
Targeting Molecular Mediators of Ferroptosis and Oxidative Stress for Neurological Disorders

By Jing Li, Bowen Jia, Ying Cheng, Yiting Song, Qianqian Li, and Chengliang Luo
Excerpt from the article published in Oxidative Medicine and Cellular Longevity, Volume 2022, Article ID 3999083, Published 22 Jul 2022, DOI: https://doi.org/10.1155/2022/3999083
Editor’s Highlights
- Oxidative stress and neurological disorders are closely related to the role of mitochondria.
- Ferroptosis may be a key mechanism to study the connection.
- Ferroptosis is an iron-dependent and nonapoptotic cell death mode, which is caused by excessive accumulation of ROS and imbalance of cell lipid oxide metabolism.
- Ferroptosis occurs earlier than apoptosis in the development of Parkinson’s disease (PD).
- Ferroptosis plays an important role in AD, in which a variety of regulatory factors are involved, and provide many potential therapeutic targets for the treatment of AD.
- There is a close connection between ferroptosis and Huntington’s disease (HD) which would provide an important potential target for the treatment of HD.
- Sigma-1 receptor (sig-1R) is one regulator that can be used to treat or relieve the symptoms of neurological disorders.
- Sigma-1 receptor is a mitochondrial endoplasmic reticulum chaperon that can regulate cell pathophysiological processes.
Abstract
As a novel mode of regulated cell death (RCD), ferroptosis was firstly proposed by Dixon in 2012 [1]. With the acceleration of population aging, nervous system diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), anxiety, depression, stroke, and traumatic brain injury (TBI) have become a huge burden on families and society. The mechanism of neurological disorders is complex, which also lacks effective treatment, so relevant research is required to solve these problems urgently. Given that oxidative stress-induced lipid peroxidation eventually leads to ferroptosis, both oxidative stress and ferroptosis are important mechanisms causing neurological disorders, targeting mediators of oxidative stress and ferroptosis have become a hot research direction at present. Our review provides a current view of the mechanisms underlying ferroptosis and oxidative stress participate in neurological disorders, the potential application of molecular mediators targeting ferroptosis and oxidative stress in neurological disorders. The target of molecular mediators or agents of oxidative stress and ferroptosis associated with neurological disorders, such as reactive oxygen species (ROS), nuclear factor erythroid 2–related factor-antioxidant response element (Nrf2-ARE), n-acetylcysteine (NAC), Fe2+, NADPH, and its oxidases NOX, has been described in this article. Given that oxidative stress-induced ferroptosis plays a pivotal role in neurological disorders, further research on the mechanisms of ferroptosis caused by oxidative stress will help provide new targets for the treatment of neurological disorders.
1. Introduction
As a novel mode of regulated cell death (RCD), ferroptosis was firstly proposed by Dixon in 2012 [1]. Ferroptosis is an iron-dependent and nonapoptotic cell death mode, which is caused by excessive accumulation of ROS and imbalance of cell lipid oxide metabolism. In 2018, the Nomenclature Committee on Cell Death (NCCD) advised defining ferroptosis as RCD caused by oxidative stress in the cell microenvironment which can be regulated by glutathione peroxidase 4 (GPX4). Moreover, ferroptosis can be inhibited by iron chelators, iron intake inhibitors, and lipophilic antioxidants [2, 3]. Recent studies have shown that ferroptosis is obviously different from traditional programmed cell death. It has distinctive morphological features and biochemical features [4], such as morphological changes of mitochondrial, accumulation of iron, and lipid reactive oxygen species (ROS). At present, it is believed that the main cause of ferroptosis to cell death is the inactivation of the cellular antioxidant system, which includes the inhibition of cystine/glutamate antiporter system (system Xc–) and GPX4, disrupted iron homeostasis, and lipid peroxidation. Ultimately, the decrease of antioxidant capacity and the accumulation of intracellular lipid ROS lead to oxidative cell death [5].
Although there are many studies about ferroptosis, the mechanism of ferroptosis is not clear yet. Ferroptosis was identified to involve in oxidative stress-induced cell death [6]. During the process of ferroptosis, cellular over-accumulation of ROS eventually generates oxidative stress and cell death. According to current research, the possible mechanisms of ferroptosis include oxidative stress, lipid peroxidation, iron metabolism, and other mechanisms to be explored. The process of oxidative stress in ferroptosis is complex that is regulated by a variety of regulatory factors.
Neurological disorders have become the major leading cause of disability and the second leading cause of death worldwide [7]. Over the past few decades, due to the lack of effective precaution and treatment, the mortality and the disabled owing to neurological diseases have increased, particularly in low-income and middle-income countries [8]. The death and disability caused by neurological disorders have caused a huge burden on families and countries. Meanwhile, it has become a serious social problem. In order to alleviate the burden, there are increasing studies on how to treat or prevent the process of neurological disorders.
Mounting evidence has shown that oxidative stress-induced ferroptosis plays a significant role in neurological disorders [3]. Oxidative stress is both an important process of ferroptosis and the pathogenesis of neurological disorders. Hence, oxidative stress has become a critical linking between ferroptosis and neurological disorders. Up to now, the inhibitors and activators of ferroptosis [9–14], as well as other involved signaling pathways [15–17], have been widely studied. Furthermore, the mediators of oxidative stress might become a potential therapeutic target for the treatment of neurological disorders by regulating the process of ferroptosis. In this review, we will briefly summarize the mechanisms of ferroptosis, and highlight the role of targeting molecular mediators of oxidative stress in neurological disorders, so as to provide an option for the therapeutic application of ferroptosis in neurological disorders.
2. The Mechanism of Ferroptosis
Ferroptosis is a unique kind of nonapoptotic-regulated cell death, which is distinct from apoptosis, autophagy, and necrosis in morphology, biochemistry, and genetics. Iron metabolism and lipid peroxidation are two essential events in ferroptosis [2]. Additionally, system Xc– and GPX4 are considered to be the primary signaling pathways [18]. Here, we summarize the main mechanisms involved in the process of ferroptosis, such as oxidative stress, lipid peroxidation, iron metabolism, and the signaling pathways (Figure 1).
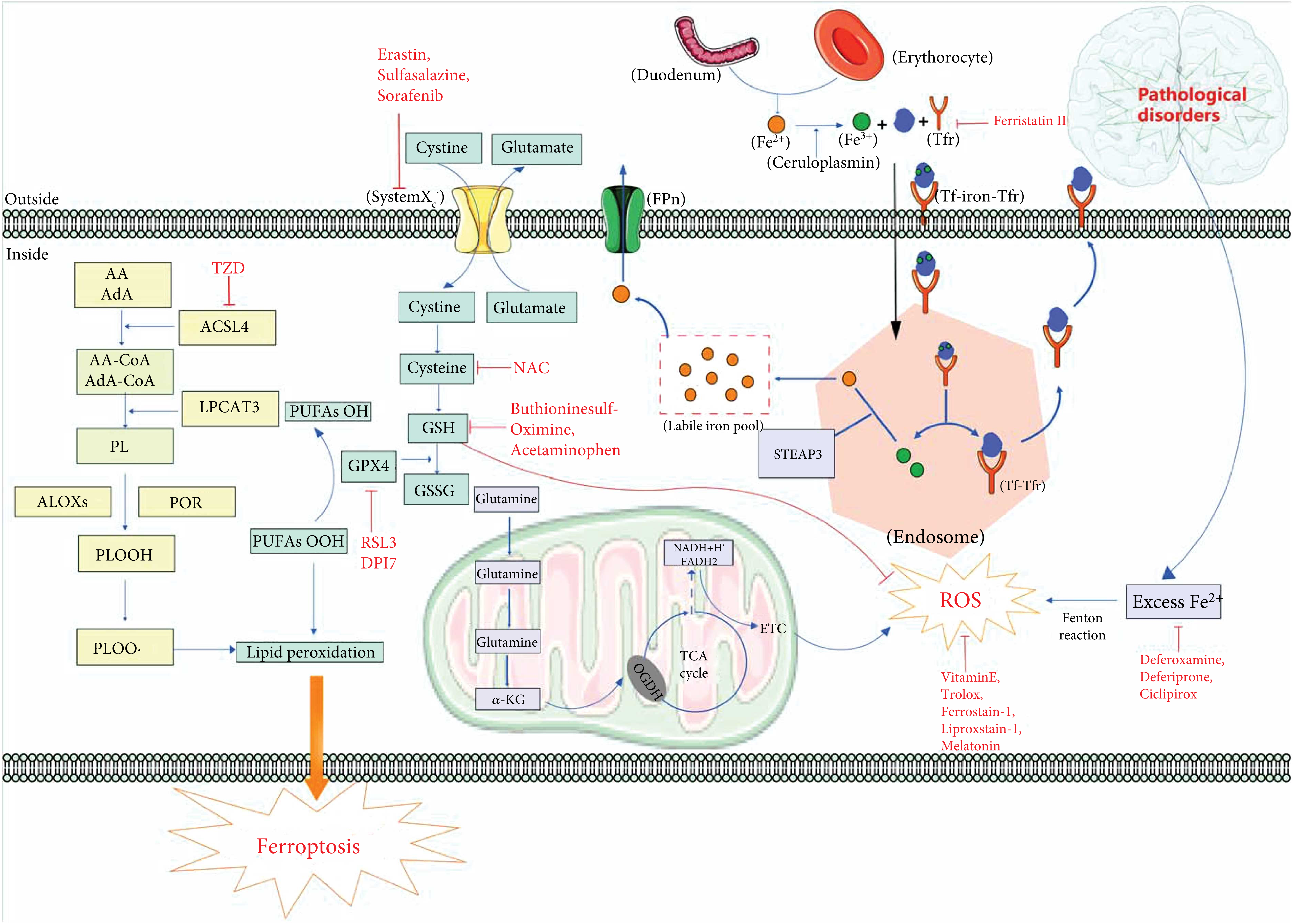
A schematic summary of ferroptosis mechanisms in neurological disorders. Lipid peroxidation and iron homeostasis are currently recognized as important mechanisms affecting ferroptosis. In cells, under the action of ACSL4, PRO, and ALOX, PUFA generates PLOOH through a series of biochemical reactions, which then generates PLOO, leading to lipid peroxidation and eventually ferroptosis. GPX4 can reduce PLOOH to PLOH to inhibit lipid peroxidation. In addition, GPX4 is regulated by the cofactor GSH. When GSH is exhausted, GPX4 will be inactivated. Glutamate and cystine generate GSH through systemXC– and GSH can be oxidized to GSSG. Mitochondria generate ROS through ETC in TCA cycle, which leads to oxidative stress and eventually ferroptosis. Fe2+ is oxidized to Fe3+ after being absorbed in the duodenum. Fe3+ enters the cell by combining with Tf and Tfr to form a complex. Iron ions decomposed from the endosome can leave the cell through FPN protein on the cell membrane, and other iron ions enter the unstable iron pool. Tf-Tfr complex leaves the cell for the next cycle. Under pathological conditions (neurological disorders), excessive Fe2+ will participate in Fenton reaction to produce a large amount of ROS, which will lead to ferroptosis. Abbreviations: AA: arachidonic acid; AdA: adrenic acid; ACLS4: acyl-CoA synthetase long chain family member 4; ALOXs: lipoxygenases; CoA: coenzyme A; DPI7: diphenyleneiodonium chloride7; ETC: electron transport chain; FPn: ferroportin; GPX4: glutathione peroxidase 4; GSH: glutathione; GSSG: oxidized glutathione; LPCAT3: lysophosphatidylcholine acyltransferase 3; OGDH: oxoglutarate dehydrogenase; PL: phospholipid; POR: cytochrome p450 oxidoreductase; PLOOH: phospholipid hydroperoxides; PUFA: polyunsaturated fatty acids; RSL3: (1S,3R)-RSL3; ROS: reactive oxygen species; STEAP3: STEAP family member 3; Tf: transferrin; Tfr: transferrin receptor; TZD: thiazolidinediones; TCA: tricarboxylic acid.
2.1. Oxidative Stress in Ferroptosis
Oxidative stress was formulated in 1985 [19], which is an important cause leading to neurological disorders that mainly arise from the imbalance between depletion of antioxidants and production of peroxides [20]. Ferroptosis is a unique, oxidative stress-induced cell death pathway characterized by glutathione depletion and lipid peroxidation. Based on current research, ferroptosis can be regulated by system Xc– [21]. System Xc–can exchange glutamate and cystine inside and outside of the cell. Glutathione is an important free radical scavenger and antioxidant in vivo, which can be categorized as either reduced (GSH) or oxidized (GSSG). Intracellular cystine can be converted into reduced GSH by a series of biochemical reactions. GPX4 is a member of the glutathione peroxidase family which can convert GSH to GSSG, and GSH/GSSG constitutes an antioxidant system and provides reducing equivalents to eliminate oxidative species [22]. GPX4 is a redox enzyme and can also inhibit ferroptosis by decreasing the level of lipid peroxides. GPX4 plays a crucial role in reducing reactive aldehydes (PUFAs-OOH) to their alcohol form (PUFAs-OH) which can reduce the content of ROS [18].
ROS are some of the most common oxidants in cells. ROS includes superoxide (O2–•), hydrogen peroxide (H2O2), lipid peroxides (ROOH), or the corresponding hydroxyl (HO•) and peroxyl radicals (ROO•) [23]. Accumulation of ROS can lead to oxidative stress, causing oxidative stress-induced lipid peroxidation. What is worse, lipid peroxidation may further generate ROS or degrade into reactive compounds capable of crosslinking DNA and proteins [24]. Oxidative stress-induced lipid peroxidation eventually leads to ferroptosis. Meanwhile, ferroptosis can be induced by oxidative stress directly (Figure 1).
2.2. Iron Metabolism in Ferroptosis
Metabolism is essential for the biochemical process in cells and goes awry in many diseases. Iron metabolism is an important mechanism of ferroptosis [2]. Iron is a significant trace element in the body [25]. Abnormal distribution and content of iron in the body can affect the normal physiological processes. Fe2+ is absorbed by the intestine or degraded by erythrocyte and then can be oxidized to Fe3+ by ceruloplasmin [26]. Fe3+ can combine with transferrin (Tf) on cytomembrane to form Fe3+-Tf, which can be endocytosed into cells to form a complex with membrane protein TF receptor (Tfr) [26]. The complex enters the endosome and is divided into TF-TFR and Fe3+ [26]. Fe3+ is then reduced to Fe2+ by the six-transmembrane epithelial antigen of the prostate 3 (STEAP3), and Fe2+ is stored in the unstable iron pool (LIP) and ferritin [26]. Nevertheless, under pathological conditions, iron homeostasis is destroyed, and the overwhelming production of Fe2+ exceeds the limit, breaking cell homeostasis [27]. Excess Fe2+ can combine with H2O2, reducing H2O2 to hydroxyl radical (OH·) by Fenton reaction [28, 29], which was first described by Fenton [30], who in 1894 reported on the oxidation of malic acid by hydrogen peroxide in the presence of ferrous ions.
2.3. Role of Mitochondria in Ferroptosis
Mitochondria, through the tricarboxylic acid cycle (TCA) and electron transport chain (ETC) activity, are the principal sources of cellular energy production [31]. Meanwhile, ROS can be produced during the process of TCA which is an important cause of ferroptosis-induced lipid peroxides [32]. In addition, mitochondrial morphological changes are considered to be an important basis for the diagnosis of ferroptosis [1]. The change of mitochondrial membrane potential is also an important discovery in the study of ferroptosis [33, 34]. To date, abnormal mitochondrial architecture including mitochondrial fragmentation, rupture of mitochondrial outer membrane, and mitochondrial shrinkage, as well as vanished mitochondrial cristae, is regarded as the typical morphological characteristic of ferroptosis [1]. In summary, mitochondria play a significant role in ferroptosis [35], and molecular research targeting mitochondria is of great significance to intervene in the development of ferroptosis (Figure 1).
2.4. Other Mechanisms
As a novel form of cell death, the mechanism of ferroptosis is complex. Ferroptosis is regulated by numerous pathways and implicated in many diseases. In addition to the mechanisms mentioned above, there are other possible mechanisms which have been proposed, such as the antioxidant mechanisms which include GPX4 pathway [36], FSP1 pathway [37], and other pathways. FSP1-NADPH-CoQ pathway [38] and nuclear factor erythroid 2-related factor 2 (Nrf2) pathway are potential regulatory pathways of ferroptosis [39]. Hence, the mechanism of ferroptosis remains to be explored.
3. Ferroptosis in Neurological Disorders
In recent years, the incidence of neurological disorders increases substantially. Because of the lack of effective treatment, sequelae caused by neurological disorders have brought an enormous burden on society. Ferroptosis has been proved to be closely related to the occurrence and development of various neurological disorders [40–43]. Therefore, figuring out the role of ferroptosis in neurological disorders will provide an important basis for the treatment of diseases (Figure 1, Table 1).
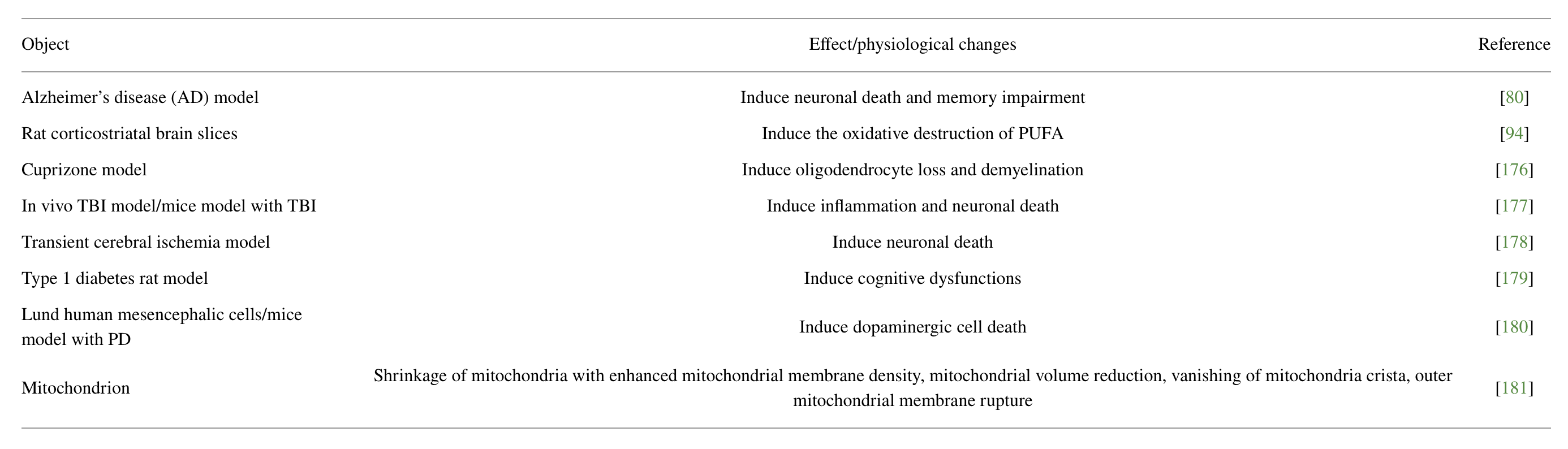
Ferroptosis-induced effects or altered physiology in neurological disorders.
3.1. Ferroptosis in Parkinson’s Disease
Parkinson’s disease (PD), also called paralysis agitans, is the second most common neurodegenerative disorder worldwide. It is typical of neuronal death in the substantia nigra pars compacta (SNpc), which adjusts motor function. PD causes clinical symptoms such as resting tremor, rigidity, bradykinesia, postural instability, and other motor symptoms [44–46]. However, its basic mechanism is not completely clear. Currently therapeutic arsenal mainly includes the application of the dopamine precursor levodopa (L-DOPA), dopamine metabolism inhibitors, and dopamine agonists [33], which only offer symptomatic relief. There still are not effective treatments for PD, so novel targets to improve therapeutic and diagnostic methods for PD patients are urgently needed. Iron is considered to be an important target of neurodegenerative diseases. Besides, iron metabolism is closely related to the pathogenesis of neurodegenerative diseases [34, 47]. Researchers found that patients with PD suffered from GSH depletion, ROS elevation, and lipid peroxidation [48–50]. Ferritin heavy chain 1 (FTH1), a main iron storage protein, can affect intracellular iron metabolism and then trigger ferroptosis. As an important ferroptosis-related protein, FTH1 is differentially expressed in rats with PD compared with normal rats. Overexpression of FTH1 can reduce the effect of ferroptosis in PD-related cells [51]. These findings indicate that ferroptosis may be used as a therapeutic target for PD [52, 53]. Neuroimaging and postmortem examination report that much iron accumulation in substantia nigra (SN) results in an increase in iron content in the residual dopaminergic neurons [54]. α-Synuclein causes iron cytotoxicity by acting on mitochondria in the neurodegeneration of PD. A study found that cyclosporine A, a blocker of mitochondrial permeability transition pore (mPTP), could prevent iron-induced mitochondrial damage and cell death. Furthermore, knocking down α-synuclein expression by siRNA can play a similar role [55]. α-Synuclein is the main component of Lewy bodies and is abundantly expressed in the nervous system, which is strongly related to PD’s pathophysiology [56]. Besides, it has been recently shown that α-synuclein causes lipid peroxidation by producing ROS, leading to increased calcium influx and consequent cell death [57]. Ferroptosis inhibitors like ferrostatin or iron chelators [58] can inhibit cell death caused by the above method, supporting the hypothesis that ferroptosis participates in this process and may harbor therapeutic potential. Studies have also shown that ferroptosis occurs earlier than apoptosis in the development of PD and ferric ammonium citrate (FAC)-induced ferroptosis was dependent on p53, not MAPK signaling pathway [54].
3.2. Ferroptosis in Alzheimer’s Disease
Alzheimer’s disease (AD) is one of the most common neurodegenerative diseases worldwide. Pathological features of AD include cerebral atrophy, intraneuronal accumulation of hyperphosphorylated tau in neurofibrillary tangles, extracellular deposition of amyloid-β peptide in senile plaques, oxidative stress, chronic inflammation, and loss of neurons and synapses [59]. There is evidence that iron excess and homeostasis disorder can lead to neurodegeneration of AD [60, 61]. The increase of brain iron content in patients with AD has been confirmed in many studies [62–67]. When brain iron regulation in AD patients is disturbed, excess Fe2+ can not only produce hydroxyl radicals in Fenton reaction but also induce neuroinflammation, resulting in oxidative stress and neurodegeneration mediated by ferroptosis [68–72]. The research found that the increase of ferritin light chain (FTL) is related to the decrease of GPX4 level in AD which indicates that dysfunctional ferritin will reduce the antioxidant capacity of brain and the level of glutathione will decrease in AD patients [73]. Increased light subunit (xCT) expression in cells of patients with AD [74] and xCT can be upregulated by Nrf2 [75, 76]. Furthermore, mice with a conditional deletion of GPX4 show cognitive impairment and hippocampal degeneration similar to patients with AD and have the neurodegenerative characteristics of ferroptosis. Simultaneously, these symptoms can be improved by ferroptosis inhibitors [77], which provide a basis for the association between AD and ferroptosis. Recently, researchers observed that tau can stabilize ferroportin (FPN1) which is the only iron export channel found in mammalian cells [78, 79]. FPN decreased significantly in the brain tissue of AD patients. According to MMSE (mini-mental state examination) scores, FPN is involved in the cognitive impairment and brain atrophy of AD [80]. In patients with AD and a mouse model of AD, NADPH oxidases 4 (NOX4) causes lipid peroxidation and promotes ferroptosis of astrocytes via the damage of mitochondrial metabolism in AD [81]. Bhatia et al. [82] summarized a variety of multitarget targeted ligands with iron chelation properties, which is potentially useful in AD. Ashraf et al. [83] summarized the clinical cases involving iron chelators, antioxidants, NAC, and selenium in the treatment of AD and provided a feasible basis for the treatment of AD. These studies have shown that ferroptosis plays an important role in AD, in which a variety of regulatory factors are involved, and provide many potential therapeutic targets for the treatment of AD.
3.3. Ferroptosis in Huntington’s Disease
Huntington’s disease (HD) is an autosomal dominant, late-onset, and fatal neurodegenerative disorder [84], which is caused by an abnormal CAG repeat in the huntingtin gene [85]. It is characterized by highly selective striatal injury, leading to dance-like movement, progressive dementia, and dystonia. In 2001, Pigeon et al. [86] first identified the association between hepcidin and iron homeostasis. Studies found that hepcidin could promote ferroptosis through iron metabolism [87] and could prevent erastin-induced ferroptosis by degrading Fpn [88]. Qian et al. [89] summarized the therapeutic potential of hepcidin in neurodegenerative diseases, including HD, indicating that there is a link between ferroptosis and HD. A study found that there is an accumulation of toxic iron in neurons of mouse model with HD and the symptoms of HD can be alleviated by deferoxamine, indicating that the accumulation of iron may contribute to the neurodegenerative process [90]. Studies have shown that HD patients will cause oxidative stress and neurotoxicity to neurons in the striatum, resulting in neuronal death, and eventually leading to motor and cognitive impairment [91]. HD patients have shown higher levels of plasma lipid peroxidation and lower GSH levels [92]. Oxidative stress and lipid peroxidation are important mechanisms leading to ferroptosis. In 2017, Cardoso et al. [93] proposed that as a key factor in regulating ferroptosis, GPX4 could provide protective mechanisms against neurodegeneration. Skouta et al. [94] found that ferroptosis inhibitor Fer-1 and its derivatives can prevent cell death in HD brain slice model. Mi et al. [85] summarized the evidence of ferroptosis involved in HD in different animal models or human patients. It revealed the close connection between ferroptosis and HD which would provide an important potential target for the treatment of HD.
3.4. Ferroptosis in Anxiety and Depression
With the development of society, the incidence rate of anxiety and depression is increasing year by year. The research on the mechanism and the treatment of anxiety and depression have also attracted extensive attention. Jiao et al. [95] found that the contents of total iron and ferrous ion in the hippocampus of chronic unpredictable mild stress (CUMS) model mice increased, and GPX4, FTH1, ACSl4, and COX-2 also changed significantly, which proved the evidence of ferroptosis in the hippocampus of the depression mouse model. Previous research reported that sodium hydrosulfide (NaHS) can alleviate the depressive and anxiety-like behaviors induced by type 1 diabetes mellitus (T1DM) [96]. Wang et al. [97] revealed that NaHS reduced ferroptosis in the prefrontal cortex (PFC) of the T1DM mouse model by reducing iron deposition and oxidative stress, increasing the expression of GPX4 and SLC7A11, thus alleviating the anxiety-like and depression-like behavior of the mouse model with T1DM. Moreover, there is increasing evidence that oxidative stress has close contact with anxiety and depression [98, 99]. As a free radical scavenger [100], Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one, EDA) can improve depression and anxiety-like behavior, as well as inhibit oxidative stress and neuroinflammation in a mouse model of depression, and knocking down of GPX4 expression in CSDs mouse model can inhibit the therapeutic effect of EDA on depression and anxiety [101]. The study has shown that GPX4 can affect the role of EDA in mouse models of anxiety and depression, which suggests that GPX4-mediated ferroptosis may be a potential mechanism affecting anxiety and depression.
3.5. Ferroptosis in Stroke
Stroke is mainly divided into ischemic stroke and intracerebral hemorrhage (ICH) stroke, the former accounting for a higher proportion [102]. Zhang et al. [103] summarized the multiple roles of ferroptosis in stroke. Jin et al. [104] reviewed some neuroprotectants that show protective effects in stroke models. These substances have been recently validated as ferroptosis inhibitors. These researches indicate that ferroptosis plays a key role in the progression and toxicity of stroke. Chen et al. [105] analyzed and identified ferroptosis-related differentially expressed genes (DEGs) in ischemic stroke by bioinformatics, which provided more evidence for the important role of ferroptosis in ischemic stroke. NAC is considered as a potential therapeutic agent of ferroptosis. Recently, studies have found that NAC can reduce neuronal death and improve functional recovery in mouse models with ICH by inhibiting ferroptosis [106]. Clinical trials and experimental studies have reported that treatment with natural compounds could reduce oxidative stress in ischemic stroke. [107]. Therefore, reducing the production of ROS during reperfusion is the key to the treatment of ischemic stroke. Guan et al. found that natural product carvacrol can increase the expression of GPX4 to inhibit ferroptosis, protecting the cognitive function of mice with ischemia-reperfusion injury [108]. In addition, Cui et al. revealed that ACSL4 can exacerbate ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation [109]. The incidence rate of ICH is low, but the deformity and mortality rates are significantly higher than ischemic stroke [110]. ICH can lead to the rupture of blood vessels in the brain and release red blood cells to produce a large amount of free iron and ROS, which result in oxidative stress and ultimately neuronal damage [111]. Iron chelator deferoxamine (DFO) can effectively alleviate ICH-induced neuronal injury in mice [112], which also provides evidence for the above process. The same characteristics of ferroptosis were observed in the animal model of ICH [113]. Furthermore, selenium can drive adaptive transcription against ferroptosis to protect neurons and provide direction for the treatment of stroke [114].
3.6. Ferroptosis in TBI
As one of the acute central nervous system (CNS) trauma [115], traumatic brain injury (TBI) is a major cause of death and disability worldwide [116]. Owing to the difficulty of treatment and serious sequelae, TBI has always been a huge problem in the medical field and caused a huge burden on families and society [41]. Analyzing the mouse TBI model [117] and clinical cases with TBI [118], abnormal regulation of ferroptosis after TBI was found both in experimental model and clinical cases. Interestingly, Xie et al. found that the content of iron in damaged cortical cells increased, the function of iron metabolism was impaired, lipid ROS accumulation was increased, and mitochondrial atrophy was enhanced. The above symptoms can be alleviated by intraventricular administration of ferroptosis inhibitor Fer-1 in a mouse model with TBI [119], which proves that ferroptosis is one of the causes of TBI. Previous studies have shown that mir-212-5p is highly expressed in the brain [120] and is associated with a variety of neurological diseases [77, 119, 121, 122]. The use of mir-212-5p in the CCI mouse model can target PTGS2 to reduce ferroptosis and protect the brain [123]. Moreover, Cheng et al. [124] found that the use of iron uptake inhibitor ferristatin II in mouse model with TBI can inhibit the formation of iron proteasomes and alleviate TBI injury in brain. For a long time, due to the lack of clear understanding, the treatment of TBI is very difficult. These studies provide a potential theoretical basis for the treatment of TBI.
4. Mediators of Oxidative Stress in Neurological Disorders
Oxidative stress can spread, which aggravates the production of ROS. Due to the high oxygen demand and high energy demand of the brain, but its antioxidant capacity is weak, it is more vulnerable to oxidative stress [125], which will lead to various neurological diseases. Therefore, inhibiting or weakening oxidative stress might effectively reduce the damage to the nervous system. Studies for targeting molecular mediators of oxidative stress are considerable and will provide effective treatment for neurological disorders (Figure 2).
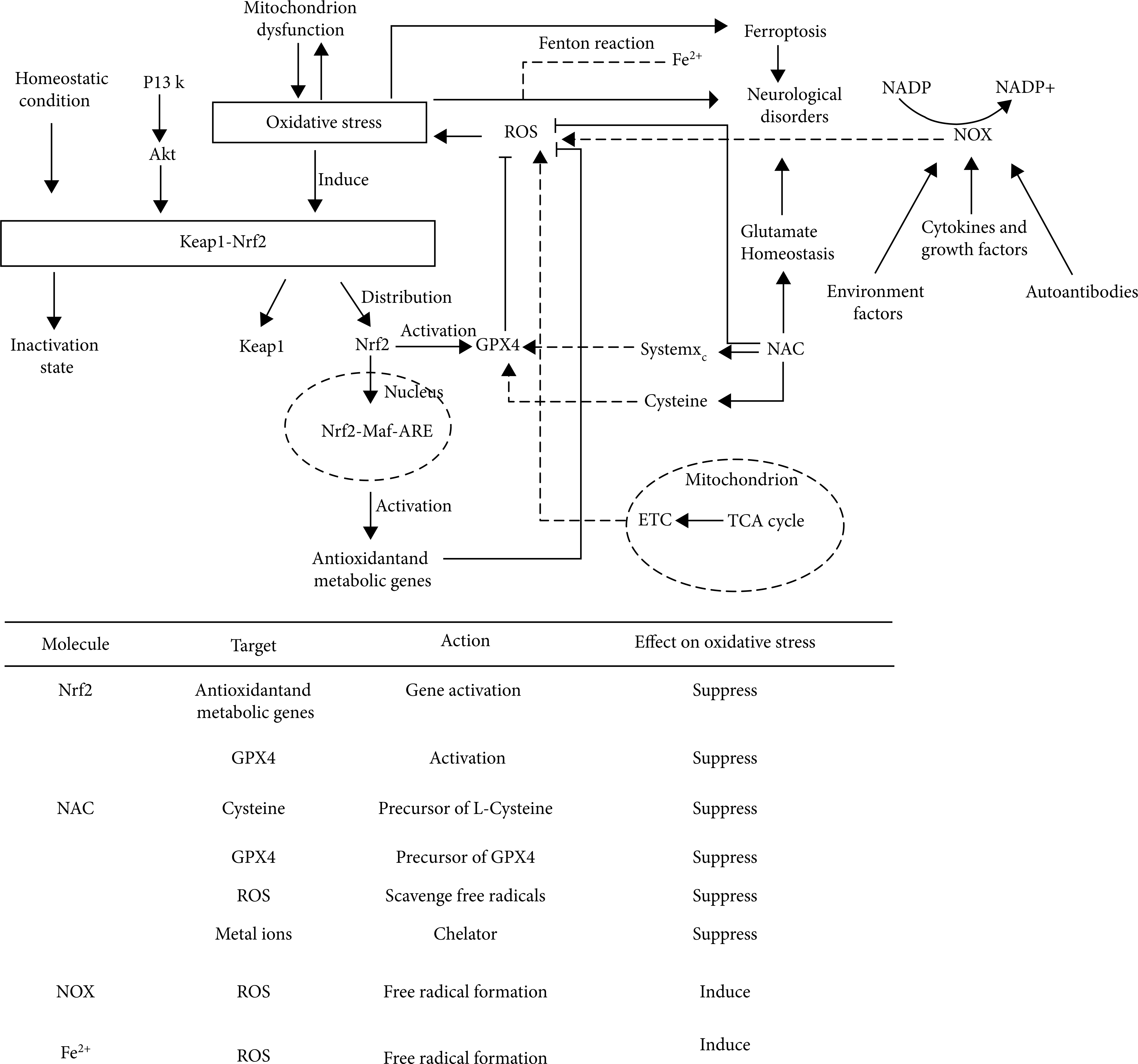
The top schematic is a summary of oxidative stress mechanisms in neurological disorders. Under homeostatic conditions, Nrf2 binds with Keap1 and remains inactive. Nrf2-keap1 is also regulated by PI3K-Akt pathway. Under pathological conditions (neurological disorders) cause oxidative stress, Nrf2 and Keap1 will be separated. Nrf2 enters the nucleus and combines with MAF and ARE to activate antioxidant metabolic genes and play an antioxidant role. In addition, Nrf2 can activate GPX4 and be inhibited by Keap1. Both TCA cycle in mitochondria and Fenton reaction involving Fe2+ can cause oxidative stress by producing ROS. NAC, as a precursor of GPX4 and L-cysteine, affects oxidative stress through systemXC– and GPX4. In addition, NAC can interfere with glutamate homeostasis and participate in the occurrence of a variety of nervous system diseases. NOX can produce ROS and cause oxidative stress under the action of a variety of regulatory factors. The bottom table is a summary of several molecules that affect oxidative stress in neurological disorders, including their action targets, action modes, and action results. Abbreviations: Akt: protein kinase B; ARE: antioxidant response element; ETC: electron transport chain; GPX4: glutathione peroxidase 4; Keap1: Kelch-like ECH-associated protein 1; Maf: musculoaponeurotic fibrosarcoma oncogene homolog; NAC: N-acetylcysteine; NOX: nicotinamide adenine dinucleotide phosphate oxidases; NADP: nicotinamide adenine dinucleotide phosphate; Nrf2: NF-E2-related factor 2; PI3K: phosphatidylinositol-3-kinase; ROS: reactive oxygen species; TCA: tricarboxylic acid.
4.1. Nrf2-ARE-Mediated Oxidative Stress
Nuclear factor erythroid 2–related factor (Nrf2) is a transcription factor belonging to the basic leucine zipper (bZIP) transcription factor family that can participate in the regulation of oxidative stress. The antioxidant capacity of Nrf2 works by inducing glutathione biosynthesis [126]. Studies found that Nrf2 can play a neuroprotective role in central nervous system (CNS) diseases [127–129]. In addition to the transcriptional regulation of Nrf2, current hot research directions for the activity of Nrf2 are Kelch-like ECH-associated protein 1 (Keap1)/Nrf2/antioxidant response element (ARE) pathway. Keap1/Nrf2/ARE can be divided into two parts, one in the cytoplasm and the other in the nucleus. Normally, Keap1 and Nrf2 bind in the cytoplasm and are in an inactive state. If they are not activated all the time, Nrf2 will be ubiquitinated and then degraded. Under certain stimulation, the binding of keap1-nrf2 will be unstable. Nrf2 will be released, transferred to the nucleus, combined with ARE, activate the transcription of downstream genes, and then translate a series of related proteins, which can induce various antioxidant enzymes, including heme oxygenase 1 (HO-1), NAD (P) H: quinone oxidoreductase 1 (NQO1), superoxide dismutase (SOD), glutathione peroxidase (GPX), and catalase (CAT). These enzymes can play physiological functions [130]. Targeted regulators of Nrf2 are of great significance in the treatment of oxidative stress-induced injury in neurological disorders. Nrf2 plays a neuroprotective role by regulating the antioxidant system of cells. The regulation will have an impact on glutathione (GSH), thioredoxin (TXN), NADPH, and various other ROS-related enzymes [131, 132].
Keap1/Nrf2/ARE pathway has been proved to be an important antioxidant target. Keap1 can perceive ROS levels through cysteine-rich regions [133–135]. In the steady state, Nrf2 in the cytoplasm can be degraded by Keap1, and Nrf2 increases rapidly under stress and transfers to the nucleus for transcriptional activity [136]. Mitoquinone (MitoQ) is a mitochondrial-targeted antioxidant and can cause a sharp decline in Keap1 in cells, prompting more Nrf2 to be transmitted to the nucleus to produce an antioxidant response. The effect was confirmed in subarachnoid hemorrhage (SAH) animal model experiments [137]. Nrf2 can also play a protective role as a therapeutic target in astrocytes. There are seven functional domains of Nrf2 (neh1-neh7) that regulate its transcriptional activity and stability [138]. Kelch-like ECH-associated protein 1 (Keap1) is an inhibitor of Nrf2 [139, 140] and interacts with Nrf2 through neh2 domain [141]. Early studies believed that Keap1 inhibition was the main mechanism to activate astrocytes. Recent studies have found that mild oxidative stress (nonlethal oxidative stress) can also activate Nrf2 independently through neh5 transactivation domain [142]. In addition to the various regulatory factors mentioned above, there are other Keap1/Nrf2 pathway regulators such as microRNA-592 (miR-592) [143], eriodictyol [144], and tripartite motif 16 (TRIM16) [145]. These regulatory factors can act on Keap1/Nrf2/ARE pathway to play the role of antioxidant stress, so as to treat or alleviate neurological disorders (Figure 2).
4.2. NADPH- and NOX-Mediated Oxidative Stress
Nicotinamide adenine dinucleotide-phosphate (NADPH) is a coenzyme, which is the product of the final electron acceptor NADP + after receiving electrons. It participates in a variety of biochemical reactions as a hydrogen transmitter in vivo. Studies have confirmed that NADPH can resist oxidative stress and improve energy metabolism, so as to play a neuroprotective role in stroke [146]. NADPH oxidases (NOXs) have 7 kinds of subtypes.
H2O2 can interact with mutant huntingtin in the brain of HD patients [147]. The activity of NOX can be measured by lucigenin-enhanced luminescence method [148]. The heterozygous huntingtin (HTT) and Cd can induce cytotoxicity which will cause NOXs-mediated oxidative stress. As an exogenous antioxidant and NOX inhibitor, apocynin can block the process of oxidative stress in the process of HD [149]. So far, there are still few studies on the role of NOX in the process of HD. As a potential therapeutic target of HD, the mechanism of NOX and its way of alleviating HD patients deserve further exploration.
Studies found that the cerebellum is an important participant in mental illness and is affected by oxidative stress. Schiavone et al. found that inhibition of NOX during brain maturation can prevent the development of psychotic behavioral dysfunction. Administration of ketamine to the postpartum mouse model can enhance oxidative stress and reduce IL-10 in this region [150]. In recent years, many compounds that inhibit NOX have been studied for the treatment of neurological disorders, and Ganguly et al. [191] reviewed the relationship between NOX and AD, and summarized drug trials of NOX inhibitors in Alzheimer’s disease and its precursor conditions. The results displayed that the activity of NOX increased in the brain of mouse models with neurodegenerative diseases and AD patients [192-194]. However, the effects of NOX inhibitors and their precursors are not satisfactory, and the therapeutic results of these naturally obtained compounds for AD are quite variable, including berberine and blueberry-derived polyphenols. Because of the complexity of neurological disorders, the study of NOX inhibitors can provide a new direction for the treatment of neurological disorders (Figure 2).
4.3. Other Mediators and Potential Therapeutic Agents
In this part, we mainly summarize three common regulators of targeted oxidative stress in neurological disorders. In addition to the above three common regulators, other regulators can be used to treat or relieve the symptoms of neurological disorders, such as PINK1, Ca2+, Sigma-1 receptor (sig-1R), peroxidase (prx), toll-like receptors (TLRs), and alarmins/c-jun N-terminal kinase (JNK) [151]. PINK1 is a mitochondrial-targeted E3 ubiquitin ligase, whose deficiency can lead to mitochondrial damage, oxidative stress, and exert neuroprotective effects by activating Parkin. Ca2+ can consume GSH to regulate cell death [152]. Sigma-1 receptor (sig-1R) [153, 154] is a mitochondrial endoplasmic reticulum chaperon that can regulate cell pathophysiological processes. Peroxidase (prx) can scavenge H2O2 to regulate redox signal transduction and play a protective role [155]. In addition, Toll-like receptors (TLRs) are also closely related to the injury of a variety of neurological disorders and can promote microglia to release neuroprotective agents through conduction [156]. Anfinogenova et al. [157] described the role of alarmins/c-jun N-terminal kinase (JNK) signaling transduction in cerebrovascular inflammation and summarized the therapeutic strategies of intracellular anti-JNK, which provided ideas for the strategies of JNK in the treatment of neurological disorders.
As an important regulator of oxidative stress, N-acetylcysteine (NAC) is a synthetic derivative of the endogenous amino acid L-cysteine and a precursor of GSH. GSH is a key factor in the clearance of ROS. Studies have shown that NAC mainly relies on GSH to exert its indirect antioxidant capacity, but this capacity is weak [158–160]. The direct antioxidant capacity of NAC depends on its nucleophilic free sulfhydryl group [158]. A large amount of preclinical evidence shows that NAC can play a therapeutic role in CNS by regulating glutamate homeostasis [161]. Glutamate is the main excitatory neurotransmitter in the nervous system. Its overexcitation will lead to neuronal damage [162]. Extracellular glutamate is regulated by glutamate transporter-1 (GLT1), glutamate aspartate transporter (GLAST), and the systemXc– to maintain homeostasis. Experiments have confirmed that NAC can activate systemXc– [163, 164] and induce the expression of GLT1 [165].
As a precursor of GSH, NAC plays a beneficial role in the pathology and sequelae of TBI. Previous studies have shown that the administration time of NAC affects the therapeutic effect of NAC on TBI. Previous studies have shown that the administration time of NAC affects the therapeutic effect of NAC on TBI. In the 3-day TBI model, NAC administration 15-60 minutes after the injury can reduce the inflammatory response [166, 167], and NAC administration 12 hours after controlled cortical impact (CCI)-induced injury can reduce the level of ROS by increasing GSH [168], and oral NAC treatment within 24 hours after injury can alleviate the sequelae of TBI, such as confusion, headache, and abnormal sleep [169]. Recent studies have also found that the use of antioxidant dual therapy targeting antioxidant stress has a significant effect on the symptoms of TBI and improves the functional outcome [170]. For example, the use of NAC and sulforaphane (SFN) dual therapy can reduce TNF, IL, and other neuroinflammatory markers. In addition, the dual therapy of NAC + SPF has also been proved to be effective in alleviating epilepsy after SE [171]. In recent studies, c57bl/6 (wild type, WT) mice and CCL5 knockout (CCL5-KO) mice were used to induce mild brain injury and establish a weight drop model [172], which is a novel TBI model that can simulate the full spectrum of human TBI, mainly focusing on simulating diffuse brain injury. In the weight drop model, NAC can significantly alleviate the symptoms of decreased memory and learning ability after trauma and increase the level of Gpx1 in the hippocampus of mice [173]. With the application of nanomaterials in biomedicine, it is found that embedding NAC eluted poly (d,l-lactide-co-glycolide) (PLGA) nanofibers into scaffolds can effectively improve the shelf life of drugs and reduce systemic side effects. At the same time, it can give full play to the neuroprotective effect of NAC and the cell proliferation of nanosystems [174], which provides a broad prospect for the application of nanosystems in nerve repair. For example, neutral hydroxyl-terminated polyamide (PAMAM) dendrimers have shown great potential as nanocarriers in multiple brain injury models [175].
5. Conclusion
In this review, we summarize the relevant mechanisms of ferroptosis and some regulators of targeted oxidative stress in the treatment of neurological disorders. In the process of searching the literature, we find that oxidative stress and neurological disorders are closely related to the role of mitochondria (Figure 2), but the connection is very complex and lacks a systematic and clear mechanism. Ferroptosis may be a key mechanism to study the connection (Figure 1). For future research on neurological disorders, the role of mitochondria and the mechanism of ferroptosis will be the focus and hotspot of research. In addition, in view of the harmfulness of neurological disorders and the complexity of oxidative stress and ferroptosis, future research on the molecular mediators targeting oxidative stress and ferroptosis should pay more attention to the underlying mechanisms, so as to provide a theoretical basis for the treatment of neurological disorders.