
Sigma-1R Protects Retinal Ganglion Cells in Optic Nerve Crush Model for Glaucoma
By Linya Li, Shaoqing He, Yang Liu, Thomas Yorio, Dorette Z. Ellis
Excerpt from the article published in Investigative Ophthalmology & Visual Science August 2021, Vol.62, 17. doi: https://doi.org/10.1167/iovs.62.10.17
Editor’s Highlights
- Degeneration of the retinal ganglion cells (RGCs), their axons, and optic nerves (ONs) are hallmarks in glaucoma.
- The Sigma-1R (σ-1r) is localized at the endoplasmic reticular (ER) membrane in some cells and at the nuclear envelope and ER in retinal cells.
- The σ-1r protects RGCs by inhibition of the apoptotic pathway involving c-Jun, p-c-Jun and caspase 3 in a time-dependent manner.
- Activation of the σ-1r results in increased ERK1/2 phosphorylation, which correlates with increased numbers of RGCs, suggesting a neuroprotective role of increased phosphorylation of ERK1/2 (members of the mitogen-activated protein-kinase) family.
- Mechanisms by which the σ-1r confer protection on RGCs are complex, and may involve the MAPK/JNK/c-Jun/caspase 3 pathway, and possible other convergent pathways.
Abstract
Purpose: The purpose of this study was to determine the effects of the Sigma-1R (σ-1r) on retinal ganglion cell (RGC) survival following optic nerve crush (ONC) and the signaling mechanism involved in the σ-1r protection.
Methods: The overall strategy was to induce injury by ONC and mitigate RGC death by increasing σ-1r expression and/or activate σ-1r activity in σ-1r K/O mice and wild type (WT) mice. AAV2-σ-1r vector was used to increase σ-1r expression and σ-1r agonist used to activate the σ-1r and RGCs were counted. Immunohistochemical and Western blot analysis determined phosphorylated (p)-c-Jun, c-Jun, and Caspase-3. Pattern electroretinography (PERG) determined RGC activity.
Results: RGC counts and function were similar in pentazocine-treated WT mice when compared to untreated mice and in WT mice when compared with σ-1r K/O mice. Pentazocine-induced effects and the effects of σ-1r K/O were only observable after ONC. ONC resulted in decreased RGC counts and activity in both WT and σ-1r K/O mice, with σ-1r K/O mice experiencing significant decreases compared with WT mice. The σ-1r transgenic expression resulted in increased RGC counts and activity following ONC. In WT mice, treatment with σ-1r agonist pentazocine resulted in increased RGC counts and increased activity when compared with untreated WT mice. There were time-dependent increases in c-jun, p-c-jun, and caspase-3 expression in ONC mice that were mitigated with pentazocine-treatment.
Conclusions: These findings suggest that the apoptotic pathway is involved in RGC losses seen in an ONC model. The σ-1r offers neuroprotection, as activation and/or transgenic expression of σ-1r attenuated the apoptotic pathway and restored RGCs number and function following ONC.
Introduction
Degeneration of the retinal ganglion cells (RGCs), their axons, and optic nerves (ONs) are hallmarks in glaucoma. Elevated intraocular pressure (IOP) is a risk factor for the development of primary open angle glaucoma (POAG)1 and current treatments for glaucoma are aimed at decreasing IOP. However, neuropathy is also observed in patients with normal tension glaucoma (IOP measurements of 6–10 mm Hg); suggesting that in addition to high IOP, there may be other common predisposing factors for the development of glaucoma. Therefore, other strategies for the treatment of glaucoma are needed, particularly those that improve or protect the health of RGCs and their axons.
The Sigma-1R (σ-1r) is a 26 kDa transmembrane, non-opioid receptor localized at the endoplasmic reticular (ER) membrane in some cells2,3 and at the nuclear envelope and ER in retinal cells.4 At the ER, the σ-1r complexes with the mitochondrion-associated ER membrane and regulates calcium transport and homeostasis between the two organelles.5,6 The mechanisms by which σ-1r modulate cell function are relatively unknown. However, recent observations suggest that the σ-1r associates with and prevents the influx of calcium by L-type voltage-gated calcium channels in RGCs.7 It has been suggested that the σ-1r provides a beneficial role in brain function. Earlier studies have suggested that σ-1r agonists are neuroprotective in Alzheimer’s disease, and serves as an effective antidepressant in mood disorders.8 In fact, the σ-1r also protect against β-amyloid exposure by inhibiting overexpression of the apoptotic protein, Bax,9 and TNF-related apoptosis inducing ligand (TRAIL) and phosphorylation of c-Jun N-terminal kinases (JNKs).10
Studies in the retina have shown that mice lacking the σ-1r are more susceptible to injury and that there are substantial losses of RGCs in σ-1r-deficient mice exposed to retinal insults.11,12 There are accelerated RGC losses in diabetes/σ-1r knock out (K/O) mice11 and accelerated RGC losses in σ-1r K/O mice subjected to ONC.12 The significant neuroprotection of RGC from apoptosis was achieved by blocking the JNK/c-Jun pathway using either JNK inhibitor, SP600125,13,14 or JNK2/3 deficient mice.15 Other studies indicate that ET-1-induced apoptosis in RGCs are associated with upregulation of c-Jun, Bax,16therefore, investigating the role of c-Jun in the σ-1r-regulation of RGC function is critical to understand the working mechanism of the σ-1r. As with the brain, these observations suggest that the σ-1r confers neuroprotection in the retina. Therefore, we will test the hypothesis that the σ-1r is neuroprotective in the retina following optic nerve injury, using an in vivo model for acute retinal neurodegeneration, the ONC model, to determine the ability of the σ-1r to protect RGCs and the signaling pathway that may be involved.
…
Results
Effects of Pentazocine on RGC Density and Function are Observable Only After ONC Injury
We tested the hypothesis that the σ-1r is activated in response to RGC stress induced by ONC. To test this hypothesis, in one set of experiments, 3-month-old WT mice were treated with and without pentazocine (0.5 mg/kg)20 and RGC numbers were counted and RGC activity measured by PERG. In another set of experiments, 3-month-old WT mice were treated with and without pentazocine (0.5 mg/kg)20 and subjected to ONC, and RGC numbers were counted and RGC activity measured by PERG. PBS served as controls for pentazocine-treatment and contralateral eyes that were not subjected to ONC served as controls for ONC treatment. Injection of pentazocine in σ-1r K/O animals served as control for off-receptor actions of pentazocine. In mice eyes that were not subjected to ONC, RGC numbers and PERG values are similar in both pentazocine-treated and PBS-treated mice (Figs. 1A, 1B). The WT mice were injected with pentazocine and then subjected to ONC, quantitative analysis demonstrated significant increases in RGC number in WT animals treated with pentazocine when compared with PBS-treated animals (Figs. 1C). Similarly, PERG of RGC activity was greater in pentazocine-treated mice subjected to ONC than in PBS-treated animals (Fig. 1D). Additionally, pentazocine failed to have a significant effect on RGC number in ONC-treated σ-1r K/O mice, when compared to non-pentazocine-treated animals, suggesting that the effects of pentazocine is occurring via the σ-1r (data not shown). Images of whole mount retinas provide qualitative evidence of decreased RGC density in mice subjected to ONC in the absence of pentazocine (Fig. 1E). These data suggest that the effects of pentazocine on RGC function and numbers are observable only after ONC insults.
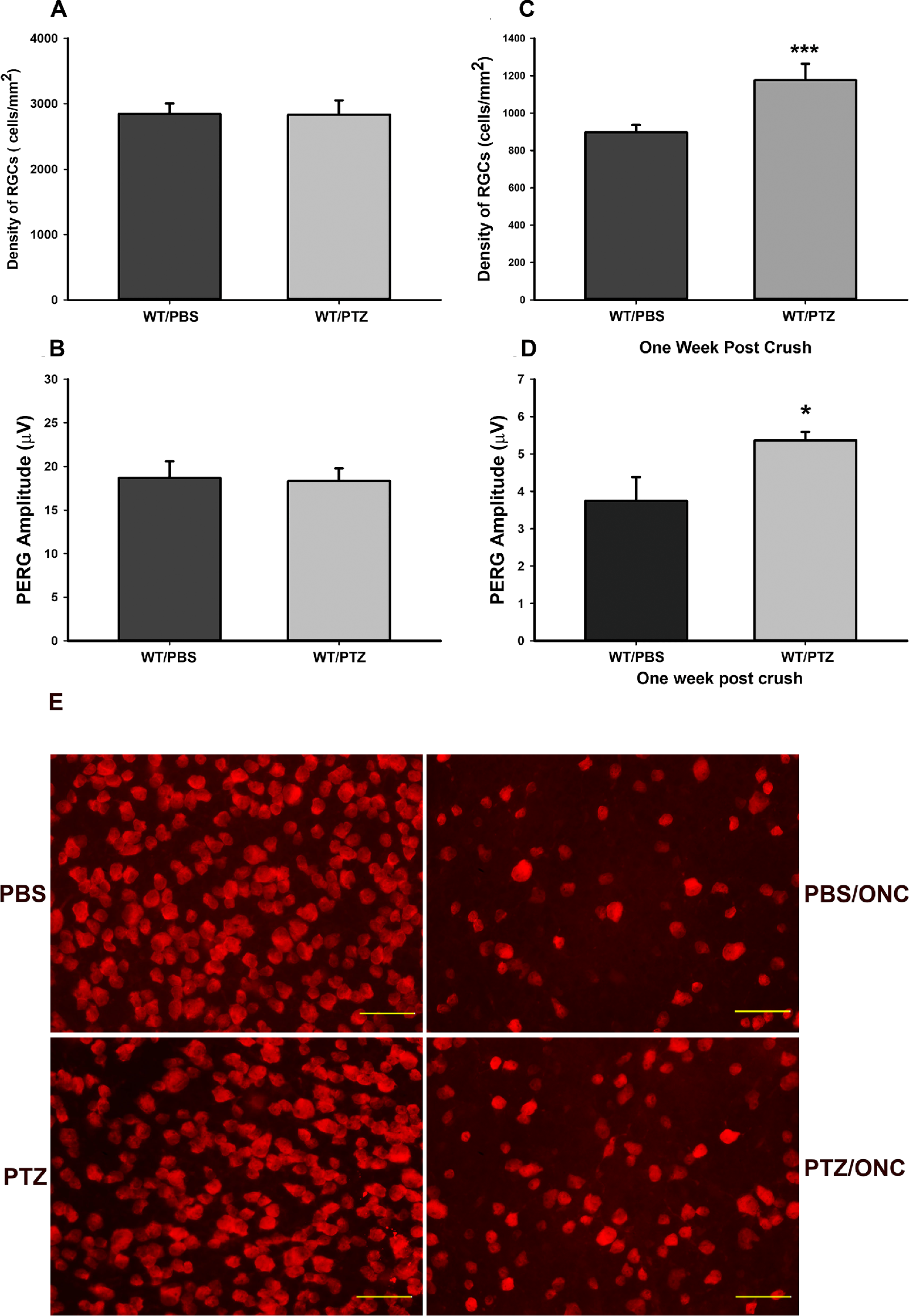
The σ-1r is activated only after injury.
(A, B) RGC cell count/density and function, as measured by pattern ERG (PERG) were similar in WT mice treated with PBS or with intraperitoneally injected pentazocine (PTZ; 0.5 mg/kg). RGC density is expressed as the number of cells per mm2, counted in a masked manner. PERG of RGC is expressed as its measurement, amplitude (µV). There were six in each group for RGC density and PERG. (C, D) Wild-type mice (WT) were treated with PTZ and subjected to ONC (Crush). PTZ-treatment increased RGC density and function when compared with untreated animals suggesting that the σ-1r is only activated in response to injury. Data are shown as mean ± SEM N = 6, each group for RGC density and PERG. ***Significantly different from control, P < 0.001, t-test), *significantly different from control, P < 0.05, t-test. (E) Flat mounts providing qualitative analysis of the density of RGCs of wild-type mice treated with PBS, PBS + ONC, pentazocine (PTZ) and PTZ + ONC that were quantified in A to 1D. Images were taken at 40 times magnification, scale bar = 50 µM.
The Effects of Knock-Out of the σ-1r on RGC
Density and Function are Observable Only After ONC InjuryTo further test the hypothesis that the σ-1r is activated in response to injury, RGCs were quantified from whole mount retinas from 3-month-old WT mice and σ-1r K/O mice. Figure 2A demonstrates similar RGC density in both WT and σ-1r K/O mice. PERG confirms that retinal function is not compromised in σ-1r K/O mice (Fig. 2B). To assess the role of the σ-1r in RGCs in response to injury, both 3-month-old WT mice and σ-1r K/O mice were subjected to ONC. The contralateral eyes served as controls. Seven days post ONC resulted in significant decreases in RGC density in both WT and σ-1r K/O animals when compared with contralateral control, however, the losses in RGCs were greater in σ-1r K/O animals than in WT animals (Fig. 2C). PERG amplitude was significantly greater in ONC-treated WT mice when compared with σ-1r K/O mice subjected to ONC (Fig. 2D). Images of whole mount retinas provide qualitative evidence of decreased RGC density in σ-1r K/O mice subjected to ONC when compared with WT mice (Fig. 2E).
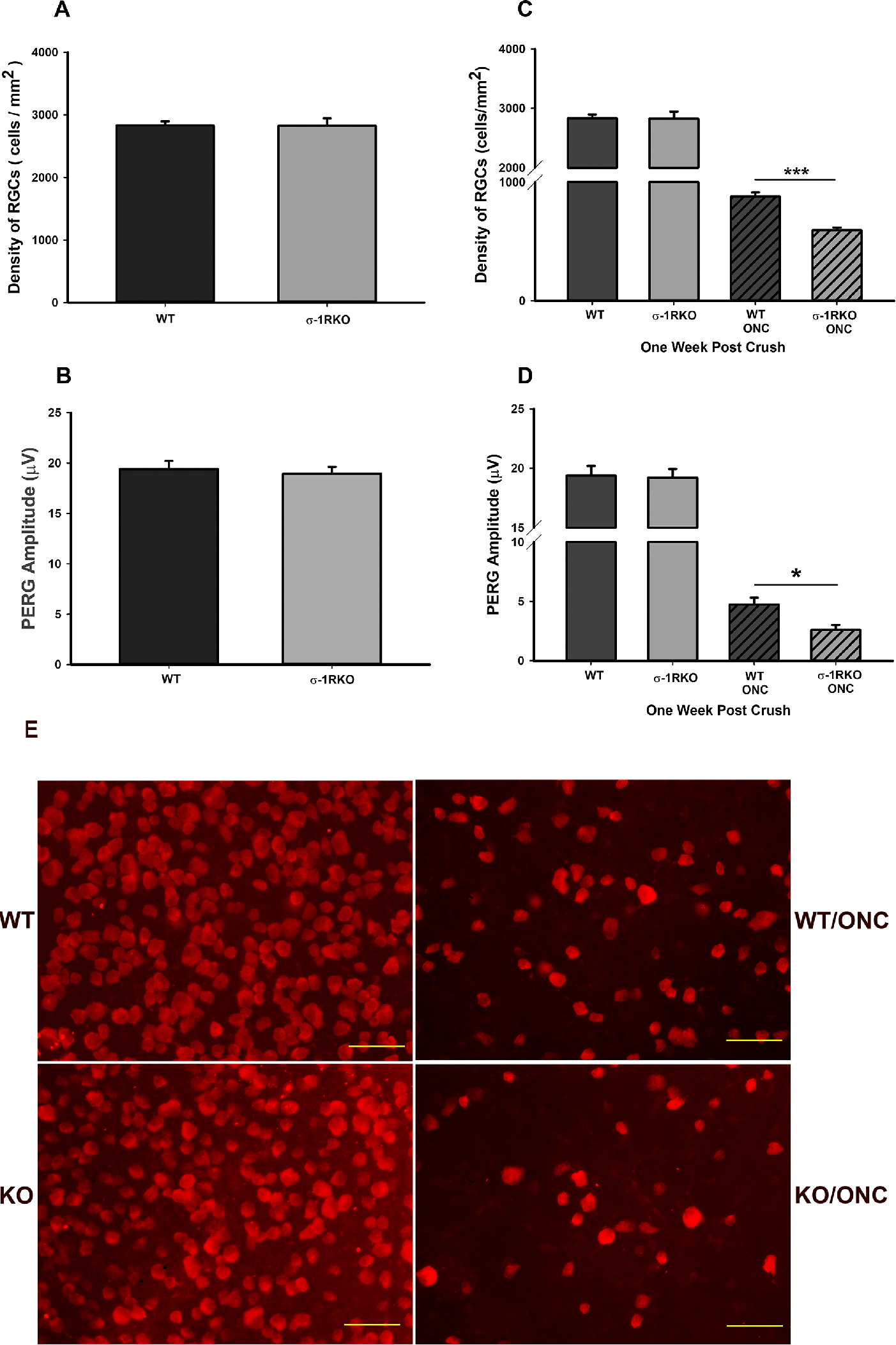
The effects of σ-1r deficit on RGC density and function is manifested only after axonal injury. Wild-type (WT) and σ-1r K/O animals were subjected to ONC and RGC cells were counted and activity measured using PERG.
(A, B) RGC density and function as measured PERG were similar in WT mice and σ-1r K/O. RGC density is expressed as the number of cells per mm2, counted in a masked manner. PERG of RGC is expressed as amplitude (µV) measured. There were eight in each group for RGC density and PERG. (C, D) Wild-type mice (WT) and σ-1r K/O mice were subjected to ONC. RGC density and function were decreased in σ-1r K/O ONC-treated mice when compared with wild-type ONC-treated mice suggesting that the σ-1r is a stress response protein and is quiescence in physiological states. Data are shown as mean ± SEM N = 8, each group for RGC density and PERG. ***Significantly different from σ-1r K/O ONC-treated mice, P < 0.001, t-test, *significantly different from σ-1r K/O ONC-treated mice, P < 0.05, t-test. (E) RGC flat mounts providing qualitative analysis of the density of RGCs of WT mice and σ-1r K/O mice exposed to ONC. Quantitative analysis was provided in A to D. Images were taken at 40 times magnification, scale bar = 50 µM.
Transfection and Expression of the σ-1r in RGCs in σ-1r K/O Mice
To determine transfection efficiency, σ-1r K/O mice were intravitreally injected with AAV2-CAG-σ-1r-GFP and the transduction efficiency of the AAV2-CAG-σ-1r-GFP in RGCs was validated using RBPMS antibody and the σ-1r antibody. GFP staining in RGCs was validated previously,21Figures 3A and 3C are immunohistochemical localization of the transfected σ-1r in RGCs 2 weeks and 6 weeks post intravitreal injection, respectively. Co-localization of RBPMS antibody with σ-1r antibody demonstrate transfection in RGCs, DAPI staining validated RGCs nuclei. Western blots (Figs. 3B and 3D) confirmed these findings; using the σ-1r antibody (σ-1R) we demonstrated that GFP/σ-1r fusion protein stained at molecular weight of 53 k/Da, whereas there was no σ-1r staining in control or empty vector containing GFP. β-Tubulin served as loading controls.
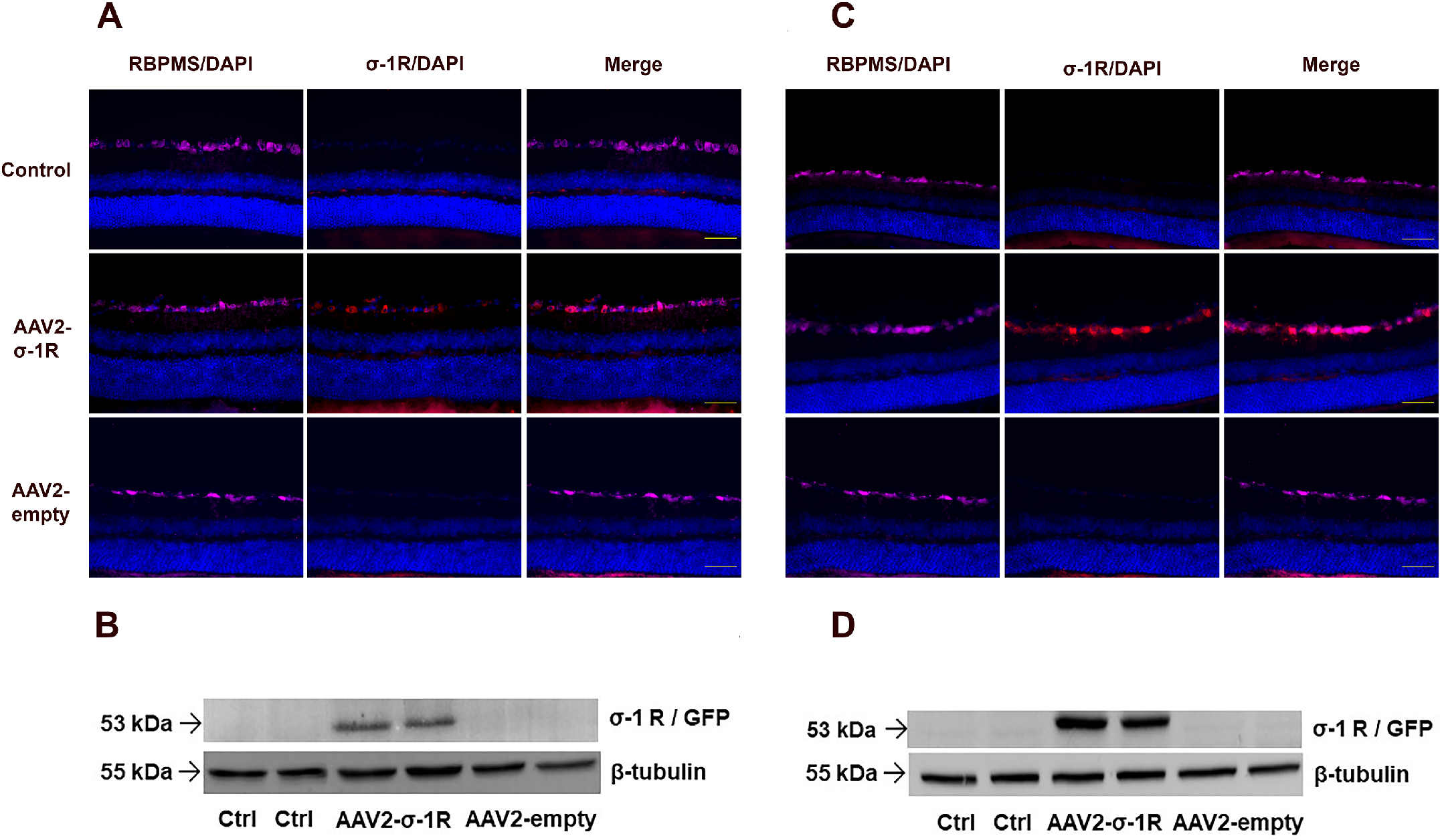
Transduction efficiency of the AAV2- σ-1r in RGCs was validated using RBPMS antibody (pink) for the detection of RGCs and the σ-1r antibody (red). Beta tubulin was used as loading control for Western blot analysis.
(A, B) Immunostaining and Western blots for 2 weeks post AAV2 vector IVT delivery. Merged images demonstrate the transgenic expression of the σ-1r in RGCs. (C, D) Immunostaining and western blots for 6 weeks post AAV2 vector IVT delivery. Merged images demonstrate increased transgenic expression of the σ-1r in RGCs, Western blots validated this. Aand C images were taken at 40 times magnification, scale bar = 50 µM. Representative of staining, N = 4.
Expression of the σ-1r Restores RGCs After ONC
To test if expression of the σ-1r in σ-1r K/O mice resulted in increased numbers of RGCs after exposure to ONC, K/O mice RGCs were transduced with AAV2- σ-1r-GFP or AAV2- empty vector for 2 weeks, and then exposed to ONC and animals were euthanized 7 days, 14 days, or 28 days post ONC. AAV2- empty vector served as control for AAV2- σ-1r-GFP and WT mice served as positive control. Quantitative assessment of RGCs demonstrated that there was a time-dependent decrease in RGC density after ONC exposure (Fig. 4A). However, RGC densities in wild type mice and AAV2- σ-1r-transduced σ-1r K/O mice were similar 7, 14, and 28 days post ONC (see Fig. 4A). RGC densities 14 days and 28 days post ONC were lower when compared with 7 days post ONC. As with RGC density post ONC, PERG demonstrated RGC function was similar in WT mice and AAV2- σ-1r-transduced σ-1r K/O mice 7 days post ONC (Fig. 4B).
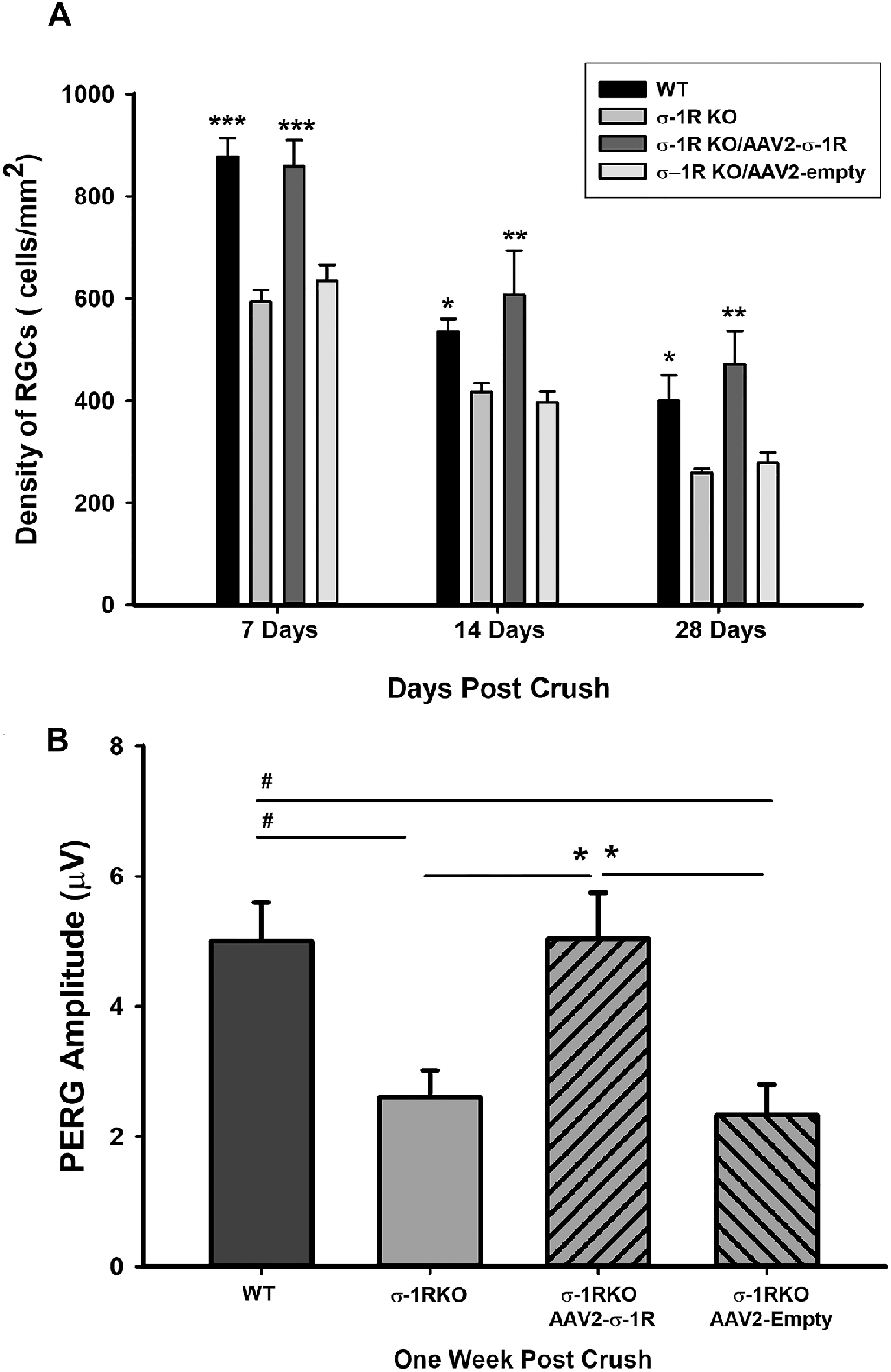
Transgenic expression of the σ-1r restores RGC density and function up to 28 days following ONC.
(A) There is significant exacerbation of RGC density loss following ONC in σ-1r K/O animals when compared to WT animals and compared with σ-1r K/O animals containing AAV2-σ-1r, demonstrating the ability of the σ-1r to attenuate RGC loss following injury. Although the density of RGCs decreased significantly 14 and 28 days post ONC, AAV2-σ-1r attenuated RGC loss following injury, similar to that of WT animals. Significantly different from σ-1r K/O and σ-1r K/O AAV2-empty vector. Data are shown as mean ± SEM N = 4, ***P < 0.001, **P < 0.01, *P < 0.05 ANOVA and Dunnett’s test. (B) Loss of RGC function was also exacerbated in σ-1r K/O animals following ONC when compared to WT animals and compared with σ-1r K/O animals containing AAV2-σ-1r. Data are shown as mean ± SEM n = 4* or #P < 0.05.
Phosphorylated (p) c-Jun, c-Jun, and Caspase 3 are Upregulated in ONC
It had previously been determined that apoptotic proteins were upregulated in isolated primary RGCs in response to oxidative stress.21–24 Therefore, in these studies, we wanted to identify proteins that were involved in the ONC-induced response. We identified phosphorylated (p) c-Jun, c-Jun, and Caspase 3 in mouse RGCs. Immunohistochemical analysis determined that there was a time-dependent upregulation of phosphorylated (p) c-Jun, c-Jun, and caspase 3 in response to ONC. Phosphorylated c-Jun and c-Jun were observed in RGCs 8 hours to 7 days post ONC (Fig. 5A). P-c-Jun was localized in the periphery and cytoplasm of the cells, whereas c-Jun was localized to the nucleus. In some instances, p-c-Jun co-localized with c-Jun. Contralateral control, without ONC, did not stain for either p-c-Jun or c-Jun. Most interestingly, caspase 3 expression was only observed 3 days and later post ONC (Fig. 5B).
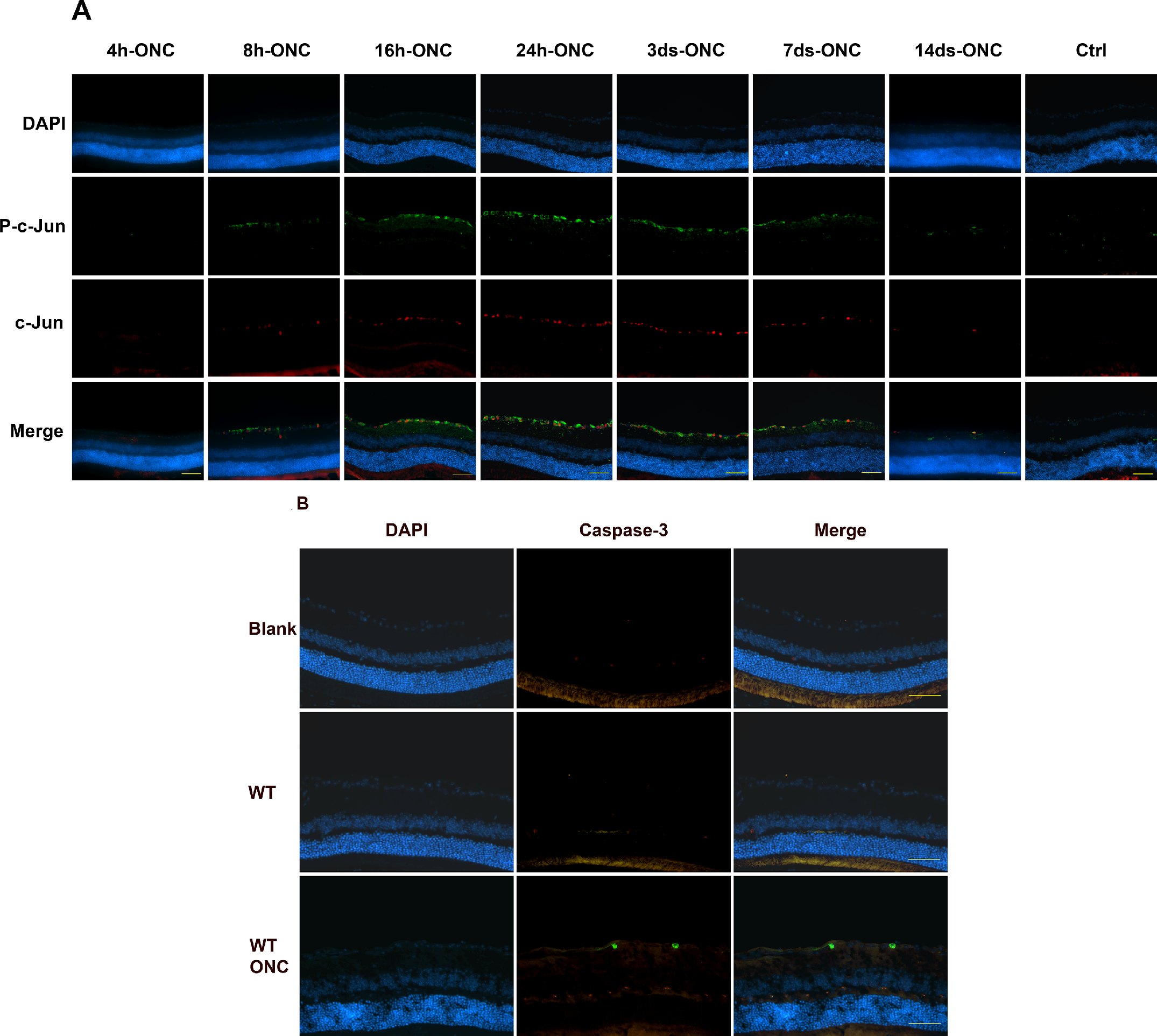
(A) Phosphorylated (p) c-Jun activity and c-Jun activity were detected as early as 8 hours post ONC, peaked at 24 hours and subsided 14 days post ONC. P-c-Jun is localized to the periphery and cytoplasm of the cell, whereas c-Jun is localized at the nucleus n = > 3 each group. Images were taken at 40 times magnification, scale bar = 50 µM. (B) Immunofluorescence of wild-type (WT) mice and wild-type mice exposed to optic nerve crush (WT/ONC) stained with caspase-3 antibody. Caspase-3 activity (green) is observable 3 days post ONC suggesting that caspase-3 is a late response gene. There were 4, images were taken at 40 times magnification, scale bar = 50 µM.
Activation of σ-1r Decreases c-Jun Expression
To determine the effects of σ-1r on c-Jun expression, mice were injected with pentazocine and then subjected to ONC, contralateral eyes that were not subjected to ONC served as controls. Figures 6A and 6B Western blots and densitometric analysis, respectively, demonstrate that exposure of the animals to pentazocine resulted in decreased expression of c-Jun suggesting that the σ-1r mitigates the apoptotic pathway activated in response to ONC.
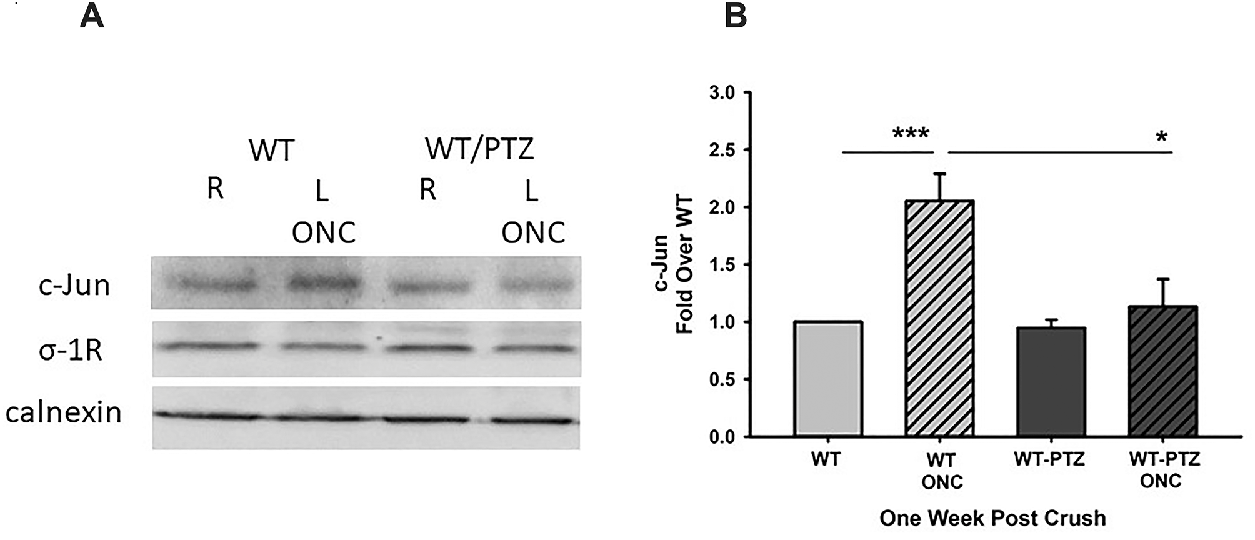
Activation of the σ-1r by pentazocine (PTZ) decreases c-Jun expression after ONC. WT mice that were treated with PTZ and exposed to ONC were stained with c-Jun and σ-1r antibodies.
(A) Western blot and (B) densitometric analyses demonstrate that PTZ-induced activation of the σ-1r resulted in significant decreases in c-Jun expression following ONC. ***Significantly different from WT animals, and *significantly different from WT ONC-treated animals, N = 4, ***P < 0.001, *P < 0.05 ANOVA and Dunnett’s test.
Discussion
Degeneration of the ON is a crucial factor in many neurological dysfunctions, including glaucoma, diabetic retinopathy, optic neuritis, hereditary optic neuropathy, and mechanical injury of the optic nerve leading to losses of RGCs and eventually blindness. Using σ-1r K/O mice, we demonstrate for the first time that transgenic expression of the σ-1r protects RGCs and RGC function in animals exposed to ONC. We have demonstrated that the σ-1r protects RGCs by inhibition of the apoptotic pathway involving c-Jun, p-c-Jun and caspase 3 in a time-dependent manner.
In this study, two sets of experiments validated the role of the σ-1r as a stress response protein in retinal disease. Under physiological conditions, treatment with pentazocine in WT mice had no significant effect on RGC cell number and RGC function when compared to WT mice that were not treated with pentazocine. However, in WT mice, activation of the σ-1r by pentazocine followed by ONC resulted in increased RGC numbers and function when compared with untreated mice. Furthermore, in physiological conditions, RGC number and activity were similar in WT mice when compared with σ-1r K/O mice. However, after ONC there were decreased RGC numbers and RGC activity, however, RGC number and activity was greater in WT animals when compared with σ-1r K/O animals. These data confirm that the σ-1r is a stress response protein and is activated in response to injury, similar to that reported previously.3,11,25 Although no functional or morphological retinal deficits are observed in σ-1r K/O mice from birth to 6 months of age, there are qualitative changes in retinal function detected first in the optic nerve head, and as time progresses, in the ganglion cell layer by 12 months of age.3,26 It is currently accepted that aging allows for increased oxidative and cellular stresses, thus, absence of the σ-1r accelerates retinal dysfunction not seen in WT mice eyes. Additionally, increased IOP is associated with RGC degeneration in glaucoma. Corroborative studies11 demonstrated that IOP was similar in 3 month old σ-1r K/O mice when compared with 3 month old WT mice. However, in response to induction of diabetes, IOP was increased in σ-1r K/O mice when compared with diabetic WT mice or non-diabetic WT mice.11 These observations, taken together, further validates the role of the σ-1r as a stress-response protein in retinal diseases.
There are several lines of evidence that demonstrate the protective role of the σ-1r in acute RGC degeneration in vivo. In WT mice, following ONC, there were significant decreases in RGCs in both pentazocine-treated and untreated mice when compared with non-ONC mice. However, the degree of RGC decrease is significantly greater in untreated animals when compared to pentazocine-treated animals. Additionally, when compared with mice lacking the σ-1r, RGC numbers in WT animals were significantly greater after ONC confirming the neuroprotective role of the σ-1r. Further evidence for the protective role of the σ-1r was demonstrated by the ability of transgenic expression of the σ-1r to restore RGC cell number and function to similar levels observed in WT mice. The significant decreases in RGC number in WT animals 7, 14, and 28 days post ONC, were observed, and these progressive decreases in RGC numbers result from the acute nature of the injury to the ON. Remarkably, transgenic expression of the σ-1r and activation of the σ-1r by pentazocine, slowed the losses of RGC cells, further demonstrating the protective effects of the σ-1r. These findings confirm our previous studies that determined that σ-1r is neuroprotective in primary RGC cultured cells that were exposed to oxidative stress.21,22
In this study, the involvement of members of MAP kinase/c- JNK pathways, c-Jun, in σ-1r neuroprotection in RGCs were examined. Immunohistochemical analysis of c-Jun and p-c-Jun demonstrated time-dependent expressions, corroborating previously published observations. Interestingly, the times of onset and duration of expression of c-Jun and p-c-Jun are not dependent on whether the insult to the tissue is chronic or acute. In acute injury by ONC, we tested and demonstrated p-c-Jun and c-Jun expression in RGCs at 8 hours to 7 days after ONC. Other studies demonstrated c-Jun expression after ONC between 195 days in rabbit retinal ganglion cell layer,27 and 6 hours after ONC in adult rats.28 In NMDA-induced neurodegenerative models, c-Jun and p-c-Jun expressions were observed 3 hours post NMDA-intravitreal injection in 8-week-old Wistar rats29 and was sustained up to 24 hours postinjection. In Sprague-Dawley rat retinas, p-c-Jun expression was increased 5 days post injected with NMDA, when compared with untreated retinas.30In chronic models of IOP elevation that result in RGC losses, c-Jun and p-c-Jun levels were increased in RGCs on the first day after IOP increases and continued to 4 weeks after IOP increases.31 Using the Morrison glaucoma model in Brown Norway rats, it was demonstrated that there was an increased c-Jun expression 2 weeks post IOP elevation.32Taken together, this suggests that p-c-Jun and c-Jun are early-to-intermediate response transcription factors.
Of interest, is that in RGCs, staining of anti-c-Jun and anti-p-c-Jun antibodies showed very little co-localization; as c-Jun expression was mostly in the nucleus, consistent with previous reports33 and p-c-Jun in the cytoplasm. Phosphorylation of c-Jun by JNK results in a more activated form p-c-Jun. Importantly, c-Jun and p-c-Jun have the ability to bind multiple targets forming complexes, thus regulating transcription and translation processes in multiple cell types.34
Although the mechanisms by which c-Jun mediates the neurodegenerative processes after ONC remains unclear, previous studies demonstrated that inhibition of calcium channel lessened axonal and RGC degeneration and inhibited c-Jun and p-c-Jun expression.35Inhibition of calcium channels also results in decreased JNK.35 These observations are of interest, as the σ-1r also associates with and prevents the influx of calcium by L-type voltage-gated calcium channels in RGCs,7 resulting in RGC protection. Additionally, activation of the σ-1r also results in decreased ONC-induced c-Jun and p-c-Jun expression. Taken together, this suggests that ONC-induced activation of the σ-1r results in decreased calcium influx, and decreased activation of the JNK/c-Jun/p-c-Jun pathway.
In our study, caspase 3 expression was not observed concurrently with c-Jun-or p-c-Jun early expression, but was expressed three days after ONC, suggest a later response in the apoptotic pathway. Interestingly, pharmacological or molecular inhibition of the JNK/c-Jun pathway resulted in decreased caspase 3 expression suggesting up-stream regulation of caspase 3 by JNK and c-Jun.36 Additionally, activation of the σ-1r by pentazocine, known to be protective in retinal neurodegeneration, obliterates the ONC-induced expression of c-Jun, p-c-Jun, and caspase 3, corroborating previous results in primary RGCs. In our previous studies, activation of the σ-1r and overexpression of the σ-1r in isolated RGCs protected RGCs by inhibiting caspase 3/7 activity in primary RGCs subjected to OGD.21
Other members of the MAPK family are ERK1/2 and the p38 kinases. Previous studies in RGC primary cultures have demonstrated that in an oxygen glucose-deprived (OGD) model, activation of the σ-1r results in increased ERK1/2 phosphorylation, which correlates with increased numbers of RGCs, suggesting a neuroprotective role of increased ERK1/2 phosphorylation.22 Interestingly, other studies have demonstrated that in the RGC-supporting glial cells, σ-1r activation protects these cells from oxidative stress via modulation of ERK1/2 activity.37 Taken together, this suggests that the mechanisms by which the σ-1r confer protection on RGCs are complex, and may involve the MAPK/JNK/c-Jun/caspase 3 pathway, and possible other convergent pathways. Additionally, this current study explored the involvement of the MAPK/JNK/c-Jun/caspase 3 pathway in the pentazocine/σ-1r-induced neuroprotection of RGCs in an ONC model for retinal disease, however, these mechanisms may have relevance in other optic neuropathy involving RGC death, including diabetic retinopathy,3 but further studies are needed.