
Sigma-1 receptor is involved in modification of ER-mitochondria proximity and Ca2+ homeostasis in cardiomyocytes
By Hideaki Tagashira, Md. Shenuarin Bhuiyan, Yasuharu Shinoda, Ichiro Kawahata, Tomohiro Numata, and Kohji Fukunaga
Introduction to the article published in Journal of Pharmacological Sciences, December 10, 2022, ISSN 1347-8613, DOI: https://doi.org/10.1016/j.jphs.2022.12.005.
Editor’s Highlights
- ER-mitochondrial Ca2+ signaling and intracellular Ca2+ homeostasis was disrupted in Sigmar knockdown NRVMs accompanied with disruption of ER-mitochondria proximity.
- Sigmar1 is involved in protecting the heart from ET-1-induced ventricular myocyte hypertrophy by maintaining of intracellular Ca2+ signaling, which may be mediated by the modification of ER-mitochondria proximity.
- Sigmar1 activation and/or upregulation can be a promising therapeutic target for the prevention of Hearth Failure.
Abstract
The Sigma-1 receptor (Sigmar1) is downregulated in heart failure model mice with mitochondrial dysfunction. However, the mechanism in detail has not been investigated. In this study, we investigated the role of Sigmar1 in ER-mitochondria proximity using Sigmar1-knockdown or -overexpressed neonatal rat ventricular myocytes (NRVMs). The endothelin-1 (ET-1)-induced cardiomyocyte hypertrophy was aggravated with the dysregulation of mitochondrial function and ER-mitochondrial junctional formation in Sigmar1-knockdown NRVMs, whereas improved in Sigmar1 overexpressed NRVMs. Our data suggests that the reduction of the cardiac Sigmar1 results in decrease mitochondrial Ca2+ influx and promotes mitochondrial fission, followed by reduced ER-mitochondria proximity, exacerbating ET-1-induced cardiomyocyte injury.
1. Main text
Heart failure (HF) is a major cause of death in the world. Although various targeted drugs such as G-protein-coupled receptors and ion channels are used to treat HF, their therapeutic effect is not satisfactory, making surgical treatment necessary.
The sigma receptor (Sigmar) was first discovered as a subclass of opioid receptors and consequently named a subtype of the opioid receptor family. Sigmar1 is highly homologous among rats, mice and humans, and it has been proposed to interact with the IP3 receptor (IP3R) type 3 in CHO cells and IP3R type 2 in cardiomyocytes as a molecular chaperone.1,2 Subsequent studies have revealed that Sigmar1 regulate different physiologic cell functions in different organs.3 However, much less is known about the molecular mechanisms of pathogenesis in Sigmar1.
Our research, along with that of other groups, previously reported that Sigmar1 is highly expressed in rodent cardiomyocytes.4,5 Our subsequent pharmacological studies have demonstrated that downregulation of Sigmar1 is involved in cardiac hypertrophy and cardiac dysfunction via dysregulation of IP3R2 and RyR2.4,6 We also observed that heart failure pathogenesis spontaneously developed with aging in Sigmar1-/- mice, and that Sigmar1-/- mice hearts showed mitochondrial swelling and decreased mitochondrial function.7 However, the mechanism of impaired mitochondrial quality control in Sigmar1-knockdown cardiomyocytes remains poorly understood. Furthermore, there have been no reports about cardiac-specific Sigmar1 transgenic mice or Sigmar1-overexpressed cardiomyocytes.
In this study, we investigated the role of Sigmar1 for ER-mitochondria proximity and mitochondrial Ca2+ signaling using Sigmar1-knockdown or overexpressed neonatal rat ventricular myocytes (NRVMs).4 We first confirmed Sigmar1 expression levels in each of the NRVMs. In the Sigmar1 siRNA-treated (si-Sigmar1) NRVMs, endogenous Sigmar1 expression decreased in approximately 50% compared to non-transfected (Cont) NRVMs, consistent with our previous results4 (Fig. 1A). In the Sigmar1/pcDNA3.1 transfected (Sigmar1) NRVMs,8 the Sigmar1 level (including endogenous Sigmar1) increased about 2.5-fold when compared to the control cells (Fig. 1A). Immunostaining confirmed that Sigmar1 knockdown or overexpressing NRVMs were found in approximately 60–70% of cells (Fig. 1B).
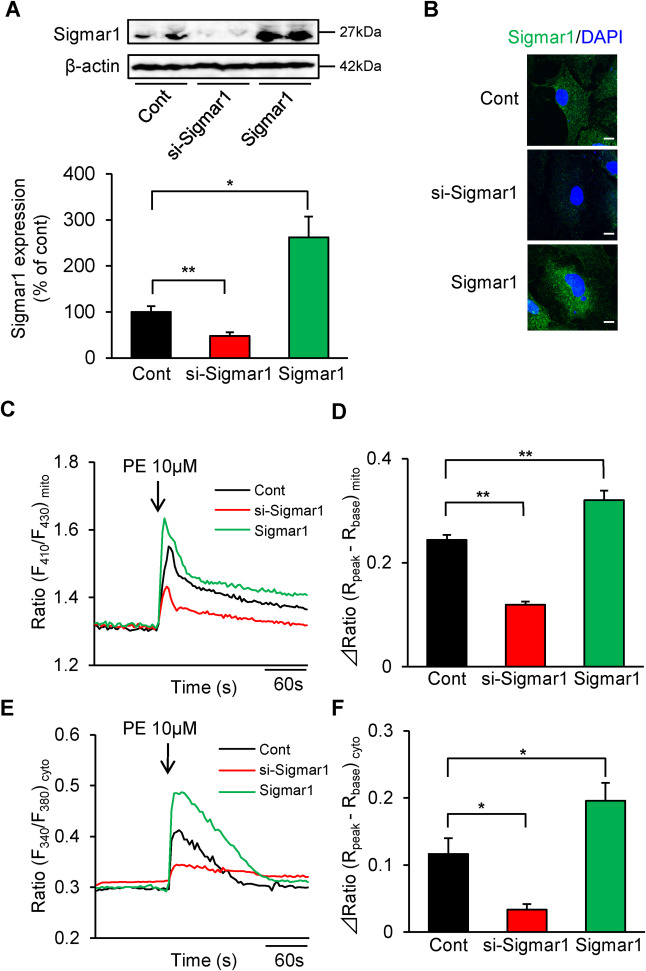
ER-mitochondrial Ca2+ signaling and intracellular Ca2+ homeostasis were disrupted in Sigma-1 knockdown cardiomyocytes.
A: Western blot analysis of Sigmar1 expression in Control (Cont), Sigmar1 knockdown (si-Sigmar1) or overexpressed (Sigmar1) NRVMs (n = 4–5). B: Confocal analysis of Sigmar1 (green) (sc-137,075, Santa Cruz, CA) and DAPI (blue) (P36962, Invitrogen, CA) in si-Sigmar1 or Sigmar1 NRVMs. Scale bar = 10 μm. Data are expressed as percentages of control cells (mean ± S.E.M.) C: Time courses of PE-induced Ca2+ influx into mitochondria detected by ratiometric-pericam-mt/pcDNA3.1 in si-Sigmar1 or Sigmar1 NRVMs.6 D: Peak increases in [Ca2+]mito induced by 10 μM PE. E: Time courses of PE-induced Ca2+ release into the cytosol using fura 2-AM in si-Sigmar1 or Sigmar1 NRVMs.6 F: Peak increases in [Ca2+]cyto induced by 10 μM PE. ΔRatio = Rpeak(peak ratio 10 s after PE treatment)-Rbase (basal ratio averaging before PE treatment). Each column consists of more than 10 cells and represents the mean ± S.E.M. ∗, P < 0.05 and ∗∗, P < 0.01 versus Cont cells. Results were evaluated for differences with Student’s t test for comparisons between two mean values. Experimental Methods in detail were described in supplemental Methods.
Because we previously reported that fluvoxamine2 and SA4503,6 a high-affinity Sigmar1 agonist, regulate IP3R-mediated Ca2+ mobilization in cardiomyocytes, we next evaluated phenylephrine (PE)-induced IP3R-mediated Ca2+ response in mitochondria and cytosol. Sigmar1 knockdown severely impaired PE-induced mitochondrial Ca2+ influx compared to the control cells (Fig. 1C and D). On the other hand, PE-induced mitochondrial Ca2+influx significantly increased in Sigmar1-overexpressed NRVMs (Fig. 1C and D). We also examined the effects of Sigmar1 expression level on the PE-induced cytosolic Ca2+response. Unexpectedly, the PE-induced Ca2+ response in the cytosol was also significantly lower in si-Sigmar1 NRVMs (Fig. 1E and F). On the other hand, it was significantly higher in Sigmar1-overexpressed NRVMs compared to the control NRVMs (Fig. 1E and F). These results indicate that Sigmar1 contributed to intracellular Ca2+homeostasis including ER and mitochondria.
As Sigmar1 overexpression promotes IP3R-mediated mitochondrial Ca2+ mobilization and Sigmar1 interacted with ER-mitochondria tethering protein complexes,1,9 we stained phospholamban (PLB), as the SR/ER marker, and MitoTracker Red CMXRos, as mitochondria marker which intake to mitochondria in a membrane potential-dependent manner, to detect mitochondrial functions and ER-mitochondria proximity in si-Sigmar1 NRVMs. In control NRVMs, PLB immunoreactivity largely merged with that of a MitoTracker Red (Fig. 2A). In untreated si-Sigmar1 NRVMs, mitochondrial fragmentation and reduction of mitochondrial membrane potential was detected, and PLB-mitochondrial merge were impaired but partially preserved (Fig. 2A). In contrast, in untreated Sigmar1-overexpressed NRVMs, more mitochondrial elongation, along with PLB-mitochondrial merge was observed (Fig. 2A). It is well established that the endothelin-1 (ET-1) is secreted from vascular endothelial cells and is involved in cardiac hypertrophy and heart failure.10 Control NRVMs exposed to ET-1 for 48 h had mitochondrial damage, and PLB-mitochondrial merge were decreased (Fig. 2A). Interestingly, in si-Sigmar1 NRVMs treated with ET-1, mitochondrial fission was markedly promoted and there was almost no observation of PLB-mitochondrial merge (Fig. 2A). Unexpectedly, the loss of mitochondrial membrane potential was not different compared to ET-1-stimulated alone. In Sigmar1-overexpressed NRVMs, ET-1-induced mitochondrial fission and reduction of PLB-mitochondrial merge was suppressed (Fig. 2A). Since it has been proposed that autophagy in cardiomyocytes promotes heart failure conditions,11 we assessed whether Sigmar1 activity governs autophagy in endothelin-1 (ET-1)-induced hypertrophic cardiomyocytes using LC-3, as the autophagy marker. In untreated si-Sigmar1 NRVMs, autophagy was only detected slightly. Similarly, control NRVMs exposed to ET-1 for 48 h had mitochondrial damage, but only slight autophagy (Fig. 2B). Interestingly, in si-Sigmar1 NRVMs treated with ET-1, punctated LC-3 was abundant (Fig. 2B). In addition, a mitophagy-like phenomenon was observed, although it was partly merged with fragmented mitochondria (Fig. 2B). On the other hand, ET-1-induced increasing autophagy was suppressed in Sigmar1-overexpressed NRVMs (Fig. 2B). Previous reports suggested that Sigmar1 knockdown reduces autophagosome clearance.12 Therefore, under Sigmar1 knockdown conditions, ET-1 stimulation exacerbates autophagosome formation, which contribute to increase the expression levels of LC-3 and mitophagy. Next, we evaluated the effects of Sigmar1 knockdown on intracellular Adenosine triphosphate (ATP) content, as the mitochondrial functions. Sigmar1 knockdown alone slightly but significantly decreased ATP content in vehicle-treated cells (Fig. 2C). As expected, ET-1 exposure for 48 h significantly decreased ATP content (Fig. 2C), and Sigmar1 knockdown exacerbated ATP reduction by ET-1 (Fig. 2C). On the other hand, Sigmar1 overexpression alone significantly increased ATP content and prevented NRVMs from ET-1-induced ATP reduction (Fig. 2C). These results suggested that Sigmar1 knockdown results in decreased mitochondrial Ca2+ influx and promotes mitochondrial fission, followed by decreased ER-mitochondrial junctional range and increased mitophagy, which contributed to ATP reduction.
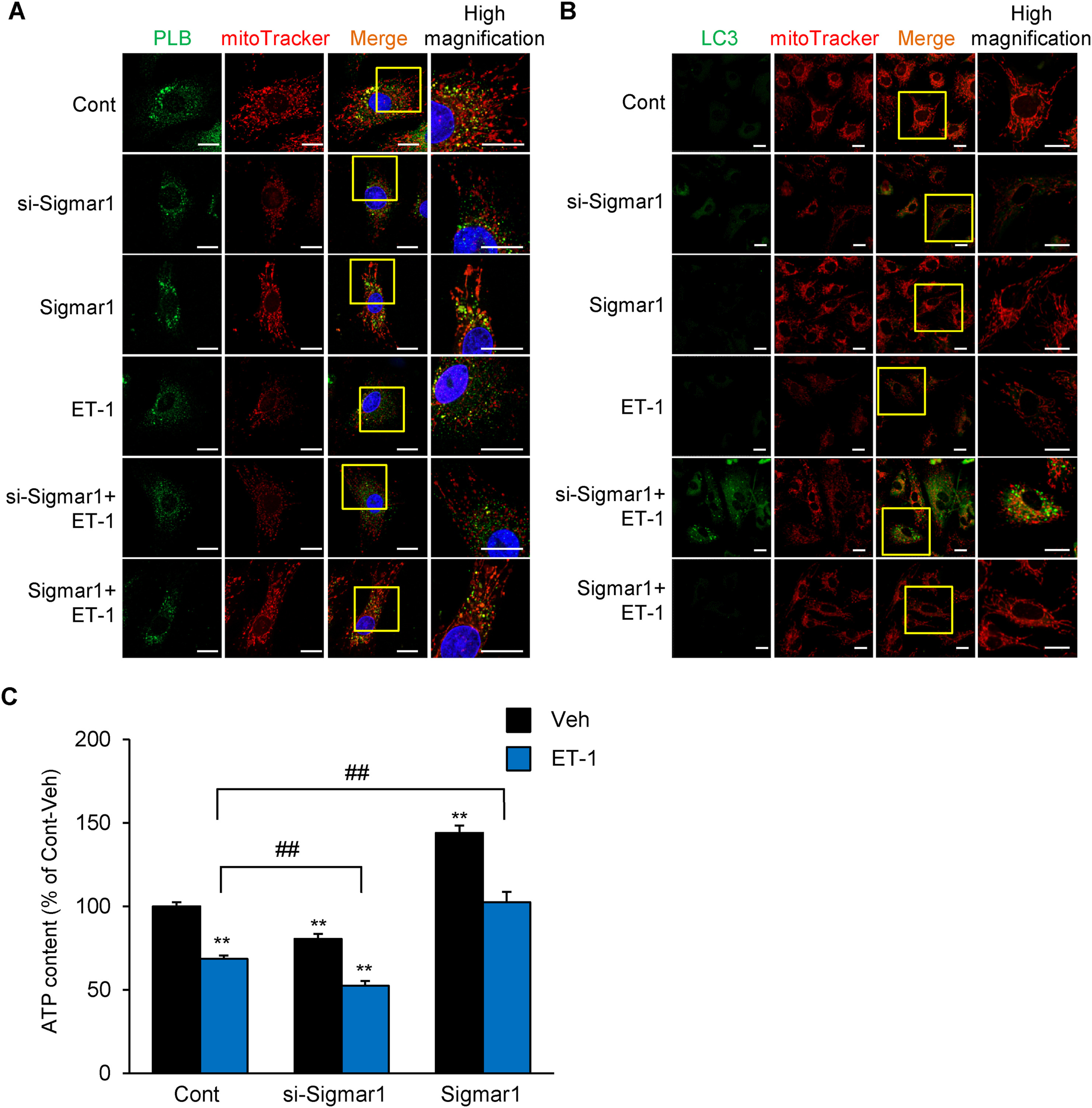
Sigmar1 knockdown aggravate ET-1-induced mitochondrial fission, mitophagy and ATP reduction in NRVMs.
A: Cultured NRVMs were processed using immunofluorescence to mark the phospholamban (PLB) (green, ER marker) (sc-393,990, Santa Cruz, CA), MitoTracker Red CMXRos (red, mitochondria marker) (M7512, Invitrogen, CA) and DAPI (blue) (P36962, Invitrogen, CA). Scale bar = 20 μm. B: Cultured NRVMs were processed using immunofluorescence to mark the LC3 (green) (PM036, MBL, Japan) and MitoTracker Red CMXRos (red, mitochondria marker) (M7512, Invitrogen, CA). High magnification indicate an enlarged view of the yellow column. Scale bar = 20 μm. C: Measurement of cellular ATP content 48 h after treatment with or without 100 nM ET-1 stimulation in Sigmar1 knockdown (si-Sigmar1) or overexpressed (Sigmar1) NRVMs (n = 10) as previously described.6 Each column represents the mean ± S.E.M. ∗∗, P < 0.01 versus the Vehicle (Veh) cells; ##, P < 0.01 versus ET-1 treated Control (Cont) cells. Results were evaluated for differences with Student’s t test for comparisons between two mean values. Experimental Methods in detail were described in supplemental Methods.
Previous studies have suggested that cardiomyocyte hypertrophy has been associated with diminished mitochondrial metabolism.13 Our group attempted to analyze the cardiac function of Sigmar1-/- mice in vivo.7 Importantly, the analyses of these mice revealed that the myocardial fibrosis and heart failure pathogenesis spontaneously develop with aging. Sigmar1–/- mice hearts showed mitochondrial swelling and decreased mitochondrial function compared to wild-type mice. We argue that the molecular mechanism of age-related cardiac mitochondrial dysfunction due to Sigmar1 deficiency is because of the disruption of Sigmar1-mediated regulation of dynamin-related protein 1 phosphorylation, which has an important role for mitochondrial fission and induction of heart failure,11 and excessive recruitment to mitochondria.7 However, there is no direct evidence of association between mitochondrial morphological change and Ca2+ signaling in Sigmar1-knockdown cardiomyocytes. Here, we demonstrated that decreasing ER-mitochondrial Ca2+ signaling and mitochondrial fragmentation was detected in Sigmar1 knockdown NRVMs, while increased mitochondrial Ca2+ influx and mitochondrial elongation was displayed in Sigmar1-overexpressed NRVMs (Figs. 1C and 2A). Consistent with our results, previous studies have shown that mitochondrial fission is associated with a decrease in the mitochondrial Ca2+ uptake capacity.14 Pennanen et al. suggested that controlling the balance between Ca2+ signaling and mitochondrial fission/fusion is sufficient to promote or prevent PE-induced cardiomyocyte hypertrophy in NRVMs.15 Our findings provide additional evidence that the suppression of mitochondrial fragmentation and maintenance of Ca2+ homeostasis play key roles in Sigmar1-mediated cardioprotection.
Our previous studies have shown that Sigmar1 is involved in AngII-induced hypertrophy.2,4,6 However, the involvement of ET-1 was unclear. In this study, we examined the anti-hypertrophic effect of the Sigmar1 protein on ET-1-induced hypertrophy. Cultured NRVMs exposed to ET-1 for 48 h indicated cardiomyocyte hypertrophy (Fig. 3A and B). Notably, Sigmar1 knockdown alone slightly but significantly increased cell surface area compared to control cells, and exacerbated the hypertrophic response induced by ET-1 (Fig. 3A and B). On the other hand, Sigmar1 overexpression significantly prevented ET-1-induced cardiomyocyte hypertrophy (Fig. 3A and B). In addition, MTT assay was performed in each cell group to detect cell death of NRVMs. No significant change in cell viability was observed between si-Sigmar1 or Sigmar1-overexpressed NRVMs. Interestingly, ET-1-induced cell death was exacerbated in si-Sigmar1 NRVMs (Fig. 3C), whereas ET-1-induced reduction of cell viability was suppressed in Sigmar1-overexpressed NRVMs (Fig. 3C). It is well known that ER stress inducer contribute to cardiac cell death, rather than cardiomyocyte hypertrophy. Therefore, these results suggested that cardiomyocyte cell death are exacerbated in si-Sigmar1 only when ET-1 induces cardiomyocyte death signals including ER stress. Transfection reagent-treated cells were used as control NRVMs, but there were no differences in ATP production, cell size and viability between non-treated (Intact), scramble RNA (scrRNA), pcDNA3.1-transfected (Mock), and control NRVMs (Fig.S1). These data demonstrate that transfection manipulation had no effects on these data, and that the reduction of cardiac Sigmar1 is involved in ET-1-induced ventricular myocyte hypertrophy, not only AngII,2 by maintaining intracellular Ca2+ signaling and ATP production, which is probably mediated by the regulation of ER-mitochondria proximity.
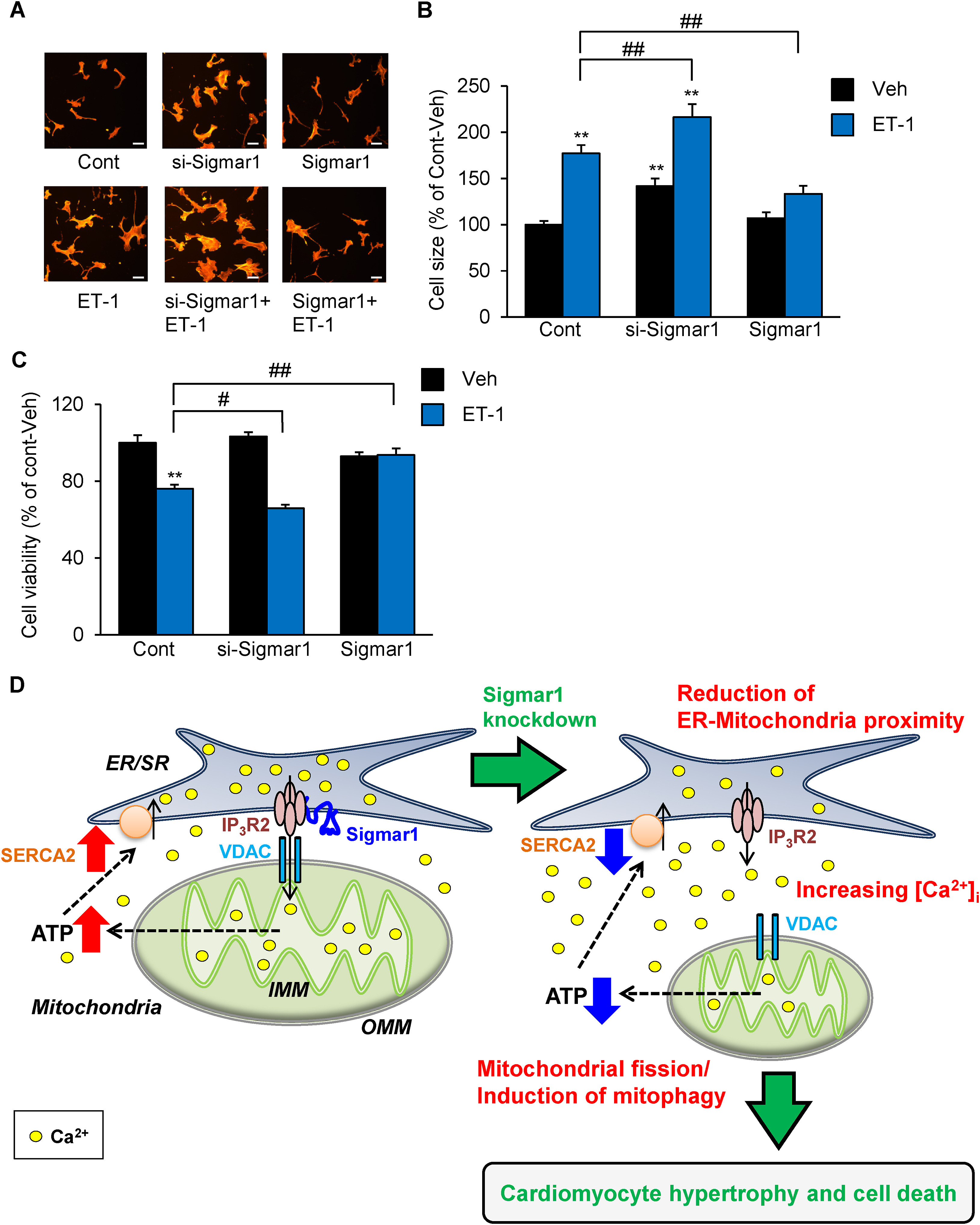
Sigmar1 overexpression prevented ET-1-induced cardiomyocyte hypertrophy.
A: Cells were fixed with 4% paraformaldehyde, stained with Rhodamine-conjugated Phalloidin, and processed for fluorescence microscopy as previously described.6 Scale bar = 50 μm. B: Cell size is expressed as a percentage of the relative surface area compared to Control (Cont)-Vehicle (Veh) cells. Each group consists of at least 50 cells. C: Cell death observed at 48 h after ET-1 treatment in Sigmar1 knockdown (si-Sigmar1) or overexpressed (Sigmar1) NRVMs (n = 5) detected by use of a calorimetric MTT assay kit (Cell count kit-8; Dojindo, Japan). Each column represents the mean ± S.E.M. ∗∗, P < 0.01 versus the Cont-Veh cells; #, P < 0.05 and ##, P < 0.01 versus ET-1 treated Cont cells. Results were evaluated for differences with Student’s t test for comparisons between two mean values. D: Schematic representation of the hypothesis tested and results obtained. Cardiac Sigmar1 is involved in protection from ET-1-induced ventricular myocyte hypertrophy by maintaining intracellular Ca2+ signaling, which is probably mediated by the modulation of ER-mitochondria contacts. Sigmar1: Sigma-1 receptor. VDAC: voltage-dependent anion channel. SERCA2: sarco (endo) plasmic reticulum Ca2+-ATPase isoform 2. IP3R2: IP3 receptor type 2. Experimental Methods in detail were described in supplemental Methods.
ET-1-induced cardiomyocyte hypertrophy and injury is well known to be associated with increased reactive oxygen species (ROS) production.16 We have previously shown that Sigmar1 agonists exert cardioprotective effects through activation of the Akt-eNOS pathway,4 suggests a ROS scavenge. In addition, Sigmar1 agonist, SA4503 and fluvoxamine show protective effects on cardiomyocyte by increasing ATP production.2,6 Indeed, ET-1-stimulated cardiomyocytes also decreased ATP production, and Sigmar1-overexpressed NRVMs increased ATP production and suppressed cardiac hypertrophy (Fig. 2, Fig. 3). Taken together, Sigmar1-mediated cardiomyocyte protection appears to involve both increased ATP production through the regulation of mitochondrial function and ROS scavenging by eNOS.
In summary, we show for the first time that ER-mitochondrial Ca2+ signaling and intracellular Ca2+ homeostasis was disrupted in Sigmar1 knockdown NRVMs accompanied with disruption of ER-mitochondria proximity. On the other hand, in Sigmar1 overexpressed NRVMs, ET-1-induced cardiomyocyte hypertrophy, ER-mitochondria proximity, and mitochondrial/intracellular Ca2+ signaling were improved. Our findings support the thesis that cardiac Sigmar1 is involved in protecting the heart from ET-1-induced ventricular myocyte hypertrophy by maintaining of intracellular Ca2+signaling, which may be mediated by the modification of ER-mitochondria proximity. Our findings provide important insight that Sigmar1 activation and/or upregulation can be a promising therapeutic target for the prevention of HF.