
Sigma non-opioid receptor 1 is a potential therapeutic target for long QT syndrome
by Song, L., Bekdash, R., Morikawa, K. et al.
Excerpt from the article published in Nat Cardiovasc Res 1, 142–156 (2022). https://doi.org/10.1038/s44161-021-00016-2
Editor’s Highlights
- Mutations in the CACNA1C gene, encoding the α2δ-1 subunit in the calcium channel CaV1.2, cause a life-threatening form of long QT syndrome (LQTS) named Timothy syndrome.
- SIGMA-1 receptor could be a therapeutic target for TS and potentially LQTS1 and LQTS2, which are caused by the G269S substitution in the KV7.1 channel and the A561V substitution in the hERG channel, respectively
Abstract
Some missense gain-of-function mutations in the CACNA1C gene, encoding calcium channel CaV1.2, cause a life-threatening form of long QT syndrome (LQTS) named Timothy syndrome with currently no clinically effective therapeutics. Here we report that pharmacological targeting of sigma non-opioid intracellular receptor 1 (SIGMAR1) can restore electrophysiological function in induced pluripotent stem cell (iPSC)-derived cardiomyocytes generated from patients with Timothy syndrome and two common forms of LQTS, type 1 (LQTS1) and type 2 (LQTS2), caused by missense trafficking mutations in potassium channels. Electrophysiological recordings demonstrate that a Food and Drug Administration (FDA)-approved cough suppressant, dextromethorphan, can be used as an agonist of SIGMAR1 to shorten the prolonged action potential in cardiomyocytes from patients with Timothy syndrome and human cellular models of LQTS1 and LQTS2. When tested in vivo, dextromethorphan also normalized the prolonged QT intervals in a mouse model of Timothy syndrome. Overall, our study demonstrates that SIGMAR1 is a potential therapeutic target for Timothy syndrome and possibly other inherited arrhythmias such as LQTS1 and LQTS2.
Main
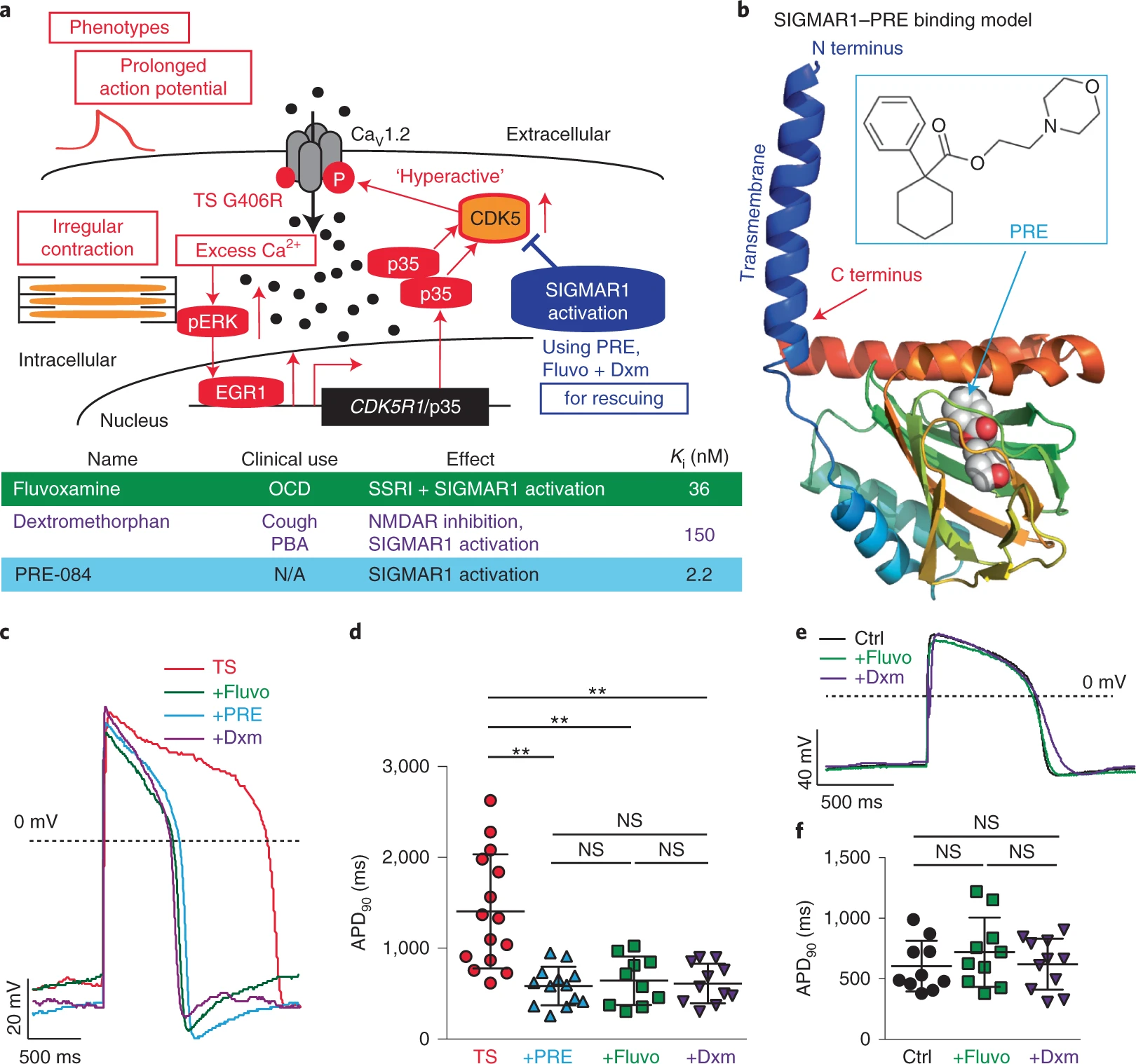
Calcium ions (Ca2+) play an essential role in cardiac excitation–contraction coupling1,2. Ca2+-handling dysfunction is associated with various cardiac arrhythmias and genetic syndromes such as LQTS3,4. Some de novo gain-of-function mutations such as the one corresponding to G406R in cardiac L-type voltage-gated Ca2+ channel CaV1.2, which is encoded by CACNA1C, cause a lethal form of LQTS (type 8, also named Timothy syndrome (TS))3,5. Previously, using human patient-specific iPSCs, we found that the increased activity of cyclin-dependent kinase 5 (CDK5) is involved in the cardiac pathophysiological basis of TS (Fig. 1a, red)6. In addition, we observed that inhibiting CDK5 using genetic approaches and chemical inhibitors, such as PHA-793887, alleviated cellular phenotypes in cardiomyocytes derived from iPSCs of patients with TS6. However, currently available inhibitors of CDK5 also show inhibitory effects on other cyclin-dependent kinases. As a result, if the inhibitors are used for clinical application for treating cardiac diseases, it may lead to adverse events7 because cyclin-dependent kinases are known to play a critical role in cell cycle regulation. Moreover, developing CDK5-specific inhibitors is challenging due to the structural similarity of the catalytic domain in different cyclin-dependent kinases8. Therefore, using small molecules to directly target CDK5 is not ideal for further translational development to fulfill unmet medical needs of patients with TS. For this reason, we decided to search for alternative approaches that may modulate CDK5 activity in human cardiomyocytes. A comprehensive literature search returned SIGMAR1 as a potential target. SIGMAR1 is an emerging target for neurodegenerative diseases and cancer9,10,11, playing an important role as a chaperone protein in endoplasmic reticulum membranes by regulating protein homeostasis, mitochondrial function, ion channel function and Ca2+ handling12. More importantly, activation of SIGMAR1 has been reported to suppress CDK5 activity in rodent neurons13, making SIGMAR1 activation an attractive alternative approach for CDK5 inhibition in TS models. Furthermore, the structure of SIGMAR1 has been uncovered14, which greatly facilitated the identification of SIGMAR1 ligands and understanding of interactions between SIGMAR1 and its ligands. Several SIGMAR1 agonists have been identified, including a potent and specific agonist, PRE-084 (ref. 15), as well as several FDA-approved drugs such as fluvoxamine16 and dextromethorphan17,18,19,20 (Fig. 1a,band Extended Data Fig. 1a). However, to our knowledge, little is known regarding the role of SIGMAR1 in cardiac ion channel regulation and pathophysiological conditions in cardiac arrhythmias. In this study, we examined the role of SIGMAR1 in the pathogenesis of LQTS as a therapeutic target and tested the hypothesis that treatment with a SIGMAR1 agonist, such as PRE-084, fluvoxamine or dextromethorphan, could rescue cardiac phenotypes in TS (Fig. 1a, blue).
Results
Effects of SIGMAR1 activation on cardiomyocytes from patients with TS
To examine whether SIGMAR1 activation modulates CDK5 activity in human cardiomyocytes as it did in neurons13, we conducted an in vitro luminescence-based CDK5 activity assay6using cardiomyocytes derived from iPSCs of patients with TS and treated them with the SIGMAR1-selective agonist PRE-084. Results indicated that treatment with PRE-084 for 2 h significantly reduced CDK5 activity in cardiomyocytes from patients with TS (Extended Data Fig. 1b). While it was reported that protein expression of a CDK5-specific activator, p35 (CDK5R1), was regulated by SIGMAR1 in neurons13, interestingly, we observed no significant change in p35 protein expression between cardiomyocytes from patients with TS with and without PRE-084 treatment (Extended Data Fig. 1c,d). Instead, protein expression of CDK5 was significantly reduced in cardiomyocytes from patients with TS treated with PRE-084, while no significant changes in CDK5 transcript levels were detected (Extended Data Fig. 1c–e). These results revealed that activation of SIGMAR1 using its selective agonist PRE-084 could modulate CDK5 activity in cardiomyocytes from patients with TS. To further explore the molecular mechanism underlying reductions in CDK5 levels induced by PRE-084, we conducted a proximity ligation assay (PLA) to investigate the potential interaction between SIGMAR1 and CDK5. We found that SIGMAR1 directly interacted with CDK5 in cardiomyocytes from patients with TS and treatment with PRE-084 significantly increased the number of SIGMAR1–CDK5 PLA signals in cardiomyocytes from patients with TS, indicating an increased interaction between SIGMAR1 and CDK5 upon SIGMAR1 activation (Extended Data Fig. 1f,g). To validate that the change induced by PRE-084 was mediated by SIGMAR1, we treated cardiomyocytes from patients with TS with a potent and selective SIGMAR1 antagonist, NE-100 (refs. 21,22,23), along with PRE-084, and used the cells in a SIGMAR1–CDK5 PLA. Results showed that cotreatment with NE-100 and PRE-084 reversed the effect of PRE-084 on the interaction of SIGMAR1 with CDK5 in cardiomyocytes from patients with TS, confirming that the effect of PRE-084 was indeed mediated by SIGMAR1 (Extended Data Fig. 1f,g). Overall, results indicated that SIGMAR1 directly interacted with CDK5 in cardiomyocytes from patients with TS and SIGMAR1 activation led to an increased interaction between SIGMAR1 and CDK5 and reduced CDK5 expression and activity in cardiomyocytes from patients with TS.
The positive effect of SIGMAR1 activation on CDK5 activity prompted us to examine the effects of the SIGMAR1 agonist PRE-084 on cellular phenotypes of cardiomyocytes from patients with TS. We first examined the effect of PRE-084 on the irregular and slow contraction phenotype in cardiomyocytes from patients with TS using a contraction assay as reported previously6. This is because, as heart beats are normally regular, irregular contractions in a cellular model indicate electrophysiological instability. Imaging results demonstrated that PRE-084 alleviated irregular contraction and increased the beating rate of the patient cardiomyocytes (Extended Data Fig. 1h–j and Supplementary Videos 1 and 2). Next, we conducted whole-cell voltage-clamp recordings to evaluate the effect of PRE-084 on the phenotype in voltage-dependent inactivation of CaV1.2 channels in cardiomyocytes from patients with TS. Results showed that PRE-084 treatment rescued the affected voltage-dependent inactivation of CaV1.2 in cardiomyocytes from patients with TS after treatment for 2 h but not after acute (<10 min) treatment (Extended Data Fig. 1k–n), supporting the beneficial effect of PRE-084 on cardiomyocytes from patients with TS. Furthermore, we examined the effect of PRE-084 on the prolonged action potential in cardiomyocytes from patients with TS and observed that PRE-084 significantly shortened the prolonged action potentials in patient cardiomyocytes (Fig. 1c,d and Extended Data Fig. 2a–c). Overall, these results demonstrated that the selective SIGMAR1 agonist PRE-084 ameliorated the phenotypes of cardiomyocytes from patients with TS. This points to SIGMAR1 activation as a potential therapeutic avenue for the cardiac phenotypes in TS.
Next, we examined the effects of fluvoxamine and dextromethorphan, which are also SIGMAR1 agonists, on the phenotypes of cardiomyocytes from patients with TS. The FDA-approved drugs fluvoxamine and dextromethorphan may have a greater translational potential for TS if the drugs are proved to be effective in a human cellular model. Similar to the selective SIGMAR1 agonist PRE-084, electrophysiological recordings revealed that the SIGMAR1 agonists fluvoxamine and dextromethorphan restored CaV1.2 voltage-dependent inactivation and action potentials in cardiomyocytes from patients with TS (Fig. 1c,d and Extended Data Figs. 1k,l and 2a–c).
At the dose that fluvoxamine and dextromethorphan rescued the electrophysiological phenotypes in cardiomyocytes from patients with TS, the two FDA-approved drugs did not significantly alter the action potential duration (APD) of normal cardiomyocytes generated from isogenic control iPSC lines in which the mutation resulting in TS was previously corrected by gene-editing technology6 (Fig. 1e,f and Extended Data Fig. 2d–f). We sought to understand the difference in sensitivity to treatment with SIGMAR1 agonists between normal cardiomyocytes and those derived from patients with TS. We found that, at baseline, there was a significant increase in levels of SIGMAR1 transcripts and SIGMAR1 protein in cardiomyocytes from patients with TS compared with those in normal cardiomyocytes (Extended Data Fig. 2g–j), which may predispose cardiomyocytes from patients with TS to respond to SIGMAR1 agonists. To further explore the upstream pathway that regulates the expression of SIGMAR1, we examined the expression of ATF4, a transcription factor that has been reported to directly regulate SIGMAR1 transcription24. Results demonstrated that there was a significant increase in ATF4 protein levels (both non-phosphorylated and phosphorylated forms) and a significant decrease in ubiquitinated ATF4 protein levels in cardiomyocytes from patients with TS compared with those in their isogenic control cardiomyocytes (Extended Data Fig. 2k–n), indicating an accumulation of ATF4 protein in cardiomyocytes from patients with TS. To confirm the regulatory role of ATF4 on SIGMAR1expression in human cardiomyocytes, we overexpressed ATF4 in human normal cardiomyocytes and conducted quantitative PCR with reverse transcription (RT–qPCR) and western blotting to examine the effect of increased ATF4 expression on SIGMAR1 mRNA and SIGMAR1 protein expression. Results showed that ATF4 overexpression significantly increased the expression of SIGMAR1 transcripts and SIGMAR1 protein in cardiomyocytes derived from human iPSCs (Extended Data Fig. 2o–r), confirming the regulation of SIGMAR1expression via ATF4. Altogether, results suggest that increased ATF4 protein levels in cardiomyocytes from patients with TS might activate the transcription of SIGMAR1 and upregulate the expression of SIGMAR1, leading to increased sensitivity of cardiomyocytes from patients with TS to treatment with SIGMAR1 agonists.
Although the observed beneficial effects of fluvoxamine and dextromethorphan on cardiomyocytes from patients with TS were both encouraging, dextromethorphan seems to hold greater therapeutic potential for translational application because of its wider age range for clinical use and favorable safety profile when used appropriately16,20,25,26,27. Thus, we decided to further characterize the effects of dextromethorphan. To examine the effect of dextromethorphan on the abnormal Ca2+-handling phenotype of cardiomyocytes from patients with TS, we used a genetically encoded fluorescent Ca2+ indicator, GCaMP6f28, for Ca2+ imaging. Compared with the use of Ca2+ dyes such as Fluo-4 AM, Ca2+ imaging using genetically encoded indicators has the distinct advantage that infected cells are intact and remain in reliable imaging condition for hours, which enables continuous monitoring of changes in Ca2+ handling after compound treatment for a longer time. Consistent with our previous findings using Fluo-4 AM for Ca2+ imaging5, Ca2+ imaging using GCaMP6f confirmed that cardiomyocytes from patients with TS demonstrated abnormal Ca2+ handling compared with isogenic control cardiomyocytes, and this phenotype could be stably monitored for at least 2 h after GCaMP6f fluorescent imaging began (Extended Data Fig. 3a–d). Using GCaMP6f-based Ca2+ imaging, we found that dextromethorphan treatment exhibited significant beneficial effects on the abnormal Ca2+-handling phenotype in cardiomyocytes from patients with TS and significantly increased Ca2+ transient frequency and shortened Ca2+ transient duration 2 h after treatment (Fig. 2a–c), which is also the time point at which we observed a significant beneficial effect of dextromethorphan on the electrophysiological phenotypes of cardiomyocytes from patients with TS (Fig. 1c,d).
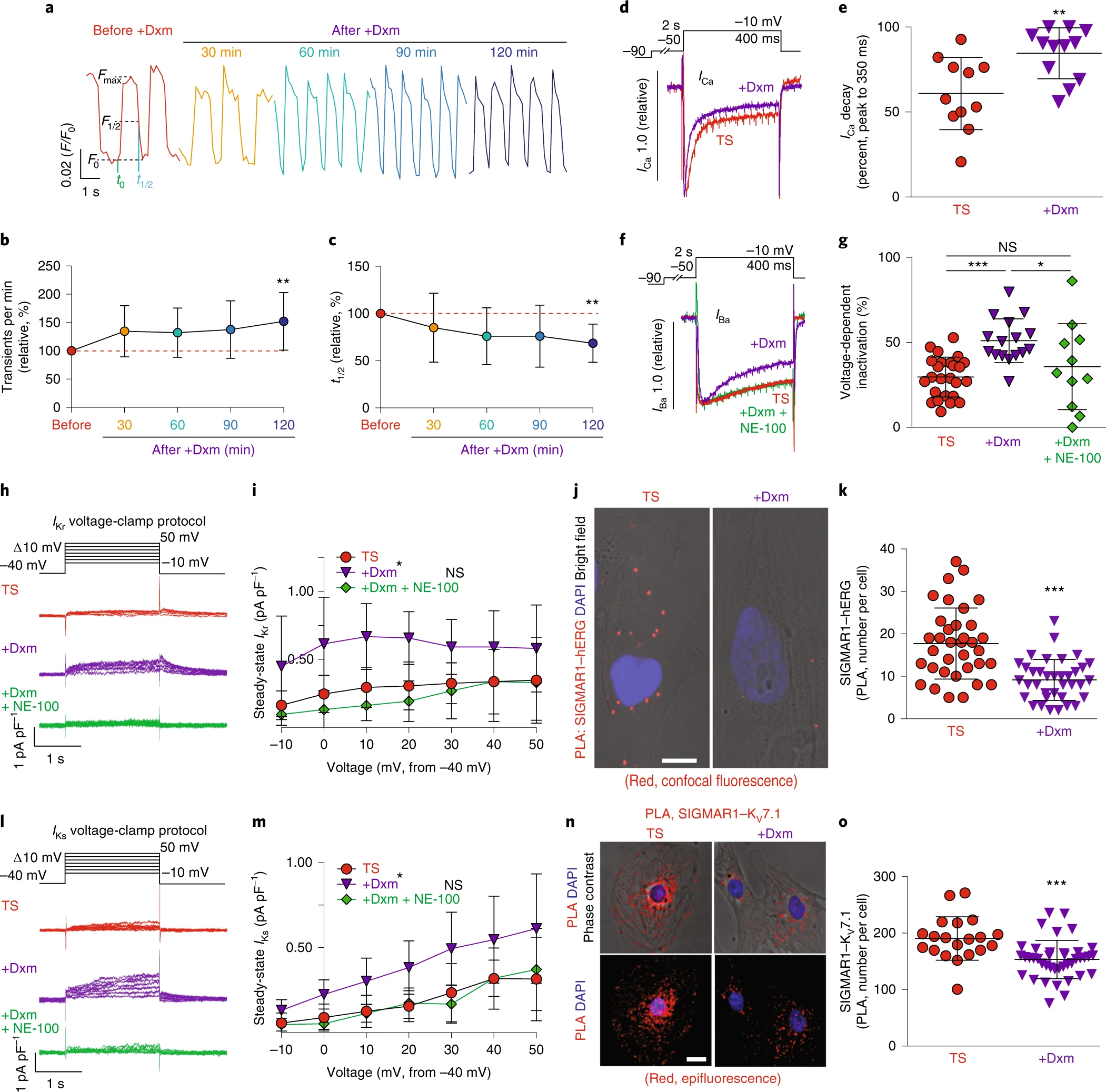
The effect of dextromethorphan on cardiac Ca2+ handling and ion channels in a human iPSC model of TS.
a, Representative traces of time-course Ca2+ imaging in spontaneously contracting cardiomyocytes from patients with TS treated with dextromethorphan (5 μM, until 120 min). b,c, Ca2+ transient frequency (b) and duration (c) analyses of cardiomyocytes from patients with TS before and after dextromethorphan treatment (n = 19). d, Representative traces of Ca2+ currents in cardiomyocytes from patients with TS with and without dextromethorphan. e, Late Ca2+ current analysis of cardiomyocytes from patients with TS with and without dextromethorphan treatment (TS, n = 11; with dextromethorphan, n = 12). f, Representative traces of Ba2+ currents in cardiomyocytes from patients with TS without treatment or treated with dextromethorphan (5 μM, 2 h, dextromethorphan) or with dextromethorphan and a SIGMAR1 antagonist, NE-100 (1 μM, dextromethorphan and NE-100). g, Voltage-dependent inactivation in cardiomyocytes from patients with TS without treatment (n = 25) or treated with dextromethorphan (n = 16) or with dextromethorphan and NE-100 (n = 11). h, Representative traces of IKr currents (E-4031 sensitive) in cardiomyocytes from patients with TS treated with dextromethorphan (5 μM) or dextromethorphan and NE-100 (each at 5 μM) or without treatment. i, IKr current amplitude analysis of cardiomyocytes from patients with TS treated with dextromethorphan (n = 9) or dextromethorphan and NE-100 (n = 10) or without treatment (n = 10) (*P < 0.05, dextromethorphan versus TS and dextromethorphan versus dextromethorphan and NE-100 at −10, 0, 10, 20 and 30 mV). j, Representative confocal fluorescent and bright-field images of cardiomyocytes from patients with TS without and with dextromethorphan treatment from the PLA (SIGMAR1–hERG, red; 4,6-diamidino-2-phenylindole (DAPI), blue). Scale bar, 10 μm. k, Quantification of SIGMAR1–hERG by the PLA in dextromethorphan-treated (n = 36) and untreated (n = 35) cardiomyocytes from patients with TS. l, Representative traces of IKs currents (chromanol 293B sensitive) in cardiomyocytes from patients with TS treated with dextromethorphan (5 μM) or dextromethorphan and NE-100 (each at 5 μM) or without treatment. m, IKs current amplitude analysis of cardiomyocytes from patients with TS treated with dextromethorphan (n = 10) or dextromethorphan and NE-100 (n = 9) or without treatment (n = 10) (*P < 0.05, dextromethorphan versus TS and dextromethorphan versus dextromethorphan and NE-100 at −10, 0, 10, 20, 30 and 40 mV). n, Representative epifluorescent and phase-contrast images of cardiomyocytes from patients with TS without and with dextromethorphan treatment from the PLA (SIGMAR1–KV7.1, red; DAPI, blue). Scale bar, 10 μm. o, Quantification of SIGMAR1–KV7.1 by the PLA in dextromethorphan-treated (n = 40) and untreated (n = 20) cardiomyocytes from patients with TS. All data are mean ± s.d. One-way ANOVA with Tukey’s multiple comparisons was used for b,c,g between groups and for i,m at each voltage step. Unpaired two-tailed Student’s t-tests were used for e,k,o. *P < 0.05, **P < 0.01, ***P < 0.001. Samples were from at least two independent differentiations.
Source data
Next, to further characterize the effect of dextromethorphan on L-type Ca2+ channel function in cardiomyocytes from patients with TS, we conducted whole-cell voltage-clamp recordings using Ca2+ as the charge carrier to examine the effect of dextromethorphan on Ca2+ channel currents. We found that dextromethorphan significantly reduced late Ca2+ currents in cardiomyocytes from patients with TS (Fig. 2d,e). This result, along with the result from electrophysiological recordings using Ba2+ as the charge carrier (Extended Data Figs. 1l and 3e), demonstrated that dextromethorphan treatment restored the inactivation of the CaV1.2 channel in cardiomyocytes from patients with TS. In addition, to validate that the effect of dextromethorphan on CaV1.2 channel inactivation was mediated by SIGMAR1, we tested the selective SIGMAR1 antagonist NE-100 (refs. 21,22,23) along with dextromethorphan in electrophysiological recordings using Ba2+ as the charge carrier. We found that the beneficial effect of dextromethorphan on the affected voltage-dependent inactivation of CaV1.2 in cardiomyocytes from patients with TS was reversed by cotreatment with NE-100, indicating that the beneficial effect of dextromethorphan on Ca2+ channel inactivation was mediated by SIGMAR1 (Fig. 2f,g). On the other hand, at the dose at which it was efficacious toward cardiomyocytes from patients with TS, dextromethorphan did not significantly alter voltage-dependent inactivation of CaV1.2 in isogenic control cardiomyocytes (Extended Data Fig. 3f,g). Furthermore, as SIGMAR1 has been reported to interact with L-type Ca2+ channels and regulate channel function in retinal ganglion cells and dorsal ganglion neurons29,30,31, we examined the potential interaction between SIGMAR1 and CaV1.2 in cardiomyocytes from patients with TS and the effect of dextromethorphan on the interaction using the PLA. Results suggest that SIGMAR1 directly interacted with CaV1.2 in cardiomyocytes from patients with TS, and there was a significant reduction in the number of SIGMAR1–CaV1.2 signals from the PLA in cardiomyocytes from patients with TS after dextromethorphan treatment (Extended Data Fig. 3h,i), indicating that dextromethorphan reduced the direct interaction between SIGMAR1 and CaV1.2 in cardiomyocytes from patients with TS. Altogether, results demonstrated that dextromethorphan restored the channel inactivation of CaV1.2 in cardiomyocytes from patients with TS through SIGMAR1 activation. The findings in aggregate indicated that SIGMAR1 might play a multifaceted role in regulating CaV1.2 channel function in human iPSC-derived cardiomyocytes by not only regulating CDK5 activity but also by directly interacting with CaV1.2.
To further investigate the effect of dextromethorphan on cardiac action potential, we performed voltage-clamp recordings to measure human Ether-à-go-go-Related Gene (hERG) currents in cardiomyocytes from patients with TS because potassium ion (K+) channels are also known to play an important role in the repolarization phase of action potential in cardiomyocytes. Interestingly, we found that there was a significant reduction in IKr currents in cardiomyocytes from patients with TS compared with isogenic control cardiomyocytes (Extended Data Fig. 3j–l). Moreover, dextromethorphan treatment significantly increased IKrsteady-state amplitude in cardiomyocytes from patients with TS, while no significant change in IKr currents was observed for isogenic control cardiomyocytes treated with dextromethorphan at the same dose (Fig. 2h,i and Extended Data Figs. 3l and 4a–c). Furthermore, the beneficial effect of dextromethorphan on IKr currents in cardiomyocytes from patients with TS was blocked by cotreatment with the SIGMAR1 antagonist NE-100 and dextromethorphan, confirming that the effect of dextromethorphan on hERG channels was mediated by SIGMAR1 (Fig. 2h,i and Extended Data Fig. 3l,m). These results suggest that SIGMAR1 activation mediated by dextromethorphan might rescue prolonged action potentials in cardiomyocytes from patients with TS by restoring K+ efflux in addition to reducing Ca2+ influx.
SIGMAR1 has been reported to directly interact with hERG channels in HEK cells32, and the direct interaction was further confirmed by atomic force microscopy33. In addition, SIGMAR1 silencing using short hairpin RNA or treatment with progesterone, which is also known to be a SIGMAR1 antagonist, decreased hERG current density and negatively regulated membrane expression of the hERG channel in leukemic cells, HEK cells and neonatal rat cardiomyocytes12,32,34, while SIGMAR1 overexpression potentiated hERG current density in Xenopus oocytes32. Thus, we decided to evaluate the potential interaction between SIGMAR1 and the hERG channel in cardiomyocytes derived from human iPSCs using the PLA and to examine the effect of dextromethorphan on their potential interaction. We found that, consistent with a previous report using HEK cells32, SIGMAR1 also directly interacted with hERG channels in cardiomyocytes derived from human iPSCs. Treatment with dextromethorphan for 2 h significantly reduced SIGMAR1–hERG signals in the PLA in cardiomyocytes from patients with TS, while no significant change in SIGMAR1–hERG signals in the PLA in isogenic control cardiomyocytes was observed with dextromethorphan treatment at the same dose (Fig. 2j,k and Extended Data Fig. 4d–g).
To investigate the molecular mechanisms underlying the effect of dextromethorphan on hERG channels further, we conducted immunocytochemistry for the hERG channel and plasma membrane with an anti-hERG antibody and the wheat germ agglutinin (WGA) fluorescent dye, respectively, using cardiomyocytes from patients with TS with and without treatment with dextromethorphan. We imaged the stained cardiomyocytes using a confocal microscope and then conducted blinded quantification for the colocalization of hERG and WGA fluorescent signals to examine the effect of dextromethorphan on the membrane localization of hERG in cardiomyocytes from patients with TS. The results suggested that there was a trend toward increased hERG–WGA colocalization in dextromethorphan-treated cardiomyocytes from patients with TS compared with that of untreated cells, but the change did not reach statistical significance (Extended Data Fig. 5a,b). In addition, we explored the possibility that SIGMAR1 activation induced by dextromethorphan treatment increased the expression of KCNH2 and hERG, resulting in the increased IKr observed in cardiomyocytes from patients with TS. Therefore, qPCR and western blotting were conducted to examine the expression of KCNH2 transcripts and hERG protein in cardiomyocytes from patients with TS with or without dextromethorphan treatment. We found that dextromethorphan significantly increased KCNH2 transcription in patient cardiomyocytes (Extended Data Fig. 5c) and did not significantly alter the protein expression of hERG (Extended Data Fig. 5d,e).
Overall, these results demonstrated that SIGMAR1 directly interacted with hERG channels in human iPSC-derived cardiomyocytes, and the SIGMAR1 agonist dextromethorphan might increase IKr currents by dissociating the interaction between SIGMAR1 and hERG channels in cardiomyocytes from patients with TS, while the exact mechanisms remain to be characterized.
The observation of an interaction between SIGMAR1 and the hERG channel in human iPSC-derived cardiomyocytes prompted us to examine whether there is a similar interaction between SIGMAR1 and another K+ channel, KV7.1, which is encoded by the KCNQ1 gene and also plays an important role in the repolarization phase of cardiac action potentials, similar to the hERG channel. Using electrophysiological recordings, we found that, similar to our observations of the hERG channel, cardiomyocytes from patients with TS showed significantly reduced IKs currents compared with isogenic controls and treatment with dextromethorphan significantly increased IKs currents in cardiomyocytes from patients with TS (Fig. 2l,m and Extended Data Fig. 5f,g). In addition, cotreatment with NE-100 and dextromethorphan blocked the beneficial effect of dextromethorphan on IKs currents in cardiomyocytes from patients with TS, confirming that the effect of dextromethorphan on KV7.1 channels is mediated by SIGMAR1 (Fig. 2l,m and Extended Data Fig. 5g). Moreover, consistent with our observations for SIGMAR1 and hERG channels, SIGMAR1 also directly interacted with the KV7.1 channel in human iPSC-derived cardiomyocytes. Similarly, dextromethorphan treatment significantly reduced SIGMAR1–KV7.1 signals in the PLA in cardiomyocytes from patients with TS (Fig. 2n,o). On the other hand, there were no significant changes in IKs currents in isogenic control cardiomyocytes after dextromethorphan treatment at the same dose, although a reduction in SIGMAR1–KV7.1 signals in the PLA was observed upon treatment (Extended Data Fig. 4h–k).
To investigate the molecular mechanisms underlying the effect of dextromethorphan on the KV7.1 channel further, we conducted immunocytochemistry for the KV7.1 channel (encoded by KCNQ1) and plasma membrane using an anti-KV7.1 antibody and the WGA fluorescent dye, respectively, and cardiomyocytes from patients with TS with and without dextromethorphan treatment. A significant increase in plasma membrane localization of the KV7.1 channel was observed upon dextromethorphan treatment (Extended Data Fig. 5h,i), indicating that SIGMAR1 activation induced by dextromethorphan treatment facilitated membrane localization of KV7.1 channels in cardiomyocytes from patients with TS. In addition, we conducted qPCR to examine the expression of KCNQ1 transcripts in cardiomyocytes from patients with TS with and without dextromethorphan treatment and observed a significant increase in the transcription of KCNQ1 isoform 1 upon dextromethorphan treatment, while transcription of KCNQ1 isoform 2 was not significantly altered by dextromethorphan treatment (Extended Data Fig. 5j). Consistent with the qPCR results, western blotting showed a significant increase in the expression of the KV7.1 isoform 1 protein but not in that of the isoform 2 protein upon dextromethorphan treatment (Extended Data Fig. 5k–m), indicating that SIGMAR1 activation induced by dextromethorphan treatment increased the expression of KV7.1 isoform 1 in addition to facilitating membrane localization of KV7.1 channels.
In summary, these results demonstrated that, in addition to the hERG channel, SIGMAR1 also directly interacted with the KV7.1 channel in human iPSC-derived cardiomyocytes, and the SIGMAR1 agonist dextromethorphan might increase IKs currents in cardiomyocytes from patients with TS by dissociating the interaction between SIGMAR1 and KV7.1 channels, increasing plasma membrane localization of KV7.1 and increasing expression of KV7.1 isoform 1.
Effects of SIGMAR1 activation on cardiomyocytes from patients with LQTS1 and LQTS2
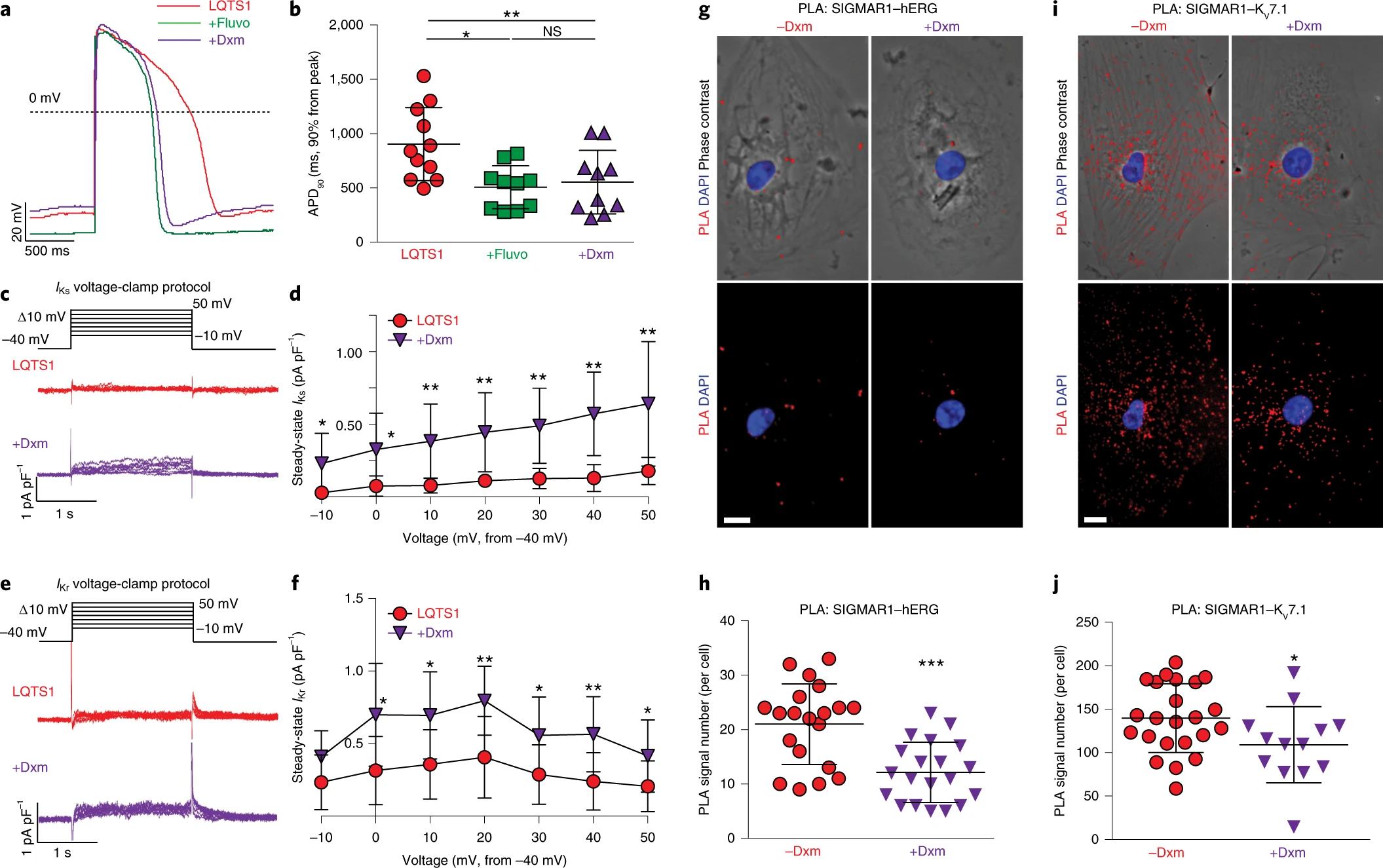
The observation that dextromethorphan increased IKr and IKs currents in cardiomyocytes from patients with TS motivated us to examine the effect of dextromethorphan on cardiomyocytes derived from human iPSCs of two common types of LQTS, LQTS1 and LQTS2, carrying missense trafficking mutations corresponding to G269S in the KV7.1 channel, and A561V in the hERG channel, respectively. The missense trafficking mutations in the KV7.1 and hERG channels were previously reported to decrease K+ currents and prolong action potentials in human iPSC-derived cardiomyocytes35,36. First, we used CRISPR–Cas9 gene-editing technology to correct the mutations in our iPSCs derived from patients with LQTS1 or LQTS2 and generated isogenic control iPSCs (Extended Data Fig. 6a–d). Next, we differentiated iPSCs from both patients with LQTS1 and LQTS2 along with their corresponding isogenic controls into cardiomyocytes and evaluated their electrophysiological phenotypes. Consistent with previous reports35,36, we found that cardiomyocytes derived from iPSCs of patients with LQTS1 or LQTS2 showed prolonged action potentials (APD, 90% from peak (APD90), mean ± s.d., LQTS1 versus the isogenic control, 904.3 ± 337.0 ms versus 522.9 ± 186.6 ms, P = 0.004; LQTS2 versus the isogenic control, 1,054.2 ± 398.9 ms versus 644.8 ± 201.9 ms, P = 0.011; unpaired two-tailed Student’s t-test) and also that cardiomyocytes from patients with LQTS1 or LQTS2 showed significantly reduced K+ currents in electrophysiological recordings compared with their corresponding isogenic control cardiomyocytes (Extended Data Fig. 6j,k,q–s). Treatment with a SIGMAR1 agonist, fluvoxamine or dextromethorphan, significantly shortened the prolonged action potential in cardiomyocytes from patients with LQTS1 or LQTS2 (Figs. 3a,b and 4a,b and Extended Data Fig. 7a–f). In addition, consistent with the effects of dextromethorphan on K+channels in cardiomyocytes from patients with TS, dextromethorphan treatment significantly increased both IKr and IKs currents in cardiomyocytes from patients with LQTS1 or LQTS2 (Figs. 3c–f and 4c–f and Extended Data Fig. 7g,h). Mechanistically, similar to what was observed in cardiomyocytes from patients with TS (Fig. 2j,k,n,o), dextromethorphan treatment significantly reduced both SIGMAR1–hERG and SIGMAR1–KV7.1 signals in the PLA in cardiomyocytes from patients with LQTS1 or LQTS2 (Figs. 3g–j and 4g–j). Moreover, at the efficacious dose, dextromethorphan did not exhibit significant effects on action potentials in the corresponding isogenic control cardiomyocytes (Extended Data Fig. 6e–i,l–p) or on the voltage-dependent inactivation of CaV1.2 in cardiomyocytes from patients with LQTS1 or LQTS2 (Extended Data Fig. 7i–n). Furthermore, a significant increase in SIGMAR1 and ATF4 expression was detected in cardiomyocytes from patients with LQTS1 or LQTS2, which may potentiate the cardiomyocytes to respond to dextromethorphan treatment, while no significant changes in CDK5 or p35 expression were observed in cardiomyocytes from patients with LQTS1 or LQTS2 compared with control cardiomyocytes (Extended Data Fig. 7o–w). Overall, these results demonstrated that the SIGMAR1 agonist dextromethorphan increased K+ currents in cardiomyocytes from patients with LQTS1 or LQTS2 by activating SIGMAR1 and that dextromethorphan treatment could be beneficial for LQTS1 and LQTS2 (caused by trafficking mutations), which are more common forms of LQTS than TS.
The effect of dextromethorphan on phenotypes in LQTS1.
a, Representative traces of action potentials in cardiomyocytes derived from iPSCs of patients with LQTS1 without treatment or treated with fluvoxamine or dextromethorphan. b, APD90 analysis of cardiomyocytes from patients with LQTS1 without treatment (n = 11) or treated with fluvoxamine (n = 10) or dextromethorphan (n = 10). Other action potential parameters are shown in Extended Data Fig. 7a,c,e. c, Representative traces of IKs currents (chromanol 293B sensitive) in cardiomyocytes from patients with LQTS1 treated with dextromethorphan or untreated. d, IKs currents (steady state) were significantly increased by dextromethorphan treatment of cardiomyocytes from patients with LQTS1 at all voltage steps (n = 10 per group). e, Representative traces of IKr currents (E-4031 sensitive) in cardiomyocytes from patients with LQTS1 treated with dextromethorphan or untreated. f, IKr currents (steady state) were significantly increased in dextromethorphan-treated cardiomyocytes from patients with LQTS1 (n = 9) at 0-, 10-, 20-, 30-, 40- and 50-mV steps compared with those of cardiomyocytes from patients with LQTS1 without treatment (n = 10). g, Representative epifluorescent and phase-contrast images of cardiomyocytes from patients with LQTS1 with and without dextromethorphan treatment from the PLA (SIGMAR1–hERG, red; DAPI, blue). Scale bar, 10 μm. h, Quantification of SIGMAR1–hERG by the PLA in dextromethorphan-treated (n = 20) and untreated (n = 20) cardiomyocytes from patients with LQTS1. i, Representative epifluorescent and phase-contrast images of cardiomyocytes from patients with LQTS1 with and without dextromethorphan treatment from the PLA (SIGMAR1–KV7.1, red; DAPI, blue). Scale bar, 10 μm. j, Quantification of SIGMAR1–KV7.1 by the PLA in dextromethorphan-treated (n = 13) and untreated (n = 23) cardiomyocytes from patients with LQTS1. All data are mean ± s.d. Treatment with fluvoxamine or dextromethorphan was carried out at 5 μM for 2 h. One-way ANOVA with Tukey’s multiple comparisons was used for b, and unpaired two-tailed Student’s t-tests were used for h,j between groups and for d,f at each voltage step. *P < 0.05, **P < 0.01, ***P < 0.001. Cardiomyocytes were from at least two independent differentiations.
Source data
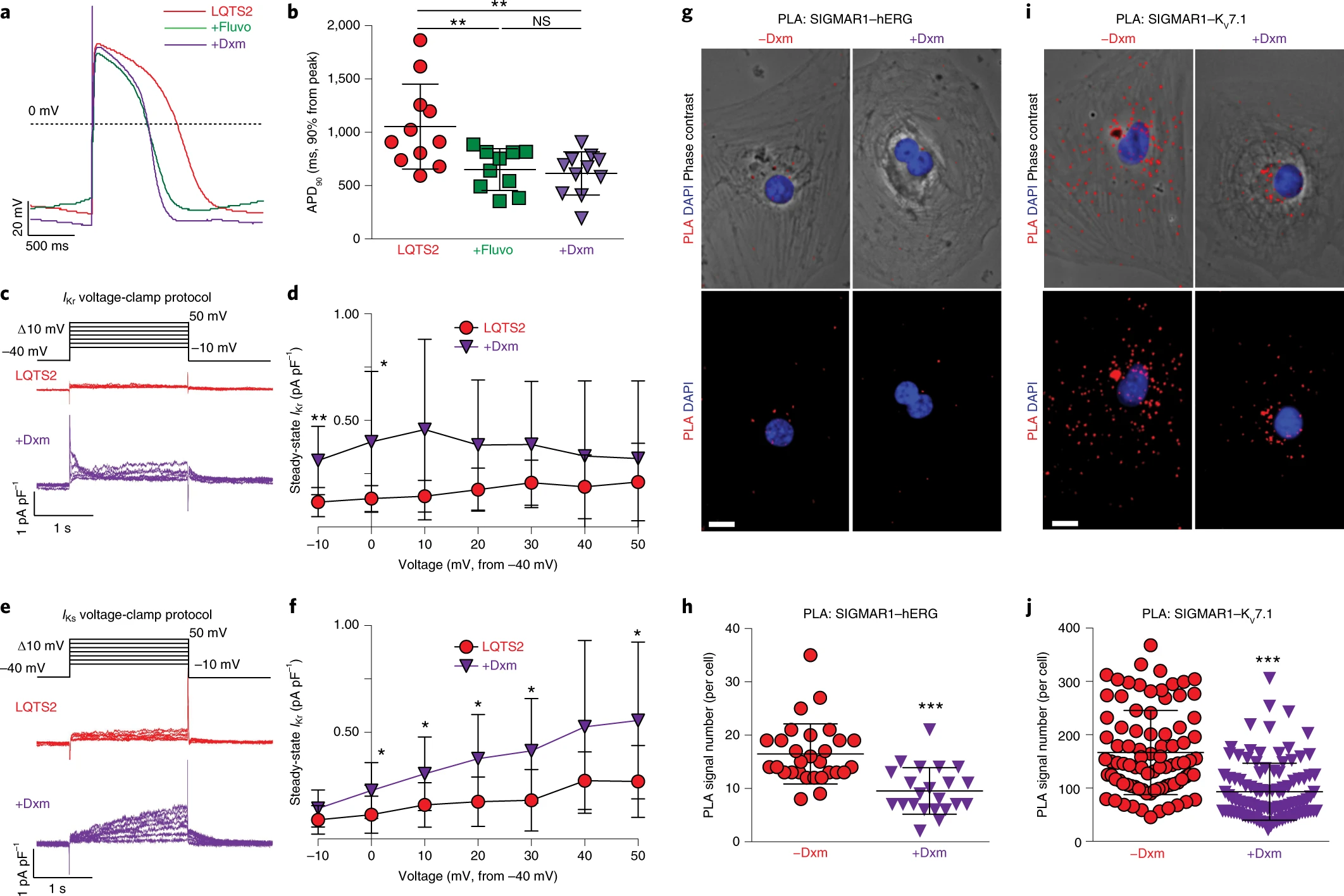
The effect of dextromethorphan on phenotypes in LQTS2.
a, Representative traces of action potentials in cardiomyocytes derived from iPSCs of patients with LQTS2 without treatment or treated with fluvoxamine or dextromethorphan. b, APD90 analysis of cardiomyocytes from patients with LQTS2 without treatment (n = 11) or treated with fluvoxamine (n = 10) or dextromethorphan (n = 11). Other action potential parameters are shown in Extended Data Fig. 7b,d,f. c, Representative traces of IKr currents (E-4031 sensitive) in cardiomyocytes from patients with LQTS2 treated with dextromethorphan or untreated. d, IKr currents (steady state) were significantly increased in dextromethorphan-treated cardiomyocytes from patients with LQTS2 (n = 9) at −10-mV and 0-mV steps compared with those of cardiomyocytes from patients with LQTS2 without treatment (n = 10). e, Representative traces of IKs currents (chromanol 293B sensitive) in cardiomyocytes from patients with LQTS2 treated with dextromethorphan or untreated. f, IKs currents (steady state) were significantly increased in dextromethorphan-treated cardiomyocytes from patients with LQTS2 at 0-, 10-, 20-, 30- and 50-mV steps compared with those in cardiomyocytes from patients with LQTS2 without treatment (n = 10 per group). g, Representative epifluorescent and phase-contrast images of cardiomyocytes from patients with LQTS2 with and without dextromethorphan treatment from the PLA (SIGMAR1–hERG, red; DAPI, blue). Scale bar, 10 μm. h, Quantification of SIGMAR1–hERG by the PLA in dextromethorphan-treated (n = 22) and untreated (n = 29) cardiomyocytes from patients with LQTS2. i, Representative epifluorescent and phase-contrast images of cardiomyocytes from patients with LQTS2 with and without dextromethorphan treatment from the PLA (SIGMAR1–KV7.1, red; DAPI, blue). Scale bar, 10 μm. j, Quantification of SIGMAR1–KV7.1 by the PLA in dextromethorphan-treated (n = 107) and untreated (n = 87) cardiomyocytes from patients with LQTS2. All data are mean ± s.d. Treatment with fluvoxamine or dextromethorphan was carried out at 5 μM for 2 h. One-way ANOVA with Tukey’s multiple comparisons was used for b. Unpaired two-tailed Student’s t-tests was used for h,j between groups and for d,f at each voltage step. *P < 0.05, **P < 0.01, ***P < 0.001. Cardiomyocytes were from at least two independent differentiations.
Source data
Effects of dextromethorphan on a mouse model of TS
Based on the intriguing in vitro results, we decided to conduct in vivo studies to examine the effect of dextromethorphan on TS further. We generated a cardiac-specific mouse model of TS, taking advantage of an inducible expression system using the Rosa26 locus, a cardiac Cre driver (α-MHC/Myf6MerCreMer) and a loxP pair with stop cassette to conditionally overexpress mutant channels from TS in the heart (Fig. 5a)37. The inducible expression system allows us to express mutant channels at the adult stage to avoid heart developmental phenotypes that may lead to early lethality. A common challenge in the field of LQTS concerning murine models is the failure to recapitulate arrhythmic phenotypes in vivo38 due to the fact that mice have different ion channel expression profiles and a remarkably higher heart rate than humans. Fortunately, our cardiac-specific mouse model of TS successfully recapitulated the LQTS phenotype and showed prolongation of the QT interval at 4 d after tamoxifen administration as revealed by electrocardiography (ECG) (Fig. 5b–d). Next, we examined the effect of dextromethorphan on the LQTS phenotype by adding dextromethorphan to drinking water for dosing. A low dose of quinidine was also added with dextromethorphan to improve the pharmacokinetic profile of dextromethorphan in vivo, as quinidine is known to competitively inhibit the metabolism of dextromethorphan to dextrorphan. Clinically, a low dose of quinidine together with dextromethorphan is used to maintain a stable increase in the plasma concentration of dextromethorphan in humans19,20. To examine the pharmacokinetic profile of dextromethorphan with the dosing strategy, blood was sampled from mice at different time points on days 4 and 11 to measure dextromethorphan concentrations in plasma. The results indicated that the dosing strategy for dextromethorphan used in the study achieved a stable increase in plasma dextromethorphan levels throughout the day (Extended Data Fig. 8a), with the plasma level of dextromethorphan in treated animals being comparable to dextromethorphan levels in human participants taking 30 mg dextromethorphan per day for 7 d (~10–60 ng ml−1)19. Next, we examined the effect of dextromethorphan on the LQTS phenotype in the mouse model of TS using ECG at day 4 and day 11. Dextromethorphan–quinidine significantly shortened the prolonged QT interval in the mouse model of TS at both time points (Fig. 5c,d and Extended Data Fig. 8b,c). Moreover, dextromethorphan–quinidine did not significantly affect QT intervals in control littermate mice (Fig. 5c,d), which is consistent with clinical reports indicating no alteration of QT intervals in human participants when dextromethorphan–quinidine was used to treat pseudobulbar affect19,20. The overall results demonstrated that dextromethorphan not only rescued the phenotypes in cardiomyocytes derived from iPSCs of patients with TS in vitro but also alleviated the LQTS phenotype in the mouse model of TS in vivo.
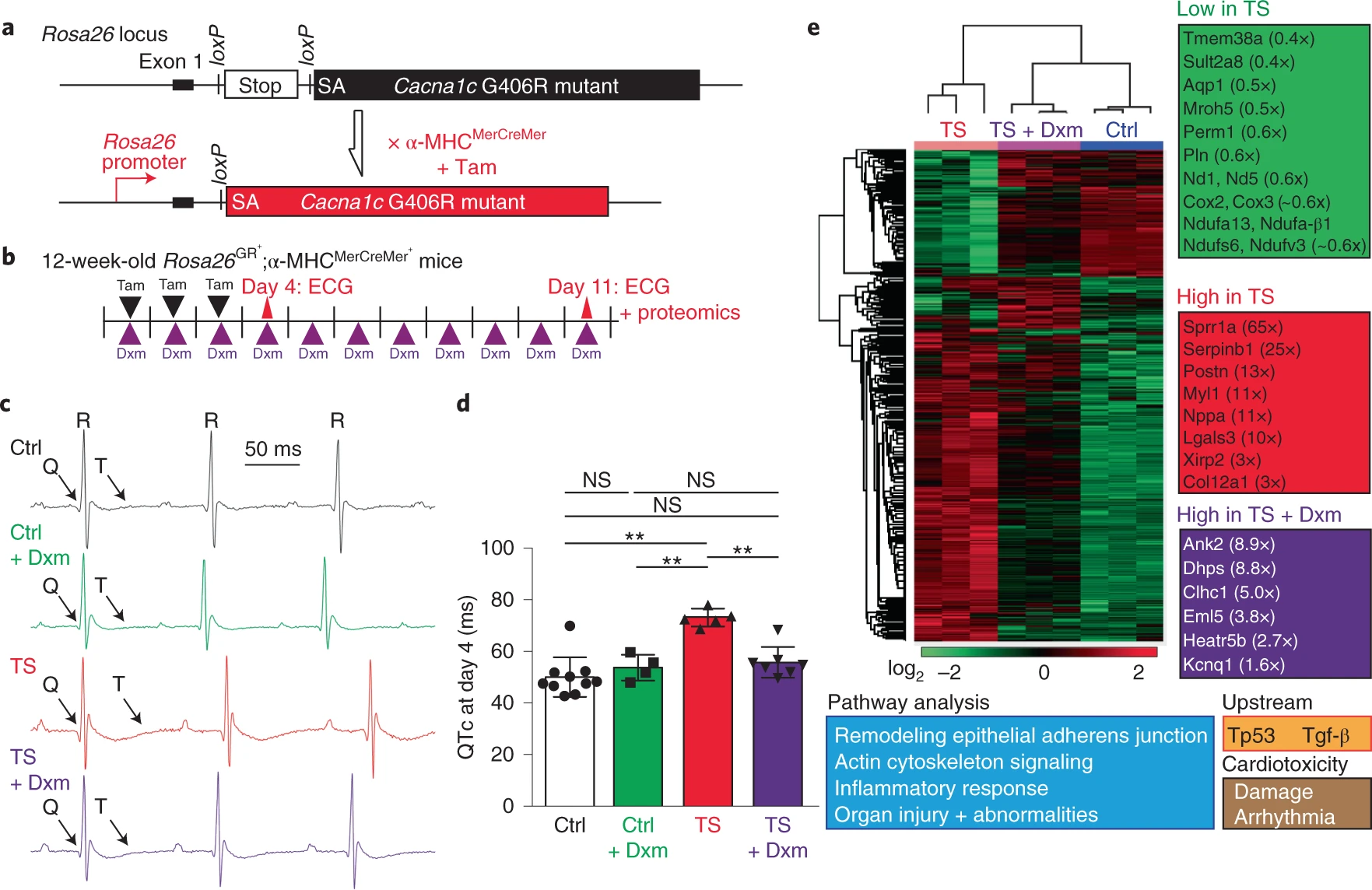
The effect of dextromethorphan on the mouse model of TS.
a, Targeting design of a TS mutant (corresponding to G406R in the Ca2+ channel Cacna1c gene) at the mouse Rosa26 locus and the breeding strategy for inducing cardiac-specific tamoxifen (Tam)-inducible expression of TS mutant channels. SA, splice acceptor. b, Experimental design for the induction of TS mutant channel expression in the heart with tamoxifen and testing dextromethorphan in vivo. Dextromethorphan was added to drinking water, and pharmacokinetic analyses of plasma dextromethorphan concentrations at day 4 and day 11 are shown in Extended Data Fig. 8a. c, Representative ECG traces in the mouse model of TS (TS) and control littermates (Ctrl) without treatment or treated with dextromethorphan at day 4. d, Prolonged QT intervals in the mouse model of TS were normalized by dextromethorphan treatment (n = 10 for control littermates, n = 4 for control littermates treated with dextromethorphan, n = 5 for TS, n = 7 for TS and dextromethorphan). QT intervals were corrected using a mouse formula (QTc, Extended Data Fig. 8b). One-way ANOVA with Sidak’s multiple comparisons was used (**P < 0.01). e, Proteomic analysis including heat map comparisons and pathway analysis at day 11 in hearts of control mice, untreated mice and dextromethorphan-treated mice in the TS model (n = 3 per group). ECG at day 11 also confirmed the beneficial effect of dextromethorphan on QT prolongation in mutant mice (Extended Data Fig. 8c). Additional analyses of several proteins with altered expression levels are shown in Extended Data Fig. 9. All data are mean ± s.d.
Lastly, to profile global changes in hearts of the mouse model of TS and the effects of dextromethorphan on mutant hearts, we performed a global proteomic analysis to profile protein expression changes in the hearts of controls, untreated and dextromethorphan-treated mice in the TS model at day 11. Proteomic results demonstrated that expression of many important molecules involved in cardiac excitation–contraction coupling, mitochondrial function, heart failure, fibrosis and cardiomyopathy (such as the TRIC channel (Tmem38a), atrial natriuretic peptide (ANP) (Nppa), periostin (Postn) and cardiomyopathy-associated gene 3 (CMYA3, Xirp2)39,40,41,42) was altered in mouse hearts in the TS model, while dextromethorphan treatment restored expression of these molecules to a level comparable to that of controls (Fig. 5e and Extended Data Fig. 9). This supports the efficacy of dextromethorphan on hearts in the TS model at the endpoint, even though dextromethorphan treatment did not restore the expression of all molecules involved in pathways related to cardiac dysfunction and arrhythmias. In addition, interestingly, the proteomic analysis indicated that dextromethorphan treatment induced a significant increase in the expression of the KV7.1 (Kcnq1) protein in hearts in the TS model (Fig. 5e and Extended Data Fig. 9), which is consistent with our observation using cardiomyocytes derived from iPSCs specific to patients with TS (Extended Data Fig. 5k–m). Because loss-of-function mutations in KV7.1 are also known to cause LQTS1 and QT prolongation, we hypothesized that the increase in KV7.1 expression might contribute to the beneficial effect of dextromethorphan on the prolonged QT interval in the mouse model of TS. To validate proteomic results, we examined the expression of KV7.1 as well as that of house-keeping molecules β-tubulin and glyceraldehyde-3-phosphate dehydrogenase (Gapdh) using western blotting and mouse heart lysates. We found that, consistent with proteomic results (Extended Data Fig. 9), the expression of β-tubulin significantly increased in mouse hearts in the TS model compared with that in other groups, while no changes in the expression of Gapdh protein between the groups were observed (Extended Data Fig. 10a,b). Also in line with proteomic results and our observation using cardiomyocytes from patients with TS, there was a significant reduction in KV7.1 protein expression in mouse hearts in the TS model, while dextromethorphan treatment restored the expression of KV7.1 to a level comparable to that of control hearts (Extended Data Fig. 10a,c). When we examined changes in Kcnq1 transcripts in mouse hearts, dextromethorphan treatment did not significantly upregulate Kcnq1transcription in hearts in the TS model, although hearts in the TS model showed a trend toward reduced Kcnq1 gene expression compared with control hearts (Extended Data Fig. 10d). Overall, the results suggest that the significant reduction in KV7.1 protein levels in hearts in the TS model may be associated with QT prolongation, although the contribution of KV7.1 to cardiac action potential repolarization in rodents is known to be relatively smaller than that in humans, and dextromethorphan might alleviate cardiac phenotypes in the mouse model of TS in part by increasing expression of KV7.1 in hearts in the TS model through a post-transcriptional mechanism.
Discussion
In summary, our results demonstrate that SIGMAR1 agonists such as dextromethorphan can rescue the phenotypes in cardiomyocytes derived from iPSCs of patients with TS and the mouse model of TS as well as cardiomyocytes derived from iPSCs of patients with LQTS1 or LQTS2 with the G269S substitution in the KV7.1 channel or the A561V substitution in the hERG channel, respectively. These results indicate that SIGMAR1 could be a therapeutic target for TS as well as LQTS1 and LQTS2, which are caused by trafficking mutations such as those corresponding to the G269S substitution in the KV7.1 channel and the A561V substitution in the hERG channel. Furthermore, we explored the underlying mechanisms of the beneficial effects of SIGMAR1 activation on cardiomyocytes from patients with TS and found that SIGMAR1 may play a multifaceted role in regulating CaV1.2 channel function in cardiomyocytes. First, SIGMAR1 directly interacts with CDK5, and SIGMAR1 activation induces a significant increase in the interaction and a significant reduction in CDK5 protein expression and activity in cardiomyocytes from patients with TS, which may restore the affected voltage-dependent inactivation of CaV1.2 in patient cardiomyocytes through the mechanism that we proposed previously6 and rescue cellular phenotypes (Fig. 1 and Extended Data Figs. 1 and 2). In addition, SIGMAR1 also directly interacts with CaV1.2 in cardiomyocytes from patients with TS, and the activation of SIGMAR1 induced by dextromethorphan treatment modulates the interaction between SIGMAR1 and CaV1.2 (Extended Data Fig. 3h,i), which may also contribute to the beneficial effects of SIGMAR1 activation on cardiomyocytes from patients with TS. Our findings provide insights into the interactions between SIGMAR1, CDK5 and CaV1.2 in human iPSC-derived cardiomyocytes, while detailed molecular mechanisms concerning the interactions remain to be fully characterized. Moreover, it is also of merit to further investigate whether other players, such as calmodulin and calmodulin-dependent protein kinase II, which play important roles in regulating Ca2+ channel function43, or the Na+/Ca2+ exchanger, which is known to modulate Ca2+ homeostasis in cardiomyocytes44, may interact with SIGMAR1 and mediate the effects of SIGMAR1 activation on Ca2+ channel and ion homeostasis.
Unexpectedly, we found that there was a significant reduction in both IKr and IKs currents in cardiomyocytes from patients with TS compared to those of isogenic control cardiomyocytes (Extended Data Figs. 3j,k and 5f), although our iPSC lines from patients with TS do not carry any mutations in KCNH2 (hERG) or KCNQ1 (KV7.1). More interestingly, we found that, in addition to its effects on the CaV1.2 channel, activation of SIGMAR1 by dextromethorphan also increased K+ currents in cardiomyocytes from patients with TS (Fig. 2 and Extended Data Figs. 3 and 5), which may further contribute to the normalization of action potentials and restoration of ion homeostasis in cardiomyocytes from patients with TS. Mechanistically, we observed that SIGMAR1 interacted with hERG and KV7.1 channels in human iPSC-derived cardiomyocytes, and dextromethorphan treatment modulated the interactions between SIGMAR1, hERG and the KV7.1 channel, increased the plasma membrane localization of KV7.1 and increased gene and protein expression of KCNQ1 and KV7.1 isoform 1 in cardiomyocytes from patients with TS. Although, to our knowledge, the interaction between SIGMAR1 and KV7.1 has not been reported previously, the direct interaction between SIGMAR1 and hERG has been studied in leukemic cells, HEK cells and neonatal rat cardiomyocytes12,32,33,34. Our results confirmed the direct interaction between SIGMAR1 and the hERG channel and SIGMAR1-mediated regulation of hERG channel function in human iPSC-derived cardiomyocytes, while detailed molecular mechanisms remain to be fully characterized. Additionally, dextromethorphan has been previously reported to inhibit hERG currents in a heterologous overexpression system45, while we found that dextromethorphan treatment significantly increased hERG currents in human patient-specific iPSC-derived cardiomyocytes. There have been notable differences in hERG currents in human iPSC-derived cardiomyocytes46 and heterologous systems potentially due to differences in cellular physiological conditions and associated molecules. Further investigations are needed to profile the effects and mechanisms of dextromethorphan and SIGMAR1 activation on hERG and KV7.1 channels.
By contrast, when we examined the effects of dextromethorphan on electrophysiological properties of isogenic control cardiomyocytes, no significant changes were observed in action potentials (Fig. 1), CaV1.2 inactivation (Extended Data Fig. 3) or K+ currents (Extended Data Fig. 4) in isogenic control cardiomyocytes treated with dextromethorphan at a dose that was efficacious for cardiomyocytes from patients with TS. We sought to understand the difference between the sensitivity of cardiomyocytes from patients with TS and isogenic control cardiomyocytes to the SIGMAR1 agonist dextromethorphan, and we found that, at baseline, there was a significant increase in the expression of SIGMAR1 transcripts and SIGMAR1 protein in cardiomyocytes from patients with TS (Extended Data Fig. 2). Furthermore, we examined the expression of ATF4, a transcription factor that was reported to directly regulate SIGMAR1 transcription24, and observed an increase in ATF4 protein levels in cardiomyocytes from patients with TS, which may contribute to the increased expression of SIGMAR1 in cardiomyocytes from patients with TS. Overall, the results suggest that cardiomyocytes from patients with TS have increased SIGMAR1 expression at baseline, which may predispose cells to respond to SIGMAR1 agonists. In addition, it is also likely that, compared to cardiomyocytes from patients with TS with highly disrupted ion homeostasis, normal cardiomyocytes without any mutations in genes encoding cardiac ion channels have a higher capacity to tolerate moderate changes in ion channel function and maintain stable ion homeostasis, which contributes to the lack of changes observed in isogenic control cardiomyocytes with dextromethorphan treatment.
The data from cardiomyocytes of patients with TS encouraged us to test SIGMAR1 agonists on cardiomyocytes from patients with LQTS1 or LQTS2 with trafficking mutations. Cardiomyocytes from patients with LQTS1 and the trafficking mutation corresponding to G269S and cardiomyocytes from patients with LQTS2 and the trafficking mutation corresponding to A561V were reported to have decreased K+ currents35,36, and we hypothesized that SIGMAR1 activation might be useful to restore K+ currents and alleviate their cellular phenotypes. Consistent with our hypothesis, cardiomyocytes from patients with LQTS1 and the G269S substitution and cardiomyocytes from patients with LQTS2 and the A561V substitution did demonstrate a decrease in K+ currents, and dextromethorphan treatment rescued the cellular phenotypes in cardiomyocytes from patients with LQTS1 or LQTS2 (Figs. 3 and 4 and Extended Data Figs. 6 and 7) by modulating interactions between SIGMAR1, hERG and the KV7.1 channel and increasing K+ currents. Our results demonstrated that SIGMAR1 activation could be beneficial for not only TS but also for LQTS1 and LQTS2, which are caused by the G269S substitution in the KV7.1 channel and the A561V substitution in the hERG channel, respectively. However, the effect of SIGMAR1 agonists and SIGMAR1 activation on LQTS1 and LQTS2 with other mutations remains to be investigated.
Intriguingly, in addition to cardiomyocytes derived from human iPSCs from patients with TS, the SIGMAR1 agonist and FDA-approved cough suppressant dextromethorphan also exhibited beneficial effects in the mouse model of TS (Fig. 5 and Extended Data Figs. 8–10). While dextromethorphan was previously reported to have a protective effect on ouabain-induced acute arrhythmias47, we reported that dextromethorphan treatment could be beneficial in a mouse model of inherited cardiac arrhythmia (TS). One of the limitations of our study is that we did not examine the effect of dextromethorphan on LQTS1 or LQTS2 mouse models due to the lack of an LQTS1 or LQTS2 mouse model recapitulating arrhythmic phenotypes in vivo38, and additional studies are necessary to evaluate the potential effect of dextromethorphan on LQTS1 and LQTS2 in vivo. Furthermore, it is also worth noting that dextromethorphan has several limitations related to clinical applications: dextromethorphan has a short half-life and induces nausea, which may limit its tolerability and long-term use, overdose of dextromethorphan could potentially lead to severe adverse cardiac events48,49, and the pharmacological profile of dextromethorphan in infants and young children has not been fully investigated, which may comprise the main demographic of patients with TS. Therefore, further investigations are absolutely required to evaluate whether dextromethorphan can be repurposed to alleviate cardiac arrhythmias in patients with TS. In addition, TS is a multiorgan disorder that features not only cardiac arrhythmias but also syndactyly, autism and hypoglycemia because of the important role of the CaV1.2 channel in various organs1,3,37,50. Therefore, the effects of dextromethorphan on the dysfunction of other affected organs remain to be investigated.
In conclusion, although the role of SIGMAR1 has emerged in various diseases including neurological disorders and cancer9,10,11 and more recently in cardiovascular diseases51,52 and coronavirus disease 2019 (refs. 53,54), our findings demonstrate that SIGMAR1 could be a therapeutic target for TS and potentially LQTS1 and LQTS2, which are caused by the G269S substitution in the KV7.1 channel and the A561V substitution in the hERG channel, respectively. Further investigations are necessary to profile global effects of SIGMAR1 activation and its agonists on different organs and tissues and to develop and optimize SIGMAR1 agonists for future clinical applications.