
The molecular role of Sigmar1 in regulating mitochondrial function through mitochondrial localization in cardiomyocytes
by Chowdhury S. Abdullah, Richa Aishwarya, Shafiul Alam, Naznin Sultana Remex, Mahboob Morshed, Sadia Nitu, Sumitra Miriyala, Manikandan Panchatcharam, Brandon Hartman, Judy King, Mohammad Alfrad Nobel Bhuiyan, James Traylor, Christopher G. Kevil, A. Wayne Orr, Md. Shenuarin Bhuiyan
Excerpt from the article published in Mitochondrion, Volume 62, 2022, Pages 159-175, ISSN 1567-7249, https://doi.org/10.1016/j.mito.2021.12.002.
Editor’s Highlights
- Sigmar1 localizes to mitochondrial membranes and is indispensable for maintaining mitochondrial respiratory homeostasis in cardiomyocytes.
- As mitochondria comprise the majority of the cell volume of cardiomyocytes and play a crucial role for the normal functioning of the heart, Sigmar1 plays a key role in mitochondria in the cardiomyocytes.
- Sigmar1 plays a key role in maintaining physiological mitochondrial fuel substrates-linked respirations through a Sigmar1-dependent mitochondrial electron transfer system (ETS)-linked respirations.
Abstract
Sigmar1 is a widely expressed molecular chaperone protein in mammalian cell systems. Accumulating research demonstrated the cardioprotective roles of pharmacologic Sigmar1 activation by ligands in preclinical rodent models of cardiac injury. Extensive biochemical and immuno-electron microscopic research demonstrated Sigmar1′s sub-cellular localization largely depends on cell and organ types. Despite comprehensive studies, Sigmar1′s direct molecular role in cardiomyocytes remains elusive. In the present study, we determined Sigmar1′s subcellular localization, transmembrane topology, and function using complementary microscopy, biochemical, and functional assays in cardiomyocytes. Quantum dots in transmission electron microscopy showed Sigmar1 labeled quantum dots on the mitochondrial membranes, lysosomes, and sarcoplasmic reticulum-mitochondrial interface. Subcellular fractionation of heart cell lysates confirmed Sigmar1′s localization in purified mitochondria fraction and lysosome fraction. Immunocytochemistry confirmed Sigmar1 colocalization with mitochondrial proteins in isolated adult mouse cardiomyocytes. Sigmar1′s mitochondrial localization was further confirmed by Sigmar1 colocalization with Mito-Tracker in isolated mouse heart mitochondria. A series of biochemical experiments, including alkaline extraction and proteinase K treatment of purified heart mitochondria, demonstrated Sigmar1 as an integral mitochondrial membrane protein. Sigmar1′s structural requirement for mitochondrial localization was determined by expressing FLAG-tagged Sigmar1 fragments in cells. Full-length Sigmar1 and Sigmar1′s C terminal-deletion fragments were able to localize to the mitochondrial membrane, whereas N-terminal deletion fragment was unable to incorporate into the mitochondria. Finally, functional assays using extracellular fluxanalyzer and high-resolution respirometry showed Sigmar1 siRNA knockdown significantly altered mitochondrial respiration in cardiomyocytes. Overall, we found that Sigmar1 localizes to mitochondrial membranes and is indispensable for maintaining mitochondrial respiratory homeostasis in cardiomyocytes.
1. Introduction
Sigmar1 is a multifunctional, ubiquitously expressed chaperone protein encoded by the SIGMAR1 gene (Aishwarya et al., 2021). The purification and cloning of Sigmar1-binding site from guinea pig liver using Sigmar1 specific probes initiated the molecular characterization of Sigmar1 (Hanner et al., 1996). Since the discovery, extensive studies have used pharmacologic approach to define Sigmar1 ligands binding stoichiometry, and determining their physiological and pharmacological profiles (Culp et al., 1992, de Costa et al., 1990, Leonardo et al., 2010, Matsuno et al., 1996, Rao et al., 1990). These studies revealed the potential roles of Sigmar1 agonists and antagonists in the diverse central nervous system (CNS)-associated disorders utilizing in vitro neuronal cell lines and in vivo rodent models. Comprehensive studies using these ligands showed Sigmar1′s biological roles in neuroinflammation, cognitive dysfunction, memory deficits, psychiatric disorders, motor neuron diseases, drug addiction, and stroke (Hayashi and Su, 2008, Penke et al., 2018, Ryskamp et al., 2019). However, most of these Sigmar1 ligands also demonstrated a binding affinity for other receptors, including serotonin receptors, serotonin reuptake transporters, and metabotropic receptors in the CNS (Fisher et al., 2016, Fontanilla et al., 2009, Hayashi and Su, 2008, Narita et al., 1996, Penke et al., 2018, Rao et al., 1990, Smith et al., 1998, Zou et al., 1998). Moreover, most of these studies were limited to dissecting the pharmacological effects of Sigmar1 ligands and lacked validation of the Sigmar1′s direct involvement by using genetic loss-of-function studies utilizing Sigmar1 null cell lines or rodent models. Thus, the precise molecular role of Sigmar1 in eliciting diverse cellular and physiological functions requires further investigation.
Despite extensive reports, the subcellular localization of Sigmar1 remains elusive. An early study demonstrated widespread expression of Sigmar1 in the CNS in rats (Alonso et al., 2000). Subsequent studies reported a diverse subcellular distribution of Sigmar1, including the plasma membrane, plasma membrane subsurface cisternae of endoplasmic reticulum (ER), ER, nuclear envelope, nucleoplasmic reticulum, mitochondria-associated ER membranes (MAM), and mitochondria (Hayashi and Su, 2003a, Hayashi and Su, 2003b, Hayashi and Su, 2004, Hayashi and Su, 2007, Mavlyutov et al., 2017b). Sigmar1 was also reported to localize at the ER and nuclear envelope in human immune cells (Dussossoy et al., 1999). Subsequently, immunoelectron microscopy (immune-EM) studies confirmed several of these reported subcellular distributions of Sigmar1 (Mavlyutov et al., 2010, Mavlyutov et al., 2011, Mavlyutov et al., 2012, Mavlyutov et al., 2013, Mavlyutov et al., 2015a, Mavlyutov et al., 2015b, Mavlyutov et al., 2016, Mavlyutov et al., 2017a, Mavlyutov et al., 2017b, Mavlyutov and Guo, 2017, Mavlyutov and Ruoho, 2007, Yang et al., 2017). However, immuno-EM examinations were unable to detect Sigmar1 at the plasma membrane (Mavlyutov et al., 2010, Mavlyutov et al., 2011, Mavlyutov et al., 2012, Mavlyutov et al., 2013, Mavlyutov et al., 2015a, Mavlyutov et al., 2015b, Mavlyutov et al., 2016, Mavlyutov et al., 2017a, Mavlyutov et al., 2017b, Mavlyutov and Guo, 2017, Mavlyutov and Ruoho, 2007, Yang et al., 2017). Sigmar1 was also detected on mitochondria isolated from rat liver (depicted as Sigmar1-like receptor) using ligand-based studies and immunostaining (Klouz et al., 2002). (+)-Pentazocine ligand binding and enzyme binding assays (using monoamine oxidases and cytochrome coxidases) showed Sigmar1 in the outer mitochondrial membrane in the isolated mitochondrial fraction and liver mitochondria have a Sigmar1 ligand binding site compared to that of brain mitochondria (Klouz et al., 2002). All these studies together suggest organ and tissue-specific Sigmar1 localization and function in the body. However, the subcellular distributions and molecular function of Sigmar1 in cardiomyocytes remain unknown.
Extensive studies from our group and others reported ubiquitous expression of Sigmar1 in the heart. Subsequent studies utilizing Sigmar1 ligands (i.e., dehydroepiandrosterone and fluvoxamine) showed activation of Sigmar1 ameliorates pressure-overload induced hypertrophy and cardiac dysfunctions in ovariectomized rats (Bhuiyan and Fukunaga, 2009, Bhuiyan et al., 2011) and transverse aortic stenosis mice (Bhuiyan et al., 2010, Tagashira et al., 2014). In line with these findings, chronic activation of Sigmar1 with fluvoxamine attenuated ventricular remodeling and arrhythmias after myocardial infarction in rats (Fo et al., 2020). We have recently reported the development of systolic cardiac dysfunction with coinciding abnormal mitochondrial ultrastructure, altered mitochondrial dynamic regulatory protein expression, phosphorylation states, and compromised mitochondrial respiration in global Sigmar1 null (Sigmar1−/−) mice hearts with ageing (Abdullah et al., 2018). However, despite these encouraging studies, there has been no study to determine subcellular localization and organelle-specific molecular functions of Sigmar1 in cardiomyocytes to pave rational drug design and discovery targeting Sigmar1. In the current study, we performed complementary biochemical, immunohistochemistry, immunocytochemistry, and ultrastructural immunostaining at the whole heart level, isolated cardiomyocytes, and isolated mitochondria to delineate Sigmar1′s distribution in cardiomyocytes. Further, we extended these findings to determine whether Sigmar1 plays any essential functions in maintaining cardiomyocyte bioenergetics and mitochondrial respirations by conducting extracellular flux analysis and high-resolution respirometry studies.
2. Results
2.1. Sigmar1 localizes to subcellular organelles in cardiomyocytes
Earlier studies reported activation of Sigmar1 using an agonist, fluvoxamine (which is also a selective serotonin reuptake inhibitor), elicits cardioprotective effects in pressure overload-induced myocardial hypertrophy and dysfunction (Bhuiyan and Fukunaga, 2009, Bhuiyan et al., 2011, Bhuiyan et al., 2010, Tagashira et al., 2014). On the contrary, pharmacologic Sigmar1 inhibition by an antagonist, haloperidol (an antipsychotic drug), aggravates cardiac pathology in pressure overload-induced heart failure in mice (Shinoda et al., 2016). Moreover, Sigmar1 ligands were tested in human clinical trials, including cutamesine (SA4503) for ischemic stroke (Phase 2) (Urfer et al., 2014), and sertraline (SADHART-CHF) for depression in patients with heart failure (Jiang et al., 2008, Serebruany et al., 2005, Xiong et al., 2015). However, all these preclinical studies are limited to assessing Sigmar1 activation and inhibition using non-specific chemical ligands having affinity to other receptors. Despite all these comprehensive studies, subcellular localization and molecular functions of Sigmar1 in the heart remain largely underexplored. In the present study, we performed fluorescence anti-Sigmar1 (green) immunostaining in human and mouse heart sections to visualize the endogenous Sigmar1′s expression pattern (Fig. 1). Sigmar1 appeared as distinct green puncta in both human and mouse hearts counterstained for cardiomyocytes (red) with cardiac Troponin I (TnI) (Fig. 1). To visualize Sigmar1′s presence and localization to subcellular compartments, we performed anti-Sigmar1 targeted quantum dot nanocrystals (Q-dots) labeling on ultra-thin adult mouse heart sections followed by transmission electron microscopy(TEM) imaging. The Q-dots TEM showed the ultrastructural localization and enrichment of endogenous Sigmar1 in the heart. Electron-dense anti-Sigmar1 labeled Q-dots (pseudocolored red dots) were observed on mitochondrial (light blue-colored) outer- (OMM) and inner-membrane (IMM) (brown arrows), lysosomes(yellow-colored), and on the sarcoplasmic reticulum/endoplasmic reticulum (SR/ER) membranes (light green-colored)-mitochondrial interface (Fig. 2A, 2B). TEM image of heart sections labeled with rabbit IgG with Q-dots was used as an Isotype control for anti-Sigmar1 rabbit primary antibody (Fig. 2A, 2B). Therefore, Q-dots TEM showed the endogenous Sigmar1 localization mostly on the mitochondrial membranes and lysosomes.
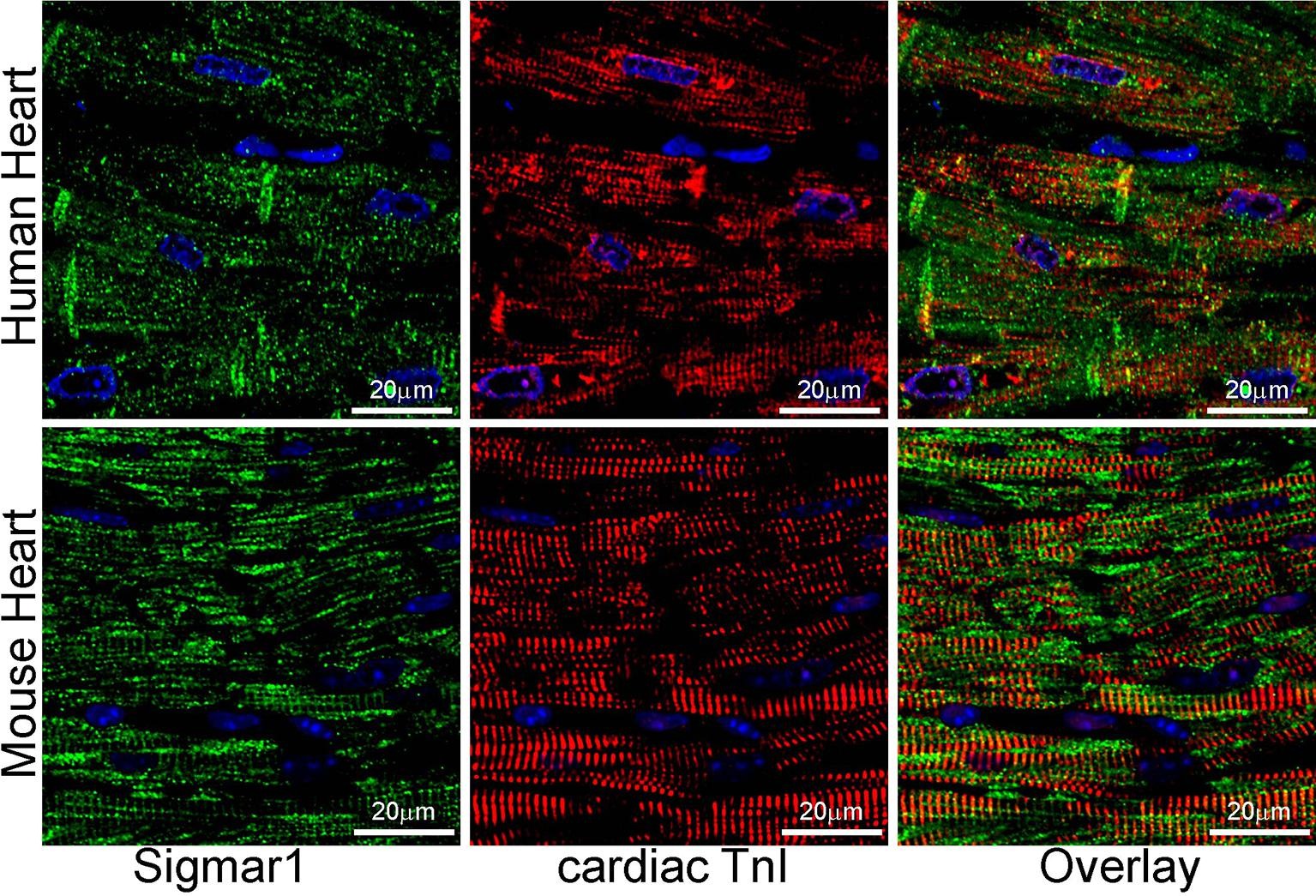
Sigmar1 expression in human and mouse ventricular heart tissues. Representative confocal microscopy images of human (top panel) and mouse (bottom panel) heart sections co immunostained with anti-Sigmar1 (green) and cardiac anti-Troponin I (cTnI, red) antibodies. Representative images showing a longitudinal view of the left ventricular sections of the human (upper panel) and mouse (lower panel) heart. Sigmar1 (green) immunostainingappeared as punctate dots in confocal fluorescence microscope images, and cTnI (red) counterstaining delineated cardiomyocytes in the heart. Representative confocal fluorescence microscope images are selected from n ≥ 3 stained human and mouse heart tissue sections. Scale bars: 20 µm. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
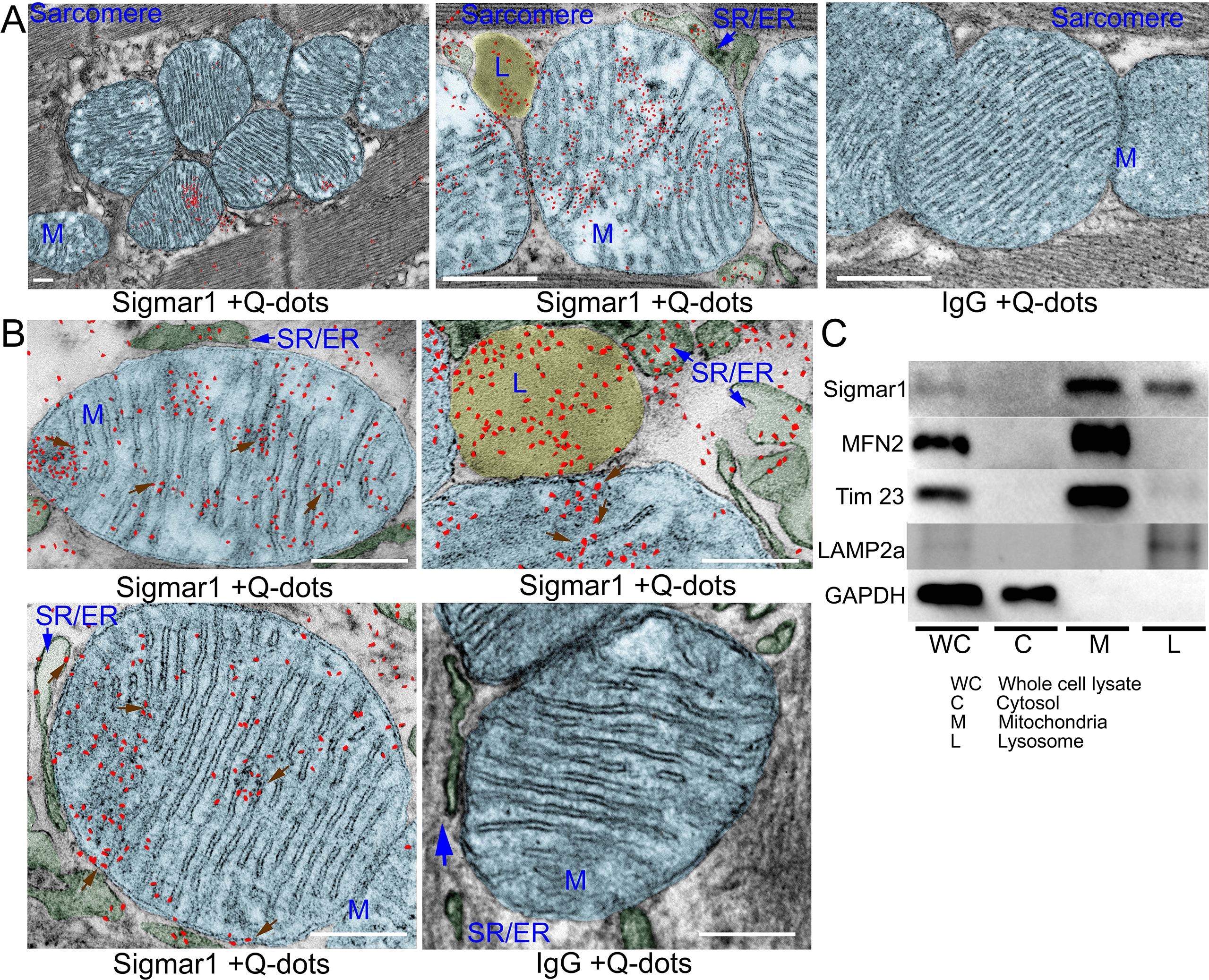
Sigmar1 immuno- labeled quantum dot (Q-dots) TEM and subcellular fractionationshowing subcellular localization in adult mice heart myocytes. (A) Representative transmission electron microscopy (TEM) image demonstrates electron-dense anti-Sigmar1 labeled Q-dots (pseudocolored in red) localization on distinct subcellular organelles in mouse heart ventricular tissues. TEM images were post-processed, and organelles were pseudocolored using GIMP 2.10 software. Mitochondria are delineated in light blue, lysosomes are in yellow, and SR/ER are in light green. TEM image of heart sections labeled with rabbit IgG and Q-dots was used as isotype control. L lysosome, M mitochondria, SR/ER sarcoplasmic reticulum/endoplasmic reticulum. Scale bar: 200 nm. (B) Representative magnified images demonstrating anti-Sigmar1 positive Q-dots localization on mitochondrial outer- and inner-membrane cristae (brown arrows). Sigmar1 positive labeled Q-dots were also detected in close proximity of mitochondria on sarcoplasmic reticulum (SR)/endoplasmic reticulum (ER) membranes (blue arrows) and lysosomes (L). TEM image of heart sections labeled with rabbit IgG and Q-dots was used as isotype control. Representative images are from n = 4 adult mouse hearts in three independent experiments in duplicates for each heart section. M mitochondria; SR/ER sarcoplasmic reticulum/endoplasmic reticulum, L lysosomes. Scale bar: 200 nm. (C) Western blot images of adult mouse heart tissue subcellular fractions consisting of whole-cell (WC) lysates, cytosolic fractions (C), lysosomal fractions (L), and mitochondrial fraction (M). Isolated subcellular fractions were subjected to Western blot analysis to assess Sigmar1′s presence and partitioning in subcellular compartments. Cytosolic protein GAPDH is used to verify isolated subcellular fractions purity. Lysosomal membrane protein LAMP2a, outer mitochondrial membrane protein MFN2, and inner mitochondrial integral membrane proteinTim23 were used to assess the successful enrichment of lysosomes and mitochondria fractions, respectively. Representative Western blots are from n > 5 subcellular fractionation-based experiments using adult mouse hearts. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To further confirm the Q-dots TEM data, we performed subcellular fractionation to purify isolated mitochondria and lysosomes from mouse heart lysates. Western blot analysis with anti-Sigmar1 antibody confirmed Sigmar1 enrichment in the purified mitochondria and lysosome fraction but not in cytosol fractions (Fig. 2C). We used cytosolic protein GAPDH to verify the purity of isolated subcellular fractions. Lysosomal membrane protein LAMP2a, outer mitochondrial membrane protein MFN2, and inner mitochondrial integral membrane protein Tim23 was used as a control to determine the successful enrichment of lysosomes and mitochondria, respectively (Fig. 2C). In summary, Q-dots TEM and subcellular fractionation data suggested abundant Sigmar1 localization in the mitochondria in mouse heart.
2.2. Sigmar1′s presence and localization to mitochondria
The heart is the most metabolically active organ in the body and possesses the highest content of mitochondria of any tissue. Therefore, we focused on Sigmar1′s role in mitochondria in subsequent studies. To corroborate our observations of endogenous Sigmar1 localization to mitochondria enriched fractions and anti-Sigmar1 labeled Q-dots appeared on mitochondrial membranes, we utilized co-immunofluorescence-based studies for anti-Sigmar1 mitochondria specific labeling in isolated primary adult mouse left ventricular cardiomyocytes (Fig. 3). We immunostained isolated primary adult mouse cardiomyocytes with anti-Sigmar1 antibody (in green) followed by co-immunostaining with mitochondrial electron transport chain supercomplexes subunit-specific cocktail antibody OXPHOS (in red) (Fig. 3A, 3B) and mitochondrial outer membrane protein Tom20 (in red) (Fig. 3C, 3D), respectively. We observed distinct yellow pixels in merged green and red channel images suggesting colocalization of anti-Sigmar1 stained green fluorescence pixels to anti-OXPHOS and anti-Tom20 stained red fluorescence pixels. Image quantifications corroborate the notable colocalization of Sigmar1-to-OXPHOS and Sigmar1-to-Tom20 stained mitochondria as evident in Mander’s colocalization coefficient and the number of colocalized voxels between green-to-red fluorescence pixels in adult mouse cardiomyocytes (Fig. 3B, 3D).
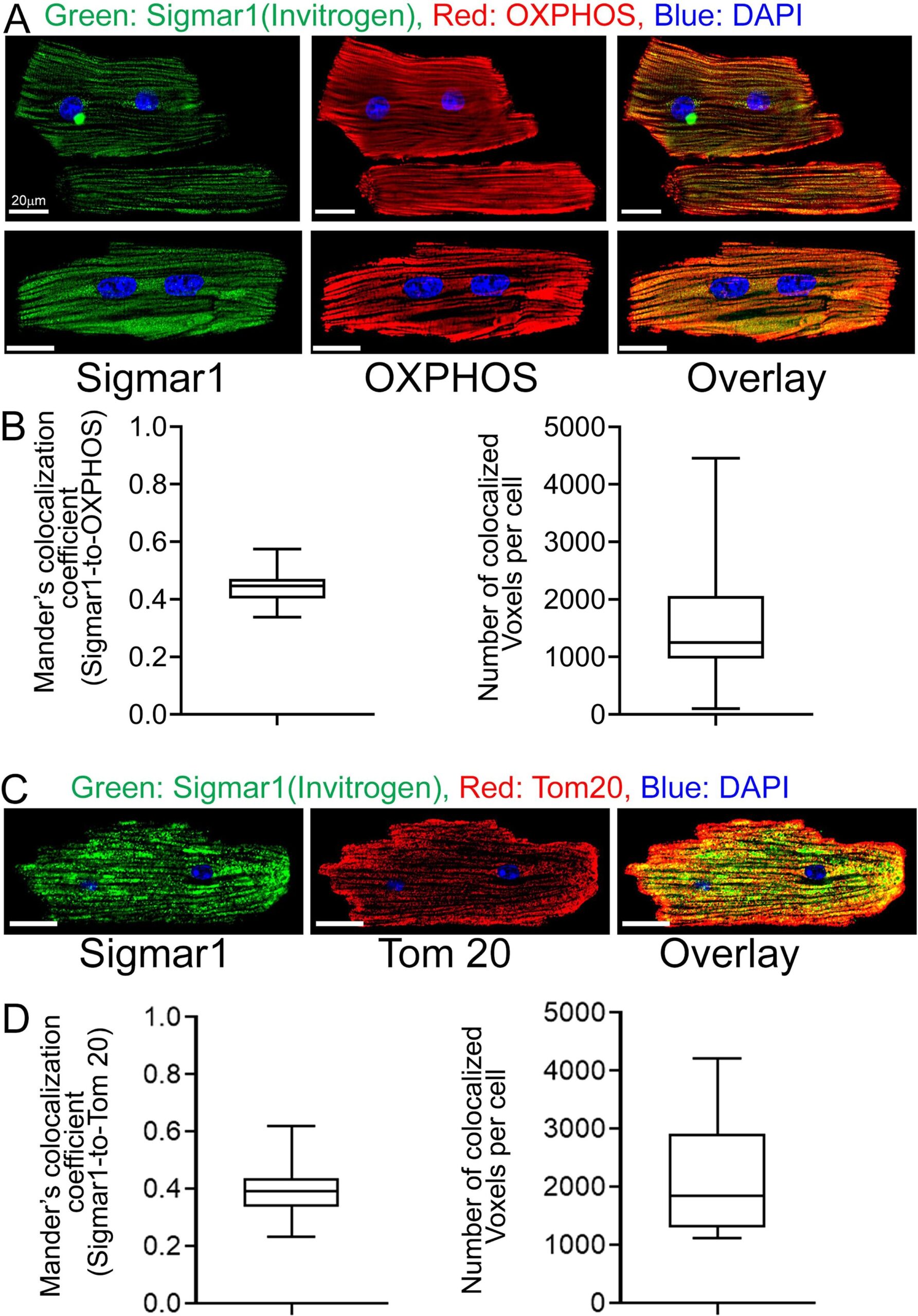
Sigmar1 localizes to mitochondria in cardiomyocytes. (A) Confocal immunofluorescence micrographs of anti-Sigmar1 (green) and anti-OXPHOS (red) co-immunostaining in isolated cardiomyocytes from adult mouse heart ventricles. Confocal microscope imaging revealed Sigmar1 puncta (green) colocalize to OXPHOS (red) stained mitochondria as evident in yellow pixels in overlay image of green and red fluorescence channel images of cardiomyocytes. Representative confocal fluorescent micrographs from two independent experiments. Scale bars: 20 µm. (B) Box plots represent Mander’s colocalization coefficient showing the extent of colocalization between Sigmar1 stained green pixels-to-OXPHOS stained red pixels and number of colocalized voxels (volume data set containing volume elements consisting of the smallest unit within an image with precise information of measured fluorescence intensity) per cardiomyocytes. Image analysis was done on sequential z-stack confocal images of N > 20 adult mouse cardiomyocytes from two independent experiments. Boxes represent interquartile ranges, lines represent medians, and whiskers represent ranges. (C) Confocal immunofluorescence micrographs of anti-Sigmar1 (green) and mitochondrial outer membrane protein anti-Tom20 (red) co-immunostaining in isolated cardiomyocytes from adult mouse heart ventricles. Anti-Sigmar1 stained puncta (green) colocalize to Tom20 (red) stained mitochondria as evident in yellow pixels in overlay images of green and red fluorescence channel images of cardiomyocytes. Representative confocal fluorescent micrographs are from three independent experiments. Scale bars: 20 µm. (D) Box plots represent Mander’s colocalization coefficient showing the extent of colocalization between Sigmar1 stained green pixels-to-Tom20 stained red pixels and number of colocalized voxels (volume data set containing volume elements consisting of the smallest unit within an image with precise information of measured fluorescence intensity) per cardiomyocytes. Image analysis was done on sequential z-stack confocal images of N > 40 adult mouse cardiomyocytes from three independent experiments. Boxes represent interquartile ranges, lines represent medians, and whiskers represent ranges. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To substantiate our observations of the presence of Sigmar1 in mitochondria in adult cardiomyocytes, we isolated mitochondria from adult mouse hearts. We labeled the active mitochondria with mitochondrial membrane potential sensitive MitoTracker Red dye (Fig. 4). Anti-Sigmar1 immunofluorescence staining reveals notable colocalization to Mitotracker red labeled mitochondria (Fig. 4A) as reflected in Mander’s colocalization coefficient values (Fig. 4C) and Pearson’s correlation coefficient (Fig. 4D) of colocalized pixels between Sigmar1-to-Mitotracker red in isolated cardiac mitochondria. Mitochondria isolated from Sigmar1 knockout mouse heart was used as a negative control and did not exhibit any distinguishable anti-Sigmar1 staining confirming the specificity of anti-Sigmar1 primary antibody staining (Fig. 4A). We used mitochondrial outer membrane protein anti-Tom20 and mitochondrial respiratory chain super complexes subunit-specific cocktail anti-OXPHOS immunofluorescence staining to confirm the isolated mitochondrial purity and entirety (Fig. 4B). Anti-OXPHOS staining showed significantly higher Mander’s colocalization coefficient to Mitotracker red-stained mitochondria than anti-Sigmar1-to-Mitotracker red (Fig. 4C). On the other hand, anti-Sigmar1 staining exhibited significantly higher Pearson’s correlation coefficient to Mitotracker red staining than anti-Tom20 and anti-OXPHOS staining, indicating the presence of similar intensity and closely existed anti-Sigmar1 stained pixels to Mitotracker red-stained pixels in isolated mitochondria (Fig. 4D). We also determined whether commercially available anti-Sigmar1 antibodies staining coincides with Mitotracker Red labeled cardiac mitochondria. We utilized a panel of anti-Sigmar1 antibodies raised against N-terminal and C-terminal amino acid sequences of Sigmar1 protein (Supplementary Fig. 1A). We used rabbit IgG as isotype control to assess the specificity of utilized primary antibodies staining (Supplementary Fig. 1B). Notably, we observed a comparatively similar degree of colocalization of all anti-Sigmar1 antibodies staining signals to Mitotracker Red labeled isolated cardiac mitochondria as evidenced in comparative Mander’s colocalization coefficient and Pearson’s correlation coefficient values except a C-terminal directed anti-Sigmar1 antibody speculatively due to antibody access to the targeted epitope of Sigmar1 against which the antibody has been developed and Sigmar1′s membrane topology(Supplementary Fig. 1C and Supplementary Fig. 1D). Altogether, our confocal immunofluorescence microscopic studies recapitulated Sigmar1′s localization to mitochondria in adult cardiomyocytes.
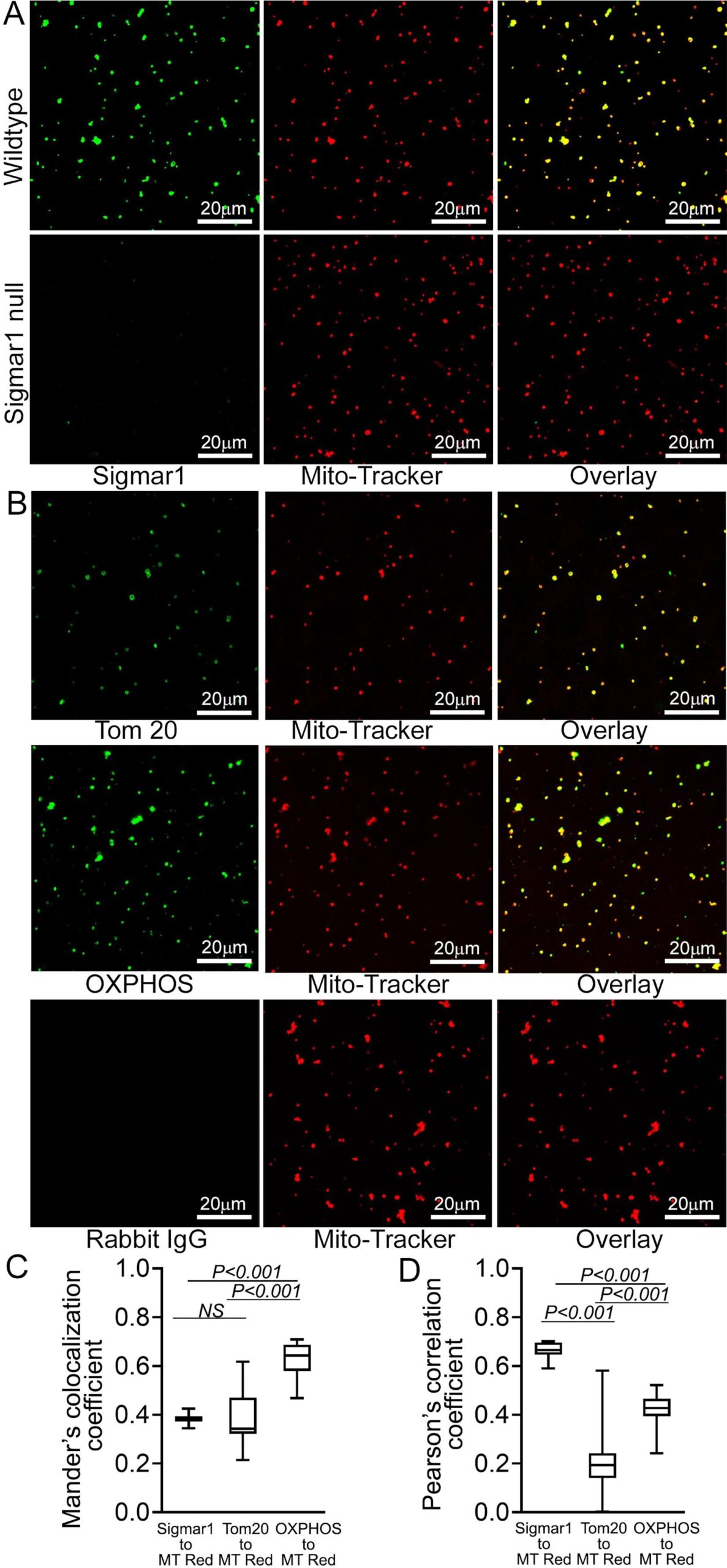
Sigmar1 localization was confirmed in isolated cardiac mitochondria. (A) Representative confocal images of anti- Sigmar1 antibody (green) and MitoTracker (red) loaded isolated mitochondria from adult mouse heart ventricles. Anti-Sigmar1 staining (green) showed high colocalization with MitoTracker red (red) staining appeared as yellow pixels in overlay images. Sigmar1 knockout mice heart ventricular tissue-derived mitochondria were used as a negative control to determine the primary antibody staining specificity. (B) Mitochondrial outer membrane integral protein anti-Tom20 and electron transport system respiratory supercomplexes subunit-specific cocktail anti-OXPHOS antibody staining are used to verify MitoTracker red labeled mitochondrial purity and entirety. Both anti-Tom20 (green) and anti-OXPHOS (green) staining show high colocalization to MitoTracker red, appearing in yellow pixels in overlay images. Rabbit IgG is used as an isotype control for anti-Sigmar1 rabbit primary antibodies. (C) Box plots representing Mander’s colocalization coefficient values illustrating fractions of anti-Sigmar1, anti-Tom20, and anti-OXPHOS (green) fluorescence colocalized towards corresponding MitoTracker Red fluorescence. (D) Box plots representing Pearson’s correlation coefficient values illustrating linearity of colocalized pixels intensity of anti-Sigmar1, anti-Tom20, and anti-OXPHOS (green) fluorescence pixels towards corresponding MitoTracker Red fluorescence. Representative images are from three independent experiments for each antibody tested. Scale bars: 20 µm. Boxes represent interquartile ranges, lines represent medians, and whiskers represent ranges. P values were determined by One-way ANOVA followed by Tukey’s multiple comparisons test. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2.3. Sigmar1 is an integral mitochondrial membrane protein
Proteins residing in the mitochondria can be either integral membrane proteins, peripheral proteins loosely bound to mitochondrial membranes, or amphitropic proteins localizing to mitochondria in response to stimuli. As anti-Sigmar1 labeled Q-dots appeared in the outer- and inner mitochondrial membranes, we further dissected the type of binding of Sigmar1 to mitochondrial membranes by alkaline extraction and dose-dependent Proteinase K enzyme digestion of isolated mitochondria. To confirm whether Sigmar1 resides as an integral mitochondrial membrane protein, we subjected the isolated cardiac mitochondria with Na2CO3(pH 11.5) to extract soluble and loosely bound peripheral proteins (Fujiki et al., 1982, Zhao et al., 2009). Sigmar1 remained in the mitochondrial pellet, whereas cytochrome c was released into the supernatant, suggesting Sigmar1 is an integral mitochondrial membrane protein (Fig. 5A). In control experiments, mitochondria treated with Triton-X 100 extracted all membrane-bound proteins to the supernatant fraction (Fig. 5A). To further dissect Sigmar1′s mitochondrial membrane localization, mitochondria were treated with increasing amounts of proteinase K (0.25 µg/ml to 2 µg/ml), which digests mitochondrial membrane proteins (Murakawa et al., 2015). Our immunoblot analysis revealed that Sigmar1 showed a dose-dependent digestion pattern similar to mitochondrial membrane proteins. We used OMM resident MAM proteins (MFN2 and DRP1), SR/ER-resident proteins (IRE1α, VCP, PDI), and cytosolic protein (Actin and GAPDH) as a control to demonstrate the purity of the mitochondrial fraction. Whole-cell lysate (60 µg protein) was used as a positive control (Fig. 5B). A high amount of proteinase K (25 µg/ml) treatment to isolated mitochondria completely digested Sigmar1 similar to mitochondrial membrane protein Tom20 (Fig. 5C). We also confirmed the enriched presence of Sigmar1 in human and rat ventricular heart tissue-derived purified mitochondrial fractions (Fig. 5D). Altogether, our complementary alkaline, detergent, and proteinase K enzyme-based digestion assays conceded that Sigmar1 is an integral mitochondrial membrane protein.
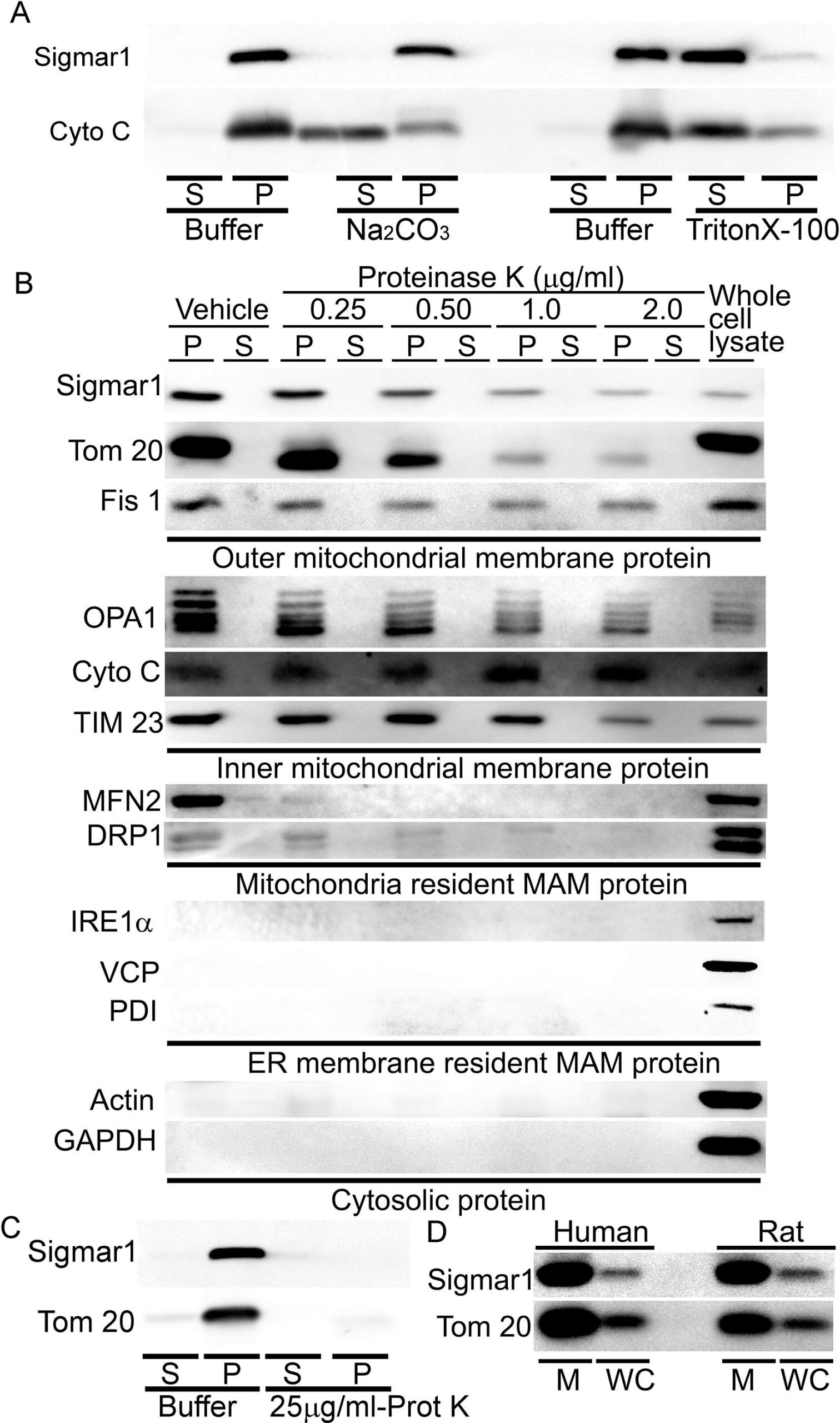
Sigmar1 is an integral mitochondrial membrane resident protein. (A) Representative Western blot images of anti-Sigmar1 and anti- cytochrome C (Cyto C) antibodies probed membranes following alkaline (Na2CO3, pH11.5) and detergent-based (10% v/v Triton X-100) extraction of mitochondrial proteins from isolated mitochondria of adult mouse hearts. S, supernatant fractions, P, mitochondrial pellet fractions. Western blot images are representative of three independent experiments using n = 3 individual adult mouse hearts. (B) Representative Western blot images of mitochondria pellet (P) and supernatant fractions (S) probed with labeled antibodies following dose-dependent Proteinase K enzyme-based digestion of isolated mitochondria from adult mouse hearts. We used OMM resident MAMproteins (MFN2 and DRP1), IMM proteins (OPA1 and TIM23), SR/ER-resident proteins (IRE1α, VCP, PDI), and cytosolic protein (Actin and GAPDH) as a control to demonstrate the purity of the mitochondrial fraction. The whole cell lysate was used as a positive control. (C) Representative western blot images of adult mouse heart-derived mitochondria pellet (P) and supernatant fractions (S) following buffer and Proteinase K (25 µg/mL) digestion. (D) Human and rat heart tissue-derived whole cell lysates and isolated mitochondria fractions immunoblotted with anti-Sigmar1 antibody. Tom20 was used to confirm the enrichment of mitochondrial fractions. M mitochondria, WC whole cell lysates. All western blot images represent n > 3 subcellular fractionation-based experiments using the biologically independent adult mouse, human, and rat hearts.
2.4. Sigmar1 N-terminal is required for incorporation to mitochondrial membranes
The crystal structure of Sigmar1 protein revealed a single transmembrane domainresiding at amino-terminal (N-terminal) with a flat membrane-associated carboxy-terminal (C-terminal) (Schmidt et al., 2016). To dissect the location of amino acid residues sequence required for mitochondrial membrane localization of Sigmar1, we conducted in silico and in vitro molecular studies (Fig. 6). Our in silico Kyte-Doolittle hydrophobicity plot showed the presence of hydrophobic amino acid (aa) residues that appeared as a prominent hydrophobic peak near the N-terminal of full-length Sigmar1 protein (Fig. 6A). Next, we exogenously expressed FLAG-tagged full length, N- and C-terminal truncation mutant Sigmar1 fragments in HEK293T cells. We confirmed the mutant construct’s expression by anti-FLAG immunoblotting in HEK293T cell lysates (Fig. 6B and 6C). Subsequently, exogenously expressed Sigmar1 fragments were detected with anti-FLAG (green) immunofluorescence staining (Fig. 6D and 6E). We found strong colocalization of full-length Sigmar1 with gradual attrition with C-terminal truncation fragments of Sigmar1 (aa1-169 and aa1-101) to Mitotracker red-stained mitochondria (Fig. 6D). Interestingly, N-terminal truncated fragments of Sigmar1 aa102-223 appeared as diffuse puncta and failed to exhibit discernible colocalization to Mitotracker red-stained mitochondria (Fig. 6D). We also observed a similar trend of colocalization between anti-FLAG stained Sigmar1 fragments-to-ER tracker staining with lesser colocalization with N-terminal truncated Sigmar1 fragment (Fig. 6E). These observations were reflected in Mander’s colocalization coefficient values of anti-FLAG stained Sigmar1 constructs to Mitotracker red and ER tracker blue fluorescence signals (Fig. 6F and 6G). Altogether, our data indicate Sigmar1′s N-terminal domain is required for mitochondrial membrane incorporation and stability.
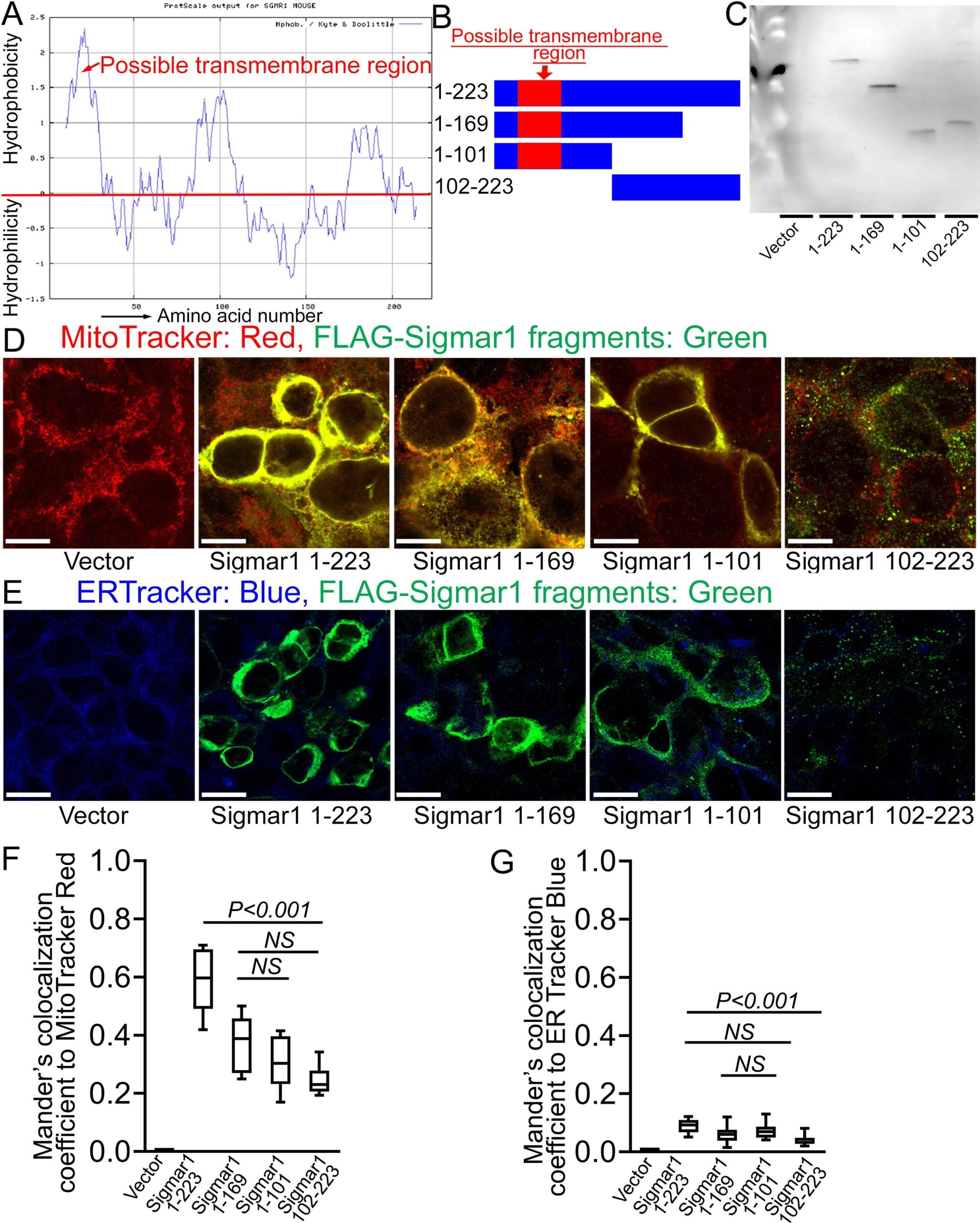
Sigmar1′s N-terminal possesses amino acid residues required for mitochondrial membrane localization. (A) Kyte and Doolittle hydrophobicity plot of full-length mouse Sigmar1 protein’s amino acid sequence (graphed using ProtScale, Expasy). The hydrophobicity plot shows a distinct hydrophobic peak residing at Sigmar1′s N-terminal (indicated with red arrow) indicative of a putative transmembrane domain. (B) Schematic diagram showing full-length and Sigmar1 fragments. Full-length Sigmar1 amino acids (aa) 1–223, C-terminal truncation fragment of Sigmar1 (aa1-169 and aa1-101) constructs containing putative transmembrane domain (amino acid residues 11–30) at Sigmar1′s N-terminal sequence. N-terminal deletion fragment of Sigmar1 aa102-223 construct lacking N-terminal residing putative transmembrane region. (C) Representative anti-FLAG antibody labeled Western blotimages demonstrating expression of FLAG-tagged full length and Sigmar1 fragment plasmid constructs in HEK293T cells. Western blot images are from n = 5 independent transfection experiments in HEK293T cells. (D) Representative confocal fluorescence images of MitoTracker red (red) and anti-FLAG antibody (green) stained HEK293T cells. Sigmar1 fragments possessing putative transmembrane domain at N-terminal (Sigmar1 aa1-223, Sigmar1 aa1- 169, Sigmar1 aa1-101) expression (anti-FLAG, green) showed significant colocalization with MitoTracker Red stained mitochondria (yellow pixels in represented merged green and red fluorescent channel images). In contrast, N-terminal lacking Sigmar1 aa102-223 construct expression showed diffused puncta (green) with lesser colocalization with MitoTracker red-stained mitochondria than full length and C-terminal truncation mutant Sigmar1 constructs. (E) Representative confocal fluorescence microscope images of ER-Tracker blue (blue) and anti-FLAG antibody (green) stained HEK293T cells. FLAG-tagged full length and Sigmar1 fragment coding plasmids showed little co-localization in anti-FLAG (green)-to-ER-Tracker (blue) overlay images. Representative images are from three independent experiments. Scale bars: 5 µm. (F)–(G) Box plots represent Mander’s colocalization coefficient illustrating fractions of anti-Sigmar1 (green) fluorescence colocalized towards corresponding MitoTracker Red and ER Tracker Blue fluorescence. Image analysis results are from N > 100 cells for each group of cells. Boxes represent interquartile ranges, lines represent medians, and whiskers represent ranges. P values were determined by One-way ANOVA followed by Tukey’s multiple comparisons test. NS not significant. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2.5. Sigmar1 is required for physiological mitochondrial respiration
To determine whether Sigmar1 plays any functional role in mitochondria, we utilized neonatal rat ventricular cardiomyocytes (NRCs), knocked down Sigmar1 by siRNA transfection, and performed mitochondrial respiration on intact cardiomyocytes. We confirmed a nearly complete ablation of Sigmar1 protein in NRCs by siRNA transfection at 5 days after siRNA transfection (Supplementary Fig. 2). Mitochondrial oxygen consumption rates (OCRs) were measured in control siRNA (negative siRNA) and Sigmar1 siRNA knockdown cardiomyocytes using the Seahorse extracellular flux analyzer (Fig. 7A). Real‐time OCRs in intact cardiomyocytes showed that basal respiration, representing the sum of all physiological mitochondrial oxygen consumption, decreased in the Sigmar1 knockdown cardiomyocytes, indicating lower respiratory function compared with control siRNA groups (Fig. 7B). The injection of oligomycin leads to a decrease in basal respiration that is reflective of oxygen consumption used to generate ATP and was not different in between the two groups (Fig. 7C). The addition of FCCP uncouples respiration from oxidative phosphorylation and allows the measurement of maximal OCR, which was lower in Sigmar1 knockdown cardiomyocytes, indicating lower overall mitochondrial activity (Fig. 7D). The extent of nonmitochondrial oxygen‐consuming processes measured by inhibiting the respiratory chain with rotenone and antimycin A, was significantly greater in Sigmar1 siRNA knockdown cardiomyocytes (Fig. 7E). Further, mitochondrial respiratory reserve capacity (Fig. 7F) as calculated by subtracting basal OCR from FCCP‐stimulated OCR and ATP turnover (Fig. 7G) measured by nonmitochondrial respiration subtracted from the ATP‐linked OCR was significantly attenuated in Sigmar1 knockdown cardiomyocytes. The maximal respiration yielded by nonmitochondrial respiration subtracted from FCCP‐stimulated OCR was also significantly reduced in Sigmar1 knockdown cardiomyocytes (Fig. 7H). The proton leak through the inner mitochondrial membrane, represented by nonmitochondrial respiration subtracted from the post-oligomycin OCR, was unchanged across the treatments (Fig. 7I). In summary, we found that the absence of Sigmar1 compromised mitochondrial bioenergetics profiles in cardiomyocytes.
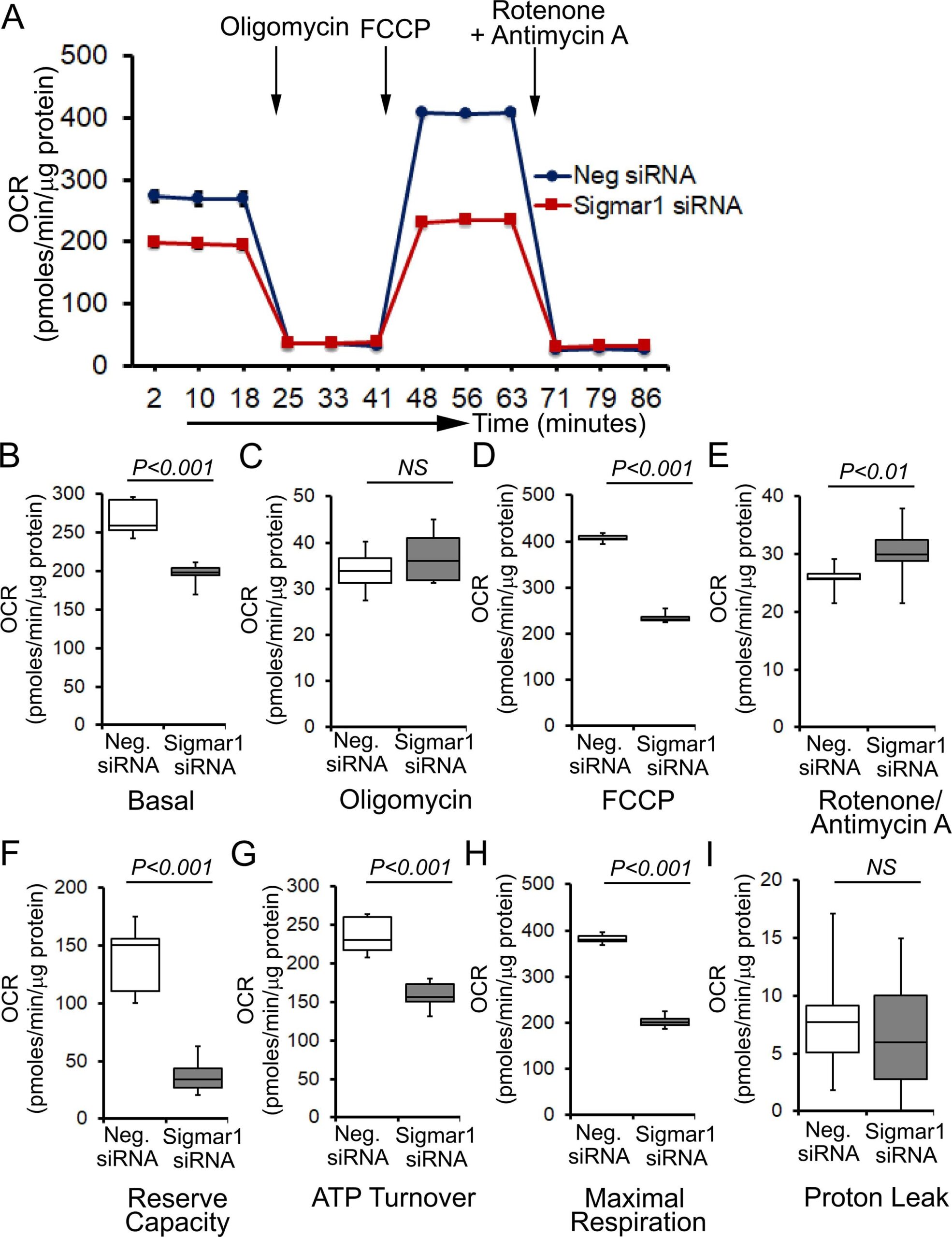
Genetic ablation of Sigmar1 suppresses mitochondrial respiration in cardiomyocytes. (A) Line graphs represent mitochondrial oxygen consumption rates (OCRs) in intact neonatal rat cardiomyocytes treated with control siRNA (Neg. siRNA) and Sigmar1 directed siRNA. Black arrows indicate sequential addition of Oligomycin (1 µM), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) (4 µM), and Rotenone (0.5 µM) plus Antimycin A (0.5 µM). OCR values are expressed as picomoles O2 per minute per µg protein. Each point represents average OCR values from five wells per treatment in two independent experiments. Box plots represent OCRs at (B) baseline as well as with the addition of (C) oligomycin, (D) FCCP, and (E) rotenone plus antimycin A addition. Key mitochondrial respiratory parameters, including (F) reserve capacity, (G) ATP turnover, and (H) maximal respiration was significantly attenuated in Sigmar1 siRNA treated cardiomyocytes compared to control siRNA treated cardiomyocytes. (I) Proton leak was unchanged between the two groups of cardiomyocytes. Boxes depict interquartile ranges, lines represent medians, and whiskers represent ranges. Pvalues were determined by the nonparametric Kolmogorov-Smirnov test. Neg. negative, siRNAsmall interfering RNA, NS not significant.
Next, we determined whether defects in physiological fuel substrates supported mitochondrial respiration were linked to the compromised bioenergetics profiles observed in Sigmar1 knocked down cardiomyocytes. We subjected Sigmar1 siRNA knockdown cardiomyocytes to high-resolution respirometry using Oroboros O2k-chamber. Control siRNA-treated cardiomyocytes served as an experimental control. To achieve convergent electron flow through complex I and complex II, NADH (N)-generating substrates (donate electrons to complex I, CI), i.e., pyruvate, malate, glutamate, and succinate (S) (activates complex II, CII), were sequentially added under saturating ADP concentration to achieve oxidative phosphorylation (OXPHOS) state (Fig. 8A). Next, Oligomycin, an ATP synthase (Complex V) inhibitor, is injected to measure complex I + II-linked leak respiration in the presence of fuel substrates and excess adenylates (Fig. 8A). FCCP is injected to uncouple mitochondrial respiration from OXPHOS, allowing measurement of maximal respiration. Mitochondrial respiratory complex I inhibitor (rotenone) and complex III inhibitor (antimycin A) were injected at the end to measure residual oxygen consumptions exclusive of mitochondrial respiration (Fig. 8A). Sigmar1 knockdown in cardiomyocytes ensues in reduced oxygen flux when supplied with NADH-linked substrates pyruvate, malate, and glutamate under both basal and OXPHOS state (in the presence of excess ADP) (Fig. 8B and 8C). The suppression of oxygen flux persisted when cardiomyocytes were supplied with succinate to activate convergent electron flow in the mitochondrial electron transfer system through NS-pathway (Fig. 8D). FCCP-stimulated uncoupled maximal respiration linked to CI + CII-linked oxygen flux, and ATP turnover rate was significantly attenuated in Sigmar1 siRNA-treated cardiomyocytes compared to control cardiomyocytes (Fig. 8E and 8F). We have not found any significant difference in non-mitochondrial residual oxygen flux between treatment groups (Fig. 8G). Overall, our data suggest the essential role of Sigmar1 in maintaining physiological mitochondrial fuel substrates-supported OXPHOS-linked respirations.
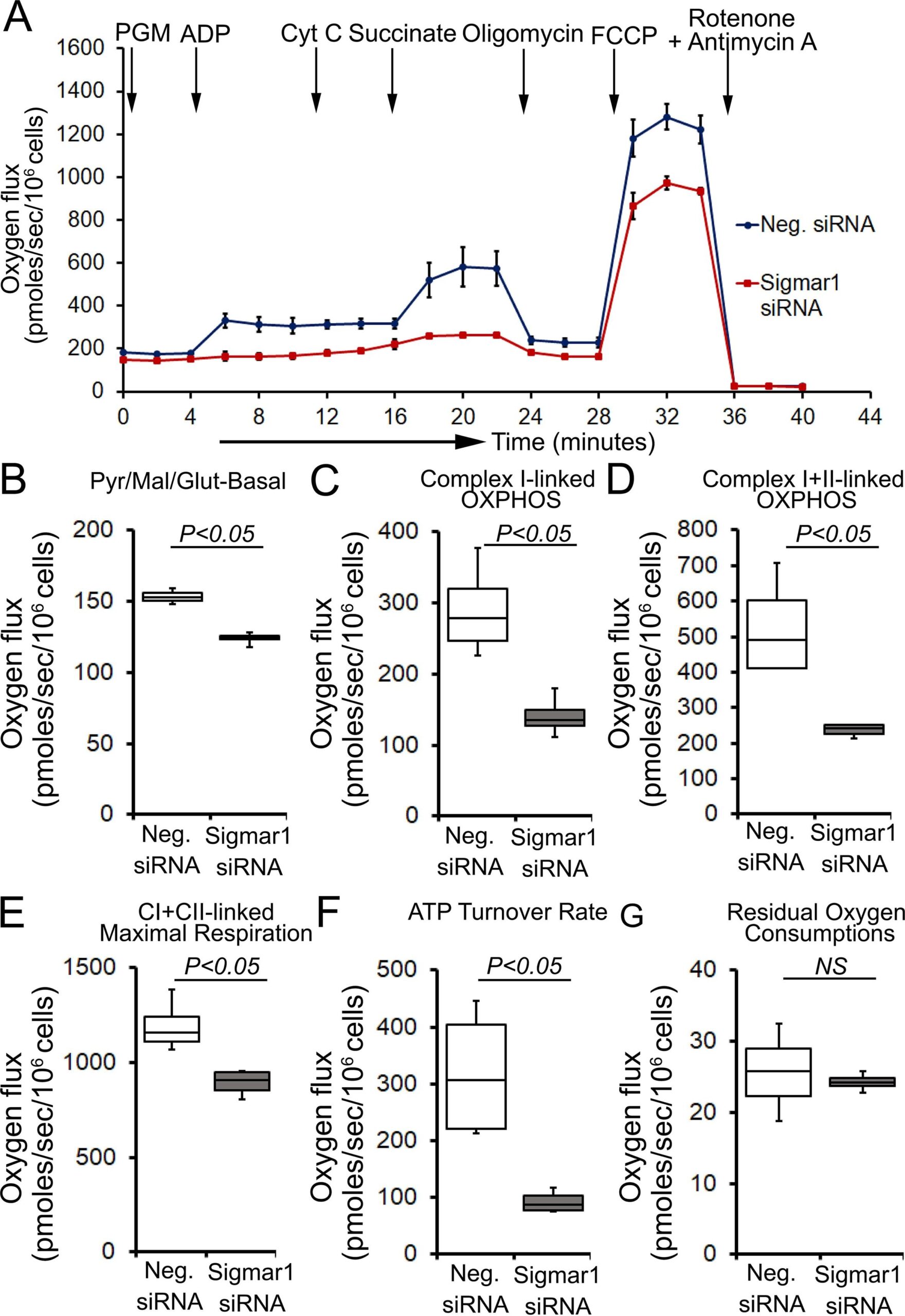
Sigmar1 knockdown attenuates fuel substrates supported OXPHOS in cardiomyocytes. (A) Representative oxygraph constituting temporal real-time oxygen flux in Saponin (15 µg/ml) permeabilized neonatal rat cardiomyocytes treated with control siRNA and Sigmar1 targeted siRNA. NADH (N)-linked substrates pyruvate (P) (4 mM), glutamate (G) (10 mM), and malate (M) (4 mM) were injected to induce electron flow at complex I of electron transfer system (ETS). ADP (2 mM) was injected to attain complex I (CI) supported oxidative phosphorylation (OXPHOS) state. Cytochrome C (10 µM) was injected to ensure outer mitochondrial membraneintegrity. Subsequently, succinate (S) (20 mM) was injected to induce convergent electron flow through the NS pathway and to measure CI + CII-linked OXPHOS. Mitochondrial proton motive force uncoupler FCCP (4 µM) was injected following oligomycin (1 µM) to measure CI + CII-linked maximal respiration. Rotenone (1 µM) and antimycin A (2 µM) were co-injected to measure non-mitochondrial oxygen fluxes. Black arrows indicate the site of substrates and inhibitors injection. Each point represents average oxygen fluxes from four independent experiments per treatment group. Oxygen fluxes are expressed as picomoles (pmoles) O2/[sec*106 cells]. Data points are expressed as mean ± SEM. (B)–(G) Box plots represent oxygen fluxes following supplementation of (B) pyruvate, malate, and glutamate (PGM) at baseline, (C) complex I-linked OXPHOS, (D) complex I + II-linked OXPHOS, (E) FCCP stimulated CI + CII-linked maximal respiration, (F) ATP turnover rate constituting oxygen flux after subtracting post-Oligomycin oxygen flux from CI + CII-linked OXPHOS state oxygen flux under available fuel substrates and excess ADP, and (G) residual oxygen consumptions (ROX) following Rotenone and Antimycin A injection. The mitochondrial respiratory parameters were calculated by subtracting ROX values in the respective experiment to attain mitochondrial respiration-specific oxygen fluxes in cardiomyocytes. Boxes represent interquartile ranges, lines represent medians, and whiskers represent ranges. P values were determined by the nonparametric Kolmogorov-Smirnov test. Neg. negative, siRNA small interfering RNA, Pyr pyruvate, Mal malate, Glut Glutamate, CI complex I, CII complex II, NS not significant.
3. Discussion
Earlier studies to characterize Sigmar1 subcellular localization are mostly limited to brain, retina, and spinal cord sections, primary neuronal cultures, and neuroblastoma cell lines. Unlike other cell systems, the heart is a special organ composed of cardiomyocytes requiring a constant energy supply for their proper functioning. Cardiac myocytes contain nearly 30% to 40% of their volume with highly organized mitochondria residing at subsarcolemmal spaces, interfibrillar spaces, and perinuclear areas (Hollander et al., 2014). In contrast, cardiomyocytes contains approximately 3.5% of sarcoplasmic reticulum (SR) (Katz, 2011). The striated muscle SR is a specialized ER subcompartment in which general ER markers (e.g. Bip and calnexin) coexist with the major SR proteins specifically responsible for Ca2+ uptake, storage, and release (Katz, 2011, Volpe et al., 1992). The SR surrounds the myofilaments and functions in collaboration with deep invaginations of the sarcolemma (transverse (t)-tubules) to activate myocyte contraction by releasing calcium from SR. Despite the well documented presence of SR in cardiomyocytes, functional relationship between the ER and SR in cardiomyocytes has remained confusing. As the literature supports Sigmar1′s localization in the ER and ER-associated mitochondrial membranes in cells containing ER-network, the lack of similar structures in cardiomyocytes provided a compelling rationale to study Sigmar1 function in the heart. Nonetheless, to our best knowledge, Sigmar1′s subcellular distribution and organelle-specific functions in cardiac myocytes have been largely overlooked in the literature.
In the present study, we demonstrated Sigmar1′s localization in the mitochondrial membranes, lysosomes and SR/ER to mitochondrial interface using Q-dots TEMand subcellular fractionation of heart cell lysates. As mitochondria comprise the majority of the cell volume of cardiomyocytes and play a crucial role for the normal functioning of the heart, we focused on elucidating the molecular role of Sigmar1 in mitochondria in the cardiomyocytes. We confirmed the Sigmar1 mitochondrial localization by quantifying the extent of endogenous Sigmar1 partitioning to mitochondria by co-immunostaining isolated adult cardiac myocytes with anti-OXPHOS, anti-Tom20, and anti-Sigmar1 antibodies followed by confocal immunofluorescent Z-stack imaging. Mander’s colocalization coefficient and the number of colocalized voxels confirmed Sigmar1 colocalizes to OXPHOS and Tom20 stained mitochondria in adult cardiac myocytes. This was further confirmed by Mitotracker red labeled isolated cardiac mitochondria to anti-Sigmar1 staining. The accuracy of the Sigmar1′s immunostaining on isolated mitochondria was validated using commercially available N-terminal and C-terminal targeted Sigmar1 antibodies. Both N-terminal and C-terminal targeted Sigmar1 antibody stained endogenous Sigmar1 exhibited comparable values of colocalization parameters (Mander’s and Pearson’s coefficients) to mitochondria with relatively smaller values when stained with C-terminal targeted antibodies. These observations indicate plausible differential membrane tropisms of Sigmar1’s N-terminal and C-terminal amino acid residues. Along with these observations, series of biochemical experiments using alkaline extraction and proteinase K treatment of purified mitochondria demonstrated Sigmar1 as an integral mitochondrial membrane protein.
Earlier studies reported Sigmar1 localization in mitochondrial membranes, vesicles, or elongated cisternae of ER in the hypothalamus and hippocampus in rats (Alonso et al., 2000). In contrast, exogenously expressed GFP-tagged Sigmar1 showed plasma membrane localization in Xenopus Oocytes and colocalized to Kv1.4 potassium channel (Aydar et al., 2002). Sigmar1 was also detected in perinuclear areas, plasmalemma regions of cell–cell contact, growth cones of neurites in mouse neuroblastoma NG-108-15 cells, focal adhesion contacts in Chinese hamster ovary (CHO)-K1 cells (Mavlyutov and Ruoho, 2007). Interestingly, Sigmar1 was detected in ER-associated detergent-insoluble lipid droplets in NG-108 cells and rat hippocampal differentiated oligodendrocytes (Hayashi and Su, 2003a, b). Notably, Sigmar1 consistently appeared in isolated mitochondria fractions and colocalized to mitochondria along with mitochondria-associated ER membranes (MAM) in CHO cells (Hayashi and Su, 2007, Mori et al., 2013). Intriguingly, Sigmar1 was found on ER and nuclear envelope (NE) and showed regulating chromatin remodelingcomplex on NE in NG-108 and Neuro-2a cells (Tsai et al., 2015). Mouse retinal photoreceptor cells exhibited predominant Sigmar1 expression on NE and sparse distribution at plasma membrane subsurface ER cisternae but not on plasma membrane itself (Mavlyutov et al., 2015a). These observations were further extended in NSC34 neuronal cells and human retinal pigment epithelial ARPE-19 cells where Sigmar1 localization was found on nucleoplasmic reticulum and ER (Mavlyutov et al., 2017b). On the other hand, Sigmar1 localization was detected on the plasma membrane, ER, and NE in mouse and rat dorsal ganglion cells (Mavlyutov et al., 2016). Subsequent studies from the same author groups reported the presence of Sigmar1 in ER, MAM, and plasma membrane subsurface ER cisternae but not at the plasma membranes in primary rat DRG neurons utilizing C-terminal tagged EGFP-APEX2-Sigmar1 fusion construct adenoviral-mediated gene delivery to rat (Mavylutov et al., 2018). Altogether, these studies indicate cell-specific diverse subcellular localization of Sigmar1 and suggest organelle-specific molecular functions of Sigmar1 under physiological and pathophysiological condition.
Accumulating studies corroborate Sigmar1 as an integral membrane protein. However, its membrane topology remains elusive due to divergent observations in literary reports. Initial studies characterized Sigmar1 with only one transmembrane domain at 80–100 amino acid (aa) sequence that reverberates in later hydrophobicity analysis, confirming a single-pass transmembrane topology of Sigmar1 (Hanner et al., 1996, Kekuda et al., 1996, Seth et al., 1997). However, another bioinformaticsTMbase platform yielded two TM domains at 10–30 aa and 80–100 aa sequence of Sigmar1 (Aydar et al., 2002). In the same study, authors reported that N- and C-terminal GFP-tagged Sigmar1 fusion protein immunofluorescence signal could only be recovered following 0.5% acetone permeabilization in Xenopus oocytes (Aydar et al., 2002). These observations indicate Sigmar1 N- and C-termini are inaccessible from outside the cells, and both termini reside at the cytoplasm. In contrast, based on endogenous Sigmar1 immunostaining using N- and C-terminal targeted antibodies in CHO cells, studies also showed that both N- and C-terminal of Sigmar1 might reside in ER lumen as immunofluorescence signals can only be observed following CHAPS or Triton X-100 mediated cell permeabilization (Hayashi and Su, 2007). These apparent discrepancies in the Sigmar1 tropism might be due to altered membrane insertion of GFP-fused Sigmar1 protein and/or cell type-specific differences in Sigmar1 localization. To this point, C-terminal EYFP-tagged Sigmar1, not N-terminal EYFP-tagged Sigmar1, exhibited a distribution pattern as seen for endogenous Sigmar1 immunostaining (Hayashi and Su, 2007). Moreover, recently reported crystal structure of purified full-length human Sigmar1 protein reconstituted into lipidic cubic phase consisting of lipid: protein at a ratio of 1.5:1.0. The crystal structure of Sigmar1 in this artificially reconstituted membrane exhibited a short single transmembrane domain residing at N-terminal facing towards ER lumen. In contrast, the C-terminal domain is oriented to the cytosolic side (Schmidt et al., 2016). The notion of a single transmembrane domain in Sigmar1 further supported based on GFP-ascorbate peroxidase 2 (APEX2)-tagged Sigmar1 fusion protein immuno-electron microscopy pattern in ND9/27 cells suggesting transmembrane domain lies between amino acids 1–80 (Mavylutov et al., 2018). However, utilizing N-terminal and C-terminal GFP-APEX2 tagged Sigmar1 fusion constructs overexpression studies in ND7/23 cells, studies also reported that electron-dense precipitates observed in the cytosol in case of N-terminal tagged Sigmar1 constructs while C-terminal tagged constructs give rise to electron-dense precipitates in the ER lumen, MAMs and plasma membrane subsurface ER cisternae in immune-electron micrographs (Mavylutov et al., 2018). These observations suggested reversed topology of Sigmar1 in contrast to its reported crystal structure model with cytosol facing N-terminal and ER lumen facing C-terminal.
To dissect out amino acid residues required for Sigmar1 mitochondrial localization, we expressed flag-tagged full-length Sigmar1, C-terminal, and N-terminal deletion mutant Sigmar1 constructs in HEK293T cells. In consort to our and earlier published in silico observations of a hydrophobic, N-terminal transmembrane domain in Sigmar1, N-terminal deletion mutant (102–223 aa) exhibited diffused dots in transfected HEK cells with significantly smaller values for Mander’s colocalization coefficient compared to C-terminal deletion mutants. These findings underscore the presence of N-terminal residing membrane anchoring transmembrane domain in 1–101 amino acid residues consistent with recent reports. We observed a similar pattern when ER was stained with ER Tracker blue in HEK293T cells, although Mander’s colocalization coefficient values were lower than Mitotracker red. While preparing the current manuscript, a recent study also reported exogenously expressed GFP-tagged Sigmar1 fusion protein colocalization to mCherry-tagged Sec61β protein in HEK293T cells, where protein transport protein Sec61 subunit beta (Sec61β) is used as ER marker protein (Zhemkov et al., 2021). Although authors used Tom20 as mitochondria marker, Mander’s colocalization coefficient of Sigmar1-to-Tom 20 stained fluorescent signals was overlooked. The differing Mander’s colocalization values of Flag-tagged Sigmar1-to-ER Tracker Blue in our study and the reported GFP-tagged-Sigmar1-to-mCherrySec61β fluorescent signals in that study might arise from multiple reasons. First, the different labeling procedure used in these studies, such as we used antibody and protein tagging free ER specific dye to label endogenous ER compartment, where as Zhemkov et al used mCherry-tagged exogenously expressed Sec61β protein to label ER. Second, physiological interactions between Sigmar1 and Sec61β protein may also contribute to colocalization signal as their endogenous interactions have not been considered. Third, intrinsic limitations of fluorescence microscopes resolution yielding slightly greater overlap coefficient values due to a large molecular mass of GFP (about 27 kDa) and mCherry (28 kDa) tagging themselves, resulting in greater spreading of fluorescence signals in digital micrographs. Nonetheless, we were able to demonstrate that Sigmar1 N-terminal possesses the transmembrane domain required for membrane localization.
Cardiac myocytes’ physiological and functional homeostasis depends on mitochondrial oxidative phosphorylation (OXPHOS) (Zhou and Tian, 2018). To determine whether Sigmar1 imparts any functional significance in mitochondrial OXPHOS, we conducted extracellular flux analysis and high-resolution respirometry analysis in neonatal rat cardiac myocytes following Sigmar1 knockdown. Mitochondrial stress test assay in intact isolated cardiac myocytes utilizing cell-permeable inhibitors and uncouplers revealed significantly reduced bioenergetic profiles in Sigmar1 knockout cardiac myocytes than the control group of cardiac myocytes. These data indicate the absence of Sigmar1 in cardiac myocytes negatively affects mitochondrial electron transport system (ETS) linked respiration. To determine specific fuel substrate linked ETS supported oxygen flux, we conducted high-resolution respirometry in permeabilized cardiac myocytes supplemented with Complex I and Complex II linked substrates in the presence of saturating ADP concentrations to achieve OXPHOS state. Notably, Sigmar1 knockdown in cardiomyocytes resulted in significant attenuation of mitochondrial Complex I and Complex II-linked OXPHOS in the presence of NADH-linked substrates, i.e., pyruvate, malate, glutamate, and succinate under saturating ADP concentrations. This compromised oxygen flux ensued in reduced maximal respirations following FCCP-induced uncoupling of mitochondrial respirationfrom intermembrane proton motive force in Sigmar1 knockdown cardiomyocytes. These data underscore the necessity of Sigmar1 in maintaining physiological mitochondrial respirations in cardiomyocytes.
Overall, our current study revealed novel subcellular localization and organelle-specific functions of Sigmar1 in cardiac myocytes. We discovered that Sigmar1 is an integral mitochondrial membrane protein in the mammalian heart through complementary biochemical, immunohistochemistry, immunocytochemistry, and immune-electron microscopic study. Through a genetic loss-of-function approach by knocking down Sigmar1 in primary cardiomyocytes, we determined the essential functions of Sigmar1 in maintaining physiological mitochondrial fuel substrates-linked respirations. Hence, our findings corroborate novel Sigmar1-dependent mitochondrial electron transfer system (ETS)-linked respirations. Altogether, our results laid the foundation of novel molecular functions of Sigmar1 in mitochondrial physiological functions. In future studies, we aim to employ biochemical and proteomics approaches to discover novel associate protein partners of Sigmar1 in regulating physiological mitochondrial structure and functions to pave rational drug design targeting Sigmar1 in cardiovascular diseases.