aβ, BSO, and Fe2+ on indicators of oxidative damage in hippocampal cells. (A) Carbonylation of cellular proteins. (B) Lipid oxidation product (TBARS) levels. (C) AOPP levels. Oxidative parameters were measured in neuronal cell lysates on the 3rd day of culture. n = 10. The error bars indicate the mean ± SD.
As stated above, the degree of lipid peroxidation was evaluated by measuring TBARS content in neuronal cell lysates. The TBARS content in control cell lysates was 5.10 ± 0.67 nmol/ml (Fig. 4B). Compared to protein oxidation (carbonylation and AOPP formation), Aβ did not significantly alter TBARS levels (5.33 ± 1.12 nmol/ml; P > 0.05). The same was not true for BSO, which significantly enhanced lipid peroxidation in neuronal cell lysates (6.97 ± 0.89 nmol/ml; P > 0.05). Aβ + BSO and BSO + Fe, but not Fe alone, significantly increased TBARS levels (Fig. 4B). Here, we also observed an unexpected decrease in lipid peroxidation in the BSO + Fe and Aβ + Fe groups compared with the Fe group (Fig. 4B).
Cellular bioenergetics analysis
To determine how Aβ, BSO, Fe, and their combination (FAB) affect cellular bioenergetics, we determined the OCR and ECAR by extracellular flux analysis. Cellular bioenergetics indicators were measured on the 3rd day of culture. The basal and ATP generation-coupled OCR and maximal respiration were significantly affected by Aβ and Fe (P < 0.05). However, the effect of BSO on OCR was negligible (Fig. 5A). BSO did not have a significant effect on the basal and ATP generation-coupled OCR (P > 0.05). Nevertheless, maximal respiration in the BSO group was almost 30% less (P < 0.05) than that in the control group. The Fe-treated groups exhibited the lowest basal OCR (Fig. 5C).
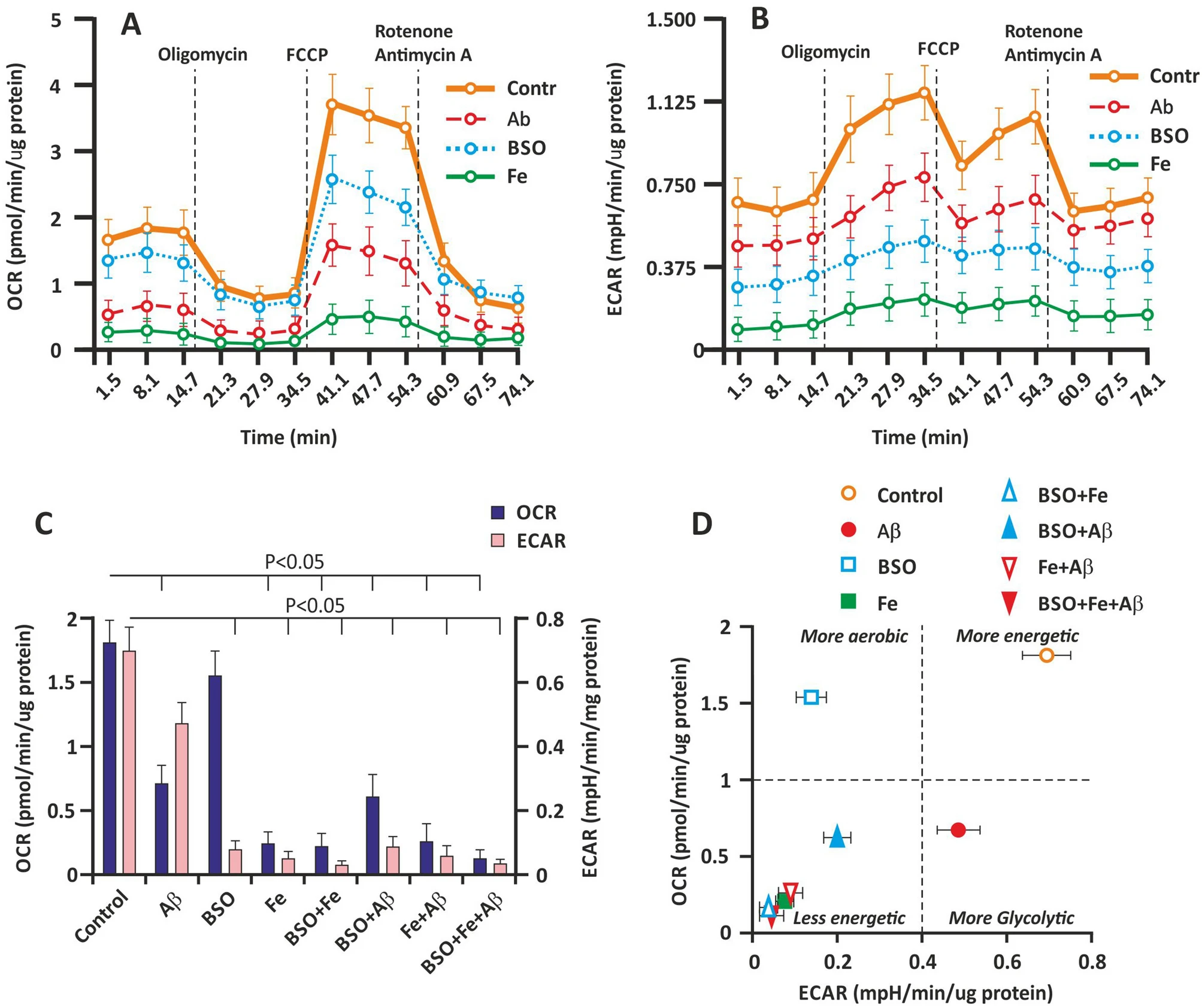
Effect of Aβ, BSO, and Fe2+ on bioenergetics parameters in hippocampal cells. (A) The OCR of cultured hippocampal cells on the 3rd day of culture. (B) The ECAR of cultured hippocampal cells on the 3rd day of culture. (C) Basal OCR and ECAR. (D) Metabolic profiles of hippocampal cells. Analyses were conducted using Wave software and XF Report Generators (Agilent Technologies).
The ECAR showed the opposite change. Using extracellular flux analysis, we found that BSO and Fe markedly decreased the ECAR; however, the effect of Aβ on the ECAR was less obvious (Fig. 5B). The BSO- and Fe-treated groups exhibited the lowest basal ECAR (Fig. 5C).
The basal ECAR and OCR were plotted, and the results are shown in Fig. 2C. The data represent the change in the metabolic profile of the cells induced by Aβ, BSO, and Fe. The different effects of Aβ and BSO on cellular bioenergetics are shown. The Aβ-treated groups showed a more glycolytic profile, and a more aerobic profile was observed in the BSO-treated groups (Fig. 5D). Aβ and BSO exerted a synergistic effect, resulting in a less energetic cellular phenotype due to a decrease in both aerobic and glycolytic flux in the cell. Fe overload more markedly inhibited cellular bioenergetics (Fig. 5D).
Discussion
In the current study, we show for the first time the effect of Aβ and the components of FAB on growth parameters, oxidative damage, and bioenergetics parameters in adult rat mixed primary hippocampal cells. First, we characterized the changes in cell number and morphology in the control group. The control group exhibited a classical growth curve: a lag phase during the first 7 days, an exponential phase (7–14 days), and a stationary phase (14–21 days of culture). Most previous studies have focused on keeping adult neurons alive for a period of time (21 days)25,29,30.
In the lag phase, hippocampal cells were round cells without outgrowths. In the exponential phase, hippocampal neurons exhibited moderate-sized apical dendrites, and early branches began to grow. In the stationary phase, despite loss of cell proliferation, extended axons and dendrites were observed. These data are in good accordance with previous reports25.
Aggregated Aβ (Aβ1-42) had a marked effect on the changes in neuronal cell growth in the exponential phase. Aβ decreased the number of cells (by 51%) on the 7th day of culture. After this, cells in the Aβ group retained moderate proliferative capability. The other experimental groups exhibited more pronounced cell loss and no proliferation. The cell number in the lag phase was robustly decreased in these groups, and no exponential phase was observed. Aβ + BSO and Aβ + Fe decreased the cell number by 82% and 84%, respectively. The BSO + Fe + Aβ group (FAB model) showed maximum inhibitory/cytotoxic activity and resulted in much more detrimental/cytotoxic consequences.
The effect of Aβ on cultured hippocampal neurons was shown previously and can be explained by Aβ-mediated neuronal cell death resulting from the loss of full-length Tau and/or the generation of toxic fragments31. The effect of chronic intracerebroventricular delivery of Aβ in combination with the prooxidant Fe2+ or the glutathione synthesis inhibitor BSO on brain histopathology and memory loss in adult rats was shown previously32. The destructive effect of Aβ in combination with oxidative stress induced by BSO and Fe most appropriately reflects AD-like symptomatology. However, there have been no studies on the effect of FAB in cultured neurons.
Using immunocytochemistry (with anti-GFAP and anti-NeuN antibodies), we assessed neuronal cell loss in the Aβ-treated groups and membrane damage/loss in the BSO- and Fe-treated groups. FAB caused total cell death. Our results were in good accordance with previous in vivo experiments33,34,35,36.
We also showed the detrimental effect of Aβ and other FAB components on metabolism. It was found that glucose consumption and lactate production were inhibited. Interestingly, unique changes in the levels of glucose and lactate were observed in the BSO-treated groups. The maximal decrease in the levels of these metabolites was observed on the 7th day of culture, after which the changes were relatively similar to those in the control group. The detrimental effect of BSO can be explained by its ability to deplete glutathione, which causes apoptotic cell death37. This phenomenon was not observed in the Aβ-treated groups and might have been due to the purification of BSO from the medium after replacement of irreversibly inhibited enzymes with new enzymes. This was not true for the Aβ-treated samples, probably because Aβ binds to cellular membranes, as presented earlier,this binding allows a constant Aβ concentration to maintained at all cultivation time points despite medium replacement38.
We analyzed the prooxidant activity of the studied analytes and found an increase in lipid peroxidation in the BSO-treated groups (high TBARS level) and oxidative damage to proteins in the Aβ-treated groups (high levels of protein carbonylation and AOPP). These findings support in the immunocytochemistry results, indicating that the membrane was likely damaged or lost in the BSO-treated groups, and are consistent with the literature39.
Finally, the difference in the effects of Aβ and BSO were assessed by extracellular flux analysis. We observed that Aβ inhibited of mitochondrial oxidative phosphorylation and that BSO inhibited glycolysis. The combination of these resulted in maximal alterations in cellular bioenergetics.
Our findings clearly show that aggregated Aβ1-42 alone is significantly less toxic to hippocampal cells. Aggregated Aβ damages neurons, and glial cells proliferate to remove Aβ from the hippocampus. External prooxidant agents (Fe2+) or inhibition of the internal antioxidant defense by BSO is more toxic to hippocampal cells than aggregated Aβ alone. Hippocampal cells fight against Aβ-induced damage more effectively than against oxidative damage. However, the combination of Aβ, external oxidative damage and inhibition of internal antioxidant defense is even more toxic, impairs cellular defense systems, and may represent the late phase of AD-associated cell damage.
Immunohistochemical analysis clearly indicated that Aβ targeted neuronal cells (NeuN positive). Glial cells proliferate and can remove toxic Aβ from the cellular environment; This explains why we observed and increase in the number of GFAP+ relative to NeuN+ cells. Aβ exerted prooxidant activity, as evidenced by increased protein carbonylation and AOPP levels. It is likely that Aβ-dependent oxidative damage might have been due to inhibition of mitochondrial oxidative phosphorylation by Aβ, which was observed in our experiments using extracellular flux analysis.
It seems that Aβ toxicity mechanism is based on ROS accumulation. All around ROS and metals with transparent valency. First of all, the whole Aβ molecule has some metal-binding sites in its first 15 amino acids constituted by the histidines 6, 13, and 14 and the tyrosine in the 10 position. These all have well-known and powerful metal-binding sites, and a nearby affinity to the best metallic chelants currently known40. The Aβ possesses the ability to reduce Cu2+ and Fe3+ towards Cu+ and Fe2+, respectively. This way, the molecular oxygen can react with reduced metals thus generating superoxide anion, which in turn combines with two hydrogen atoms to form hydrogen peroxide that may later react with another reduced metallic ion and then forming the hydroxyl radical by Fenton reaction. The Aβ (in its radical form) can extract protons from the neighboring lipids or proteins, thus generating lipid peroxides and carbonyls, respectively41. Above it, using data achieved by extracellular flux analysis, we clearly demonstrated Aβ induced mitochondrial dysfunction. The later can be considered as a next cause of ROS overproduction in neurons. Growing evidence suggests that Aβ has deleterious effects on mitochondrial function and contributes to energy failure, neuronal apoptosis and production of ROS in AD brain42. However, in all probability, well-functioning antioxidant defense system of the organism is able to compensate prooxidant consequences of Aβ alone. Thus, as a real and powerful cause of Alzheimer’s disease-like neurodegeneration, we consider simultaneous damage of pro/antioxidant homeostasis with the accumulation of Aβ peptides in neuronal cell.
Conclusion
In conclusion, the results of this study show that the combination of aggregated Aβ, an antioxidant-depleting agent (BSO) and a prooxidant (Fe2+) (FAB) more completely recapitulates neuronal cell damage in vitro. This strongly indicates a critical role for the combination effect of Aβ and oxidative stress in the development of neurodegeneration.