
Sigma 1 receptor regulates ERK activation and promotes survival of optic nerve head astrocytes

By Jing Zhao, Barbara A. Mysona, Jing Wang, Graydon B. Gonsalvez, Sylvia B. Smith, and Kathryn E. Bollinger
Excerpt from the article published in PLOS ONE, September 12, 2017, DOI: https://doi.org/10.1371/journal.pone.0184421
Editor’s Highlights
- Neuronal and glial cell types throughout the retina and optic nerve contain sigma 1 receptor (S1R), and its agonists display robust neuroprotective properties both in vitro and in vivo.
- A common link between S1R and cell survival mechanisms is the extracellular signal-regulated protein kinase (ERK1/2) cascade.
- The effect of S1R activation on the status of the ERK1/2 pathway might be dependent on the cell type.
- S1R inhibition is a potential therapeutic option for neoplastic disorders including cancer of the breast and prostate
- S1R agonists, including (+)-pentazocine, have been shown to protect retinal ganglion cells (RGCs) from death under conditions of metabolic and oxidative stress.
- S1R activation protects ONHAs via a mechanism that likely involves modulation of the ERK1/2 signaling pathway.
Abstract
The sigma 1 receptor (S1R) is a unique transmembrane protein that has been shown to regulate neuronal differentiation and cellular survival. It is expressed within several cell types throughout the nervous system and visceral organs, including neurons and glia within the eye. S1R ligands are therapeutic targets for diseases ranging from neurodegenerative conditions to neoplastic disorders. However, effects of S1R activation and inhibition within glia cells are not well characterized. Within the eye, the astrocytes at the optic nerve head are crucial to the health and survival of the neurons that send visual information to the brain. In this study, we used the S1R-specific agonist, (+)-pentazocine, to evaluate S1R activation within optic nerve head-derived astrocytes (ONHAs). Treatment of ONHAs with (+)-pentazocine attenuated the level and duration of stress-induced ERK phosphorylation following oxidative stress exposure and promoted survival of ONHAs. These effects were specific to S1R activation because they were not observed in ONHAs that were depleted of S1R using siRNA-mediated knockdown. Collectively, our results suggest that S1R activation suppresses ERK1/2 phosphorylation and protects ONHAs from oxidative stress-induced death.
Introduction
Sigma 1 receptor (S1R) is a small (25kDa), transmembrane protein that is highly conserved but has no known homology to existing mammalian proteins[1]. It is expressed throughout organs and cell types of the central and peripheral nervous, immune, endocrine and reproductive systems[2–5]. Previous studies indicate that S1R functions as a “pluripotent modulator” that interacts with several ion channels and signaling pathways including calcium channels, inositol phosphates, and protein kinases[6]. Despite extensive research, understanding of the molecular cascades triggered by S1Rs is rudimentary. However, the effects of several agonists and antagonists of S1R have been well characterized within models of neurodegenerative disease and cancer therapeutics[7–12]. Overall, these studies indicate that agonists of S1R are pro-survival whereas antagonists of S1R inhibit tumor cell proliferation and induce apoptosis[13]. Therefore, published findings suggest possible applications of sigma ligands in diverse fields including oncology, and treatment of neurodegenerative diseases.
Neuronal and glial cell types throughout the retina and optic nerve contain S1R, and its agonists display robust neuroprotective properties both in vitro and in vivo[7, 14–17]. Recent studies indicate that S1R agonists may offer a treatment option for degenerative eye diseases including photoreceptor dystrophies, and glaucoma[18–21]. Conversely, S1R inhibition is a potential therapeutic option for neoplastic disorders including cancer of the breast and prostate[22, 23]. Understanding the molecular mechanisms that underlie S1R activity is critical to establishing better treatments for eye diseases. Furthermore, evaluation of S1R within ocular tissues contributes to knowledge of how ligands for this receptor would affect the eye, if used systemically as either anti-neoplastic or neuroprotective agents.
A common link between S1R and cell survival mechanisms is the extracellular signal-regulated protein kinase (ERK1/2) cascade. The ERK1/2 molecular signaling pathway regulates basic cell functions, including proliferation, differentiation and survival[24]. Several analyses indicate that S1R interacts with the ERK1/2 pathway, but the mechanism and intracellular consequences of this interaction are not well characterized[20, 21, 25–27]. For example, studies show that the prototypic S1R agonist, (+)-pentazocine (PTZ), protects retinal ganglion cells (RGCs) from hypoxia-induced cell death by increasing activation of ERK1/2[21]. In addition, our recent work describes enhancement of retinal ERK1/2 phosphorylation concomitant with robust protection from excitotoxic injury within retinas of (+)-pentazocine-treated mice[20]. These recent results are in contrast with findings of (+)-pentazocine-mediated decreased ERK1/2 activation within primary microglia cell cultures derived from retina[28]. These findings raise the possibility that the effect of S1R activation on the status of the ERK1/2 pathway might be dependent on the cell type.
S1R agonists, including (+)-pentazocine, have been shown to protect retinal ganglion cells (RGCs) from death under conditions of metabolic and oxidative stress[14, 21]. These protective properties make (+)-pentazocine a potential treatment for optic neuropathies, in which vision loss is caused by degeneration of RGCs[29]. In glaucoma, the most common optic neuropathy, the optic nerve head (ONH) is considered the initial site of disease[30, 31]. It is the most anterior portion of the optic nerve, and the site where RGC axons converge as they leave the eye and pass into the brain. The optic nerve head astrocytes (ONHAs) are the most common glial cells within the ONH. They are vital to the health of the RGCs. Thus, evaluating their response to S1R activation and inhibition is critical to understanding ophthalmic treatment effects of S1R agonists and antagonists.
In the present study, we examined the effects of S1R activation and inhibition in primary cultures of ONHAs. We induced S1R activation using the S1R-specific agonist, (+)-pentazocine. Treatment with this compound attenuated ERK phosphorylation and protected ONHAs from oxidative stress-induced death. In addition, inhibition of S1R using siRNA-mediated knockdown blocked both (+)-pentazocine-induced prosurvival effects, and (+)-pentazocine-induced effects on ERK1/2 activation. Our results therefore suggest that S1R activation protects ONHAs via a mechanism that likely involves modulation of the ERK1/2 signaling pathway.
…
Results
Culture of ONHA and expression of S1R
Previous work indicated that S1R is expressed within the RGCs and glial cells of the optic nerve, but it has not been evaluated in ONHAs [16]. We cultured ONHAs from rats between postnatal day 3 and 5 using previously described methods[32, 33]. We used rats for these analyses because many mechanistic studies involving S1R have utilized rodents. In addition, due to its relative size, the optic nerve head of the rat is more easily isolated from surrounding tissues than that of the mouse. Our cultured cells expressed S1R, GFAP and NCAM, but did not express markers for oligodendrocytes or microglia (Fig 1).
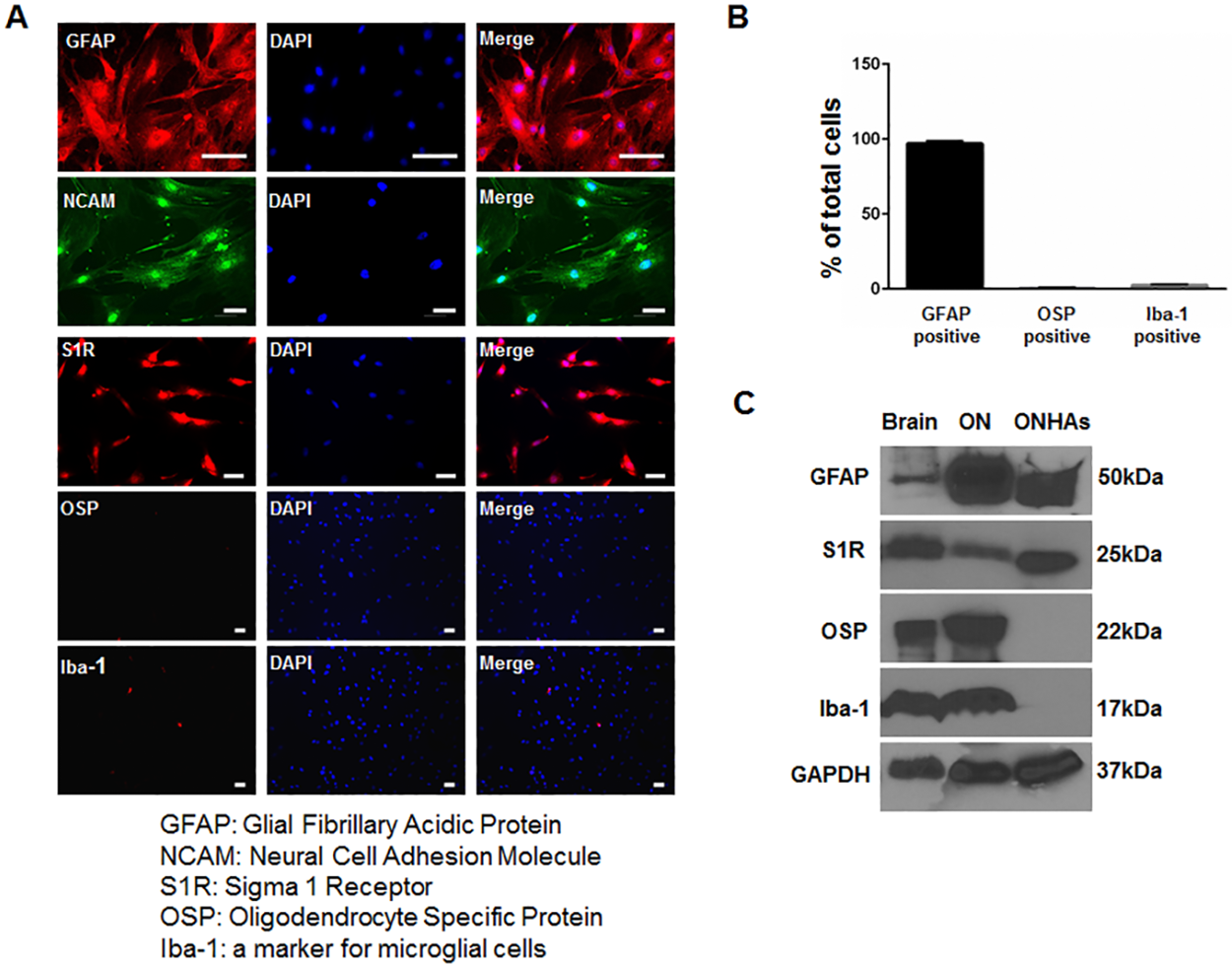
Characterization of cultured primary rat optic nerve head astrocytes (ONHA).
(A) ONHAs were fixed and probed with antibodies against GFAP (red) and NCAM (green), S1R (red), OSP (red), and Iba-1 (red). The cells were counterstained with DAPI to label DNA (blue) as a marker for nuclei. Scale bar: 50μm. (B) Quantitative analysis shows that more than 95% of the cells in culture express GFAP. (C) The cell lysates from ONHAs (lane 3) were positive for GFAP, a marker for astrocytes and S1R, but negative for Iba-1, a marker for microglial cells, and OSP, a marker for oligodendrocytes. The protein extract from rat brain (lane 1) and from rat optic nerve tissue (lane 2) were used as positive controls.
Protection of ONHAs from oxidative stress-induced cell death by the S1R agonist, (+)-pentazocine
We exposed ONHAs to increasing doses of (+)-pentazocine. Cell viability was then measured using the MTT assay. (+)-Pentazocine did not cause a significant change in percentage viability compare to the untreated control (Fig 2A). This result indicates that the S1R agonist, (+)-pentazocine, is minimally toxic to ONHA. We then exposed ONHA cultures to progressively increasing levels of oxidative stress by applying increasing concentrations of the chemical oxidant, hydrogen peroxide (H2O2). Results showed a concentration-dependent decrease in cell viability when cultures were exposed to increasing levels of oxidative stress (Fig 2B). The ONHAs were then subjected to death-inducing levels of H2O2 while undergoing treatment with the S1R agonist, (+)-pentazocine. We chose to expose cells to a 10μM concentration of (+)-pentazocine because previous studies indicate that this dosage is neuroprotective of retinal ganglion cells in vitro [21]. In addition, this concentration of (+)-pentazocine has been shown to attenuate oxidative stress in retinal Muller glial cells [35]. When cell cultures received (+)-pentazocine for one hour prior to and during the H2O2 application, the oxidative stress-induced cell death was significantly mitigated (Fig 2C).
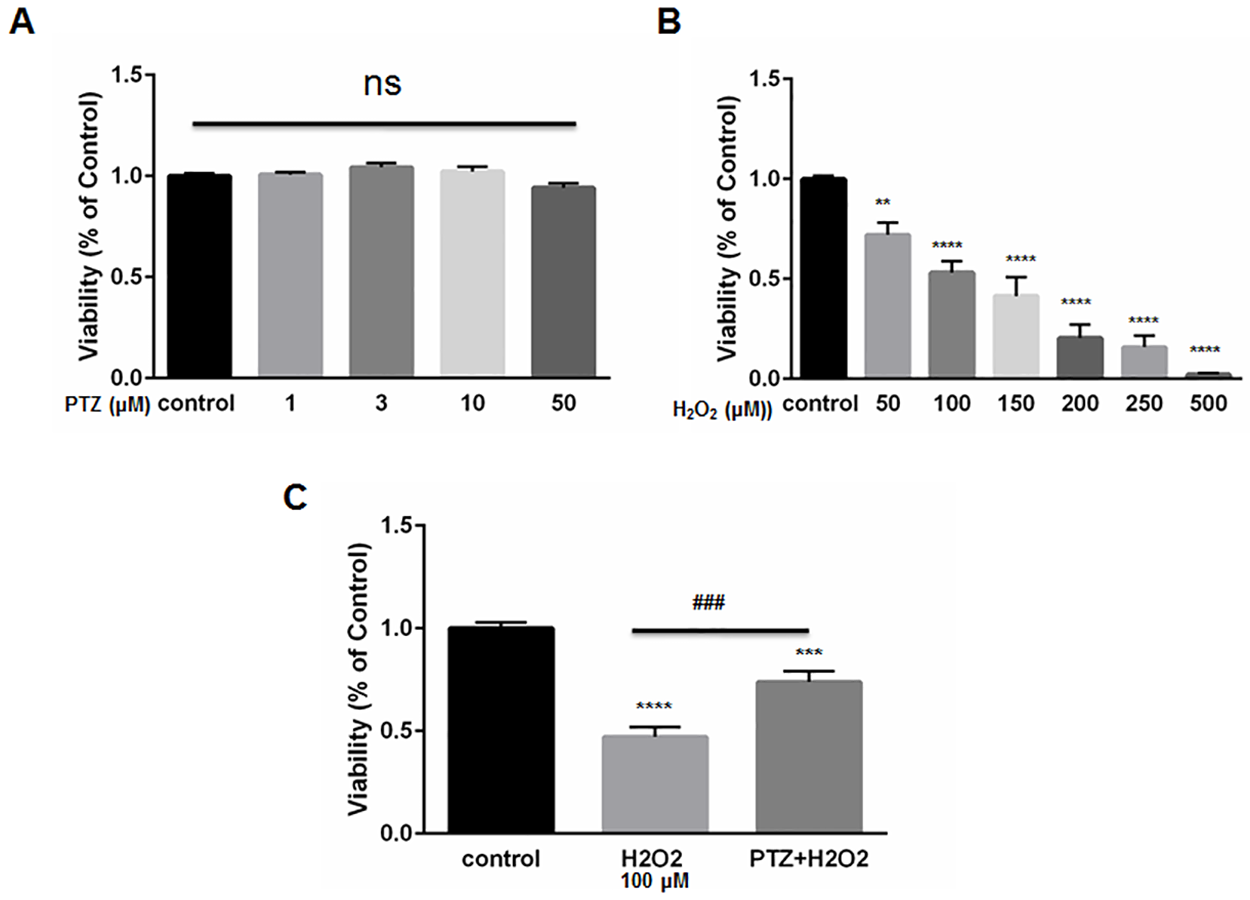
The effect of (+)-pentazocine and H2O2 on ONHAs viability.
(A) ONHAs were treated with (+)-pentazocine (PTZ) at varying concentrations (1,3,10 or 50μM) for 24 hours. MTT assay was performed to assess viability. Treatment with PTZ did not cause a significant change in percentage viability compared to the untreated control cells. At the highest concentration of PTZ (50μM), there was a trend toward decrease in viability that was not significant. (B) ONHAs were exposed to various H2O2concentrations (50,100,150, 200, 250, 500μM) for 24 hours. H2O2 induced ONHA death in a dose dependent manner. (C) ONHAs were treated with 100μM H2O2 for 24 hours in the presence or absence of PTZ (10μM, 1 hour pretreatment followed by co-treatment). Exposure to H2O2 significantly decreased viability compared to non-exposed cells. Compared to H2O2-exposure with no PTZ treatment, the H2O2-exposed, PTZ-treated ONHAs showed significantly increased viability. Data were analyzed using one-way ANOVA followed by Tukey-Kramer post hoc test for multiple comparisons. Significantly different from control *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Significantly different between groups ###P<0.001. Experiments were performed in quadruplicate and repeated 3 times.
S1R agonist, (+)-pentazocine inhibits ROS generation induced by oxidative stress in ONHAs
Studies indicate that increased intracellular reactive oxygen species (ROS) will affect the functional capacity of astrocytes, independent of their viability[36]. For example, an increase in intracellular ROS leads to altered astrocytic glutamate uptake and impaired glioprotection of associated neurons[37]. Therefore, we measured intracellular ROS generation within ONHAs, with and without (+)-pentazocine, in the presence of oxidative stress. Intracellular ROS detection was performed using CellROX Green Reagent. We found that exposure of ONHAs to H2O2 caused a significant increase in intracellular ROS (Fig 3). When the oxidative stress-exposed cell cultures were treated with (+)-pentazocine, intracellular ROS generation was significantly suppressed (Fig 3). Treating control ONHAs with (+)-pentazocine in the absence of oxidative stress caused no significant change in intracellular ROS generation (Fig 3).
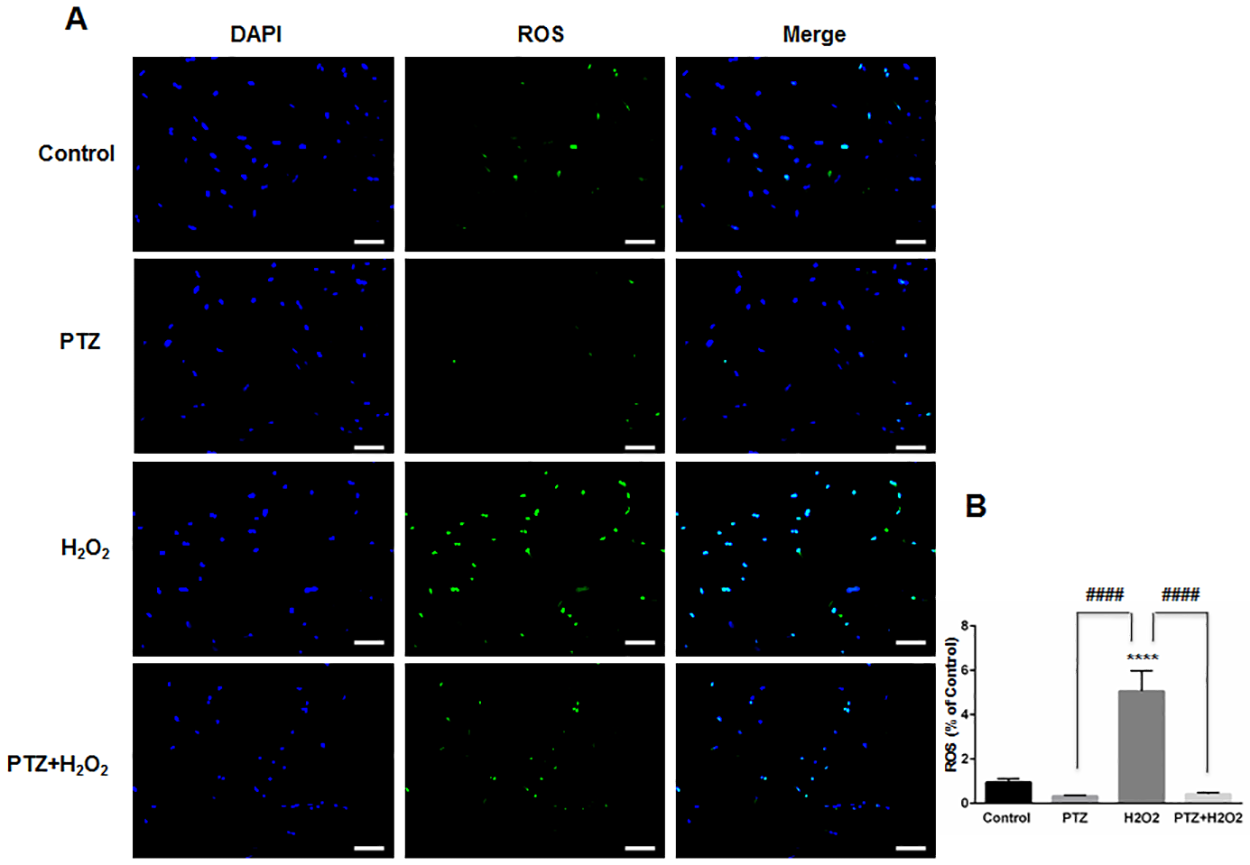
Effect of PTZ on ROS generation when ONHAs exposed to H2O2.
(A) Representative images of ONHAs treated with 100μM H2O2 for 24 hours in the presence or absence of PTZ (10μM, 1 hour pretreatment followed by co-treatment). ROS generation was visualized using CellROX Green reagent. Scale bar: 100μm. (B) Quantitative analysis of intracellular ROS. For each group, three coverslips were quantified, and eight images were taken from each coverslip. Mean signal intensity was quantified by ImageJ. Intracellular ROS generation increased when ONHAs were exposed to H2O2. The ROS generation was inhibited by PTZ. Data were analyzed using one-way ANOVA followed by Tukey-Kramer post hoc test for multiple comparisons. Significantly different from control ****P<0.0001. Significantly different between groups ####P<0.0001. Experiments were repeated 3 times.
(+)-Pentazocine-mediated effects on ERK1/2 activation in oxidative stress-exposed ONHAs
Several recent studies have linked the pro-survival effects of S1R agonists to changes in ERK1/2 phosphorylation status[20, 21, 26]. For example, ERK activation, induced by stimulation of S1R, leads to protection of purified RGCs under conditions of oxygen-glucose deprivation[21]. To examine the effect of (+)-pentazocine on ERK1/2 activation within ONHAs, we first analyzed ERK1/2 phosphorylation within ONHA cultures exposed to H2O2 (100μm) at a concentration previously shown to cause severe cellular stress (Fig 2C). Cultures were treated with H2O2 (100μM) for 15 minutes, 30 minutes, 1 hour, 3 hours, or 24 hours. The cells were then harvested and western blot analysis was performed to assay for the phosphorylated fraction of total ERK1/2 protein (pERK1/2). Consistent with previous studies of oxidative stress-exposed astrocytes, we found that exposure of ONHAs to H2O2 led to increased levels of pERK1/2 at time points of 15 minutes to 24 hours following application, with peak activation occurring at the 30 minute time point (Fig 4A)[38]. Cultures were then treated with (+)-pentazocine for one hour prior to and during the H2O2 exposure. Resultant analyses showed that the level and duration of ERK1/2 phosphorylation within (+)-pentazocine-treated cells was reduced in comparison to the control (Fig 4A and 4B). In addition, the bell-curve of ERK1/2 phosphorylation was shifted to earlier time points. In contrast to control cells, peak phosphorylation of ERK1/2 was observed as early as the 15 minute time point in pentazocine-treated cells (Fig 4A and 4B). Despite this temporal shift, the total level of ERK1/2 phosphorylation at time points beyond 30 minutes was significantly reduced in pentazocine-treated cells in comparison to the control (Fig 4A and 4B).
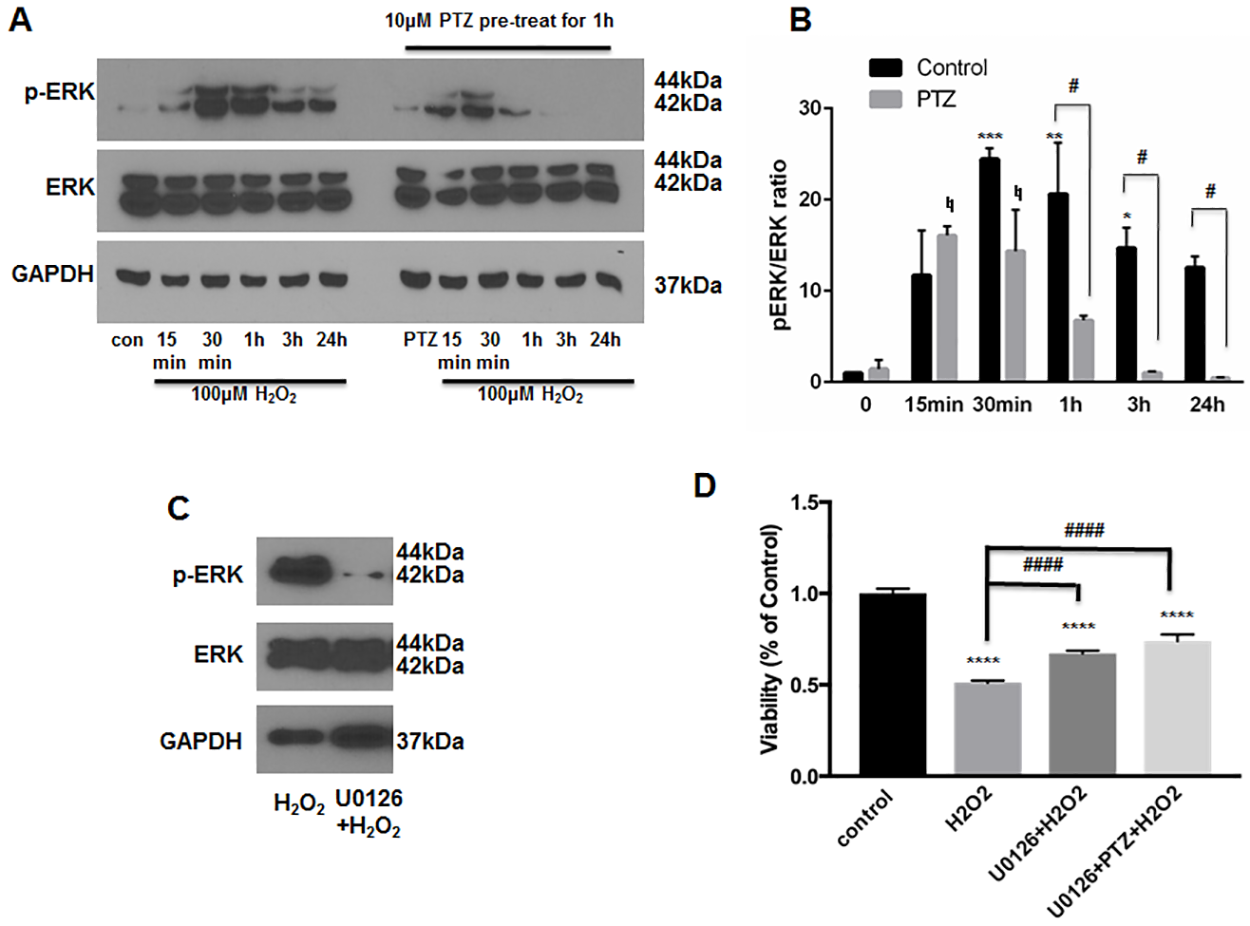
Inhibition of ERK1/2 phosphorylation in ONHAs.
(A) ONHAs were incubated with 100μM H2O2 at 37°C for 15 minutes, 30 minutes, 1 hour, 3 hours and 24 hours in the presence or absence of PTZ (10μM, 1 hour pretreatment followed by cotreatment). Western blot analysis of cell lysates is shown. Phosphorylation of ERK was increased at the 15 minute time point following H2O2application, and peaked between 30 minutes and 1 hour. Lysates derived from H2O2-exposed cells treated with PTZ showed decreased ERK phosphorylation at 1 hour, 3 hour and 24 hour time points. (B) Quantitative analysis of pERK levels represented as pERK normalized to total ERK. Results are presented as fold change of the pERK/total ERK ratio derived from H2O2 exposed cells versus non-H2O2 exposed cells. Experiments were repeated 3 times. Data were analyzed using two-way ANOVA followed by Tukey-Kramer post hoc test for multiple comparisons. Significance levels for pERK change in the untreated (no PTZ) control group (black columns) are as follows: *p<0.05 **p<0.01, ***p<0.001. Significance level for pERK change in the PTZ-treated group (gray columns) is as follows: ♮p<0.05. Significance between the untreated control group (black columns) and PTZ-treated group (gray columns) at the 1 hour, 3 hour and 24 hour time points is represented as follows: #p<0.05 Note that the change in pERK between these groups was not significant at the 0 minute, 15 minute, and 30 minute time points. (C) ONHAs were exposed to 100μM H2O2 for 24 hours in the presence or absence of U0126 (10μM, 1hour pretreatment followed by co-treatment). Western blot showed decreased ERK phosphorylation levels under conditions of U0126 treatment compared with no treatment. (D) ONHAs were treated with 100μM H2O2 for 24 hours in the presence or absence of U0126 (10μM, 1hour pretreatment followed by co-treatment). MTT assay was performed to assess viability. Exposure to H2O2 significantly decreased ONHA viability and treatment with U0126 increased viability compared to the non-treated, H2O2-exposed cells. Combined treatment with PTZ and U0126 did not show a significant additive effect on cell viability. Data were analyzed using one-way ANOVA followed by Tukey-Kramer post hoc test for multiple comparisons. Significantly different from control: ****p<0.0001. Significantly different between groups: ####p<0.0001. Experiments were performed in quadruplicate and repeated 3 times.
Inhibition of ERK1/2 phosphorylation is pro-survival in ONHAs
As described above, treatment of oxidative stress-exposed ONHAs with (+)-pentazocine lead to increased survival and attenuation of ERK1/2 activation. To determine whether decreased ERK1/2 phosphorylation could be a mechanism through which (+)-pentazocine promotes survival, we analyzed the effects of inhibiting ERK1/2 activation in ONHAs. To do this, we treated ONHA cultures with U0126, an inhibitor of ERK phosphorylation, for one hour prior to and during application of oxidative stress. Viability of the U0126-treated ONHAs was assayed after 24 hours of exposure to H2O2. Cell cultures treated with U0126 showed reduced cell death and ERK1/2 phosphorylation compared with untreated cultures (Fig 4C and 4D). These results suggest that inhibition of ERK1/2 activation promotes survival of ONHAs under conditions of oxidative stress.
Knockdown of S1R within ONHAs blocks (+)-pentazocine-mediated pro-survival effects
Results presented in Fig 2. suggest that the S1R specific agonist, (+)-pentazocine, protects ONHAs from oxidative stress-induced cell death. To test the hypothesis that (+)-pentazocine-mediated cellular survival occurs through an S1R-dependent mechanism, we depleted S1R within ONHAs using small interfering (si)RNAs.
Initial knockdown experiments were performed using a human cervical cancer cell line (HeLa cells), because preliminary conditions for transfection were more easily determined using a cell line versus primary ONHAs. Cell cultures were transfected with scrambled siRNA or with S1R siRNA targeting human S1R. Western blot analysis was used to measure the level of S1R protein at 3 days and 6 days following transfection. These assays showed no change in the level of S1R protein in scrambled siRNA-transfected cultures. However, at 3 days and 6 days following transfection with S1R siRNA, we found significant decreases in S1R protein levels (Fig 5A–5D). Using MTT assay, we then measured HeLa cell viability at time points of 3 days and 6 days following siRNA transfection. Cells transfected with scrambled siRNA showed no change in viability compared with non-transfected cultures (Fig 5G and 5H). However, knockdown of S1R resulted in significant baseline HeLa cell death, without exposure to oxidative stress (Fig 5E and 5F). These results are consistent with previous reports showing that S1R inhibition restricts growth and causes death within rapidly dividing cell cultures[22, 23].
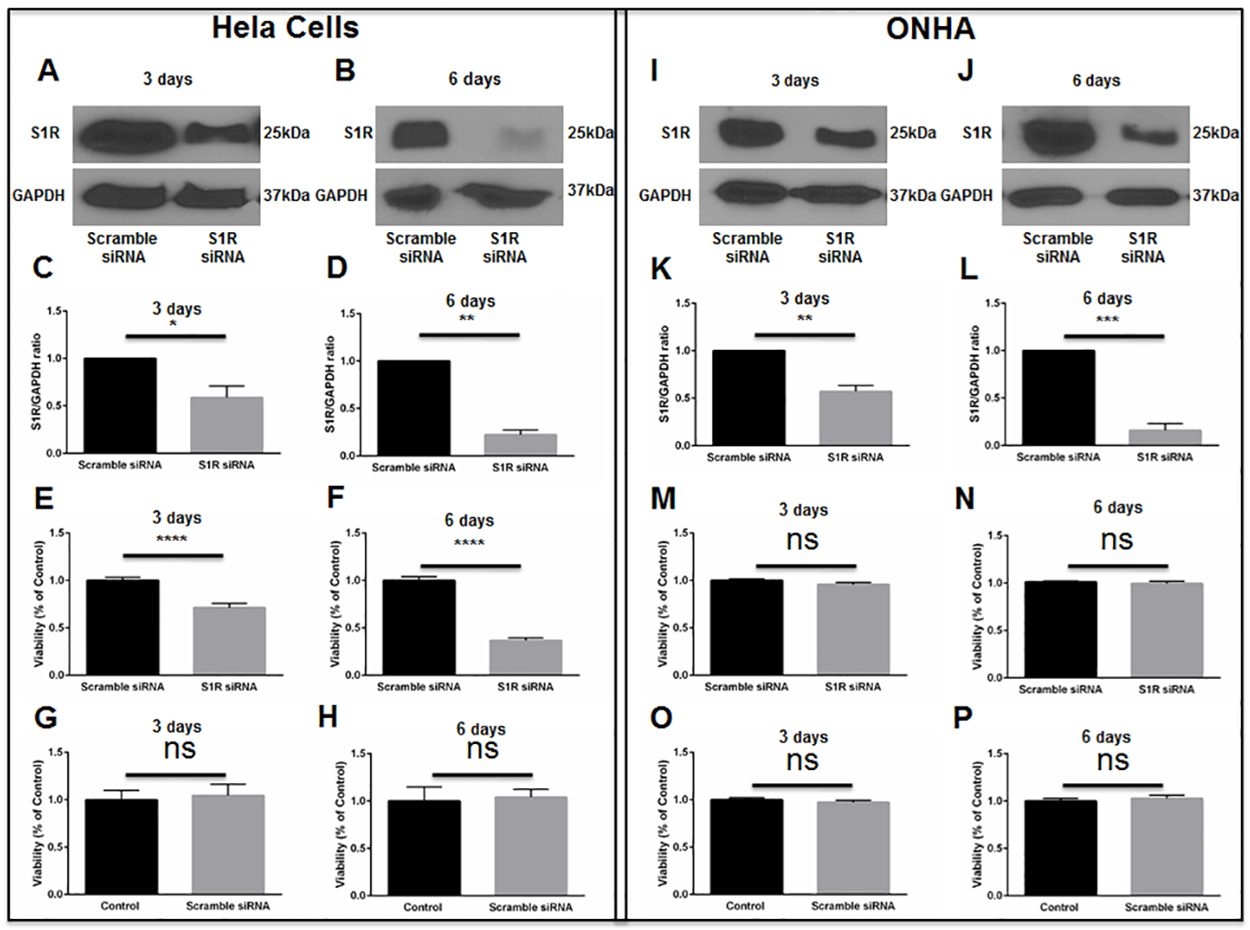
S1R knockdown in HeLa cells and ONHAs.
HeLa cells were transfected with human scrambled siRNA or with human S1R siRNA. Western blot analysis of S1R levels at 3 days (A) and 6 days (B) following transfection is shown. Quantitation of western blot results shows S1R levels normalized to GAPDH as the internal control (C, D). Results are presented as fold change of S1R levels derived from the S1R siRNA-transfected cells compared to S1R levels derived from scrambled siRNA-transfected cells. Data were analyzed using t-test. Significantly different from control *p<0.05, **p<0.01. Experiments were repeated 3 times. MTT assay was performed to assess viability at 3 days (E) and 6 days (F) after scrambled or S1R siRNA transfection. Viability was significantly decreased in S1R siRNA-transfected HeLa cells compared to scrambled siRNA-transfected cells. Transfection with scrambled siRNA did not cause significant HeLa cell death compared to non-transfection control (G, H). Data were analyzed using t-test. Significantly different from control ****p<0.0001. Experiments were performed in quadruplicate and repeated 3 times. ONHAs were transfected with rat scrambled siRNA or with rat S1R siRNA. Western blot analysis of S1R levels at 3 days (I) and 6 days (J) following transfection is shown. Quantitation of western blot results shows S1R levels normalized to GAPDH as the internal control (K, L). Results are presented as fold change of S1R levels derived from the S1R siRNA-transfected cells compared to S1R levels derived from scrambled siRNA-transfected cells. Data were analyzed using t-test. Significantly different from control: **p<0.01, ***p<0.001. Experiments were repeated 3 times. MTT assay was performed to assess viability at 3 days (M) and 6 days (N) after scrambled or S1R siRNA transfection. Viability was not significantly changed in S1R siRNA-transfected ONHAs compared to scrambled siRNA-transfected cells. Transfection with scrambled siRNA did not cause significant ONHA death compared to non-transfection control (O, P). Experiments were performed in quadruplicate and repeated 3 times.
We then performed knockdown of S1R within primary ONHAs. Cultures were transfected with scrambled siRNA or S1R siRNA targeting rat S1R. Consistent with results obtained in HeLa cells, S1R knockdown efficiencies were significant at 3 and 6 days following transfection of ONHAs (Fig 5I–5L). In contrast to results observed in HeLa cells, knockdown of S1R within ONHAs did not significantly affect cellular viability (Fig 5M–5P).
We next determined whether (+)-pentazocine could protect S1R-knockdown ONHAs from oxidative stress-induced cell death. Therefore, we subjected knockdown cell cultures to 100μM H2O2 in the presence or absence of (+)-pentazocine treatment (Fig 6A). Non-treated knockdown cultures exposed to 100μM H2O2 showed significant cell death at 6 days post-transfection with S1R siRNA (Fig 6A). When identically-exposed S1R knockdown cultures were treated with (+)-pentazocine, no significant protection from cell death was observed (Fig 6A).
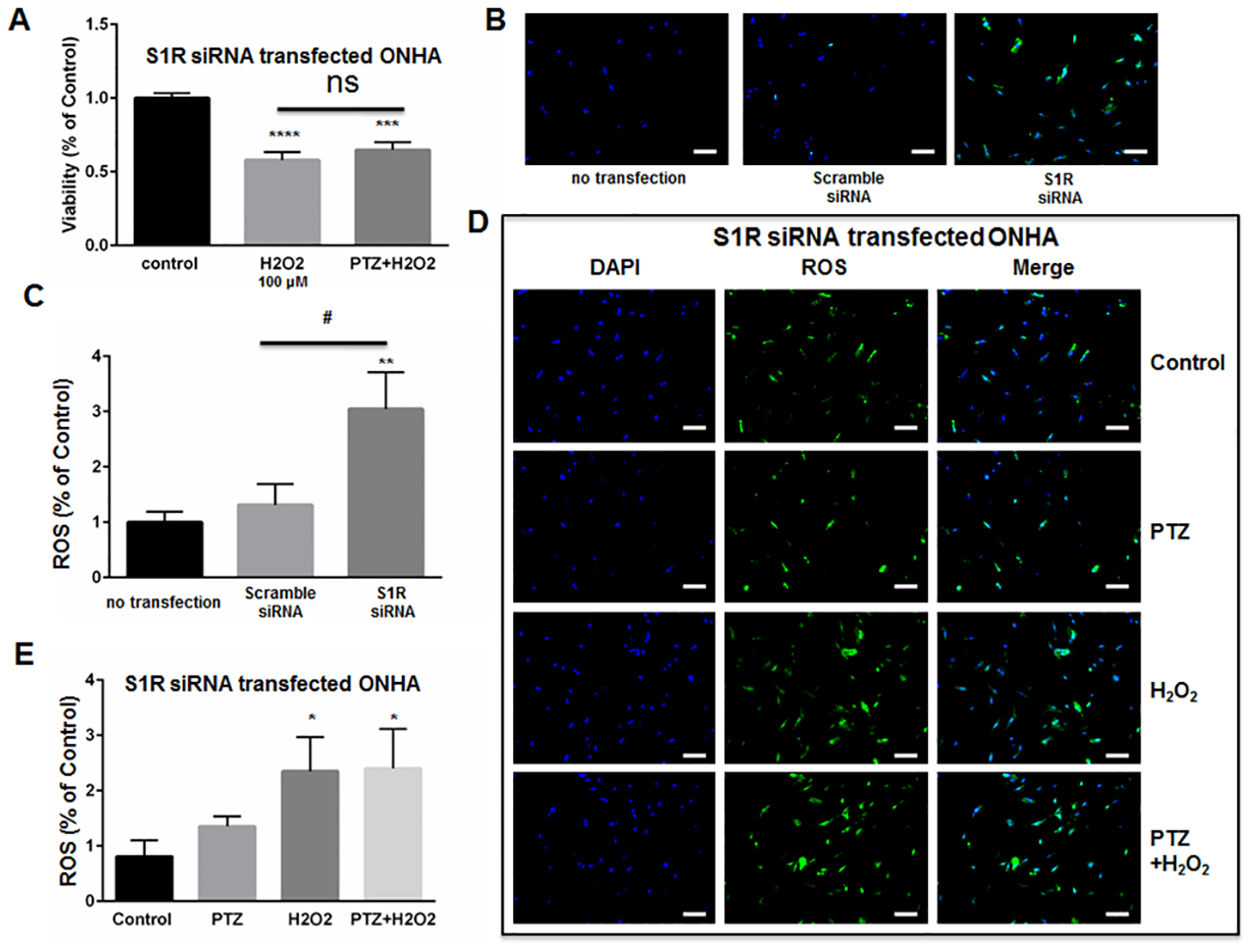
Knockdown of S1R within ONHAs blocks (+)-pentazocine-mediated suppression of ROS generation.
(A) Five days following transfection with S1R siRNA, ONHAs were treated with 100μM H2O2 for 24 hours in the presence or absence of PTZ (10μM, 1 hour pretreatment followed by co-treatment). MTT assay was performed to assess viability.100μM H2O2induced 50% cell death, and PTZ treatment did not significantly increase cell viability. (B-C) Effect of S1R knockdown on ROS generation in ONHAs. Data were analyzed using one-way ANOVA followed by Tukey-Kramer post hoc test for multiple comparisons. Significantly different from control: ***p<0.001, ****p<0.0001. (B) Representative images of ONHAs six days following transfection with either scrambled siRNA or S1R siRNA. ROS generation was visualized using CellROX Green reagent. Scale bar: 100μm. (C) Quantitative analysis of intracellular ROS. For each group, three coverslips were quantified, and eight images were taken from each coverslip. Mean signal intensity was quantified by ImageJ. Transfection with scrambled siRNA did not significantly increase ROS generation compared with non-transfected control cells. S1R siRNA-transfected ONHAs showed increased intracelluar ROS compared with scrambled siRNA-transfected ONHAs. Data were analyzed using one-way ANOVA followed by Tukey-Kramer post hoc test for multiple comparisons. Significantly different from control: **p<0.01. Significantly different between groups: #p<0.05. (D) Representative images of S1R siRNA-transfected ONHAs treated with 100μM H2O2 with or without PTZ for 24 hours. Scale bar: 100μm. (E) Quantitative analysis of intracellular ROS. For each group, three coverslips were quantified, and eight images were taken from each coverslip. Mean signal intensity was quantified by ImageJ. ROS generation increased when cells were incubated with H2O2. The ROS generation was not inhibited by PTZ treatment. Data were analyzed using one-way ANOVA followed by Tukey-Kramer post hoc test for multiple comparisons. Significantly different from control *p<0.05. Experiments were repeated 3 times.
As described above (Fig 3), we previously observed decreased ROS generation when H2O2-exposed ONHAs were treated with (+)-pentazocine. To determine whether the (+)-pentazocine-mediated inhibition of ROS generation occurred through S1R, we measured reactive oxygen species (ROS) generation in S1R knockdown ONHAs. Measurements were performed under conditions of H2O2 exposure, in the presence and absence of (+)-pentazocine. Intracellular ROS levels were assayed as described in Fig 3. In contrast to scrambled siRNA–transfected cells, the S1R-knockdown ONHAs showed high baseline ROS generation (Fig 6B and 6C). Exposure of S1R siRNA-transfected ONHAs to H2O2 resulted in a significant increase in intracellular ROS generation (Fig 6D and 6E). Treatment of the H2O2-exposed, S1R-knockdown cultures with (+)-pentazocine did not significantly inhibit ROS generation (Fig 6D and 6E).
Overall, these results suggest that knockdown of S1R blocks (+)-pentazocine-induced mitigation of ROS generation and inhibits (+)-pentazocine-mediated pro-survival effects in oxidative stress-exposed ONHA. Thus, S1R appears to be necessary for the survival-promoting effects of (+)-pentazocine on ONHA.
Knockdown of S1R within ONHAs increases baseline ERK1/2 phosphorylation and blocks (+)-pentazocine-mediated suppression of ERK1/2 phosphorylation
As described above, our results indicate that treatment of ONHAs with (+)-pentazocine inhibits oxidative stress-induced ERK1/2 activation (Fig 4). To address whether suppression of ERK1/2 activation can occur through S1R, we performed knockdown of S1R in ONHAs, followed by western blot analysis of pERK1/2 levels. S1R-knockdown cell cultures were treated with or without oxidative stress (H2O2), in the presence or absence of (+)-pentazocine. Measurement of ERK1/2 phosphorylation was performed at time points of 15 minutes to 24 hours following application of H2O2. In contrast to scrambled siRNA-transfected cells, the S1R-knockdown ONHAs showed a high baseline level of pERK (Fig 7A–7D), with a trend toward increased ERK1/2 activation in the presence of oxidative stress. Treatment with (+)-pentazocine did not significantly suppress ERK1/2 phosphorylation (Fig 7E and 7F). Based on these results, we conclude that S1R functions to decrease pERK1/2 levels at baseline in ONHAs. In addition, our results also suggest that (+)-pentazocine-mediated suppression of oxidative stress-induced ERK1/2 activation occurs through S1R.
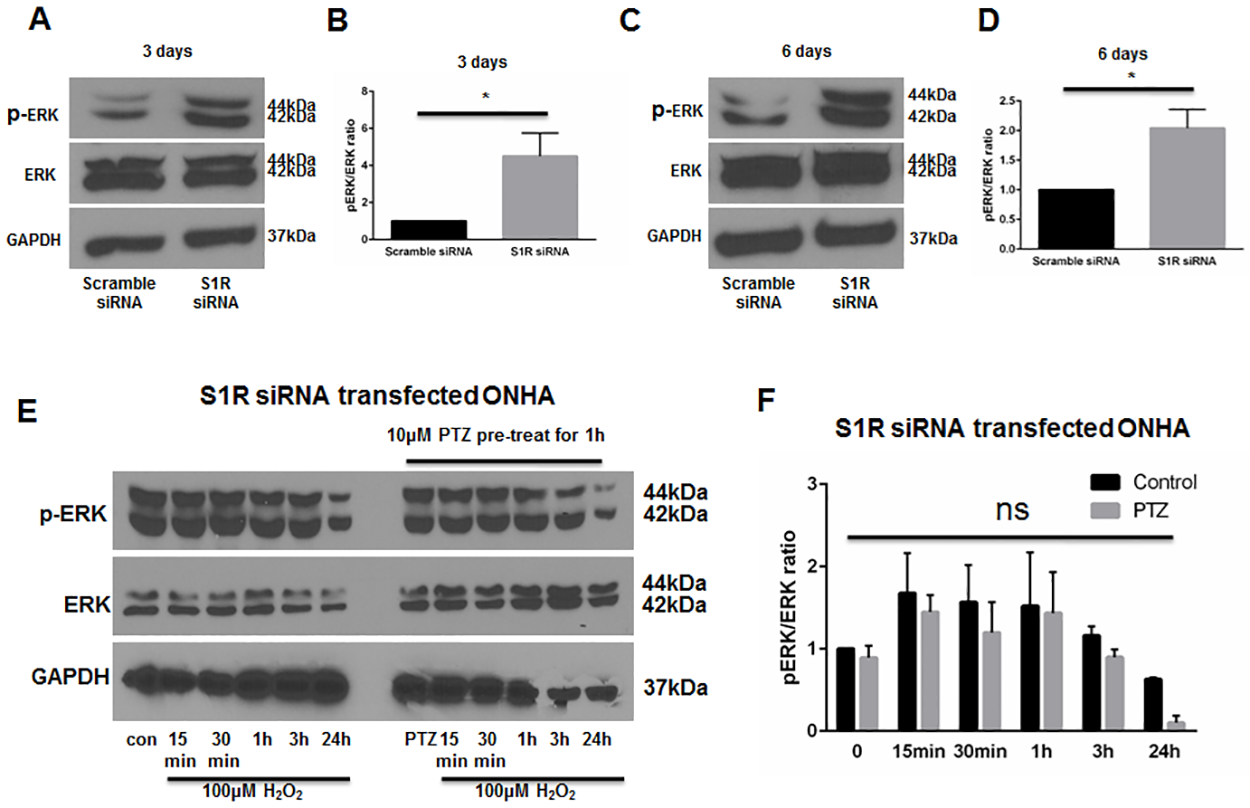
Knockdown of S1R within ONHAs blocks the (+)-pentazocine-mediated suppression of ERK1/2 phosphorylation.
Phophorylation of ERK was detected after 3 days (A) and 6 days (C) following transfection of scrambled or S1R siRNA in ONHA. At 3 days (A) and 6 days (C) following S1R siRNA transfection, pERK was increased compared to scrambled siRNA control. (B, D) Quantitative analysis of pERK levels represented as pERK normalized to total ERK. Results are presented as fold change of the pERK/total ERK ratio derived from S1R siRNA transfected cells versus scrambled siRNA transfected control cells. Data were analyzed using t-test. Significantly different from control: *p<0.05. Experiments were repeated 3 times. (E) Effect of PTZ on H2O2–exposed S1R siRNA transfected ONHA. 5 days after S1R siRNA transfection, ONHA were incubated with 100μM H2O2 at 37°C for 15 minutes, 30 minutes, 1 hour, 3 hours and 24 hours in the presence or absence of PTZ (10μM, 1 hour pretreatment followed by cotreatment). Western blot analysis is shown. (F) Quantitative analysis of pERK levels represented as pERK normalized to total ERK. Results are presented as fold change of the pERK/total ERK ratio derived from H2O2 exposed, S1R siRNA transfected cells versus control non-H2O2 exposed, S1R siRNA transfected cells. Data were analyzed using two-way ANOVA followed by Tukey-Kramer post hoc test for multiple comparisons.
Discussion
Agonists and antagonists of S1R might offer novel treatment options for a broad range of disorders including neurodegenerative diseases and cancer[7, 8, 12, 39]. Stimulation of S1R protects purified neurons, including RGCs, from cell death under conditions of oxidative, metabolic, and excitotoxic stress[7, 14, 21]. However, S1Rs are expressed in glia as well as neurons, within the brain, spinal cord, and optic nerve[16, 40, 41]. Previous work using brain-derived astrocytes indicates that inhibition of S1R leads to decreased astrocyte reactivity[42]. However, the effects of activation or inhibition of S1R on astrocytes derived from ocular tissues are not well characterized. Multiple studies indicate that glia are critical to the health and survival of neurons[43, 44]. Consistent with these findings, astrocytes can modulate the function of associated RGCs, and changes within ONH astrocytes are among the earliest signs of glaucomatous optic neuropathy[45–47]. Therefore, the results of these analyses are critical to evaluation of S1R as a therapeutic target.
Our results show that stimulation of S1R using the specific agonist, (+)-pentazocine, protects ONHAs from death under conditions of oxidative stress. In addition, we found that (+)-pentazocine treatment suppresses ROS generation and modulates ERK activation within ONHAs. Several studies indicate that (+)-pentazocine acts specifically through the S1R[48, 49]. However, some reports describe non-specific (+)-pentazocine-mediated effects[50]. Our results show that (+)-pentazocine-associated pro-survival responses are blocked by knockdown of S1R. Therefore, we conclude that S1R mediates these effects.
Initial transfections of S1R siRNA were performed using the HeLa cell line. Interestingly, knockdown of S1R within these cultures caused significant cell death at baseline, independent of oxidative stress exposure. Comparison of S1R protein levels between transfected HeLa cells and ONHAs showed similar efficiency of S1R knockdown. However, in contrast to HeLa cells, suppression of S1R expression within ONHAs did not cause significant baseline cell death. Several studies have evaluated the pro-apoptotic and growth-inhibitory effects of S1R inhibition within neoplastic cell types[12, 51]. Reported mechanisms that underlie these effects include direct regulation of apoptosis signaling pathways, ER and oxidative cellular stress responses[22, 23, 52]. Our results support the hypothesis that the S1R may be a cancer-specific therapeutic target. However, knock down of S1R in ONHAs was associated with increased levels of ROS and a higher baseline level of ERK phosphorylation. Thus, although survival of ONHAs was not affected upon knock down of S1R, their in vivo functions might still be compromised. Thus, the effects of S1R inhibition on these and other cell types must be considered if S1R inhibition is used systemically in cancer treatment.
Recent reports indicate that S1R agonists, including (+)-pentazocine, enhance ERK1/2 phosphorylation within neurons. For example, studies using (+)-pentazocine-treated cultures of primary RGCs have found an S1R-dependent increase in ERK1/2 phosphorylation following 6 hours of oxygen-glucose deprivation[21]. In addition, studies have shown that the S1R agonist, 4-PPBP, enhances ERK1/2 phosphorylation in primary cultures derived from mixed cortical and hippocampal neurons[25].
Our data, showing S1R-mediated suppression of ERK1/2 activation in optic nerve head astrocytes would seemingly conflict with studies reported from neuronal cultures, as discussed above. Our results indicate that stimulation of S1R using (+)-pentazocine suppresses ERK1/2 activation within optic nerve head-derived astrocytes. We have also shown that (+)-pentazocine inhibits ERK1/2 phosphorylation within primary retinal microglia[28]. Taken together, results suggest that agonist-mediated S1R stimulation leads to differential modulation of ERK activity within primary neurons versus glia.
Overall, results from multiple studies indicate that agonists for S1R function to increase ERK1/2 phosphorylation in some circumstances, but act to decrease ERK1/2 phosphorylation in others. This may seem counter-intuitive, but many drugs, including S1R agonists, can show opposite effects under some circumstances [53, 54]. In addition to cell-type differences, the physio-pathological conditions under which the drug is used, as well as drug concentration at the site of action, can determine the signaling cascades that are engaged.
The activities of basic protein kinase signaling pathways, including ERK1/2, do vary depending upon the cellular model analyzed. For example, within the oligodendroglial cell line, CG4, pharmacological blockade of the ERK1/2 pathway prevents H2O2-induced cell death[55]. In addition, inhibition of ERK1/2 within glia cells blocks production of neuro-toxic nitric oxide and protects midbrain-derived neurons from degeneration[56]. However, within neurons, glutamate-induced, transient activation of ERK1/2 elicits a prosurvival response, while oxidative stress-induced sustained ERK1/2 activation promotes neuronal death[57]. Overall it is likely that the magnitude and duration of ERK1/2 activation within a given cell type, determines the life or death-promoting capacity of this protein kinase[58]. Our results show that sustained inhibition of ERK1/2 phosphorylation increases the viability of oxidative stress-exposed ONHAs. Therefore, inhibition of ERK phosphorylation is likely one mechanism through which S1R mediates pro-survival effects within ONHAs. However, S1R has been shown to interact with a multitude of intracellular targets. Further studies are needed to address which of the many S1R-mediated effects contributes to promotion of ONHA survival upon treatment with the S1R agonist, (+)-pentazocine.
Despite the apparently opposing cell-type specific effects of S1R on ERK1/2 activity, the S1R agonists are pro-survival in multiple primary cell cultures[21, 25, 27, 59] Moreover, S1R stimulation within optic nerve head astrocytes and retinal microglia results in decreased intracellular ROS generation and decreased release of neurotoxic inflammatory cytokines. Therefore, although S1R agonists may variably modulate ERK1/2 activity within glial cells and RGCs, the overall effect of S1R stimulation within these cell types may be to promote neuroprotection. For example, a multitude of studies indicate that neuroinflammation, mediated by microglia and astrocytes, is a key contributing factor to optic nerve degeneration in glaucoma[60, 61]. S1R agonists might act within both astrocytes and microglia to mitigate neurotoxic inflammatory responses while simultaneously augmenting pro-survival pathways within visual system neurons themselves. If this is the case, activation of S1R would offer a powerful, pluripotent, therapeutic option for neuroprotective treatment in glaucoma. Furthermore, antagonists of S1R generally suppress cellular growth and survival[12, 22, 62]. Our results are consistent with these paradigms.