
Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1
By Christina M. Maher; Jeffrey D. Thomas; Derick A. Haas; Charles G. Longen; Halley M. Oyer; Jane Y. Tong; and Felix J. Kim
Excerpt from the article published in Molecular Cancer Research, Volume 16, Issue 2; 1 February 2018; 243–255; DOI: https://doi.org/10.1158/1541-7786.MCR-17-0166
Editor’s Highlights
- Sigma1 modulators can be used to sequester and degrade secretory pathway proteins by a potentially novel form of selective autophagy, and the first demonstration of autophagic degradation of PD-L1.
- IPAG (1-(4-Iodophenyl)-3-(2-adamantyl) guanidine) induces autophagosomal degradation of nascent, ER-localized PD-L1, thus preventing surface expression of PD-L1.
- Control of PD-L1 through Sigma1 supports a role for Sigma1 in antitumor immunity and provides insight into the mechanism(s) by which Sigma1 compounds elicit their immunomodulatory effects.
- Selective small-molecule Sigma1 ligands may be used as novel modulators of the tumor immune microenvironment.
- The use of Sigma1 modulators to pharmacologically promote antitumor immunity presents an opportunity for novel drug combinations with checkpoint inhibitor agents to expand the population of patients that respond to PD-L1/PD-1–targeted therapies.
Abstract
Emerging evidence suggests that Sigma1 (SIGMAR1, also known as sigma-1 receptor) is a unique ligand-regulated integral membrane scaffolding protein that contributes to cellular protein and lipid homeostasis. Previously, we demonstrated that some small-molecule modulators of Sigma1 alter endoplasmic reticulum (ER)–associated protein homeostasis pathways in cancer cells, including the unfolded protein response and autophagy. Programmed death-ligand 1 (PD-L1) is a type I integral membrane glycoprotein that is cotranslationally inserted into the ER and is processed and transported through the secretory pathway. Once at the surface of cancer cells, PD-L1 acts as a T-cell inhibitory checkpoint molecule and suppresses antitumor immunity. Here, we demonstrate that in Sigma1-expressing triple-negative breast and androgen-independent prostate cancer cells, PD-L1 protein levels were suppressed by RNAi knockdown of Sigma1 and by small-molecule inhibition of Sigma1. Sigma1-mediated action was confirmed by pharmacologic competition between Sigma1-selective inhibitor and activator ligands. When administered alone, the Sigma1 inhibitor decreased cell surface PD-L1 expression and suppressed functional interaction of PD-1 and PD-L1 in a coculture of T cells and cancer cells. Conversely, the Sigma1 activator increased PD-L1 cell surface expression, demonstrating the ability to positively and negatively modulate Sigma1 associated PD-L1 processing. We discovered that the Sigma1 inhibitor induced degradation of PD-L1 via autophagy, by a mechanism distinct from bulk macroautophagy or general ER stress–associated autophagy. Finally, the Sigma1 inhibitor suppressed IFNγ-induced PD-L1. Our data demonstrate that small-molecule Sigma1 modulators can be used to regulate PD-L1 in cancer cells and trigger its degradation by selective autophagy.
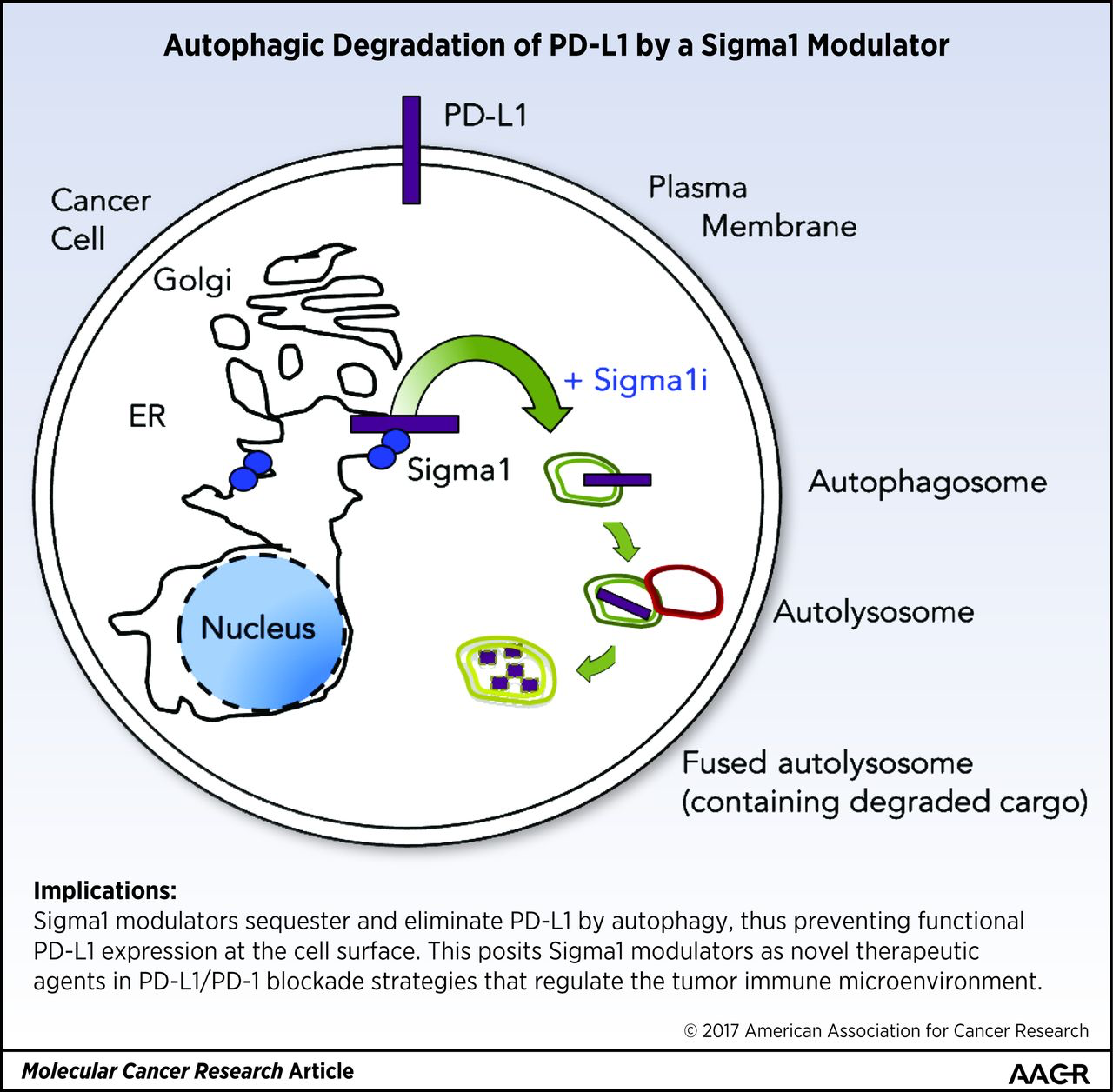
Implications: Sigma1 modulators sequester and eliminate PD-L1 by autophagy, thus preventing functional PD-L1 expression at the cell surface. This posits Sigma1 modulators as novel therapeutic agents in PD-L1/PD-1 blockade strategies that regulate the tumor immune microenvironment.
Visual Overview
Introduction
Programmed death-ligand 1 (PD-L1, also known as B7-H1 or CD274) is a type I integral membrane glycoprotein that is cotranslationally inserted into, posttranslationally modified in, and transported through the secretory pathway of a range of cell types, including tumor cells (1–4). In this pathway, the endoplasmic reticulum (ER) is the primary site of synthesis, folding, and assembly of secreted and integral membrane proteins. Protein homeostasis in the ER relies on the timely convergence of multiple mechanisms that detect protein–concentration thresholds, provide quality control, and regulate the ebb and flow of ER proteins (4–6). Integral membrane proteins are intrinsically dependent on ER protein homeostasis machinery to reach the cell surface.
PD-L1 expressed at the surface of tumor cells can act as a T-cell inhibitory checkpoint molecule and can inactivate tumor-infiltrating immune cells that express cell surface programmed death-1 (PD-1; also known as CD279; refs. 1, 3, 7). Immune checkpoint inhibitors that block PD-L1/PD-1 interactions are promising therapeutic agents in strategies that exploit antitumor immune responses. However, a relatively small percentage of cancer patients respond to anti–PD-L1/PD-1 monotherapy, which thus far includes subsets of patients with melanoma, non–small cell lung cancer, metastatic bladder cancer, and renal cell carcinoma (7–10). Although high PD-L1 expression has been associated with poor prognosis in a range of cancers (11–13), high PD-L1 expression levels do not necessarily correspond with response to anti–PD-L1/PD-1 therapeutic agents (7, 14). For instance, anti–PD-L1/PD-1 agents to treat prostate and triple-negative breast tumors with high levels of PD-L1 have shown minimal efficacy in early clinical trials (7–10, 12–15). Therefore, the search for predictive biomarkers of response to PD-L1/PD-1 agents is an active area of investigation (14, 16), as is the search for novel agents and combination therapy approaches that modulate the tumor microenvironment to promote antitumor immunity (7, 14–17).
Full-length PD-L1 comprises 229 amino acids leading with an N-terminal signal sequence, IgV- and IgC- extracellular domains, which engage PD-1 on infiltrating immune cells, a single transmembrane domain, and a relatively short 31 residue cytoplasmic tail, which has no defined functional motifs and whose role in PD-L1 biology has not been determined (1–3, 18). The cellular factors and processes engaged in the synthesis, maturation, and transport of PD-L1 are also not well defined, and few PD-L1–regulatory proteins have been identified (1–3, 18).
Sigma1 is a unique ligand-operated integral membrane chaperone or scaffolding protein that is highly expressed in the ER of a range of cancer cell lines (19). Initially thought to be an opioid receptor (19), Sigma1 lacks homology with any known mammalian protein and is a distinct protein, unrelated to any traditional receptor (20, 21). A growing body of evidence demonstrates that inhibition of Sigma1 can suppress growth, proliferation, and induce apoptosis in multiple cancer cell lines (19) and demonstrates the regulation of cell-intrinsic properties of cancer cells by Sigma1; however, antitumor immunity activity induced by Sigma1 inhibition has not been determined (19). Emerging data suggest Sigma1 is a multifunctional chaperone (22) or scaffolding protein (19, 23) involved in maintaining ER protein homeostasis and supporting the increased demand for secretory pathway protein synthesis associated with tumor growth (19, 24). In this regard, we have shown that selective small-molecule Sigma1 modulators can be used to regulate protein translation and activate the unfolded protein response (UPR) and autophagy in a pharmacologically controllable manner (24, 25).
Autophagy describes a set of cellular sequestration and degradation mechanisms by which cells can maintain energy levels under conditions of metabolic stress as well as a mechanism by which large aggregates of misfolded proteins and cellular components are sequestered into membrane-bound vesicles called autophagosomes and subsequently targeted for lysosomal degradation (26, 27). Multiple types of autophagy have been identified corresponding to distinguishing mechanistic features, autophagic cargo proteins, and organelles involved. These include bulk macroautophagy, chaperone-mediated autophagy, secretory autophagy, ubiquitin-selective autophagy, lipophagy, mitophagy, and most recently ER-phagy (26–32). Emerging reports demonstrate that components of the secretory pathway membrane remodeling and protein trafficking machinery contribute to autophagy (28–32).
We previously reported a role for Sigma1 in autophagy in cancer cells and the ability to pharmacologically control autophagy with small-molecule Sigma1 modulators (24). In this study, we asked whether the ER protein homeostasis regulating properties of small-molecule Sigma1 modulators could be exploited to regulate the transport and stability of PD-L1 in cancer cells and thus suppress the surface expression of PD-L1 and potentially abrogate PD-L1/PD-1–mediated inactivation of immune cells.
…
Results
Sigma1 physically associates with PD-L1
PD-L1 is a type I integral membrane glycoprotein that is synthesized in, posttranslationally modified in, and transported through the cellular secretory pathway to reach the plasma membrane of tumor cells. The MDA-MB-231 triple-negative breast cancer cell line has been reported to express high levels of PD-L1 under standard cell culture conditions (11). By confocal microscopy, we confirmed that PD-L1 is enriched in the cytoplasm of MDA-MB-231 cells as well as present on the surface of these cells (Fig. 1A). Sigma1 is a unique integral membrane chaperone or scaffolding protein in the secretory pathway and is enriched in the ER of cancer cells. We performed isopycnic centrifugation to demonstrate that Sigma1 cofractionates with PD-L1 in MDA-MB-231 cells. We show that 50% of PD-L1 and 78% of Sigma1 coisolated in density gradient fractions 5, 6, and 7 (Supplementary Fig. S1). This suggests that Sigma1 and PD-L1 occupy membrane domains with similar density and thus similar cellular compartments. Therefore, we asked whether Sigma1 could physically interact with PD-L1.
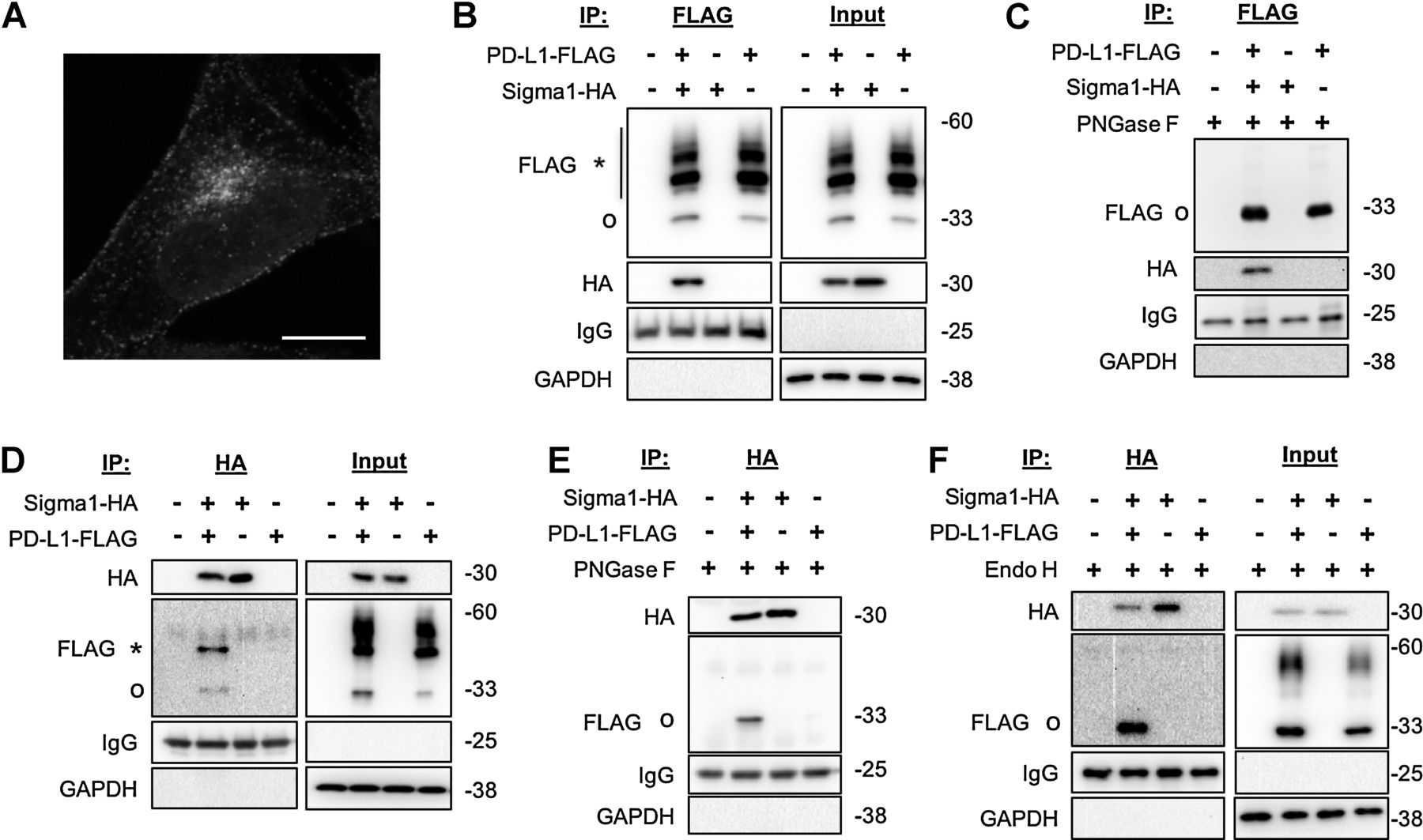
PD-L1 association with Sigma1.
A, Confocal micrograph showing cytoplasmic and cell surface staining of PD-L1. MDA-MB-231 cells in standard culture medium, cells fixed and immunostained 72 hours after seeding onto poly-d-lysine–coated coverslip. Scale bar, 10 μm. B, Coimmunoprecipitation of Sigma1-HA with PD-L1-FLAG from transiently transfected MDA-MB-231 cells. Glycosylated (*) and nonglycosylated (o) PD-L1-FLAG indicated. Immunoglobulin (IgG) and GAPDH loading controls for immunoprecipitated and input fractions, respectively. C, Anti-FLAG immunoprecipitated samples treated with PNGase F. Nonglycosylated (o) PD-L1-FLAG. D, Coimmunoprecipitation of PD-L1-FLAG with Sigma1-HA from transiently transfected MDA-MB-231 cells. Glycosylated (*) and nonglycosylated (o) PD-L1-FLAG. E and F, Anti-HA immunoprecipitated samples treated with PNGase F (E) and endoglycosidase H (Endo H; F). Nonglycosylated (o) PD-L1-FLAG.
Using carboxy-terminal (C-terminal) FLAG-tagged PD-L1 (PD-L1-FLAG) and C-terminal HA-tagged Sigma1 (Sigma1-HA), we found that Sigma1 coimmunoprecipitates with PD-L1 (Fig. 1B). The PD-L1-FLAG protein was detected as multiple bands, a predicted approximately 33-kDa unmodified species as well as a number of higher molecular weight (MW), slower migrating bands. PD-L1 is glycosylated as it is processed and matures through the secretory pathway, which is reflected in the heterogeneous, smeared migration pattern of PD-L1 bands on the immunoblot. We performed a peptide:N-glycosidase F (PNGase F) digestion of immunoprecipitated PD-L1-FLAG eluate and found that the PD-L1 smear was reduced to a single band of approximately 33 kDa, confirming that the higher MW, slower migrating bands are glycosyl–PD-L1 (Fig. 1C).
Interestingly, in the reciprocal co-IP, only the approximately 33-kDa nonglycosylated species and an approximately 45 kDa PD-L1-FLAG species coimmunoprecipitated with Sigma1-HA (Fig. 1D). PNGase F digestion of the immunoprecipitated samples reduced the higher MW PD-L1 band to approximately 33 kDa, confirming that the approximately 45 kDa band is a glycosylated form of the protein (Fig. 1E). Digestion of co-IP eluates with Endo H also reduced the approximately 45 kDa species to 33 kDa (Fig. 1F). Whereas PNGase F cleaves nearly all types of N-linked (Asn-linked) glycosylation, including complex glycans, Endo H cleaves only high mannose and some hybrid types of N-linked carbohydrates but not complex glycans. Thus, oligosaccharide structures beyond enzymatic modification by Golgi alpha-mannosidase II in the secretory pathway are resistant to Endo H cleavage. This differential sensitivity to PNGase F and Endo H is used to track the maturation of glycoproteins. The pattern observed here suggests that Sigma1 physically associates primarily with glycosyl-PD-L1 formed early in the proximal secretory pathway, ER, and early Golgi compartments (Fig. 1F).
PD-L1 protein levels are suppressed by RNAi-mediated knockdown and pharmacologic inhibition of Sigma1
We propose that Sigma1 is a ligand-operated scaffolding protein that promotes the stability, processing, assembly, and trafficking of specific proteins in the secretory pathway of cancer cells. In support of this hypothesis, we found that siRNA-mediated knockdown of Sigma1 resulted in a significant decrease in PD-L1 protein levels in triple-negative MDA-MB-231 breast cancer and androgen-independent PC3 prostate cancer cells (Fig. 2A). We also confirmed this effect with two distinct Sigma1 shRNAs, which produced similar trends in both of these cell lines (Fig. 2B). These data demonstrate that Sigma1 is required to sustain PD-L1 protein levels in these cancer cells. It is important to note that Sigma1 knockdown affects only a specific subset of proteins; shRNA-mediated knockdown of Sigma1 did not result in a significant decrease in HSP90 or Akt protein levels (Supplementary Fig. S2).
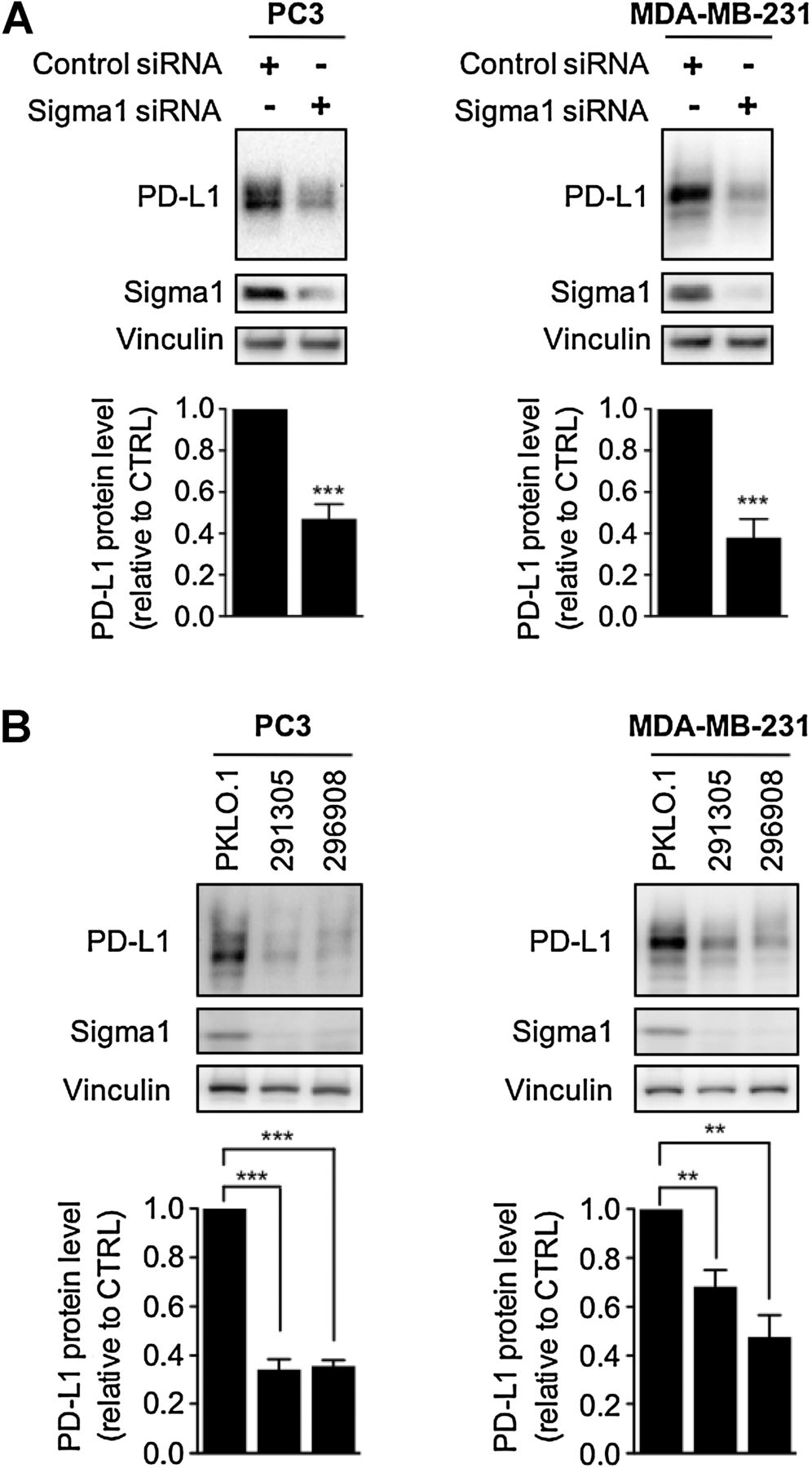
Sigma1 knockdown results in decreased PD-L1. A and B, Knockdown of Sigma1 by siRNA (A) and by two distinct shRNAs (291305, 296908) compared with the control shRNA (PKLO.1; B) in PC3 and MDA-MB-231 cells and corresponding decrease in PD-L1 protein levels. For all blots, bands were quantified by densitometry, and data are presented as levels of PD-L1 relative to control siRNA or shRNA (pKLO.1). Bars represent mean and SEM from at least three independent determinations. **, P< 0.01; ***, P < 0.001.
Subsequently, we asked whether a small-molecule inhibitor of Sigma1 could mimic the effects of Sigma1 knockdown. Here, we used IPAG, a prototypic selective small-molecule inhibitor of Sigma1 that we previously have shown engages the UPR, the ubiquitin proteasome system, and autophagy (23, 24). We found that IPAG induced a significant dose-responsive decrease of total PD-L1 protein in PC3 and MDA-MB-231 cells (Fig. 3A). In support of the selectivity of proteins affected/targeted by Sigma1 modulator treatment, we determined that MHC-I, a related secretory pathway associated membrane protein, was not eliminated under standard cell culture conditions in MDA-MB-231 cells and was only slightly decreased in PC3 cells (Fig. 3B). Thus, both RNAi knockdown and pharmacologic inhibition of Sigma1 selectively suppressed PD-L1 protein levels in cancer cells.
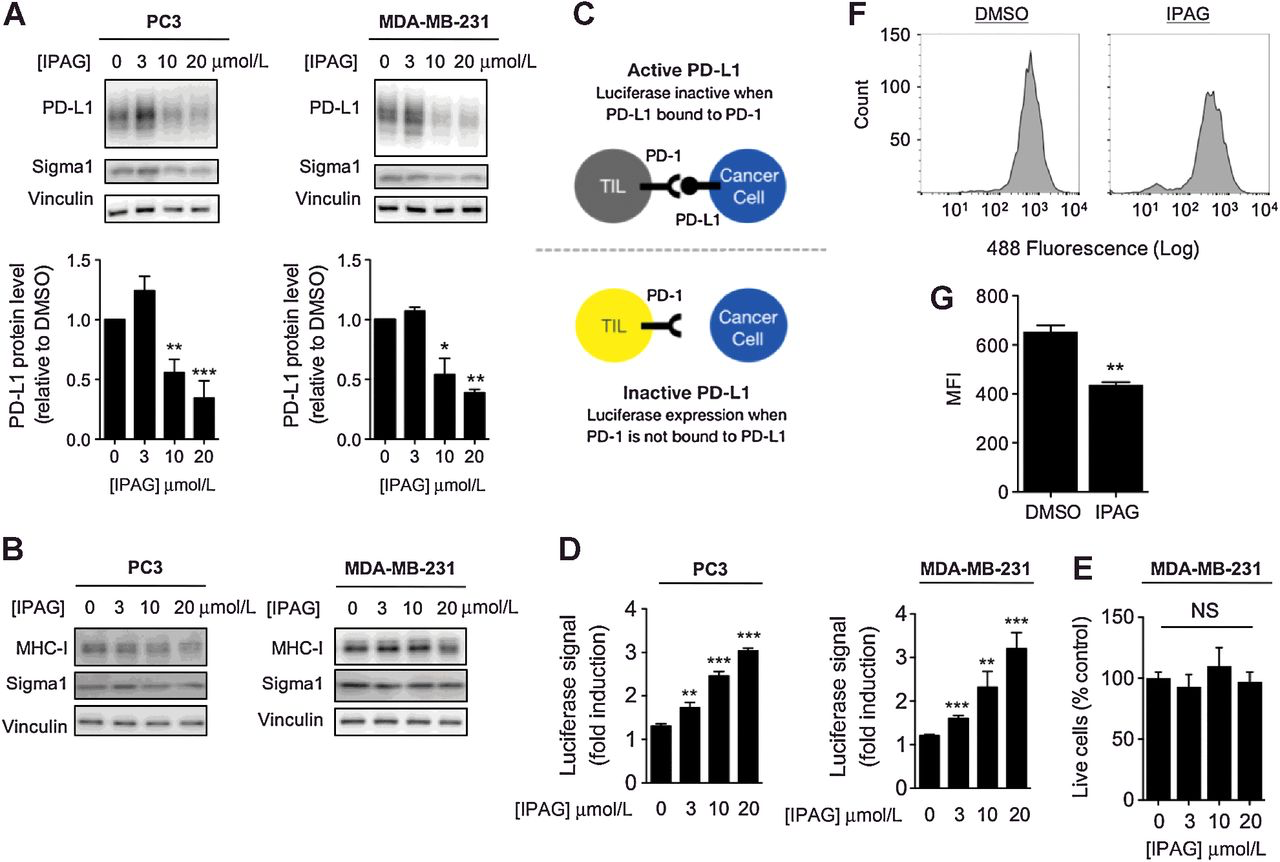
Suppression of PD-L1 protein levels in cancer cells and corresponding activation of cocultured T cells in response to small-molecule inhibition of Sigma1. A, Immunoblot of PD-L1 from whole-cell protein extracts from MDA-MB-231 and PC3 cells treated for 16 hours with DMSO (0), 3, 10, and 20 μmol/L IPAG (Sigma1 inhibitor). Immunoblots were quantified by densitometry for each cell line. Data represent mean values from at least three independent determinations, and error bars represent SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001. B, Immunoblot of MHC-I protein levels with DMSO (0), 3, 10, and 20 mmol/L IPAG treatment for 16 hours in PC3 and MDA-MB-231 cells, highlighting Sigma1 modulator specificity. C, Schematic illustration of PD-L1/PD-1 blockade assay. D, PD-L1/PD-1 blockade assay performed with PC3 and MDA-MB-231 cells treated with 3, 10, and 20 μmol/L IPAG for 16 hours. Jurkat NFAT-luciferase reporter cells (10,000 cells/well) were added, and cells were cocultured for 6 hours. Data are presented as fold induction over nontreated control cocultures. **, P < 0.01; ***, P < 0.001. E, Evaluation of cell death by Trypan blue exclusion following treatment with DMSO (0), 3, 10, and 20 μmol/L IPAG for 16 hours. No significant (NS) cell death was observed under these conditions. F, Cell surface expression of PD-L1 in response to treatment with small-molecule Sigma1 inhibitor. Flow cytometry of PD-L1 on the surface of MDA-MB-231 cells treated with IPAG (20 μmol/L) for 16 hours. G, Quantification of MFI of PD-L1 cell surface expression from three independent determinations. **, P < 0.01.
To determine the operative mechanism by which the Sigma1 modulator eliminated PD-L1, we evaluated the potential and relative contribution of protein synthesis and degradation by treating with IPAG in the presence of the translation inhibitor, cycloheximide. Whereas translation arrest by cycloheximide alone did not result in a decrease in PD-L1 protein levels over 8 hours, the combination of cycloheximide and IPAG resulted in significant decreases in PD-L1 protein levels after 4 and 8 hours of treatment (Supplementary Fig. S3). These data suggest that although IPAG-mediated translation arrest may contribute to decreased PD-L1 levels in response to long-term IPAG treatment, protein degradation plays a significant role in the elimination of PD-L1.
Pharmacologic inhibition of Sigma1 decreases PD-L1 cell surface expression and immune checkpoint activity in vitro
Mature PD-L1 expressed at the surface of tumor cells binds its cognate receptor PD-1 on the surface of infiltrating T cells and thus contributes to suppression of antitumor immunity. Blockade of PD-L1 binding to PD-1, that is, immune checkpoint inhibition, prevents inactivation of T cells and promotes antitumor immunity. Here, we utilized an NFAT transcriptional response element luciferase reporter assay (NFAT-luciferase) to evaluate the effects of Sigma1 inhibition on PD-L1/PD-1 interaction blockade (Fig. 3C). This assay involved coculture of PD-L1–expressing MDA-MB-231 or PC3 cancer cells with PD-1–expressing NFAT-luciferase transfected Jurkat T-cells. PD-L1/PD-1 binding results in T-cell inactivation and the abrogation of luminescent signal, whereas blockade of PD-L1/PD-1 binding prevents inactivation of genes regulated by the NFAT response element and results in luminescent signal (Fig. 3C).
MDA-MB-231 and PC3 cells were treated with multiple concentrations of IPAG (3, 10, and 20 μmol/L) for 16 hours, removed drug-containing media, and then cocultured with Jurkat NFAT-luciferase reporter T cells in the absence of drug. The transcriptionally induced bioluminescent signal increased in a concentration related manner in both MDA-MB-231 and PC3 cell cultures, indicating that IPAG disrupted PD-L1′s checkpoint activity (Fig. 3D). Importantly, we confirmed that the concentrations of IPAG and 16-hour treatment time used in these experiments did not induce cell death (Fig. 3E). On the basis of these data, we performed all subsequent mechanism-focused experiments well within the 16-hour time point to minimize potential confounds associated with cell death.
We next assessed how Sigma1 inhibition affects the expression of PD-L1 at the cell surface. Using flow cytometry, we demonstrated that treatment with IPAG (20 μmol/L) for 16 hours significantly decreased cell surface expression of PD-L1 on MDA-MB-231 cells (Fig. 3F and G). These data suggest that Sigma1 inhibition effectively disrupts PD-L1 function at least in part by decreasing its functional expression on the plasma membrane.
We also performed a biochemical membrane fractionation assay, which separates and isolates the cell surface plasma membrane from intracellular membranes, such as the ER and the Golgi, based on the affinity of membranes to distinct polymer phases (36). We used this technique to determine the extent to which mature PD-L1 at the plasma membrane and intracellular PD-L1 is suppressed by IPAG. IPAG treatment for 16 hours at a concentration of 10 μmol/L resulted in a salient decrease in the intracellular membrane PD-L1 and a more modest decrease in plasma membrane PD-L1 (Supplementary Fig. S4). These data suggest that IPAG treatment decreases nascent PD-L1 protein stability, thus preventing the trafficking of functional PD-L1 complexes to the cell surface.
A selective positive modulator of Sigma1 blocks the IPAG-mediated decrease of PD-L1 and promotes its cell surface expression
SA4503 is a Sigma1-selective putative agonist or activator (positive modulator) (37). Cotreatment of SA4503 with IPAG blocked the decrease in PD-L1 protein levels, demonstrating Sigma1-specific pharmacologic activity (Fig. 4A). Interestingly, by confocal microscopy, treatment with SA4503 alone resulted in increased cell surface expression of PD-L1 (Fig. 4B and C), suggesting that PD-L1 stability and trafficking can be differentially promoted and inhibited by positive and negative modulators of Sigma1, respectively.
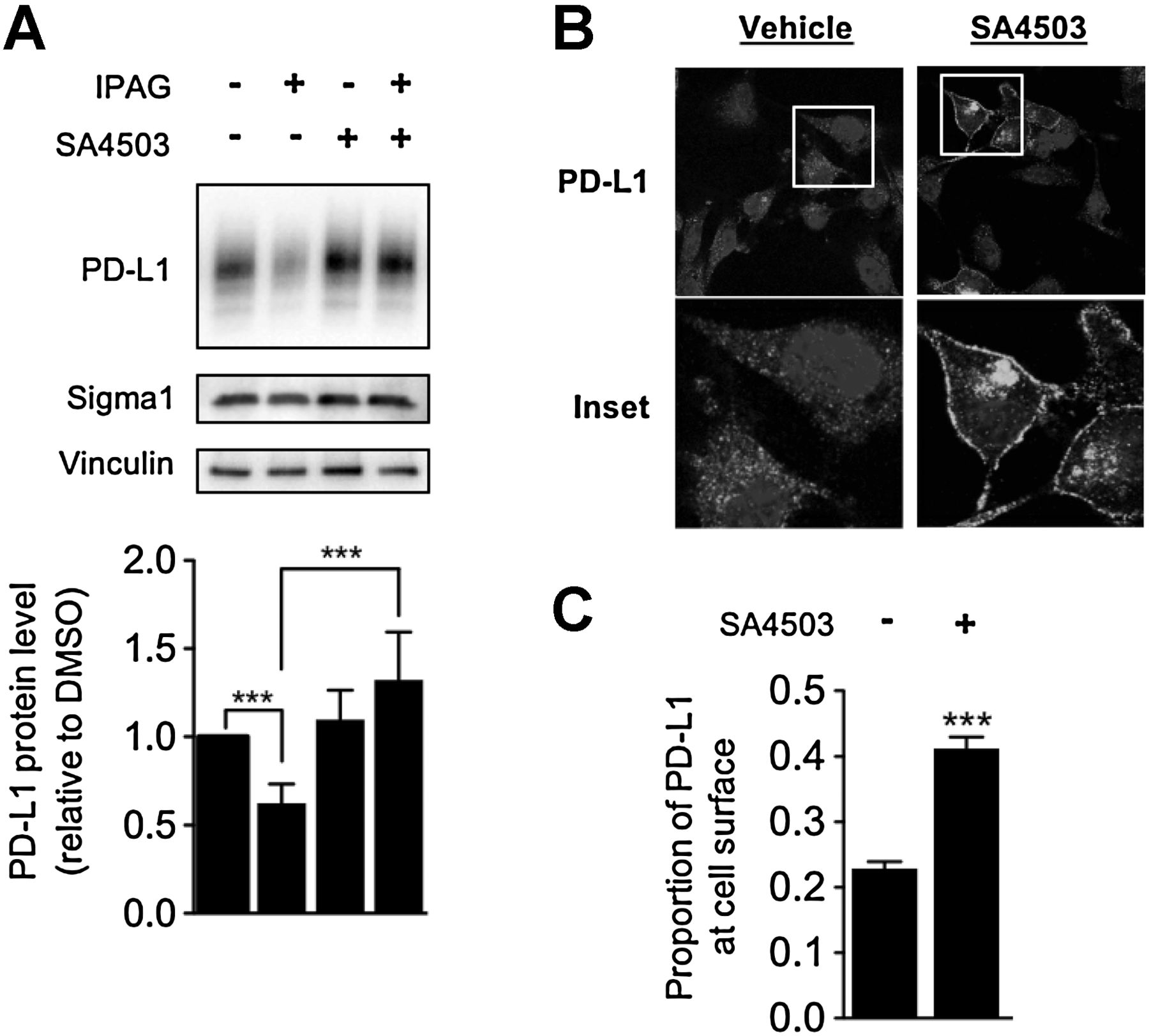
Opposing effects of small-molecule Sigma1 activator and inhibitor on PD-L1 protein levels and cell surface expression. A, Immunoblot of whole-cell lysates from MDA-MB-231 cells treated with IPAG (10 μmol/L), SA4503 (20 μmol/L), or cotreated with IPAG (10 μmol/L) and SA4503 (20 μmol/L) for 16 hours. Immunoblot bands were quantified by densitometry, and data are presented as levels of PD-L1 relative to nontreated control. B, Confocal micrographs showing PD-L1 and DAPI in MDA-MB-231 cells treated with SA4503 (10 μmol/L) for 48 hours. C, Quantification of the proportion of PD-L1 expressed on the cell surface compared with total cellular PD-L1. Data represent mean and SEM from at least three independent determinations. ***, P < 0.001.
In some cells, SA4503 treatment was associated with intense localized intracellular clusters of PD-L1 staining. The nature of this clustered staining pattern is unclear.
Treatment with a small-molecule Sigma1 inhibitor induces selective autophagic degradation of PD-L1
We previously demonstrated that IPAG could induce autophagy in cancer cells (24). Therefore, we asked whether autophagy contributes to the decrease in PD-L1 protein levels in this study. An IPAG treatment time course (0, 4, 8, and 16 hours) experiment revealed increased LC3B corresponding with decreased PD-L1 levels (Fig. 5A). Thus, autophagosome formation coincides with decreasing PD-L1, suggesting a role for autophagy.
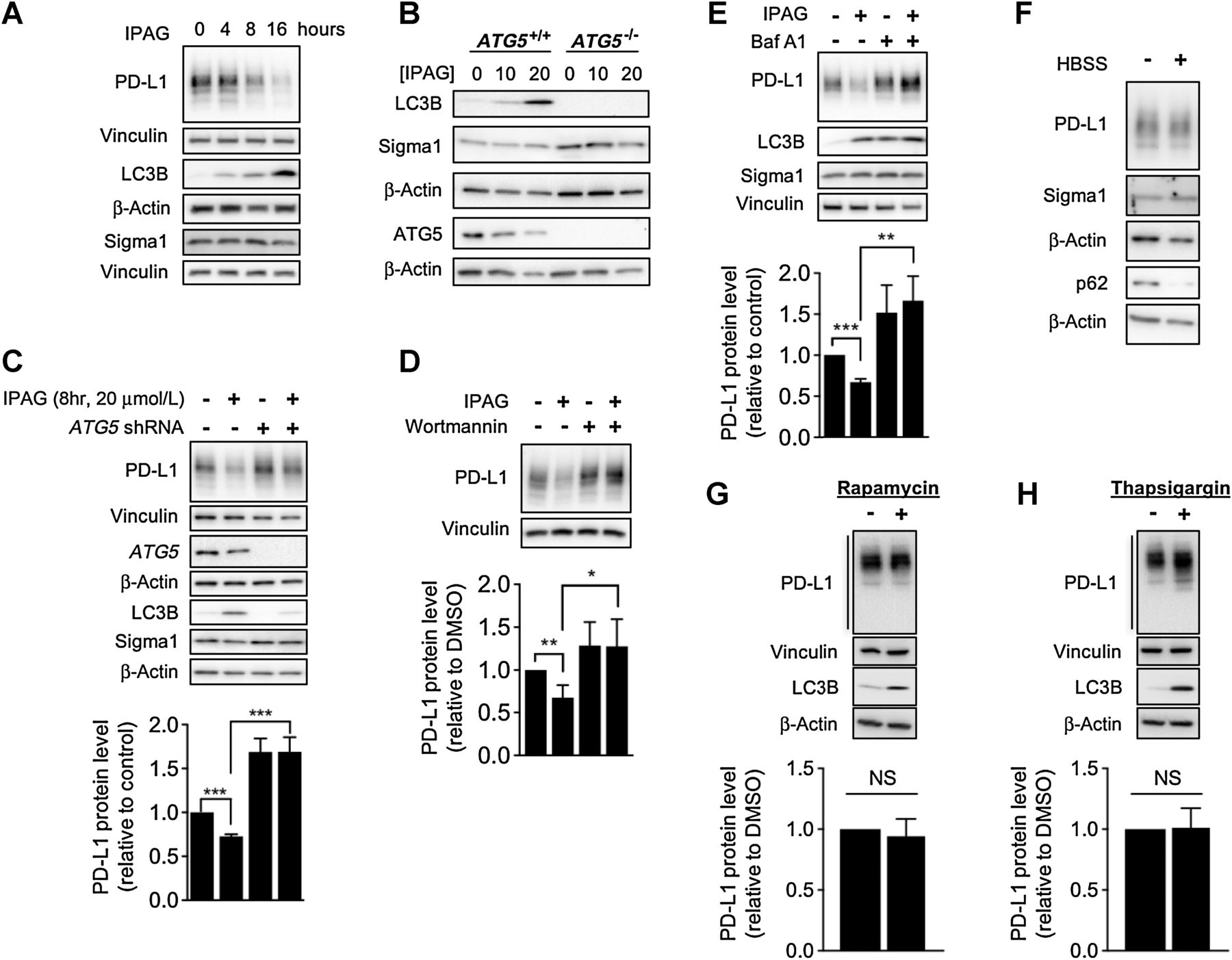
Inhibition of Sigma1 results in the degradation of PD-L1 via selective autophagy. A, Immunoblot of whole-cell protein extracts from MDA-MB-231 cells treated with 20 μmol/L IPAG for 4, 8, and 16 hours. B, Immunoblot of proteins from WT and ATG5 knockout (KO) MEFs treated with 10 and 20 μmol/L IPAG for 16 hours. C, Immunoblot showing PD-L1 protein levels in IPAG-treated cells following shRNA knockdown of ATG5 (initial stages of autophagy, autophagosome formation). ***, P < 0.001. D and E,IPAG combined with 50 nmol/L wortmannin (D; early stages of autophagy) or IPAG with 10 nmol/L bafilomycin A1 (E; Baf A1; late stages of autophagy, autolysosomal degradation) for 8 hours. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Data represent mean and SEM from at least three independent determinations. Bulk macroautophagy induced by culturing cells in HBSS (F) for 8 hours or treatment with rapamycin (G; 100 nmol/L) for 16 hours. H, ER-stress associated autophagy induced by thapsigargin (0.3 μmol/L) for 16 hours. Immunoblot bands were quantified by densitometry, and data are presented as levels of PD-L1 relative to nontreated control. Data represent mean and SD from at least two independent determinations.
We confirmed that IPAG induced autophagy by treating WT and ATG5 KO MEFs with IPAG and demonstrating that LC3B was induced in the WT but not in the ATG5 KO MEFs (Fig. 5B). Subsequently, we confirmed that autophagy contributes to Sigma1 modulator induced PD-L1 degradation by blocking the process in MDA-MB-231 cells by RNAi knockdown of ATG5 (Fig. 5C; Supplementary Fig. S5A and S5B), of ATG7 (Supplementary Fig. S5C), and by pharmacologically inhibiting autophagy with wortmannin and bafilomycin A1 (Baf A1; Fig. 5D and E). First, we performed shRNA-mediated knockdown (KD) of ATG5, the product of an essential autophagy gene required for the formation of autophagosomes (26, 27, 31). Knockdown of ATG5 in MDA-MB-231 cells blocked IPAG-mediated degradation of PD-L1 (Fig. 5C; Supplementary Fig. S5A and S5B). This experiment showed that autophagosome formation is required for IPAG-induced degradation of PD-L1. We replicated the results of this study with two distinct ATG5 shRNAs (data shown for TRCN0000330392 in Fig. 5C and in Supplementary Fig. S5A, and similar results were obtained with TRCN0000151474, shown in Supplementary Fig. S5B). Furthermore, we confirmed this effect with another essential autophagy gene, ATG7. ATG7 shRNA knockdown also mitigated PD-L1 degradation by IPAG when compared with the control (Supplementary Fig. S5C), confirming the fundamental role of autophagy in IPAG-mediated degradation of PD-L1.
Furthermore, we showed that pharmacologic inhibition of early and late stages of autophagy prevented PD-L1 decrease during IPAG treatment. Wortmannin is a Vps34 inhibitor that blocks autophagosome formation (26, 27, 38). Consistent with ATG5 knockdown, cotreatment with IPAG and wortmannin abrogated the PD-L1–decreasing effect of IPAG (Fig. 5D). Bafilomycin A1 (Baf A1) inhibits vacuolar H+ATPase, which blocks fusion of autophagosomes with lysosomes, and thus blocks autolysosomal degradation or autophagic flux (26, 27). We previously showed that IPAG induces autophagic flux in a number of cancer cell lines (24) and confirmed that IPAG induces autophagic flux here as well (Supplementary Fig. S6). Baf A1 blocked the decrease in PD-L1 in IPAG-treated MDA-MB-231 cells (Fig. 5E).
Next, we tested whether PD-L1 protein levels would be suppressed by other inducers of autophagy. Interestingly, induction of bulk macroautophagy by cell starvation in Hank’s buffered saline solution (HBSS) or treatment with rapamycin (26, 27) did not alter PD-L1 protein levels (Fig. 5F and G). As IPAG induces ER stress–associated autophagy (24), we asked whether this was a general mechanism by which PD-L1 could be degraded. In contrast to IPAG, we found that a widely used chemical inducer of ER stress–associated autophagy, thapsigargin (TG), which blocks the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA; refs. 26, 27), did not induce PD-L1 degradation under the experimental conditions evaluated herein (Fig. 5H). Altogether, these data suggest that Sigma1 modulators induce a form of selective autophagy.
Finally, we performed confocal microscopy to visualize autophagic sequestration and degradation of PD-L1 during IPAG treatment (Fig. 6). We treated MDA-MB-231 cells stably expressing a GFP-LC3 construct with IPAG alone or in the presence of Baf A1 (Fig. 6A). The formation of GFP-LC3 punctae indicates autophagosome formation. Baf A1 blocks autophagic flux and causes accumulation of autophagosomes and their cargo. This is detected as increased numbers and in some cases size of GFP-LC3 punctae. Consistent with the results described above, IPAG treatment produced GFP-LC3 labeled autophagosomes and induced their colocalization with PD-L1 (Fig. 6A). In contrast, treatment with Baf A1 alone resulted in increased GFP-LC3 autophagosomes but did not induce the same colocalization (Fig. 6A). Cotreatment with IPAG and Baf A1 resulted in increased numbers of GFP-LC3–positive punctae that colocalized with PD-L1, indicating an accumulation of PD-L1–containing autophagosomes (Fig. 6A–C). Under conditions of treatment with IPAG alone and cotreatment of IPAG and Baf A1, PD-L1 appears to be engulfed in autophagosomes (Fig. 6B and C). Altogether, these data suggest that autophagy is the operative mechanism by which IPAG induces intracellular degradation of PD-L1.
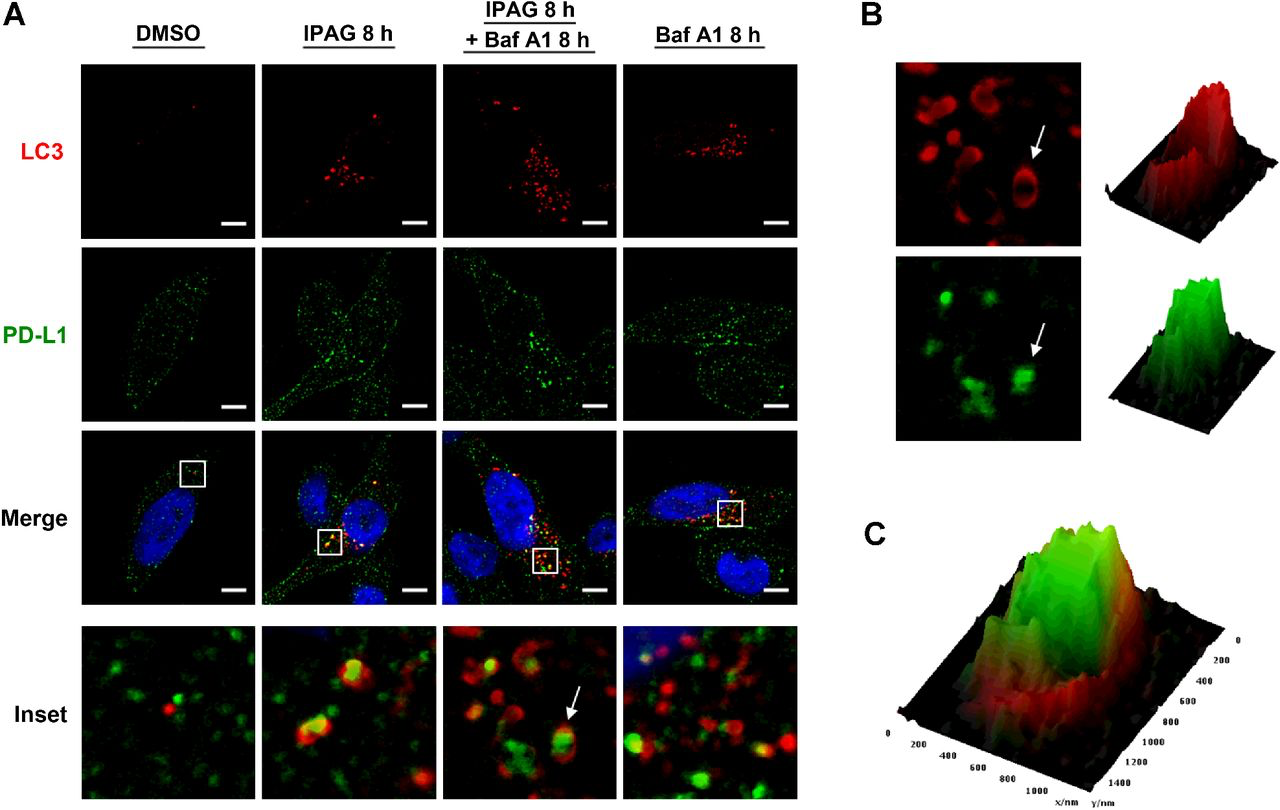
PD-L1 colocalizes with autophagosome marker GFP-LC3 following Sigma1 inhibition. A, Confocal micrographs showing colocalization of PD-L1 (green) and GFP-LC3 (red) in MDA-MB-231 cells treated for 8 hours with 20 μmol/L IPAG alone or combined with 10 nmol/L bafilomycin A1 (Baf A1). Scale bar, 5 μm. B, Magnified image of autophagosome (red) and PD-L1 (green) identified by white arrow in inset (A). Interactive surface plot generated from pixel maps of autophagosome (red) and PD-L1 (green). C,Overlay of autophagosome and PD-L1 interactive surface plots in B.
IFNγ induction of PD-L1
IFNγ is an immunostimulatory cytokine that has long been of interest for its potential in promoting antitumor immunity (39, 40). However, exposure to IFNγ also induces PD-L1 expression in cancer cells, thereby suppressing the antitumor immune response through adaptive immune resistance (39–41). As IFNγ is present in the tumor microenvironment, we asked whether small-molecule Sigma1 inhibitors could suppress/eliminate PD-L1 induction by IFNγ treatment. In our model, we found that IFNγ (100 ng/mL for 24 hours) induced PD-L1 protein levels (Fig. 7A–D) and transcripts (Fig. 7E and F) in MDA-MB-231 and PC3 cells. Cotreatment with IPAG (10 μmol/L for the final 16 hours of the 24-hour IFNγ treatment) decreased PD-L1 protein levels back to levels similar to or less than DMSO-treated conditions (Fig. 7A–D), with a slight decrease and a slight increase in PD-L1 transcripts in MDA-MB-231 and PC3 cells, respectively (Fig. 7E and F). It is important to note that, under IFNγ combined with IPAG conditions, PD-L1 transcript levels remain statistically significantly higher than DMSO control, whereas the protein level decreases to similar levels as the control. Therefore, under the experimental conditions used here, that is, 16-hour treatment with IPAG, the decrease in PD-L1 protein can be attributed to posttranslational effects, likely protein degradation.
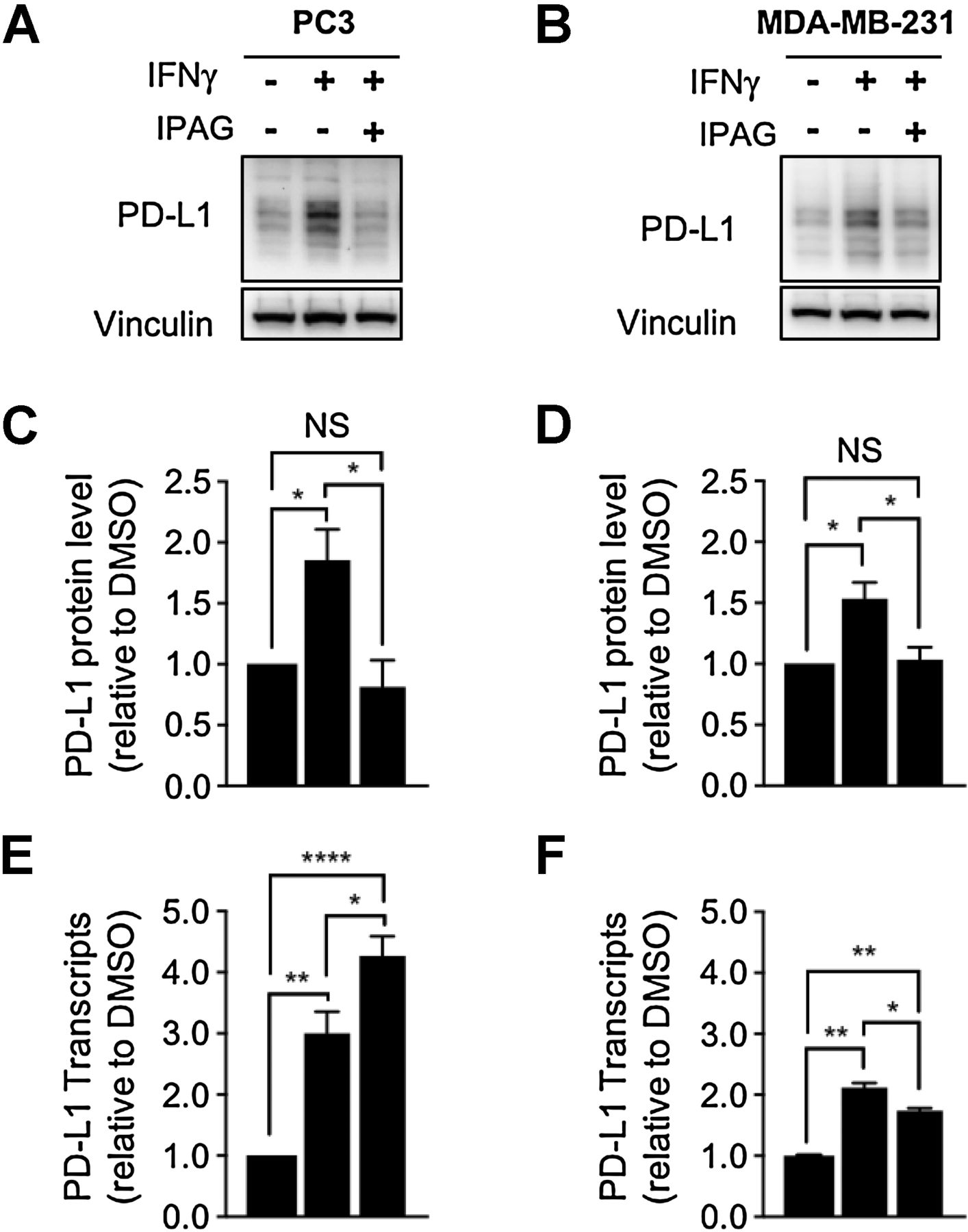
Small-molecule inhibition of Sigma1 suppresses IFNγ-induced upregulation of PD-L1 protein levels in cancer cells. Immunoblot and quantification of PD-L1 protein levels from whole-cell protein extracts from PC3 (A) and MDA-MB-231 (B) cells treated with 100 ng/mL IFNγ for 24 hours alone or in combination with 10 μmol/L IPAG for the last 16 hours. C and D, Immunoblots were quantified by densitometry for each cell line, and data are presented as levels of PD-L1 relative to nontreated control. Data represent mean values from at least three independent determinations and error bars represent SEM. *, P < 0.05. E and F, qRT-PCR results for PD-L1 transcript levels for PC3 (E) and MDA-MB-231 cells (F) treated with the same drug conditions used in A and B. Data are presented as fold induction over nontreated control. *, P < 0.05; **, P < 0.01; ****, P < 0.0001. Data represent mean values and error bars represent SEM.
Discussion
To our knowledge, this is the first demonstration of a direct physical and functional interaction between Sigma1 and PD-L1, the first demonstration that Sigma1 modulators can be used to sequester and degrade secretory pathway proteins by a potentially novel form of selective autophagy, and the first demonstration of autophagic degradation of PD-L1. These data suggest that Sigma1 modulators can be used to regulate PD-L1 transport and stability through regulation of pharmacologically responsive protein complexes.
Sigma1 modulator concept: positive and negative modulators, inhibitors, and activators
Traditionally, small molecules with affinity for Sigma1 have been classified as agonist or antagonist based primarily on data from rodent behavior assays (19). Differences between putative Sigma1 antagonists and agonists, however, have remained unresolved at the molecular and cellular level. Sigma1 does not have the properties of a traditional receptor, and therefore, designation of Sigma1 selective compounds as classically defined receptor antagonists or agonists may be inaccurate. Emerging evidence suggests that Sigma1 may function either as an allosteric cofactor protein associated with bona fide receptor systems or as a novel chaperone (19) or scaffolding protein. Therefore, it may be more appropriate to use the term Sigma1 modulator, and not agonist or antagonist, for compounds that pharmacologically alter Sigma1 activity. Here, we provide evidence in support of this concept with opposing actions of the putative Sigma1 antagonist IPAG (negative modulator or inhibitor) and the putative agonist SA4503 (positive modulator or activator). The Sigma1-selective actions of these compounds have been demonstrated elsewhere (24, 37, 42). In these studies, we also show that SA4503 blocked IPAG-mediated decreases in PD-L1 protein levels, pharmacologically demonstrating Sigma1-specific actions (Fig. 4A).
Insight into the role of Sigma1 in cancer
A number of publications report the cancer cell growth and survival inhibiting effects of compounds with affinity for Sigma1 (19); however, the specific mechanisms underlying these actions and the physiologic role of Sigma1 in cancer cells have remained unclear. Our data suggest that Sigma1 may play a role in regulating the components or compartments of the secretory pathway of cancer cells. Modulation of PD-L1 stability, transport, and activity by Sigma1 is consistent with such a physiologic role. Although the precise biochemical and molecular mechanisms governing Sigma1 actions in the secretory pathway of cancer cells remain to be determined, we posit that Sigma1 modulators coopt components of the ER protein homeostasis machinery to regulate the transport of PD-L1 transiting the secretory pathway to the plasma membrane. Demonstration that Sigma1 inhibitors can be used to sequester secretory pathway proteins into autophagosomes supports this notion. These results raise questions regarding what other secretory pathway-dependent proteins are regulated by Sigma1.
Sequestration and degradation of PD-L1 by pharmacologically induced selective autophagy
The biochemical and molecular mechanisms that govern PD-L1 transcription, translation, processing, assembly, transport, and functional binding partners (the intracellular proteins and cellular factors with which PD-L1 associates throughout its life cycle) remain poorly defined. Our data show that Sigma1 can physically interact with intracellular PD-L1 in pharmacologically responsive protein complexes that can be driven to autophagic sequestration and degradation of PD-L1. Our previous discovery that Sigma1 ligands can induce autophagy (24) and our observation that PD-L1 sequesters in punctate cytoplasmic clusters (Fig. 6) led us to investigate this process as a potential mechanism involved in Sigma1 inhibitor–induced PD-L1 degradation. By high-resolution confocal microscopy, we demonstrated that IPAG induces the sequestration and autophagic degradation of PD-L1 (Fig. 6). Inhibition of IPAG-mediated PD-L1 degradation by cotreatment with bafilomycin A1 (Baf A1) or with wortmannin (Fig. 5D and E) supports our hypothesis of autophagic degradation. Baf A1 blocks late-stage autophagic degradation, while wortmannin is a small-molecule inhibitor of Vps34, which blocks formation of autophagosomes (early-stage autophagy; refs. 27, 38).
ATG5 and ATG7 are essential autophagy genes required for autophagosome assembly and formation (31, 43). We confirmed that IPAG-induced autophagy requires ATG5 with ATG5 knockout MEFs (Fig. 5B). RNAi-mediated knockdown of ATG5 and ATG7 in MDA-MB-231 cells both prevented the elimination of PD-L1 in response to treatment with IPAG, thereby confirming autophagosomal degradation of PD-L1 (Fig. 5C; Supplementary Fig. S5).
Interestingly, treatment of cells with HBSS or rapamycin, both of which are commonly used to induce bulk macroautophagy (27, 44), did not result in decreased PD-L1 protein levels (Fig. 5F-G), nor did treatment with thapsigargin, which induces general ER stress–associated autophagy (Fig. 5H; refs. 27, 44). These data suggest that the Sigma1 modulator induces degradation of PD-L1 by a potentially novel form of selective autophagy.
In addition, because rapamycin, thapsigargin, wortmannin, and IPAG all suppress protein synthesis (25, 27, 44, 45), inhibition of protein translation may contribute to the suppression of PD-L1 levels during long-term treatment. However, it appears as though protein degradation is the operative mechanism by which PD-L1 protein levels are suppressed following IPAG treatment; this is supported by several lines of evidence demonstrating that whereas 16-hour treatment with IPAG results in decreased PD-L1 protein levels, treatment with rapamycin and thapsigargin did not (Fig. 5). This was further supported by the accelerated decrease in PD-L1 in IPAG-treated cells in which protein translation has been blocked by cycloheximide (Supplementary Fig. S3).
Our data suggest that IPAG induces autophagosomal degradation of nascent, ER-localized PD-L1, thus preventing surface expression of PD-L1. Salient elimination of the internal membrane fraction of PD-L1 and coimmunoprecipitation of Sigma1 with the lower MW species of N-glycosylated PD-L1 suggest that these proteins associate in the early/proximal secretory pathway, likely the ER. It is important to note, however, that the ER expands throughout the cytoplasmic space and associates with essentially all organelles within the cell (5). We do not rule out the possibility that Sigma1 may also bind to the cytoplasmic domain of plasma membrane–expressed PD-L1 and influence the levels and activity of cell surface PD-L1. Indeed, Sigma1 has been reported to bind to the cytoplasmic domains of other plasma membrane proteins (19) and plasma membrane components that interact with and contribute to autophagy as well as endosomal/endolysosomal internalization and degradation of cell surface proteins (31, 32).
The negative modulator of Sigma1, IPAG, suppresses PD-L1/PD-1 interaction associated signaling and transcriptional activity at 3 μmol/L, a concentration that does not alter PD-L1 levels (Fig. 3A and D), suggesting that Sigma1 regulates potential PD-L1 cofactors or interacting proteins required for its activity or modifies PD-L1 in a way that inhibits its function prior to or distinct from its destabilization and degradation. Emerging data suggest that Sigma1 associates with and can regulate multiple secretory pathway resident proteins in cancer cells. We have shown previously that Sigma1 modulators regulate ErbB2/HER2 and ErbB3/HER3 levels and activity in prostate and breast cancers (ref. 23 and our unpublished data, respectively).
Sigma1, PD-L1, and antitumor immunity
This study addresses the intracellular processes regulated by Sigma1 modulators, and it provides evidence in support of the notion that the pharmacologic mechanism of action of Sigma1 modulator compounds involves modulation of Sigma1-associated functional protein interactions. Sigma1 engages in protein–protein interactions with multiple partner and client proteins. It is likely that ligand-induced changes in some of these protein associations may contribute to suppression of PD-L1. This may occur through direct regulatory mechanisms or indirectly through feedback regulation mechanisms. For example, Sigma1 has been implicated in immunomodulation (reviewed in ref. 19), and a role for Sigma1 in antitumor immunity has been proposed (reviewed in ref. 19). Sigma1 activators (agonists) have been shown to induce the production and secretion of IL10 and TGFβ and promote tumor growth in a syngeneic mouse lung cancer model (46). Interestingly, in this model, IL10 was only induced at the tumor site and increased TGFβ only occurred in tumor-bearing mice, suggesting increased cytokine production in response to the tumor or increased cytokine production by the tumor cells (46). Sigma1 inhibitors (putative antagonists) abrogated the production of IL10 and suppressed Sigma1 activator–induced tumor growth (46). Sigma1 modulators have also been reported to regulate the production of IFNγ (46). PD-L1 production and activity through modulation of cell surface expression are regulated in part by cytokines released in the tumor microenvironment during antitumor immune responses, including IFNγ, TNFα, TGFβ, IL17, and IL10 (7, 13, 14, 47, 48). Taken together, these data suggest that pharmacologic modulation of Sigma1 can regulate PD-L1 production and activity via immune response induced cytokine-mediated extracellular feedback loops as well as directly via protein–protein associations. These mechanisms regulate the tenor of the tumor immune microenvironment (49).
There is compelling rationale for and reemerging interest in combination therapies to sustain antitumor immune responses induced by stimulatory cytokines such IFNγ (39–41). The ability of Sigma1 modulators to suppress PD-L1 in cancer cells treated with immunostimulatory cytokines, such as IFNγ (Fig. 7), reveals potential therapeutic approaches involving cotreatment with IFNγ and Sigma1 modulators to prolong/sustain antitumor immune responses.
PD-L1 also costimulates IL10 secretion (50). Sigma1 activators (agonists) have been shown to block antitumor immunity, in part by induction of IL10 (46). Thus, Sigma1 regulation of PD-L1 may contribute to antitumor immunity through checkpoint inhibition as well as cosuppression of anti-inflammatory cytokines. Sigma1 inhibitors may promote antitumor immunity in part by eliminating PD-L1 as well as by modulating the production and secretion of cytokines from cancer cells.
Thus, control of PD-L1 through Sigma1 supports a role for Sigma1 in antitumor immunity and provides insight into the mechanism(s) by which Sigma1 compounds elicit their immunomodulatory effects. Our data suggest that selective small-molecule Sigma1 ligands may be used as novel modulators of the tumor immune microenvironment. The use of Sigma1 modulators to pharmacologically promote antitumor immunity presents an opportunity for novel drug combinations with checkpoint inhibitor agents to expand the population of patients that respond to PD-L1/PD-1–targeted therapies.