
The sigma-1 receptor curtails endogenous opioid analgesia during sensitization of TRPV1 nociceptors
By M. Carmen Ruiz-Cantero, Elsa Cortés-Montero, Aakanksha Jain, Ángeles Montilla-García, Inmaculada Bravo-Caparrós, Jaehoon Shim, Pilar Sánchez-Blázquez, Clifford J. Woolf, José M. Baeyens, and Enrique J. Cobos
Excerpt from the article published in British Journal of Pharmacology, 1– 20, 07 December 2022, DOI: https://doi.org/10.1111/bph.16003
Editor’s Highlights
- Sigma-1 receptors limit peripheral opioid analgesia during sensitization of peptidergic C nociceptors.
- Sigma-1 antagonists are able to harness neuronally derived endogenous opioids to reduce hyperalgesia at the pain site, by promoting TRPV1 desensitization and increasing μ receptor activity.
- The modulation of endogenous opioid analgesia by sigma-1 receptors might have potential clinical application for pain treatment.
Abstract
Background and Purpose: Peripheral sensitization contributes to pathological pain. While prostaglandin E2 (PGE2) and nerve growth factor (NGF) sensitize peptidergic C-nociceptors (TRPV1+), glial cell line-derived neurotrophic factor (GDNF) sensitizes non-peptidergic C-neurons (IB4+). The sigma-1 receptor (sigma-1R) is a Ca2+-sensing chaperone known to modulate opoid analgesia. This receptor binds both to TRPV1 and the μ opioid receptor, although the functional repercussions of these physical interactions in peripheral sensitization are unknown.
Experimental Approach: We tested the effects of sigma-1 antagonism on PGE2-, NGF-, and GDNF-induced mechanical and heat hyperalgesia in mice. We used immunohistochemistry to determine the presence of endomorphin-2, an endogenous μ receptor agonist, on dorsal root ganglion (DRG) neurons. Recombinant proteins were used to study the interactions between sigma-1R, μ- receptor, and TRPV1. We used calcium imaging to study the effects of sigma-1 antagonism on PGE2-induced sensitization of TRPV1+ nociceptors.
Key Results: Sigma1 antagonists reversed PGE2- and NGF-induced hyperalgesia but not GDNF-induced hyperalgesia. Endomorphin-2 was detected on TRPV1+ but not on IB4+ neurons. Peripheral opioid receptor antagonism by naloxone methiodide or administration of an anti-endomorphin-2 antibody to a sensitized paw reversed the antihyperalgesia induced by sigma-1 antagonists. Sigma-1 antagonism transfers sigma-1R from TRPV1 to μ receptors, suggesting that sigma-1R participate in TRPV1-μ receptor crosstalk. Moreover, sigma-1 antagonism reversed, in a naloxone-sensitive manner, PGE2-induced sensitization of DRG neurons to the calcium flux elicited by capsaicin, the prototypic TRPV1 agonist.
Conclusion and Implications: Sigma-1 antagonism harnesses endogenous opioids produced by TRPV1+ neurons to reduce hyperalgesia by increasing μ receptor activity.
What is already known
- Sigma-1 antagonists enhance analgesia induced by morphine and other opioid drugs.
What does this study add
- Sigma-1 antagonism harnesses the analgesic potential of opioid peptides released from sensitized TRPV1+ sensory neurons.
What is the clinical significance
- Sigma-1 antagonism induces opioid analgesia exclusively at the painful site without administration of opioid drugs.
1 INTRODUCTION
The sigma-1 receptor (sigma-1R) is a ligand-operated chaperone that, in response to the increase in the intracellular Ca2+ concentration, physically interacts with several different receptors and channels (Su et al., 2016). N-methyl-D-aspartate receptors (NMDAR) are major protein targets of sigma-1R (Sánchez-Fernández et al., 2017) and sigma-1 antagonism decreases NMDAR activity in the spinal cord to inhibit central sensitization and ameliorate neuropathic pain (Zamanillo et al., 2013). Neuropathic pain is an intended primary indication of the selective sigma-1R antagonist S1RA, which has been successfully tested in a phase IIa clinical trial on chemotherapy-induced neuropathic pain (Bruna et al., 2018), after phase I studies which demonstrated its safety and tolerability in healthy people (Abadias et al., 2013).
The secondary intended indication of S1RA is the enhancement of opioid analgesia (Vela et al., 2015), as there is overwhelming preclinical evidence for the increase of the antinociceptive effect induced by several clinically relevant opioid drugs (such as morphine) by sigma-1 antagonism (Sánchez-Fernández et al., 2017). Sigma-1R participates in the crosstalk between the μ-opioid receptor and NMDAR to modulate opioid analgesia at the CNS. Ca2+-activated calmodulin (CaM) is a negative regulator of NMDAR and sigma-1R competes with CaM for the binding to NMDAR; therefore, the binding of the sigma-1R to NMDAR reduces CaM-induced NMDAR inhibition, which decreases μ receptor actions. In the presence of a sigma-1 antagonist, sigma-1Rs dissociate from the NMDAR and transfer to the μ receptor, allowing CaM to bind NMDAR, and enhancing μ receptor activity (Rodríguez-Muñoz, Cortés-Montero, et al., 2015; Rodríguez-Muñoz, Sánchez-Blázquez, et al., 2015). Modulation of μ receptor-mediated analgesia by the sigma-1R has been classically attributed to actions in the CNS (Mei & Pasternak, 2007); however, we more recently reported that sigma-1 antagonism also enhances peripheral antinociception induced by opioid drugs, including not only opioid analgesics such as morphine and fentanyl, but also the peripheral μ receptor agonist loperamide, used clinically as an antidiarrheal drug (Sánchez-Fernández et al., 2013 and 2014). In fact, sigma-1 antagonism is even able to induce peripheral antihyperalgesic effects by the potentiation of endogenous opioid peptides derived from immune cells in peripheral inflamed tissue (Tejada et al., 2017).
Transient receptor potential vanilloid-1 (TRPV1) is another more recently identified protein target of sigma-1Rs (Cortés-Montero et al., 2019; Ortíz-Rentería et al., 2018). In adult mice, TRPV1 receptors are mostly concentrated in peptidergic C-nociceptors, which can express neuropeptides such as calcitonin gene-related peptide (CGRP) or substance P (Priestley, 2009; Renthal et al., 2020), and play an important role in peripheral sensitization in response to algogenic ligands such as prostaglandin E2 (PGE2) (Moriyama et al., 2005) and nerve growth factor (NGF) (Zhang et al., 2005). Non-peptidergic C-nociceptors do not express TRPV1 but can be labelled with Isolectin B4 (IB4) and sensitized by other factors, such as glial cell line-derived neurotrophic factor (GDNF) (Álvarez et al., 2012). These peripheral sensitizers are produced in a variety of pathological pain states and play a pivotal role in pain generation (Ji et al., 2016; Kotliarova & Sidorova, 2021). We and others have recently reported that sigma-1Rs are present in every DRG neuron (Bravo-Caparrós et al., 2020; Montilla-García et al., 2018; Shin et al., 2020), although the role of these receptors in the periphery has not been well studied. We aimed here to test whether sigma-1 antagonists ameliorate the hyperalgesia induced by peripheral sensitizers. As we found that the antihyperalgesic effect of sigma-1 antagonists on sensitizers of TRPV1+ nociceptors involves activation of the endogenous opioid system, we sought to determine the endogenous opioid peptide involved and its cellular source, and also tested whether there might be crosstalk between μ receptors and TRPV1 with the participation of sigma-1Rs, similar to the one described for the NMDAR in the CNS.
…
3 RESULTS
3.1 Involvement of TRPV1+ nociceptors in the hyperalgesia induced by PGE2, NGF and GDNF
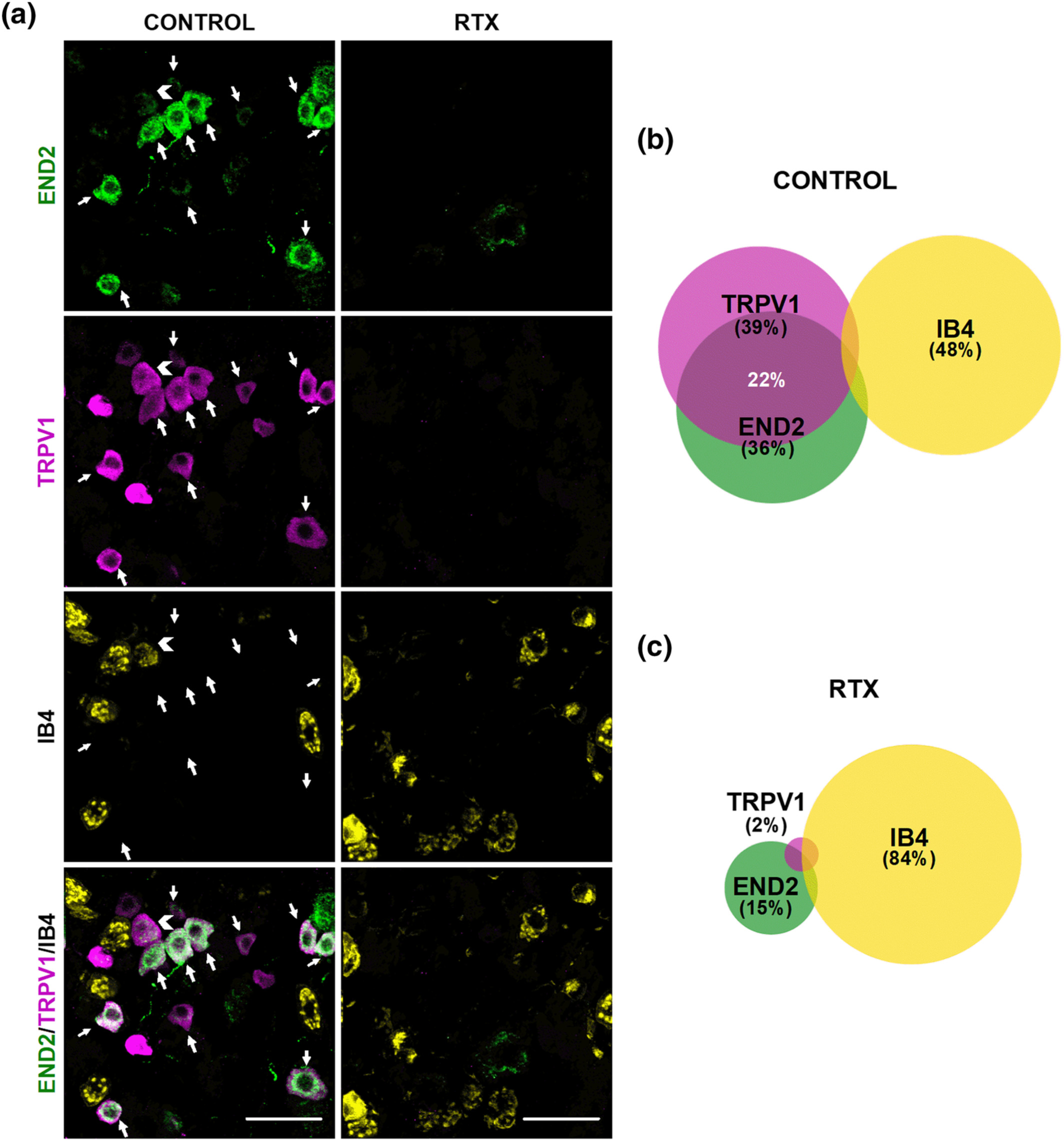
We first studied the distribution of several neuronal markers in the DRG from intact female mice. Specifically, we stained for sigma-1R, CGRP, TRPV1, and IB4. The sigma-1R stained numerous cells with neuronal morphology (Figure 1a); in fact, the double labelling of sigma-1Rs with the pan-neuronal marker PGP9.5 showed that sigma-1Rs were present in most, if not all, DRG neurons (PGP9.5+ cells) (Figure S2). CGRP+ cells accounted for 28% of sigma-1R+-neurons (Figure 1a,b). Double labelling of CGRP and TRPV1 showed that both neuronal populations markedly overlap, as most CGRP+ neurons express TRPV1, and most TRPV1+ neurons express CGRP (Figure 1c,d). On the other hand, staining for TRPV1 and IB4 showed minimal overlap among DRG neurons, and each population constituted about one-third of sigma-1R+ cells (see top panels of Figure 2a for representative images, and Figure 2b). Treatment with RTX virtually abolished TRPV1 labelling, but IB4 staining was still readily detectable (Figure 2a, bottom panels). In fact, the proportion of IB4+ neurons in the remaining sigma-1R+ cells was even increased, since when TRPV1+ population is ablated, the remaining IB4 neurons represent a higher percentage considering the number of surviving neurons as the 100% (compare Figure 2b,c). These results confirm the specificity of the ablation of TRPV1+ neurons by RTX.
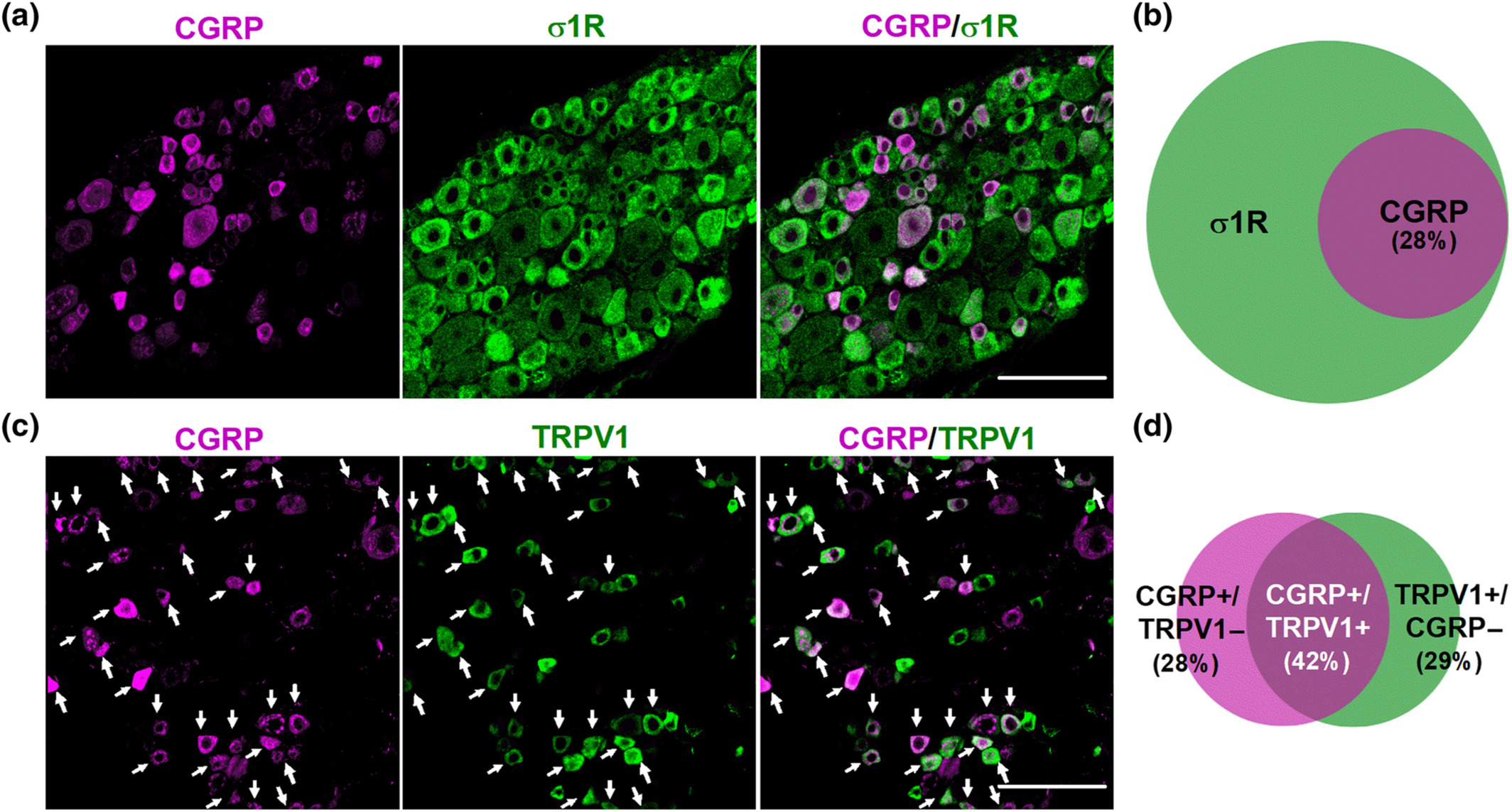
Double labelling of CGRP in combination with the sigma-1 receptor (σ1R) and TRPV1. Immunostaining was performed in the L4 dorsal root ganglion (DRG) from female mice. (a) Representative images from the double labelling of CGPR (magenta) and σ1R (green). Scale bar 50 μm. (b) Venn diagram showing the overlap between CGRP+ and σ1R+ neurons. (c) Representative images from the double labelling of CGPR (magenta) and TRPV1 (green). White arrows indicate co-localization of CGRP and TRPV1 markers. Scale bar 50 μm. (d) Venn diagram displaying the percentage of CGRP+, TRPV1+ and CGRP+/TRPV1+ neurons among the total number of neurons labelled with any of these markers. Samples from five mice were used to construct the Venn diagrams.
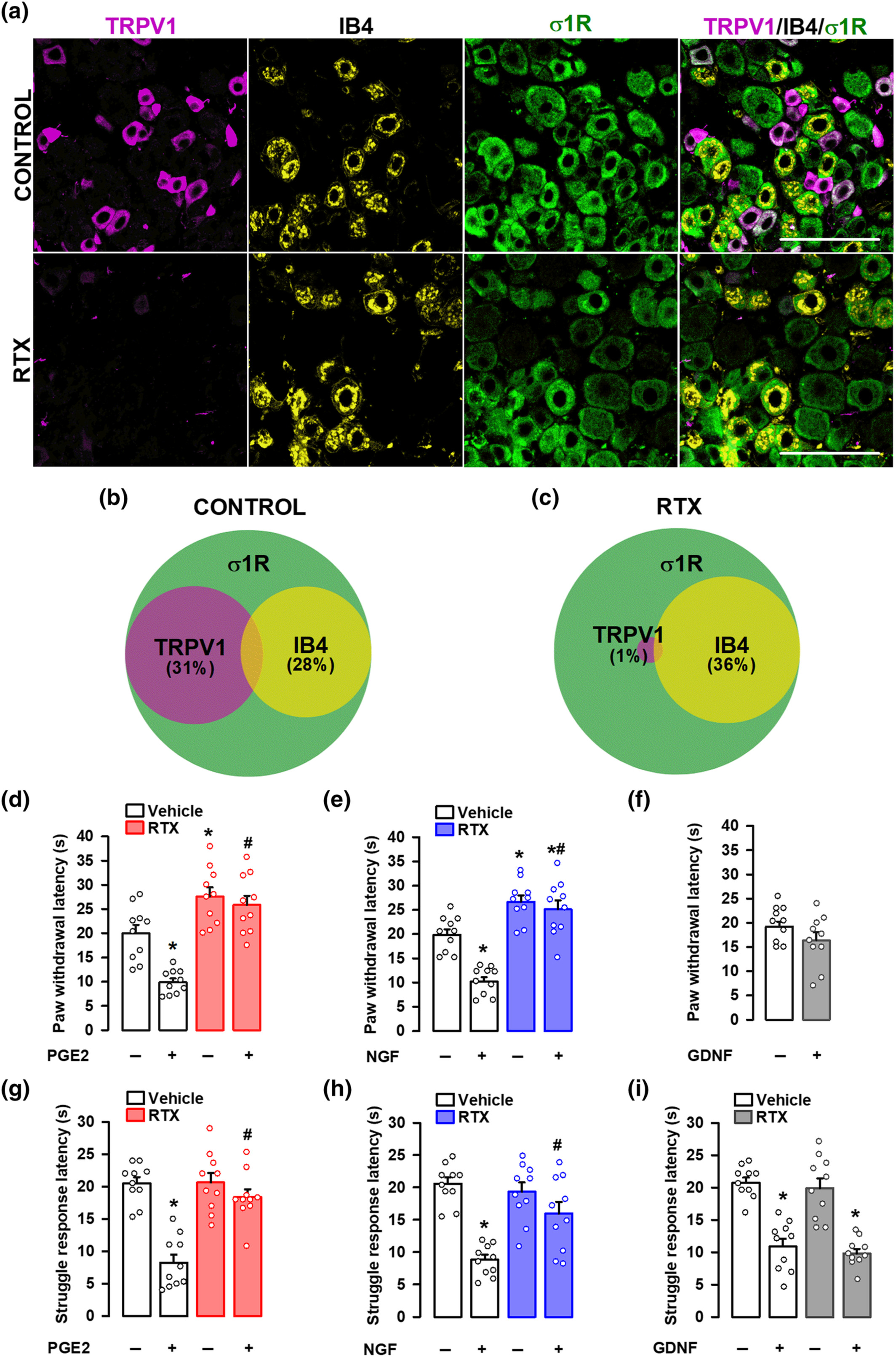
Effect of the ablation of TRPV1-expressing neurons on mechanical hyperalgesia induced by PGE2, NGF and GDNF. Female mice were treated intraperitoneally (i.p.) with resiniferatoxin (RTX, 25 μg kg−1 for two consecutive days) or its vehicle 5 days before obtaining samples or performing the behavioural experiments. (a) Triple labelling of TRPV1 (magenta), isolectin B4 (IB4, yellow) and sigma-1 receptor (σ1R, green) in the L4 dorsal root ganglion (DRG). Top panels: samples from vehicle-treated mice (control). Bottom panels: samples from mice treated with resiniferatoxin (RTX). Scale bar, 100 μm. (b,c) Venn diagrams showing the overlap between TRPV1+, IB4+ and σ1R+ neurons in samples from control mice (b) and from mice treated with RTX (c). Samples from five mice per group were used to construct the Venn diagrams. (d–i) the behavioural results represent the latency to paw withdrawal evoked by a heat stimulus of 42 ± 1°C (d–f), and the latency to struggle response evoked by a mechanical stimulus of 100 g (g–i) in mice treated intraplantarly (i.pl.) with PGE2 (0.5 nmol) (d,g), NGF (1 μg) (e,h), GDNF (40 ng) (f,i), or their solvents. Values are the mean ± SEM (10 animals per group). Statistically significant differences between the values obtained in control non-sensitized animals and mice treated with the peripheral sensitizer, *P < 0.05; and between the values obtained in animals sensitized with PGE2 or NGF, and administered with RTX or its vehicle, #P < 0.05 (one-way ANOVA followed by Bonferroni test). Data shown in H and I were log-transformed to meet the ANOVA assumptions.
We next aimed to study the effects of the in vivo ablation of TRPV1-expressing neurons by RTX on behavioural responses to sensory stimulation after the administration of several peripheral sensitizers, as well as in non-sensitized animals. PGE2 and NGF induced a marked decrease in paw withdrawal latency to a contact heat stimulus (42 ± 1°C) in comparison to saline-injected mice, denoting development of heat hyperalgesia (Figure 2d,e). RTX increased the response latency in non-sensitized animals as well as in mice sensitized with PGE2 or NGF (Figure 2d,e). We also tested the effect of GDNF on heat sensitivity. In our experimental conditions, this neurotrophin (in contradistinction to the other peripheral sensitizers tested) was unable to induce heat hyperalgesia (Figure 2f).
PGE2 and NGF also induced mechanical hyperalgesia, decreasing the struggle latency to paw pressure in comparison to saline-injected mice (Figure 2g,h). RTX treatment did not affect the responses to mechanical stimulation in non-sensitized animals but abolished PGE2- and NGF-induced mechanical hypersensitivity (Figure 2g,h). Therefore, while TRPV1-expressing neurons are dispensable for mechanical nociceptive pain, they are essential for the mechanical hyperalgesia induced by these two chemical algogens. GDNF also induced significant mechanical hypersensitivity, which remained in spite of the ablation of TRPV1+ neurons by RTX treatment (Figure 2i). Therefore, although mechanical hypersensitivity can be triggered by PGE2, NGF and GDNF, only that induced by PGE2 and NGF is dependent on TRPV1+ nociceptors.
3.2 The antihyperalgesic effects of sigma-1 antagonism and the peripheral opioid system
As all three peripheral sensitizers induced mechanical hyperalgesia, we first tested the effects of sigma-1 antagonists on this sensory modality, in female animals. The systemic (subcutaneous, s.c.) administration of the sigma-1 antagonists S1RA and BD-1063 did not modify the struggle latency to mechanical stimulation in non-sensitized animals (Figure S3), but induced a dose-dependent increase in the response latency in PGE2- or NGF-treated mice, reaching values similar to control animals (i.e., a full antihyperalgesic effect) at the highest dose tested (Figure 3a,b). However, neither S1RA nor BD-1063, at doses that fully reversed hyperalgesia induced by PGE2 or NGF, were able to induce any effect on GDNF-induced mechanical hyperalgesia (Figure 3c). The s.c. administration of the sigma-1 agonist PRE-084 did not alter the struggle response to mechanical stimulation in non-sensitized animals (Figure S3), but reversed the effect of both S1RA and BD-1063 on PGE2-induced hyperalgesia (Figure 3d,e, respectively). These results support the selectivity of the effects induced by the sigma-1 antagonists on the receptor. Mirroring the results in female mice, S1RA induced a full antihyperalgesic effect to the mechanical stimulus in male mice sensitized with PGE2, and this effect was reversed by PRE-084 (Figure S4A). In addition, S1RA was unable to induce any effect on GDNF-induced mechanical hyperalgesia in male mice (Figure S4B). These results suggest that the overall effect of sigma-1 antagonism on peripheral sensitization is preserved in both sexes in the mouse.
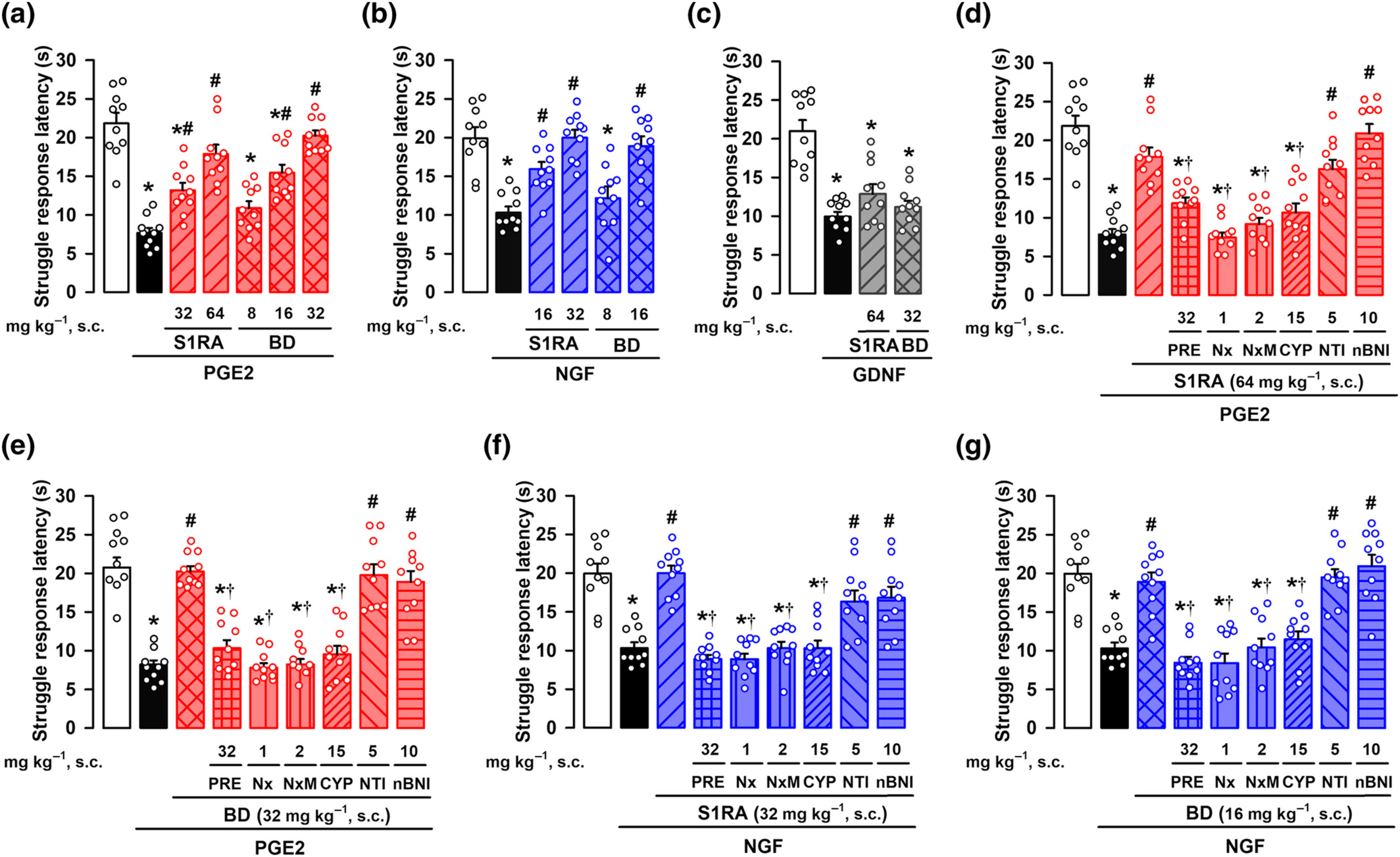
Effects of the systemic administration of the sigma-1 antagonists S1RA and BD-1063 (BD) on mechanical hyperalgesia induced by PGE2, NGF, or GDNF. The results represent the latency to struggle response evoked by a mechanical stimulus of 100 g in female mice administered subcutaneously (s.c.) with S1RA, BD or saline, and intraplantarly (i.pl.) with (a) PGE2 (0.5 nmol), (b) NGF (1 μg), (c) GDNF (40 ng), or their solvents. Animals treated with S1RA or BD or their solvent also received s.c. administration of PRE-084 (PRE), naloxone (Nx), naloxone methiodide (NxM), cyprodime (CYP), naltrindole (NTI), nor-binaltorphimine (nBNI), or saline and were tested on mechanical hyperalgesia induced by PGE2 (d,e) and NGF (f,g). Values are the mean ± SEM (10 animals per group). Statistically significant differences between the values obtained in control non-sensitized animals (white bars) and the other experimental groups, *P < 0.05; between the values obtained in PGE2-, NGF-, or GDNF-treated animals administered with S1RA, BD or their solvent, #P < 0.05; and between PGE2- or NGF-treated animals administered with S1RA or BD alone or their association with PRE, Nx, NxM, or CYP, †P < 0.05 (one-way ANOVA followed by Bonferroni test). Data shown in (c) and (e) were log-transformed to meet the ANOVA assumptions.
The ameliorative effects induced by S1RA or BD-1063 on PGE2-induced hyperalgesia in female mice were also reversed not only by the opioid antagonist naloxone but also by its peripherally restricted analogue naloxone methiodide (Figure 3d,e). Similarly, the effects induced by S1RA on PGE2-induced hyperalgesia in male mice were also fully reversed by naloxone methiodide (Figure S4A). These results suggest the involvement of the peripheral opioid system in the effects induced by sigma-1 antagonism in mice from both sexes. To identify which opioid receptor subtype was participating in the antihyperalgesic effects induced by S1RA and BD-1063 in female mice, we used antagonists with selectivity for the opioid receptor subtypes. The antihyperalgesic effect induced by the sigma-1 antagonists was abolished by the μ receptor antagonist cyprodime, but not the δ receptor antagonist naltrindole or the κ receptor antagonist nor-binaltorphimine (Figure 3d,e). We also tested the effects of S1RA and BD-1063 on NGF-induced hyperalgesia, with equivalent results (i.e., the effects of the sigma-1 antagonists were reversed by PRE-084, naloxone, naloxone methiodide and cyprodime, but not by naltrindole or nor-binaltorphimine) (Figure 3f,g). These results suggest that the effect of systemically administered sigma-1 antagonists on PGE2- and NGF-induced hyperalgesia involves the activation of peripheral μ receptors, but not other opioid receptor subtypes. The s.c. administration of S1RA and BD-1063 also induced robust antihyperalgesic effects to a heat stimulus in mice sensitized with PGE2 and NGF, and these were reversed by both the sigma-1 agonist PRE-084 and the peripheral opioid antagonist Nx-M (Figure S5), mirroring the peripheral opioid effects induced by sigma-1 antagonism on mechanical hyperalgesia induced by these sensitizers of TRPV1+ neurons.
We also tested the effects of PRE-084 and the opioid antagonists on the antihyperalgesic effect induced by morphine in female mice, the prototypic opioid agonist. Systemic (s.c.) administration of morphine induced a dose-dependent antihyperalgesic effect in animals sensitized with PGE2 and tested with the mechanical stimulus (Figure S6A). The antihyperalgesic effect of morphine was not modified by the sigma-1 agonist PRE-084 (Figure S6B) (at the same dose that reverses the antihyperalgesic effect induced by the sigma-1 antagonists). The antihyperalgesic effect of morphine was fully reversed by the opioid antagonist naloxone and its quaternary derivative naloxone methiodide (Figure S6B), indicating that these effects were mediated peripherally. Morphine effects were also dose-dependently and fully reversed by cyprodime (at the same dose used to reverse the effect of sigma-1 antagonists), but not by naltrindole or nor-binaltorphimine (Figure S6B).
We also tested the effect of morphine on GDNF-induced hyperalgesia in female mice. This opioid also induced a dose-dependent antihyperalgesic effect (Figure S6C), which was reversed by naloxone but not by PRE-084 (Figure S6D). In contrast to the results on PGE2-induced hyperalgesia, naloxone methiodide did not modify the antihyperalgesic effect of morphine after sensitization with GDNF (Figure S6D), pointing to central actions of morphine as responsible for this antihyperalgesic effect. These results highlight the differences of opioid effects depending on the peripheral sensitizer used, which might be related to the different neuronal populations sensitized by each algogen.
In summary, both the systemic administration of sigma-1 antagonists and morphine induced antihyperalgesic effects, which are mediated by peripheral μ receptor activation, in animals sensitized with PGE2 or NGF. The opioid mediated antihyperalgesic effects of sigma-1 antagonism were absent though on GDNF-induced hypersensitivity. Moreover, morphine’s antihyperalgesic effect on GDNF-induced hyperalgesia does not depend on peripheral opioid receptors.
3.3 Sigma-1 antagonism and endogenous opioid peptides
We hypothesized that sigma-1 antagonists might induce peripheral opioid effects at the peripheral terminal sensitized site, where the animals receive sensory stimulation. The i.pl. administration of the sigma-1 antagonists S1RA or BD-1063 dose-dependently fully reversed mechanical hyperalgesia induced by either PGE2 (Figure 4a) or NGF (Figure 4b) in female mice, without altering the response latency of non-sensitized animals (Figure S7).
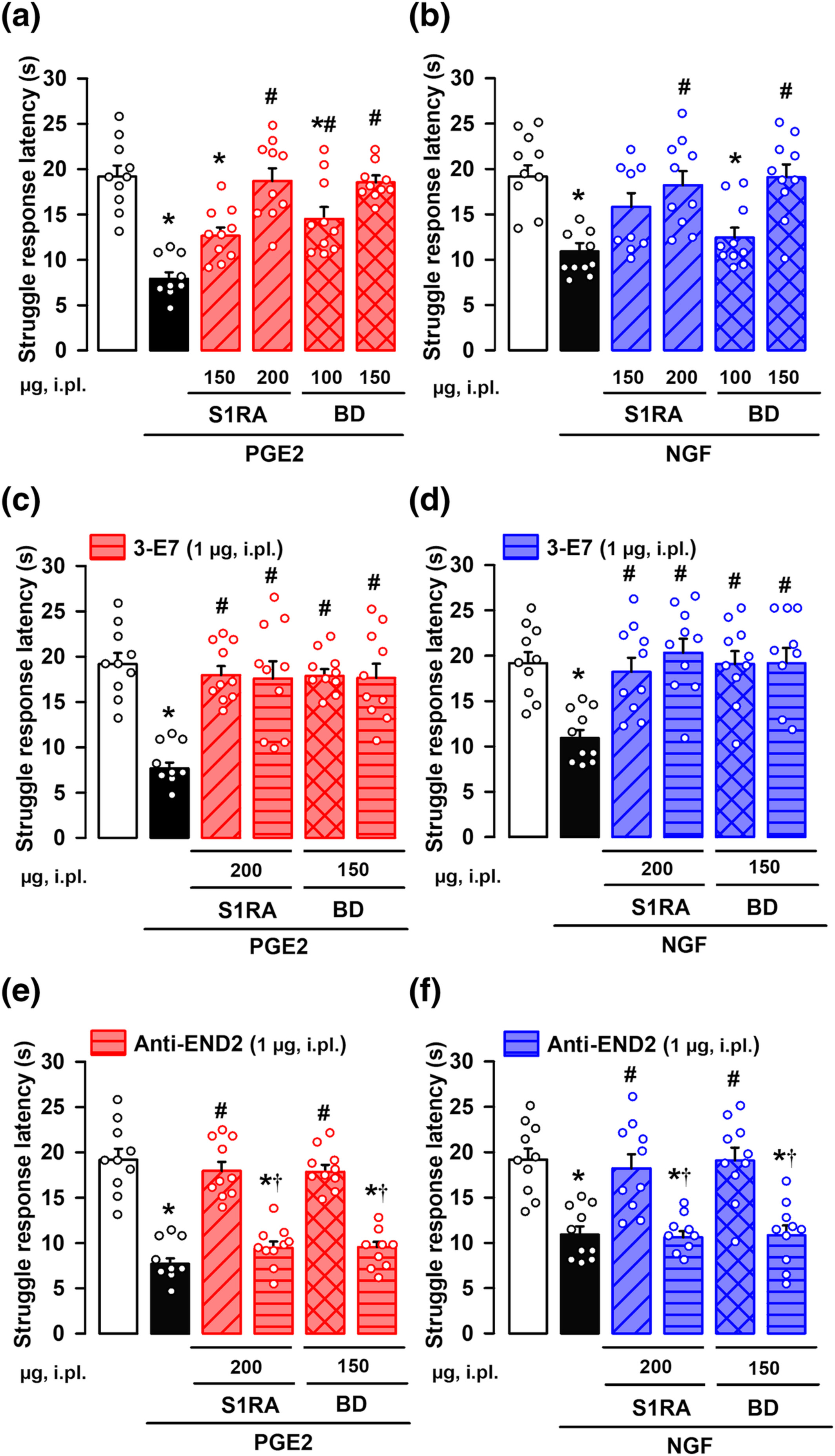
Effects of the local administration of the sigma-1 antagonists S1RA and BD-1063 (BD) on mechanical hyperalgesia induced by PGE2 or NGF. The results represent the latency to struggle response evoked by a mechanical stimulus of 100 g in female mice administered intraplantarly (i.pl.) with S1RA, BD or saline, and with (a,c,e) PGE2 (0.5 nmol), (b,d,f) NGF (1 μg) or their solvent. Animals i.pl. treated with S1RA or BD were also coadministered with antibodies for endogenous opioid peptides: 3-E7 (c,d) and anti-endomorphin-2 (anti-END2) (e,f), and tested on mechanical hyperalgesia induced by PGE2 (c,e) and NGF (d,f). Values are the mean ± SEM (10 animals per group). Statistically significant differences between the values obtained in control non-sensitized animals (white bars) and the other experimental groups, *P < 0.05; between the values obtained in PGE2- or NGF-treated animals administered with S1RA, BD or their solvent #P < 0.05; and between PGE2- or NGF-treated animals administered with S1RA or BD alone or coadministered with the anti-END2 antibody, †P < 0.05 (one-way ANOVA followed by Bonferroni test). Data shown in C were log-transformed to meet the ANOVA assumptions.
To neutralize the actions of endogenous opioid peptides at the sensitized site, we administered i.pl. a monoclonal antibody, 3-E7, which recognizes the pan-opioid sequence Tyr-Gly-Gly-Phe at the N-terminus of most opioid peptides (see Methods). The i.pl. administration of 3-E7 did not modify the antihyperalgesic effect of S1RA or BD-1063 on the mechanical hyperalgesia induced by either PGE2 (Figure 4c) or NGF (Figure 4d), suggesting that opioid peptides containing the target sequence of the 3-E7 do not mediate the effect observed. The administration of this antibody did not alter the response latency of non-sensitized animals (Figure S7).
Taking into account that the antihyperalgesic effect of sigma-1 antagonism appeared to be mediated exclusively by peripheral μ receptor activation (as described in the preceding section), that endomorphins have a high affinity and selectivity for μ receptors, and that these endogenous opioid peptides differ from most opioid peptides in their N-terminal sequence (Horvath, 2000; Machelska, 2011) (and therefore are not susceptible to the neutralizing effects of 3-E7), they are candidates for the antihyperalgesic effects induced by sigma-1 antagonists. Supporting this, i.pl. administration of a monoclonal antibody against endomorphin-2 did not alter the response latency of non-sensitized animals (Figure S7), but fully reversed the local antihyperalgesic effect of both S1RA and BD-1063 after sensitization with either PGE2 (Figure 4e) or NGF (Figure 4f).
We then aimed to identify the source of the endomorphin-2 responsible for the antihyperalgesic effects induced by sigma-1 antagonism, and tested immune cells and peripheral sensory neurons as possible sources of this endogenous opioid peptide.
We first tested in female mice whether the i.pl. administration of the peripheral sensitizers recruited immune cells to the injected site. We used samples from mice administered with carrageenan, a pro-inflammatory agent, as a positive control. Although we found a prominent increase in immune cells (stained with the pan-haematopoietic cell marker CD45) in paw tissue after carrageenan injection, we did not find any accumulation of these cells after the injection of PGE2, NGF or GDNF (Figure 5a,b). As neutrophils are known to be recruited early to the inflamed site (Rittner et al., 2001), we also tested for possible increases of this specific immune cell population. We found prominent neutrophil (CD45+ CD11b+ Ly6G+ cells) recruitment in paw tissue after carrageenan injection, but we did not find any accumulation of these immune cells after the injection of any of the algogenic compound tested (Figure 5c,d). Therefore, it is unlike that immune cells would constitute the cells harbouring the endogenous opioid peptides responsible of the peripheral opioid effects induced by sigma-1 antagonism against PGE2- and NGF-induced hyperalgesia.
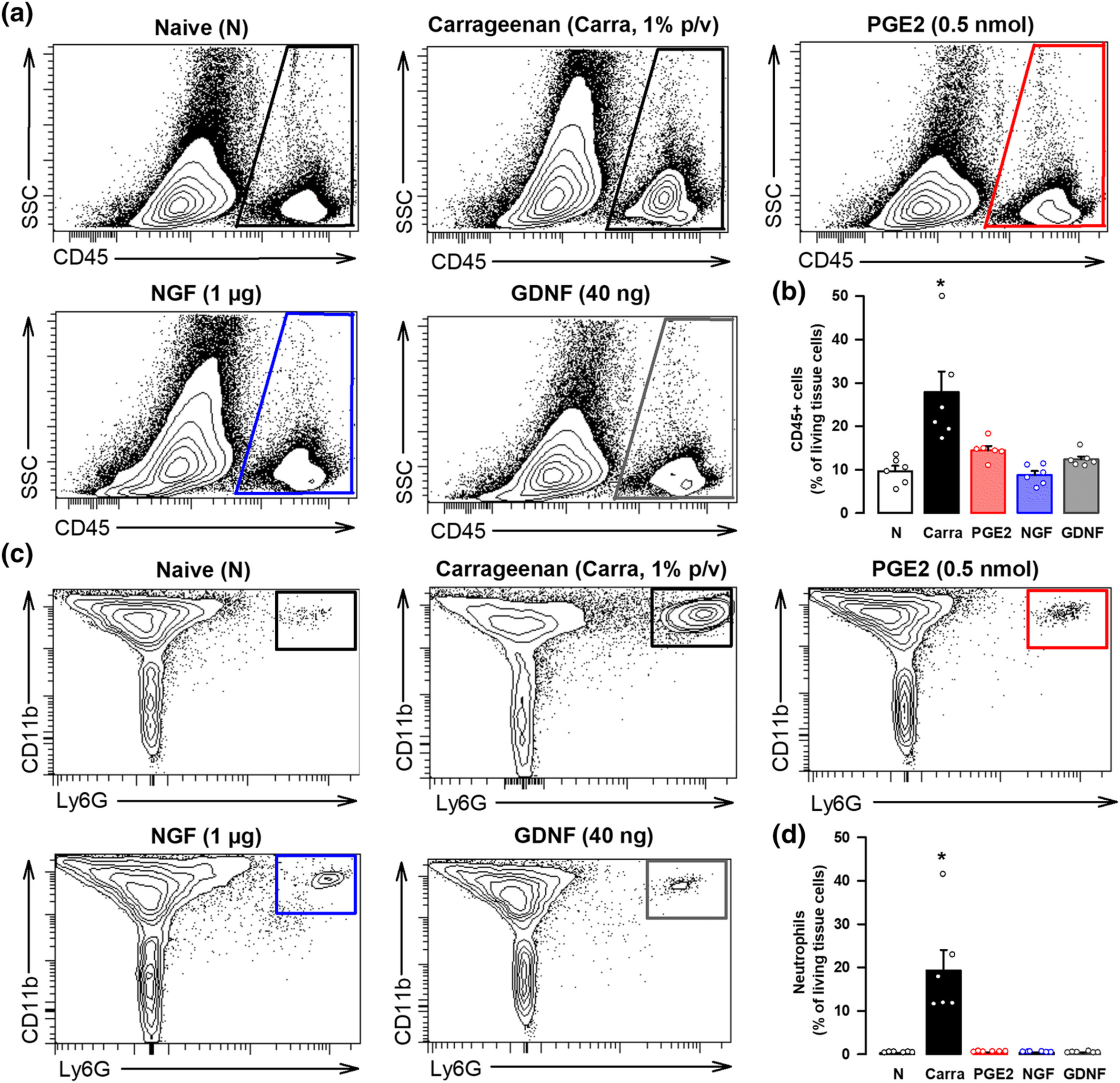
The in vivo treatment with PGE2, NGF, or GDNF does not produce significant immune cell recruitment at the injection site. (a) Representative side scatter (SSC) versus CD45 plots showing that cells from haematopoietic lineage (CD45+ cells) do not increase in the paw from female mice after intraplantar (i.pl.) administration of PGE2 (0.5 nmol), NGF (1 μg) or GDNF (40 ng), in comparison to naïve (N) mice, whereas this cell population greatly increases after the i.pl. administration of a solution containing 1% carrageenan (carra). Gating for CD45+ cells is shown as a trapezoid in the right side of each FACS diagram. (b) Quantification of CD45+ cells with respect to the number of living cells in the paw from naïve mice and after the i.pl. treatments. (c) Representative FACS diagrams, gated from CD45+ cells, showing that neutrophils (CD11b+ Ly6G+ cells) greatly increase in the paw from female mice after the i.pl. administration of carra but not after the injection of any of the three peripheral sensitizers tested. Gating for neutrophil quantification is shown as a square in the right corner of each FACS diagram. (d) Quantification of neutrophils with respect to the number of living cells in the paw from naïve mice and after the i.pl. treatments. (b,d) values are the mean ± SEM (six animals per group). Statistically significant differences between naïve and carrageenan-treated animals: *P < 0.05. There were no significant differences between naïve animals and those treated with PGE2, NGF, and GDNF (one-way ANOVA followed by Bonferroni test). Data shown in (b) and (d) were log-transformed to meet the ANOVA assumptions.
We then tested whether endomorphin-2 was present in peripheral sensory neurons from female mice. Using the same antibody which administered in vivo was able to abolish the antihyperalgesic effects of sigma-1 antagonists, we found endomorphin-2 immunoreactivity in DRG samples, and interestingly, the majority of this labelling was found in TRPV1+ nociceptors, with a virtual absence of endomorphin-2 staining on IB4+ neurons (see left panels of Figure 6a and Figure 6b). A few additional larger cells with neuronal morphology (TRPV1-/IB4- cells) were also found to be endomorphin-2+ (see left panels of Figures 6a and S8 for representative images of high and low magnification, respectively). As a proof of the specificity of the expression of endomorphin-2 by TRPV1+ nociceptors, we performed immunostaining experiments after RTX administration. Treatment with this toxin completely ablated TRPV1 staining and most of endomorphin-2 immunoreactivity (Figure 6c), which remained only in some larger neurons, whereas IB4 labelling was globally preserved (see right panels of Figures 6a and S8), and in fact constituted most of the labelled (CGRP+, IB4+ or TRPV1+) neurons after the ablation of TRPV1+ nociceptors (Figure 6c). These results suggest that most of the neurons which express endomorphin-2 correspond to peptidergic (TRPV1+) nociceptors. This pattern for the expression of endomorphin-2 agrees with the naloxone-sensitive effect of sigma-1 antagonists on hyperalgesia induced by sensitizers of TRPV1+ nociceptors, such as PGE2 or NGF.
Endomorphin-2 (END2) is present in TRPV1+ but not in IB4+ neurons. (a) Triple labelling of endomorphin-2 (END2, green), TRPV1 (magenta), and isolectin B4 (IB4, yellow) in L4 DRG from female mice. Left panels: samples from solvent-treated mice (control). Right panels: samples from mice treated with resiniferatoxin (RTX). White arrows indicate co-localization of END2 and TRPV1 markers. White arrowhead indicates co-localization of TRPV1 and IB4 markers. Scale bar 50 μm. (b,c) Venn diagrams displaying the percentage of TRPV1+, IB4+, and END2+ neurons among the total number of neurons labelled with any of these markers in samples from control mice (b) and from mice treated with RTX (c). Samples from five mice per group were used to construct the Venn diagrams.
3.4 Sigma-1R: A link between TRPV1 and the μ receptor
As previously commented, the modulation of μ receptor antinociception by sigma-1R at the CNS involves the participation of NMDARs (see the Introduction section). Taking into account that the opioid-dependent antihyperalgesic effect of Sigma-1 antagonism appears to involve peptidergic (TRPV1+) nociceptors, we hypothesized that the central NMDAR mechanism might have a peripheral analogue based on interactions of TRPV1, sigma-1Rs, and μ receptor.
Using recombinant proteins, we found that the sigma-1R and CaM each strongly interact with the C-terminal segment of TRPV1 in the presence of calcium (see lanes one and seven in Figure 7a). However, when the sigma-1R and CaM are present together, sigma-1R binds to TRPV1 and this does not allow CaM to bind the C-terminal domain of TRPV1 (Figure 7a, lane six). This CaM binding site is important for TRPV1 desensitization (Numazaki et al., 2003). The presence of either S1RA or BD-1063 hinders the interaction between sigma-1R and TRPV1 (Figure 7a, lanes two and four), allowing CaM to bind to the TRPV1 channel (Figure 7a, lanes three and five). We also examined the influence of μ receptors on the interaction between sigma-1Rs and TRPV1 and found that the presence of the C-terminus of the μ receptor, which contains the binding site for sigma-1Rs (Rodríguez-Muñoz, Cortés-Montero, et al., 2015; Rodríguez-Muñoz, Sánchez-Blázquez, et al., 2015) enhanced the dissociation of sigma-1Rs from the C-terminal domain of TRPV1 that is induced by S1RA (Figure 7b). In the presence of S1RA, the interaction between the sigma-1R and the C-terminal domain of the μ receptor is markedly increased, in spite of the presence of the C-terminus of TRPV1 (Figure 7c). In other words, sigma-1 antagonism promotes the transfer of sigma-1Rs from the C-terminal domain of TRPV1 to the C-terminus of the μ receptor, and this facilitates the binding of CaM to the C-terminus of TRPV1.
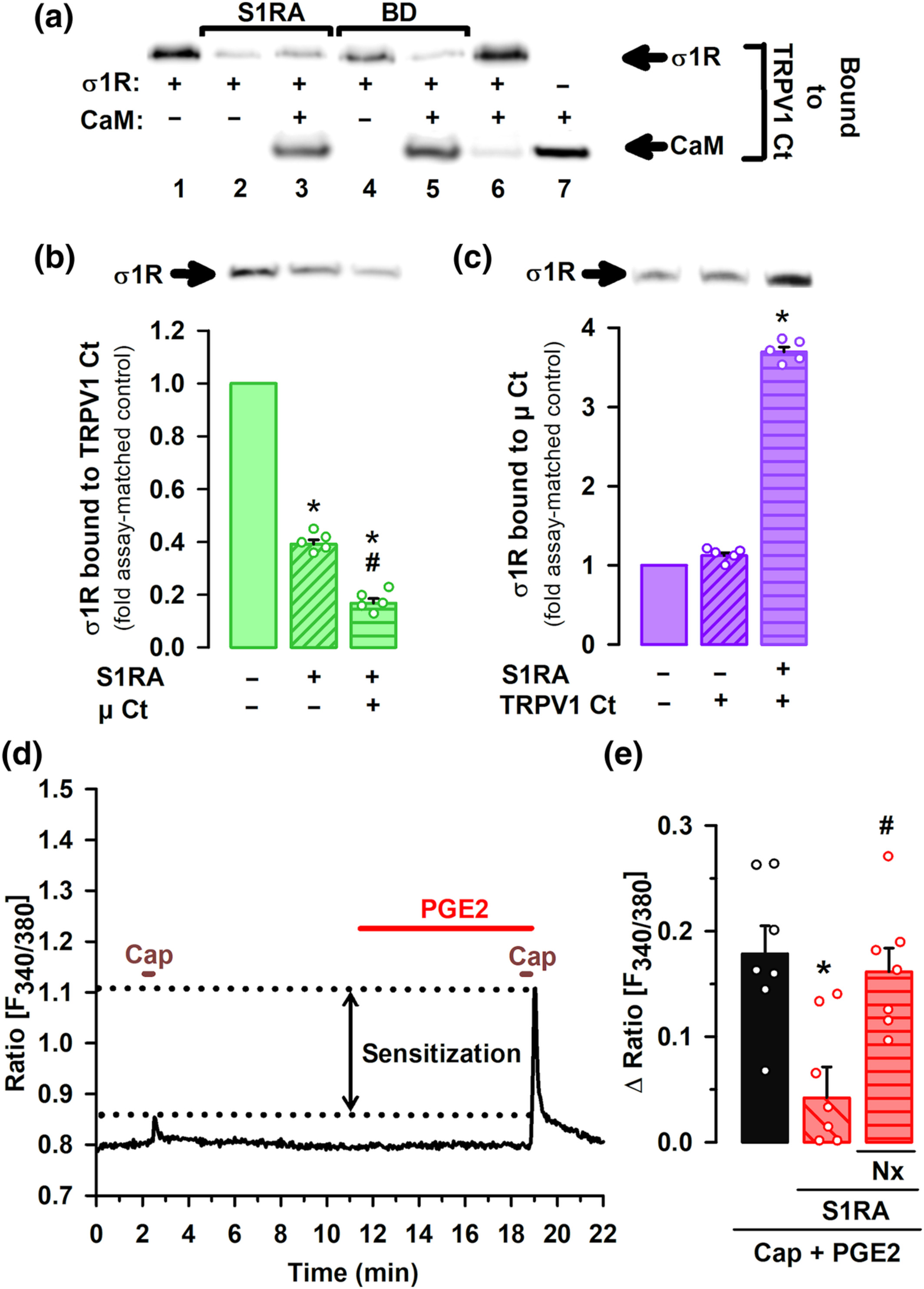
Influence of sigma-1 antagonism on the interaction of the sigma-1 receptor (σ1R) with TRPV1 and μ opioid receptors (μ), and the effect on PGE2-induced sensitization of TRPV1 neurons. (a–c) Experiments were performed in the presence of CaCl2. Blots shown are representative of three experiments. Gels were cropped to show bands under investigation only and full-length gels are provided in Figure S9A–C. (a) Effect of S1RA and BD-1063 (BD) on the in vitro interaction of the σ1R and calmodulin (CaM) with the C-terminus (Ct) of TRPV1. σ1R and CaM were incubated with TRPV1 Ct immobilized in N-hydroxysuccinimide (NHS)-activated Sepharose. The bands represent σ1R and CaM that remained bound to the TRPV1 Ct after incubation with the sigma-1 antagonists or their solvent. (b) Immobilized TRPV1 Ct was incubated with σ1R with and without μ receptor Ct. The blots represent σ1R that remained bound to the TRPV1 Ct after incubation with S1RA or its solvent. (c) Immobilized μ receptor Ct (μ Ct) was incubated with σ1R with and without TRPV1 Ct. The blots represent σ1R that remained bound to the μ receptor Ct after incubation with S1RA or its solvent. (b,c) The signals from the blots were expressed as the change relative to the controls, which were assigned an arbitrary value of 1. Values are the mean ± SEM (five determinations per group). Statistically significant differences between the values obtained in the control (solid bars) and the other experimental groups (striped bars), *P < 0.05; and between the values of the group incubated with S1RA alone or with μ receptor Ct, #P < 0.05 (Kruskal–Wallis test followed by Student–Newman–Keuls test). (d) Representative calcium imaging recording (ratio F340/380) of a cultured mouse DRG neuron treated with 0.05-μM capsaicin (Cap) before and after treatment with PGE2. Sensitization of calcium flux by PGE2 is shown between dotted lines. (e) Mean response amplitudes for the increase in the ratio F340/380 in response to capsaicin in cultured neurons from female mice, sensitized with PGE2 and incubated with S1RA, naloxone (Nx) or their solvents. Values are the mean ± SEM of the values found in seven different culture dishes, each obtained from a different mouse. Statistically significant differences between the capsaicin response in PGE2-sensitized neurons incubated with S1RA or its solvent, *P < 0.05; and between the responses from sensitized neurons treated with S1RA alone or associated with Nx, #P < 0.05 (one-way ANOVA followed by Bonferroni test).
Our results indicate that sigma-1Rs are a key player in the crosstalk between the μ receptor and TRPV1. We then explored the functional consequences of the interactions between sigma-1Rs, TRPV1 and opioid receptors for nociceptor sensitization. We performed calcium-imaging experiments on cultured capsaicin-sensitive DRG neurons from adult female mice, sensitized with PGE2. The application of a low concentration of capsaicin (0.05 μM) produced a hardly measurable increase in intracellular calcium concentration. However, after application of PGE2, these neurons showed a robust increase in intracellular calcium in response to the same low concentration of capsaicin (see Figure 7d for a representative recording), indicating the sensitization of TRPV1+ nociceptors by this algogen. We then measured the effect of S1RA on the PGE2-induced sensitization, and found that application of the sigma-1 antagonist greatly decreased the peak amplitude of PGE2-sensitized neurons responses to capsaicin (Figure 7e). This effect was reversed by the opioid antagonist naloxone (Figure 7e), which indicates that S1RA acts through opioid activation to reverse TRPV1+ neuron sensitization.
4 DISCUSSION
TRPV1+ nociceptors can be sensitized by PGE2 and NGF to express the endogenous μ receptor agonist endomorphin-2. We show that sigma-1Rs are also expressed by these neurons and participate in the crosstalk between TRPV1 and the μ receptor, tonically limiting the antihyperalgesic effect of the endogenous opioid peptide.
Most CGRP+ DRG neurons express TRPV1 and vice versa, as shown here and in previous studies (Priestley, 2009). On the other hand, TRPV1+ and IB4+ neurons constitute separate cellular populations (peptidergic and non-peptidergic C-nociceptors, respectively) with only occasional overlap, as shown in the current and previous studies using several mouse strains (Sheehan et al., 2019; Woodbury et al., 2004; Zwick et al., 2002). The in vivo ablation of peptidergic (TRPV1+) C neurons by RTX increased the response latency to heat stimulus in non-sensitized animals and in mice sensitized with PGE2 or NGF, in agreement with the known role of TRPV1+ neurons in the coding of heat nociception or hypersensitivity (Cavanaugh et al., 2009). We also show that RTX-sensitive neurons are dispensable for mechanical nociceptive pain, in agreement with previous studies (Montilla-García et al., 2018; Zhang et al., 2013), but they are essential for the mechanical hyperalgesia induced by PGE2 or NGF. These latter results can be explained by the fact that both algogenic compounds induce mechanosensitivity in those nociceptors, which are normally mechanically insensitive (Emery et al., 2016; Prato et al., 2017), and this might be dependent on a phenotypic switch of peptidergic C-nociceptors (Prato et al., 2017).
The systemic administration of the sigma-1 antagonists S1RA and BD-1063 reversed mechanical and heat hyperalgesia induced by PGE2 and NGF. Interestingly, the opioid antagonist naloxone and its peripherally restricted analogue naloxone methiodide abolished the effects of not only morphine, used as a reference opioid analgesic, but also those of the sigma-1 antagonists, indicating that these effects were opioid in nature and mediated peripherally. These peripheral opioid-dependent effects induced by sigma-1 antagonism are seen in mice from both sexes. In addition, the antihyperalgesic effects of sigma-1 antagonists and morphine to the mechanical stimulus involved the activation of the μ receptor, but not other opioid receptor subtypes, as they were fully reversed by cyprodime, but not by naltrindole or nor-binaltorphimine (at doses known to inhibit μ, δ, and κ opioid receptor responses, respectively) (Baamonde et al., 2005; Hutcheson et al., 1999). Although it seems evident to attribute the effect of morphine to direct actions on the μ receptor (Matthes et al., 1996), S1RA or BD-1063 lack any affinity for the μ receptor (Sánchez-Fernández et al., 2013). Since sigma-1 antagonism is known to potentiate opioid analgesia (Sánchez-Fernández et al., 2017), we hypothesized that the peripheral opioid-like effects of sigma-1 antagonists on PGE2- and NGF-induced hyperalgesia might be the result of the potentiation of endogenous opioid peptides released at the sensitized site. The local administration of sigma-1 antagonists abolished PGE2- and NGF-induced mechanical hyperalgesia, and this was not reversed by the administration of 3-E7, a monoclonal antibody which recognizes the N-terminus of most endogenous opioid peptides. However, the antihyperalgesic effects of sigma-1 antagonists were reversed by an antibody against endomorphin-2, which lacks the consensus N-terminus of other opioid peptides and, in agreement with the previously commented μ-opioid selectivity of the effects induced by sigma-1 antagonists, is a selective μ-opioid agonist (Horvath, 2000; Machelska, 2011).
Immune cells can produce endomorphins (Labuz et al., 2006; Mousa et al., 2002), and we recently described that sigma-1 antagonism enhances the opioid analgesia induced by opioid peptides released by immune cells during inflammation (Tejada et al., 2017). We show here that PGE2 or NGF did not recruit immune cells which might account for the antihyperalgesic effect of sigma-1 antagonists. However, we found endomorphin-2 immunoreactivity in mouse DRG neurons, in agreement with previous studies in the rat which show that endomorphin-2 (but not endomorphin-1) is produced by peripheral sensory neurons (Fichna et al., 2007; Sanderson et al., 2004; Scanlin et al., 2008). Importantly, we show that endomorphin-2 is mostly expressed by peptidergic (TRPV1+) nociceptors, a result which also agrees with the previously described distribution of this endogenous opioid peptide in the rat DRG, as it was shown to colocalize with substance P and CGRP (Sanderson et al., 2004). Therefore, the sensory neurons required for PGE2- and NGF-induced hyperalgesia are the same neuronal subtype which expresses endomorphin-2. In spite of the well-known analgesic actions of endomorphin-2 (Fichna et al., 2007), the production of this endogenous opioid peptide by TRPV1+ nociceptors seems not enough to counterbalance sensitization by PGE2 or NGF, except when potentiated by sigma-1 antagonists. Our results might be explained by the contribution of an autocrine mechanism, in the peripheral terminal of the nociceptor, in the antihyperalgesic effect of sigma-1 antagonism.
The modulation of μ receptor-mediated analgesia by sigma-1Rs in the CNS relies on the binding of the sigma-1R to the NMDAR, physically preventing the binding of CaM to the NMDAR, and hence reducing the inhibition of channel activity. Sigma-1 antagonism dissociates Sigma-1Rs from NMDAR and transfer them to the μ receptor. In this situation, CaM gains access to NMDARs to curtail channel activity and consequently enhancing μ receptor actions (Rodríguez-Muñoz, Cortés-Montero, et al., 2015 and b). TRPV1 is another protein partner of sigma-1Rs (Cortés-Montero et al., 2019; Ortíz-Rentería et al., 2018), and similar to NMDARs, it is a Ca2+ channel regulated by CaM, and binding of CaM to the C-terminus of TRPV1 promotes the desensitization of the channel (Numazaki et al., 2003). We showed here that sigma-1 antagonism promotes the transfer of sigma-1Rs from the C-terminal domain of TRPV1 to the C-terminus of the μ receptor, and this facilitates the binding of CaM to the C-terminus of TRPV1. Therefore, we show for the first time that the mechanism for the modulation of opioid analgesia by sigma-1Rs based on the crosstalk between NMDARs and μ receptors has an analogue on peptidergic C neurons that uses TRPV1 instead of NMDARs. TRPV1 has multiple known protein partners able to alter channel function (Zhao & Tsang, 2016). It may be worth testing in future studies whether sigma-1R influence other components of the interactome of TRPV1 in addition to CaM binding.
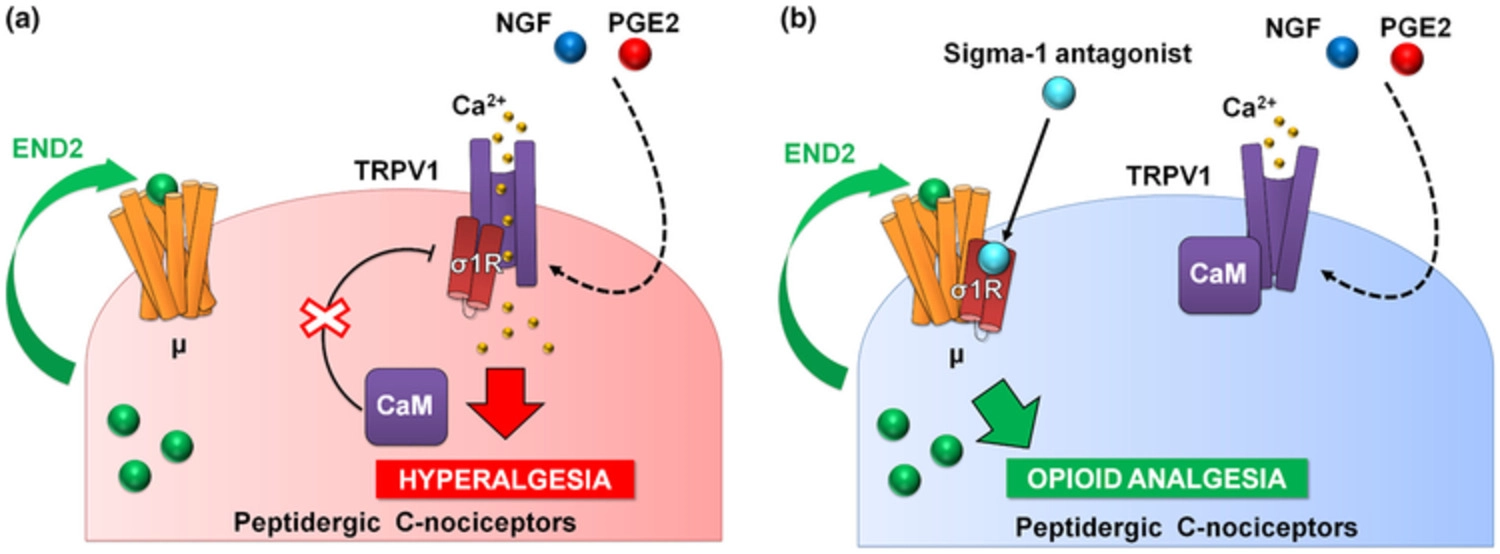
PGE2 increased calcium flux induced by capsaicin, the prototype TRPV1 agonist (Wainger et al., 2015), and this was fully reversed by S1RA, and in a naloxone-sensitive manner. It is known that endomorphin-2 is released by DRG neurons in response to intracellular calcium increases (Scanlin et al., 2008), and this might account for the naloxone-sensitive effect of S1RA that we recorded. Altogether, our data and previous literature show that sensitization of TRPV1 results in enhanced Ca2+ influx, which promotes the release of endogenous opioid peptides (endomorphin-2) with the potential to induce analgesia through μ receptor activation. However, this analgesia through neuronally derived endogenous opioids might be curtailed by the binding of sigma-1R to TRPV1 (Figure 8a). Sigma-1R antagonists trigger the transfer of sigma-1Rs from the TRPV1 to the μ receptor, decreasing Ca2+ flux and enhancing the action of endomorphin-2 to induce opioid analgesia in the sensitized peripheral terminal, in the absence of an exogenous opioid drug (Figure 8b).
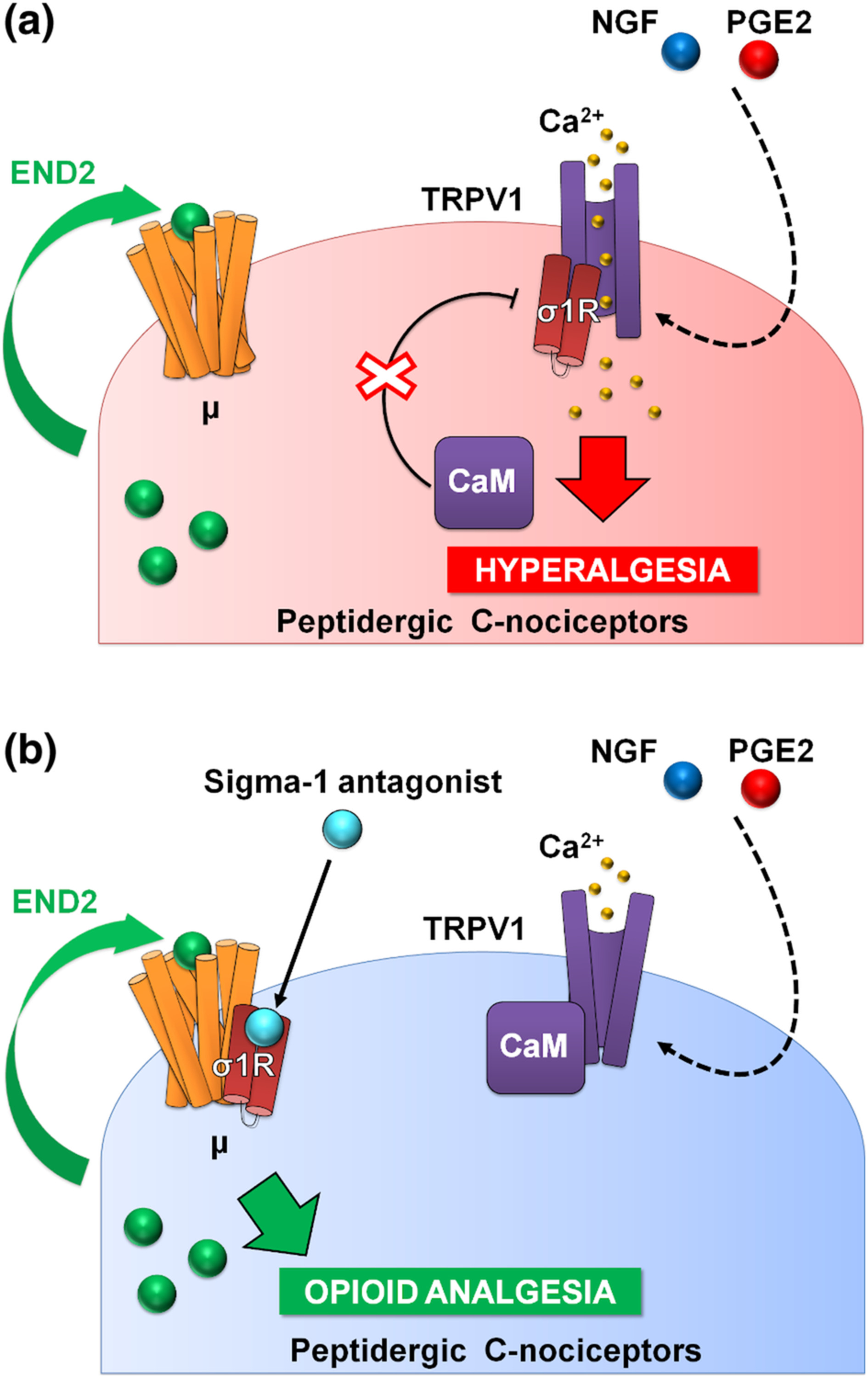
Proposed mechanism of action for the effects of sigma-1 antagonism on hyperalgesia induced by sensitization of TRPV1+ neurons. (a) Sensitization by algogenic chemicals (such as PGE2 and NGF) favours Ca2+ influx through TRPV1. In response to Ca2+, the sigma-1 receptor (σ1R) binds to TRPV1 preventing calmodulin (CaM) binding (and therefore preventing desensitization of the channel). TRPV1+ neurons produce the endogenous opioid peptide endomorphin-2 (END2), whose effects are not sufficient to relieve hyperalgesia. (b) Sigma-1 antagonists transfer σ1R from TRPV1 to μ opioid receptors (μ), and this facilitates the interaction of CaM with the desensitization site of TRPV1 and the enhancement of the effects of END2, producing opioid-mediated antihyperalgesic effects during nociceptor sensitization.
Although extracellular calcium influx through TRPV1 after capsaicin activation is the expected primary responsible for the increase in intracellular calcium we observed, other channels in the plasma membrane and in intracellular locations can also be activated after TRPV1 stimulation and participate in calcium flux (DuBreuil et al., 2021; Shah et al., 2020). Therefore, the modulation of TRPV1 by sigma-1 receptors may merit further study using electrophysiological recordings as a more direct approach to study channel functioning.
TRPV1 are relevant for both pain and itch (Roberson et al., 2013). As sigma-1 antagonism decreases sensitization of TRPV1+ neurons, it could be hypothesized that these drugs might induce antipruritogenic effects in addition to the antihyperalgesic effects showed here. This possibility will be addressed in future studies.
We also tested the effects of sigma-1 antagonism on the hyperalgesia induced by a different peripheral sensitizer: GDNF. This algogenic compound induced RTX-insensitive mechanical hyperalgesia without inducing significant heat hypersensitivity. The cellular targets of GDNF are non-peptidergic C-nociceptors (IB4+ neurons) (Álvarez et al., 2012), a neuronal population resistant to RTX (Montilla-García et al., 2018; Zhang et al., 2013) that although relevant for mechanical hypersensitivity, is dispensable for heat sensitivity (Cavanaugh et al., 2009), which explains the behavioural effects observed. We also show that sigma-1 antagonism was absolutely devoid of effect on GDNF-induced hyperalgesia in either female or male mice. It is relevant to note that IB4+ neurons do not express endomorphin-2. In addition, similar to PGE2 or NGF, GDNF failed to recruit immune cells at the site of injection that could harbour endogenous opioid peptides to be potentiated by sigma-1 antagonism. Therefore, our results suggest that sigma-1 antagonism requires the presence of an opioid agonist that can be potentiated in order to relieve hyperalgesia from peripheral sensitization, and point to the specificity in the modulation of the sensitization of TRPV1 neurons because of their content on endomorphin-2.
TRPV1+ nociceptors constitute a relatively small population of neurons in the mouse, but most human nociceptors express TRPV1 (Middleton et al., 2021). Therefore, it would be expected that the effects of sigma-1 antagonism on peripheral sensitization, which appear to be restricted to TRPV1+ neurons in the mouse, would be broader in humans.
In summary, sigma-1 receptors limit peripheral opioid analgesia during sensitization of peptidergic C nociceptors. Sigma-1 antagonists are able to harness neuronally derived endogenous opioids to reduce hyperalgesia at the pain site, by promoting TRPV1 desensitization and increasing μ receptor activity. The modulation of endogenous opioid analgesia by sigma-1 receptors might have potential clinical application for pain treatment.