
Sigma-1 receptor chaperones rescue nucleocytoplasmic transport deficit seen in cellular and Drosophila ALS/FTD models
By Pin-Tse Lee, Jean-Charles Liévens, Shao-Ming Wang, Jian-Ying Chuang, Bilal Khalil, Hsiang-en Wu, Wen-Chang Chang, Tangui Maurice, and Tsung-Ping Su
Excerpt from the article published in Nature Communications 11, 5580 (2020). https://doi.org/10.1038/s41467-020-19396-3
Editor’s Highlights
- Amyotrophic lateral sclerosis (ALS) or frontotemporal dementia (FTD), either sporadic or familial, share a (G4C2)-RNA hexanucleotide repeat expansion (HRE) upstream of the start codon of the C9orf72 gene.
- The HRE causes the nucleolar stress resulting in the diffusion of an essential component of nucleoli, nucleolin, to disperse throughout the nucleus.
- The (G4C2)-RNA repeat expansion binds to the Ran-activating protein (RanGAP) at the nuclear pore, resulting in nucleocytoplasmic transport deficit and accumulation of Ran in the cytosol.
- The nuclear pore that Ran passes through between cytosol and nucleus is called nuclear pore complex (NPC) that is a megadalton structure at the nuclear membrane.
- The sigma-1 receptor (Sig-1R) colocalizes with RanGAP and nuclear pore proteins (Nups) and stabilizes the latter.
- Sig-1R exists at the NPC where it counteracts the aberrant nucleocytoplasmic distribution of Ran, caused by the (G4C2)-RNA repeats, by chaperoning Nups and by sponging away the toxic (G4C2)-RNA repeats.
- The overexpression of Sig-1R-E102Q mutant per se causes a gain-of-toxicity related to its action on the IP3R at the MAM important for ATP production.
- This explanation on the action of the Sig-1R-E102Q at IP3R at the MAM, together with current result showing the (G4C2)30 co-expression exacerbating the toxic effect of the mutant in the eye, render support to the above notion that (G4C2)-RNA repeats may lead to dysfunctional mitochondria and reduced cellular bioenergetics.
ABSTRACT
In a subgroup of patients with amyotrophic lateral sclerosis (ALS)/Frontotemporal dementia (FTD), the (G4C2)-RNA repeat expansion from C9orf72 chromosome binds to the Ran-activating protein (RanGAP) at the nuclear pore, resulting in nucleocytoplasmic transport deficit and accumulation of Ran in the cytosol. Here, we found that the sigma-1 receptor (Sig-1R), a molecular chaperone, reverses the pathological effects of (G4C2)-RNA repeats in cell lines and in Drosophila. The Sig-1R colocalizes with RanGAP and nuclear pore proteins (Nups) and stabilizes the latter. Interestingly, Sig-1Rs directly bind (G4C2)-RNA repeats. Overexpression of Sig-1Rs rescues, whereas the Sig-1R knockout exacerbates, the (G4C2)-RNA repeats-induced aberrant cytoplasmic accumulation of Ran. In Drosophila, Sig-1R (but not the Sig-1R-E102Q mutant) overexpression reverses eye necrosis, climbing deficit, and firing discharge caused by (G4C2)-RNA repeats. These results on a molecular chaperone at the nuclear pore suggest that Sig-1Rs may benefit patients with C9orf72 ALS/FTD by chaperoning the nuclear pore assembly and sponging away deleterious (G4C2)-RNA repeats.
Introduction
Amyotrophic lateral sclerosis (ALS) or frontotemporal dementia (FTD), either sporadic or familial, is a devastating neurological disease that currently has no cure. The exact molecular mechanisms which lead to this disease remain to be fully clarified. Two studies in 2011 discovered that the (G4C2)-RNA hexanucleotide repeat expansion (HRE) upstream of the start codon of the C9orf72 gene plays a critical role in the familial ALS1,2 and FTD3. The (G4C2)-RNA repeats in normal subjects range between 3 and 20. In ALS/FTD patients those repeats can be up to hundred or thousand. It is known that HRE can form G-quadruplex structures through the intermolecular hydrogen bonding between guanines4. Exactly how the HRE causes ALS/FTD is a very active area of research5,6,7,8,9,10.
A study demonstrated that the HRE causes the nucleolar stress resulting in the diffusion of an essential component of nucleoli, nucleolin, to disperse throughout the nucleus11. This result suggests an interaction between nucleolin and HRE in C9orf72 ALS patients and suggests the nucleolar stress as an underlying mechanism of this disease. Another study12 indicated that the HRE binds to RanGAP and in doing so impedes the activation of RanGTP (i.e., Ran-GTPase (referred to as Ran in this report) in the form of GTP). Ran is a small Ras‐related GTPase that mediates the nucleocytoplasmic exchange of macromolecules across the nuclear envelope. Normally, RanGTP needs to be activated by RanGTP-activating protein (RanGAP) at the cytosolic side of the nuclear pore before it can be hydrolyzed to RanGDP to firstly provide energy for an effective transport of cargos from cytosol into nucleus and secondly to allow itself to be transported into nucleus. Once inside the nucleus, RanGDP is converted by guanine exchange factor into RanGTP which is then transported back to cytosol so that it can be activated by RanGAP to initiate a new round of cycle to facilitate cargo entries into the nucleus12,13. In C9orf72 patients, as a result of this action of HRE on RanGAP, Ran is heavily accumulated in the cytoplasm, reflecting a pathological nucleus/cytoplasmic gradient of Ran as well as a deficient nucleocytoplasmic transport in ALS12,13. The nuclear pore that Ran passes through between cytosol and nucleus is called nuclear pore complex (NPC) that is a megadalton structure14,15 at the nuclear membrane. The proteins that make up the NPC are called nucleoporins (Nups) that is composed of at least 34 distinct constituent proteins14,15. Some of Nups are facing cytosol, at the midportion of pore, or facing the nucleus internal14,15. The stability of Nups may relate to the integrity of the nucleocytoplasmic transport. It has to be mentioned that the nucleocytoplasmic transport deficit as seen in the maldistribution of Ran is also an important factor in frontotemporal dementia (FTD)3. Thus understanding the fundamental mechanisms controlling the transport is critical not only to ALS but also to FTD.
The Sig-1R16,17,18,19,20,21,22,23,24,25 is a ligand-regulated molecular chaperone that resides mainly at the mitochondria-endoplasmic reticulum (ER) interface, referred to as the mitochondria-associated ER membrane (MAM), where it chaperones IP3R3 to ensure proper Ca2+ signaling from the endoplasmic reticulum into mitochondria26,27. The Sig-1R exists at other parts of cell as well and has been proposed to be an important protein for cellular survival28,29,30,31,32,33,34 as a dynamic pluripotent modulator in living systems35.
Different type of cells has been shown to exhibit different subcellular localization of Sig-1Rs. For example, electron-microscopic studies show that while the Sig-1R exists on the plasma membrane in the dorsal root ganglia36, it exists however only inside of retinal neurons37. Interestingly, the Sig-1R exists at the nucleoplasmic reticulum38 as well as at the nuclear envelope of neurons39 where the Sig-1R binds emerin to recruit chromatin-remodeling molecules and regulates gene transcription39.
The Sig-1R has been reported to relate to the familial ALS40,41. In animal model, a study suggested that a loss of function of Sig-1Rs at the MAM might lead to dysfunctional ER-mitochondrion crosstalk and thus the ALS42. Expression of Sig-1R with E102Q mutation recapitulates ALS pathology in Drosophila43. A lack of Sig-1R has been shown to exacerbate ALS progression in a mouse model of ALS44,45. A drug targeting Sig-1R has been shown to be effective in an animal model of ALS30. Other potential mechanisms, if any, underlying the role of Sig-1Rs in ALS remain to be fully established.
Here we found that in cellular models the Sig-1R exists at the NPC where it counteracts the aberrant nucleocytoplasmic distribution of Ran, caused by the (G4C2)-RNA repeats, by chaperoning Nups and by sponging away the toxic (G4C2)-RNA repeats. Further, we extend the functional readouts of the biochemical and cellular biological data obtained in cell lines to a Drosophila model and validate that the Sig-1R but not its E102Q mutant reverses morphological, behavioral, and electrophysiological deficits caused by the (G4C2)-RNA repeats. Those results are presented in this report.
Results
The Sig-1R exists at the nuclear pore
Our previous report demonstrated the existence of Sig-1Rs at the nuclear envelope in proximity to RanBP239. We examined here if the Sig-1R may exist at the nuclear pore in HeLa cells. Immunocytohistochemistry indicates the colocalization of HA-tagged Sig-1Rs with endogenous RanGAP and nuclear pore proteins Nup62 and Nup358 (i.e., RanBP2) (Fig. 1a). In immunoprecipitation (IP) assay, GFP-tagged-Sig-1Rs co-IP with RanGAP and Ran (Fig. 1b). With mAb414 as the blotting antibody, which is well-known to recognize FG-repeat-Nups, Nups co-IP with V5-tagged Sig-1R (Fig. 1c). Anti-Nup50 antibody also co-IPs the endogenous Sig-1R (Fig. 1d). Note: similar to that seen with HA-Sig-1R, the endogenous Sig-1R colocalizes with RanGAP1, Nup62, and NuP358 (Supplementary Fig. S1).
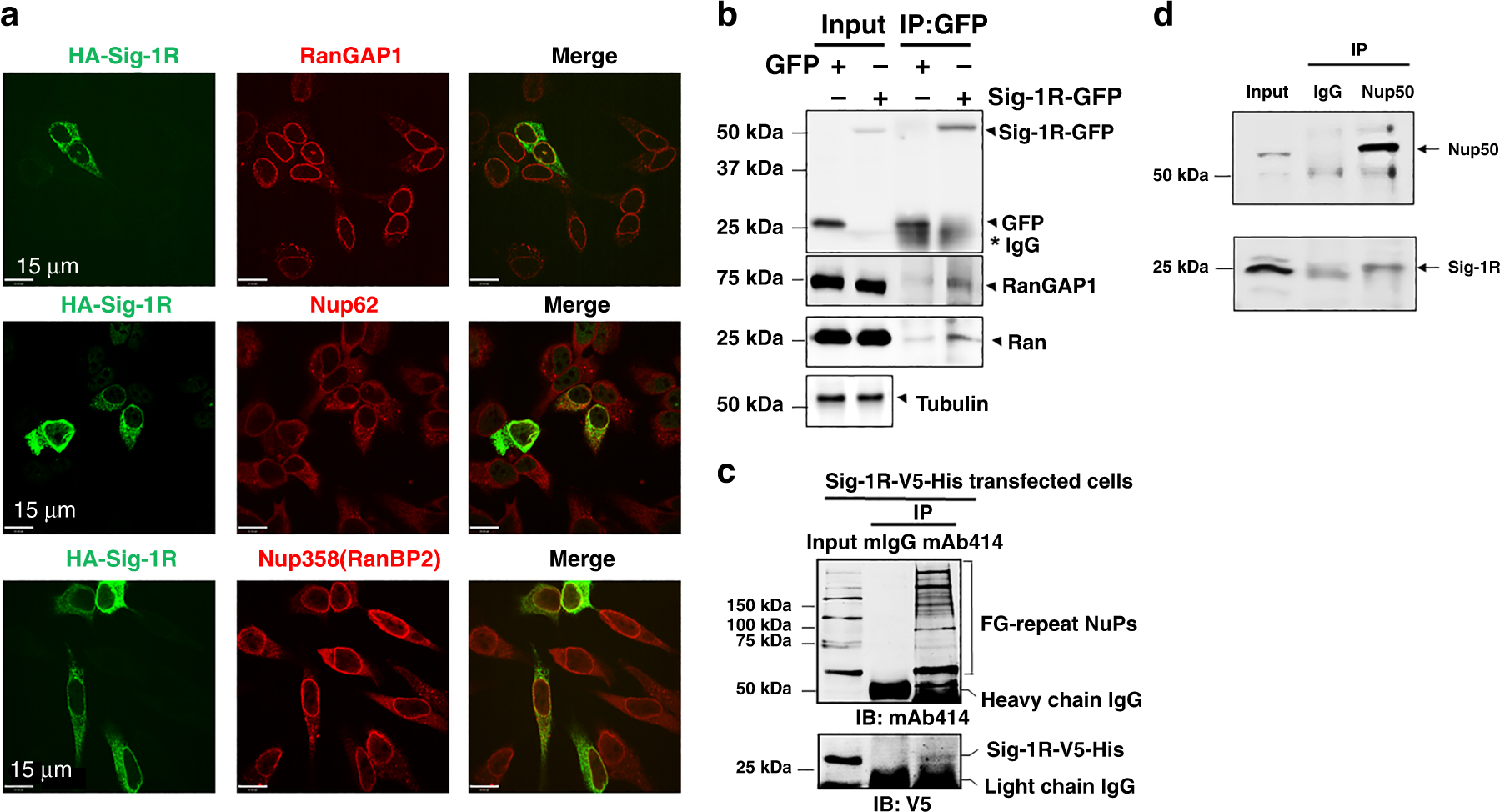
a Immunohistochemistry followed by confocal microscopic examination indicates perinuclear colocalizations of immunoreactive HA-tagged Sig-1R (green) with RanGAP (red), Nup62 (red) and Nup358 (red). HeLa cells transiently transfected with human HA-Sig-1R vectors were used. bCoimmunoprecipitation (Co-IP) of GFP-tagged Sig-1R with RanGAP and Ran. HeLa cells were transfected with GFP or GFP-Sig-1R vectors for 24 h before the co-IP experiment. Proteins interacting with GFP (control) or GFP-Sig-1R were detected by western blot. c NuPs’ interaction with Sig-1R in a co-IP experiment. The mAb414 pulled down FG repeats-containing Nups together with Sig-1R-V5-His which was transfected into HeLa cells. d Nup50 antibody co-IPed with endogenous Sig-1R which in this experiment was detected by the Santa Cruz B5 anti-Sig-1R antibody (sc137075). Note: all other endogenous Sig-1Rs in western blot in the cell line portion of this study was detected by custom-made anti-Sig-1R antiserum #5460 (see Methods section). The two Sig-1R antibodies have been used interchangeably in the lab to reserve #5460 which is custom-made polyclonal and is limited in quantity (see Methods section). Note: Santa Cruz B5 anti-Sig-1R is monoclonal, thus almost unlimited. Note: colocalization of endogenous Sig-1R with RanGAP1 and Nup62 in HeLa cells is shown in Supplementary Fig. S1. Sig-1R, Sigma-1 receptor, RanGAP RanGTP-activating protein, Nup nucleoporin. n = 4 (a), n = 4 (b), n = 3 (c), and n = 3 (d) independent experiments with similar results each from biologically independent cells or cellular preparations.
NSC-34 motor neuron-like cells (spinal × neuroblastoma hybrid cells) are often used as a bona fide cellular model to investigate the physiopathological mechanisms of ALS46,47. Indeed, HA-Sig-1Rs colocalize with RanGAP and Nup62 at the nuclear membrane in NSC-34 cells (Fig. 2). HA-tagged Sig-1Rs colocalize with endogenous RanGAP and Nup62 at the nuclear membrane in NSC-34 cells (Fig. 2a). Also, three-dimensional (3D) images acquired by sequentially capturing a series of 2D sections were performed to further confirm the nuclear envelope localization of HA-Sig-1Rs in NSC-34 cells (Fig. 2b, c). Results indicated that HA-Sig-1Rs colocalize with RanGAP in sections 3–20 (Fig. 2b) and Nup62 in sections 3–18 (Fig. 2c), specifically indicated as such at section 11 in the cross-sectional intensity scanning (the central panels of Fig. 2b, c).
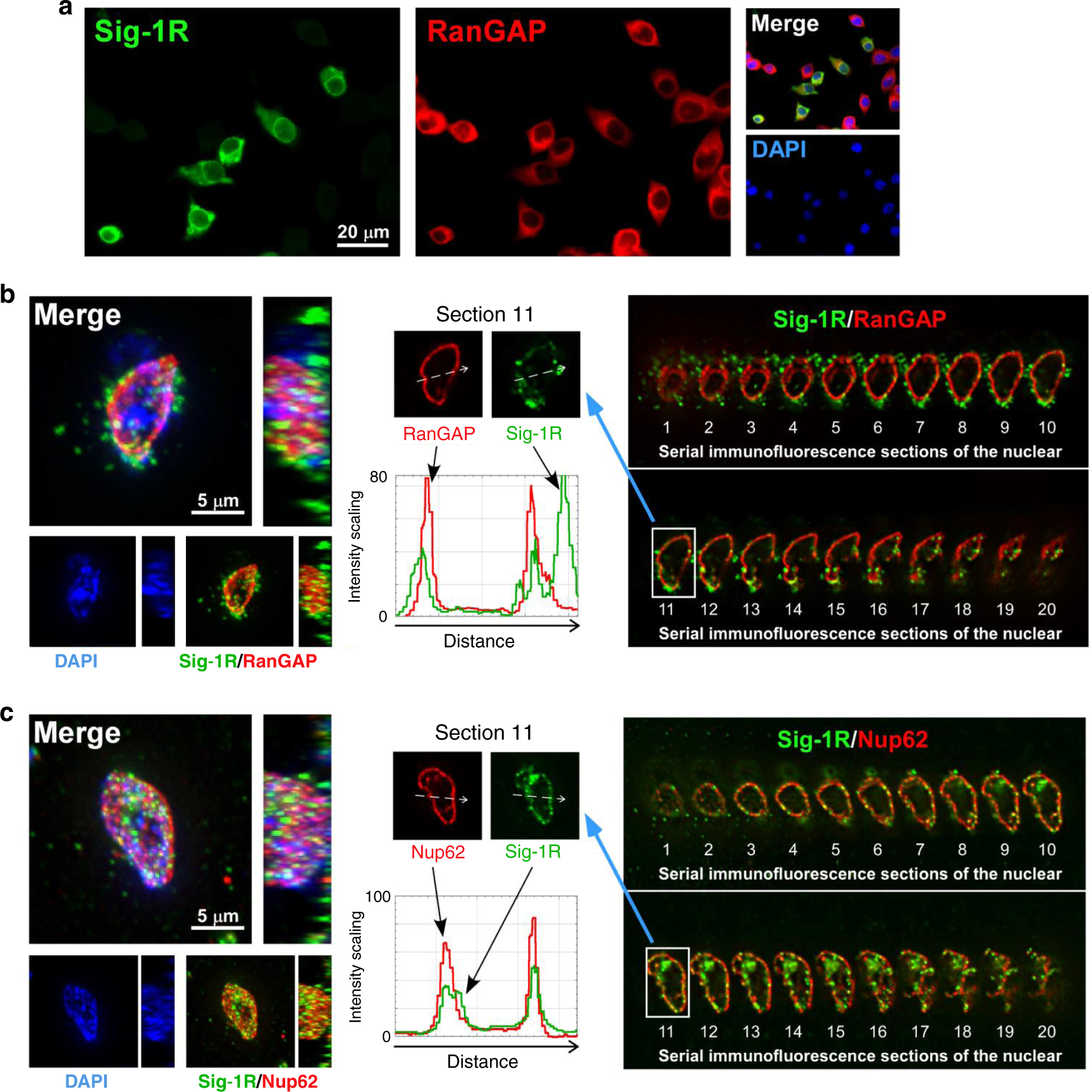
a Colocalization of HA-Sig-1R with RanGAP. Cells were transiently transfected with pcDNA-HA-Sig-1R, using Lipofectamine 2000, which provided ~50% of transfection efficiency in NSC-34 cells (https://www.thermofisher.com). Two days after transfection, cells were double-labeled with anti-HA and anti-RanGAP antibodies and examined by confocal microscopy. HA-Sig-1R, green; endogenous RanGAP, red; DNA, blue. b, c Multiple focal planes (Z sections) of the whole nuclear volume were acquired by the DeltaVision microscopy imaging systems. Results of the whole-nucleus image analysis of Sig-1R and RanGAP (b) or Nup62 (c) are shown. On left panels of b and c, square images are the top-down view (z-axis), and rectangle panels are a side view (x-axis) of the 3D reconstruction of images. On right panels of b and c, 20 sections were obtained from a cell. Number 1 is the Z-start at the top surface of the cellular nucleus; number 20 is Z-end at the bottom layer of the nucleus which was near the attachment of the cell to the coverslip. On central panels, white dotted arrows in images of the two sections 11 indicate the track of fluorescence intensity profiles (ImageJ: Plot Profile command) along the arrows. A shift of each focal plane in the Z-axis is 0.25 μm. Three-dimensional reconstructions were made from the Z-series images. Again: HA-Sig-1R, green; endogenous RanGAP/Nup62, red; DNA, blue. Sig-1R, Sigma-1 receptor, RanGAP RanGTP-activating protein, Nup62 nucleoporin 62. n = 3 independent experiments with similar results from biologically independent cells.
Those results suggest that the Sig-1R exists at the nuclear pore where it interacts with RanGAP and certain Nups.
Sig-1R stabilizes the nuclear pore proteins
Inasmuch as the Sig-1R is a molecular chaperone in chaperoning IP3 receptor26 as well as IRE-148, we examined if the Sig-1R may influence the stability of de novo Nups in HeLa cells. Indeed, cells treated with shSig-1R for 48 h to knockdown Sig-1Rs show a reduction of Nup358, Nup214, Nup62 (Fig. 3a) and Nup50 (Fig. 3b). Turnover of Nups was then examined in a time-lapsed manner in cycloheximide-treated cells in which cycloheximide was used to stop the de novo synthesis of proteins. Cycloheximide is known to interfere with the translation step in protein synthesis, thus blocking translational elongation. In the presence of cycloheximide, all proteins detected by western blots are existing proteins waiting to be degraded without the presence of newly de novo synthesized proteins. Thus, western blots typically show a time-dependent decrease of that protein of interest. Western blotting indeed indicates a time-dependent decrease of Nup358 and Nup214 between 4 and 8 h after cycloheximide (100 µg/ml) treatment (Fig. 3c). A representative western blot result from the Nup50 turnover is shown in supplemental information (Supplemental Information, Fig. S2). Note that 150 µg/ml of cycloheximide was needed to see the decrease of de novo synthesized Nup50 (Supplemental Information, Fig. S2), suggesting a relatively stable nature of Nup50 when compared to other Nups. Summarized results from multiple independent time-lapsed experiments indicate that the Sig-1R knockdown significantly decreases the stability of Nup358, Nup214, and Nup50 (Fig. 3d–f). PCR (Supplemental Information, Fig. S3a) and Real-time PCR (Supplemental Information, Fig. S3b) indicate that mRNA levels of those Nups are not affected by the knockdown of Sig-1Rs. Those results suggest that the Sig-1R chaperone stabilizes Nups at the post-translational level.
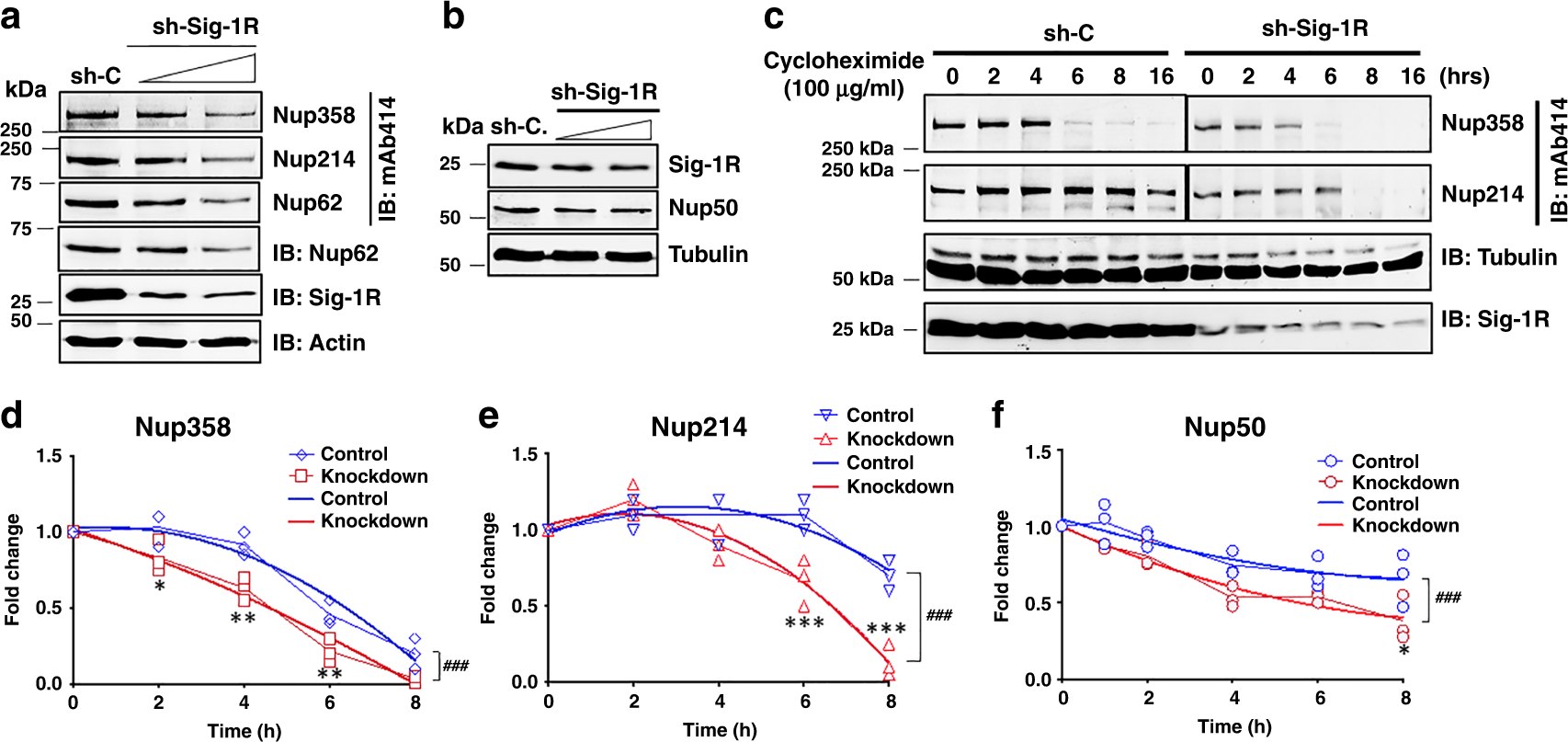
a Knockdown of sigma-1 receptor (Sig-1R) by shRNA (shSig-1R) dose-dependently caused a reduction of nucleoporins (Nups). HeLa cells were transiently transfected with either shRNA control (sh-C) or different doses of shSig1R vector. Forty-eight hours after transfection, western blot was performed to detect protein expression levels of Sig-1R, Actin, and Nups. In the western blot, Nup62 was probed by mAb414 or a specific Nup62 antibody. b Sig-1R knockdown dose-dependently reduced the level of Nup50. c Faster turnover of Nup358 or Nup214 in Sig-1R knockdown cells. Cycloheximide (100 µg/ml) was added to stop de novo synthesis of proteins. Time-lapsed levels of Nups were examined by western blot probed by mAb414. d–f Summarized results from three sets of independent turnover studies on Nup358, Nup214, and Nup50. Data were subjected to two-way ANOVA followed by Sidak’s multiple comparisons test (Graphpad Prism version 8.2). For d Nup358, p values are 0.022, 0.001, 0.0045, and 0.0801 for 2 h, 4 h, 6 h, and 8 h, respectively; for e Nup214, p values are 0.7858, 0.1538, 0.0004, and <0.0001 for 2 h, 4 h, 6 h, and 8 h, respectively; For f Nup50, p values are 0.2869, 0.583, 0.0602, 0.2899, and 0.0074 for 1 h, 2 h, 4 h, 6 h, and 8 h, respectively. *p < 0.05; **p < 0.01; ***p < 0.001. Data were also subjected to non-linear regression for best fit. For d Nup358, F (3,24) value: 11.47; for e Nup214, F (3, 24) value: 20.21; for f Nup50, F (3, 30) value: 9.754; ###p < 0.001. Blue lines: non-linear regression curve for wild-type cells. Sig-1R, Sigma-1 receptor, Nup nucleoporin. n = 3 (a), n = 2 (b), and n = 3 independent experiments with similar results from biologically independent preparations. Note: data from c were analyzed to yield results for d–f.
Purified Sig-1R binds the (G4C2)-RNA repeats
The Sig-1R has been shown to bind proteins23,35 and lipids49,50, suggesting that the Sig-1R may accommodate diverse chemical natures of its binding partners. Thus, it is not unreasonable to speculate that the Sig-1R might bind RNA as well.
Firstly, we used immunostaining to examine if Sig-1Rs might colocalize with (G4C2)31-RNA5that were transfected into HeLa cells. The RNA fluorescence in situ hybridization (RNA FISH) technique was used to detect the (G4C2)31-RNA5. Indeed, the HA-Sig-1R detected by the HA antibody colocalizes with (G4C2)31-RNA, especially in the perinuclear area (Fig. S4a). The specificity of the RNA signal in FISH was verified by the DNase and RNase in that only the RNase abolished the FISH signal (Fig. S4b).
Next, we examined if purified Sig-1Rs might bind biotinylated (G4C2)10-RNA in an assay carried out in test tube containing purified molecules. The assay conditions are shown in supplementary data (Supplemental Information, Fig. S5a). Purified human GST-Sig-1R and GST are also shown (Supplemental Information, Fig. S5b, c). Results indicated that purified GST-human Sig-1R (lane 4, Fig. 4a) but not GST (lane 3, Fig. 4a) directly binds Biotin-(G4C2)10-RNA. Much clearer result was obtained when rat microsomes, which are highly enriched in Sig-1Rs, were solubilized and incubated with Biotin-(G4C2)10-RNA for the assay (Fig. 4b). This is because of the mass action law governing that the increased concentration of Sig-1Rs in liver microsomal preparation will bind more to the same concentration of Biotin-(G4C2)10-RNA. When scrambled RNA (i.e., Biotin-(A2U2GC)10-RNA) was used as control, it failed to bind to the mouse Sig-1R-YFP (Fig. 4c, lane 8, note: compared to lane 4). Single amino acid mutation of Sig-1R at amino acid 102 from glutamic acid to glutamine was reported to relate to familial ALS41. We examined if this mutant of Sig-1R (Sig-1R-E102Q-YFP) may have an altered ability to bind Biotin-(G4C2)10-RNA. Results showed that this mutant of Sig-1R shows a reduced affinity for the (G4C2)10-RNA (Fig. 4d; lane 5, wild type; lane 6, mutant).
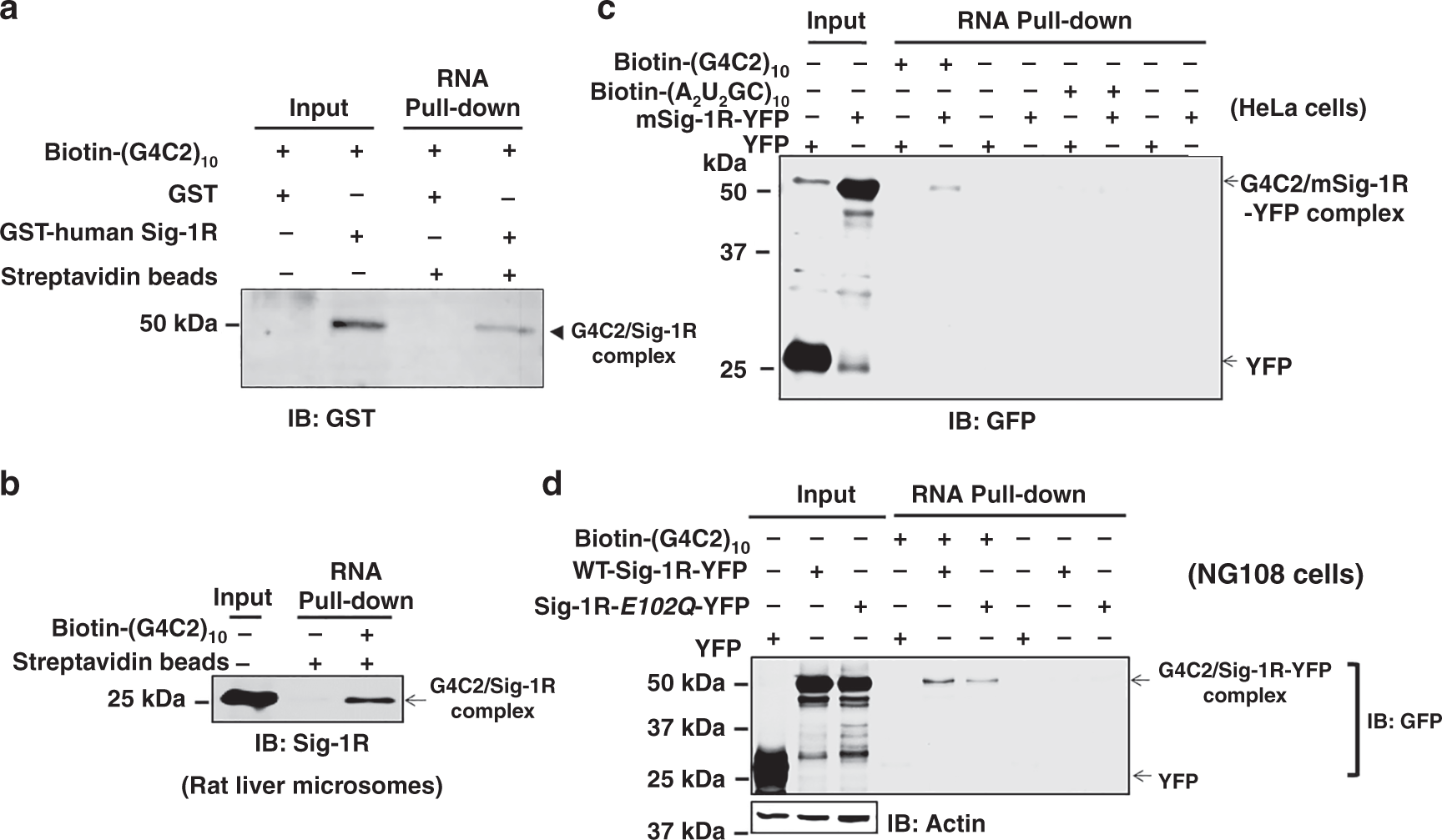
a Purified human sigma-1 receptor (Sig-1R) directly binds (G4C2)10-RNA in a chemical reaction in test tube. The biotin pull-down assay plus western-blot analysis were performed to detect the association of biotin-labeled (G4C2)10-RNA and GST-tagged human Sig-1R. b (G4C2)10-RNA binds the endogenous Sig-1R in rat liver microsomal preparation. Biotin-labeled (G4C2)10-RNA was incubated with lysates from rat liver microsomes followed by the biotin pull-down assay plus western-blot to detect the association of biotin-labeled (G4C2)10-RNA and endogenous Sig-1R. c Scrambled RNA repeats failed to bind Sig-1R. Biotin-labeled (A2U2GC)10-RNA was chosen as the scrambled RNA control. HeLa cells were transfected with mouse Sig-1R-YFP (mSig-1R-YFP) vectors for 24 h before the biotin pull-down assay plus western blot. Note: compare lane 4 vs lane 8. d Sig-1R mutation from glutamic acid to glutamine at amino acid 102 (i.e., Sig-1R-E102Q) has a lower affinity for (G4C2)10-RNA. NG-108 cells were transiently transfected with wild-type human Sig-1R-YFP or Sig-1R-E102Q-YFP vector for 24 h before the biotin pull-down assay plus western blot. Note: compare lane 5 vs lane 6. Sig-1R, Sigma-1 receptor. n = 4 (a), n = 3 (b), n = 3 (c), and n = 3 (d) independent experiments with similar results from biologically independent cellular preparations.
Thus, the Sig-1R can bind directly the (G4C2)31-RNA. The amino acid 102 of Sig-1R seems to play an important role in the interaction.
Sig-1R effects on (G4C2)-RNA repeats-induced Ran gradient across nuclear membrane in HeLa cells
Since Sig-1Rs exist at the nuclear pore close to RanGAP (Figs. 1a and 2b) and are, as shown above, able to bind (G4C2)-RNA repeats, the possibility exists that Sig-1Rs might absorb away some of the toxic (G4C2)-RNA repeats from RanGAP in a manner like a molecular sponge. We examined therefore in this section if Sig-1Rs may affect the aberrant nucleocytoplasmic Ran gradient imposed by (G4C2)-RNA repeats in HeLa cells. Two studies were carried out in this section as follows, i.e., immunocytochemistry and subcellular fractionation followed by western blot.
The (G4C2)31-RNA5 was used to transfect cells. After the transfection, cells were examined by immunocytochemistry for the nucleocytoplasmic gradient (N/C ratio) of Ran. In (G4C2)31-RNA-transfected cells, immunoreactive Ran increases significantly in the cytoplasm when compared to controls (Fig. 5).

The (G4C2)31-pcDNA5 or empty vector (Ctrl.) was transiently transfected into HeLa cells. Distribution of endogenous ras-related nuclear protein (Ran; Green) was detected by immunostaining and confocal microscopy. The semi-quantification of cytosolic or nuclear Ran was performed by using NIH Image J. (version 1.51b; right panel). Data are presented as means ± SEM for control cells (n = 29) and for cells (n = 12) overexpressing (G4C2)31-RNA. Two-tailed unpaired Student’s t test, p = 0.0003, ***p < 0.001. Ran: ras-related nuclear protein. n = 3 independent experiments with similar results from biologically independent cells.
We next examined if the knockdown of Sig-1Rs might affect the pattern of immunoreactive Ran under the influence of (G4C2)31-RNA. Results showed that there is apparently an increase of cytoplasmic Ran in cells treated with shSig-1R when compared to the control shRNA-treated cells (Fig. 6a, b). Successful knockdown of Sig-1Rs by transfection of shSig-1R is shown in a western blot (Fig. 6c).
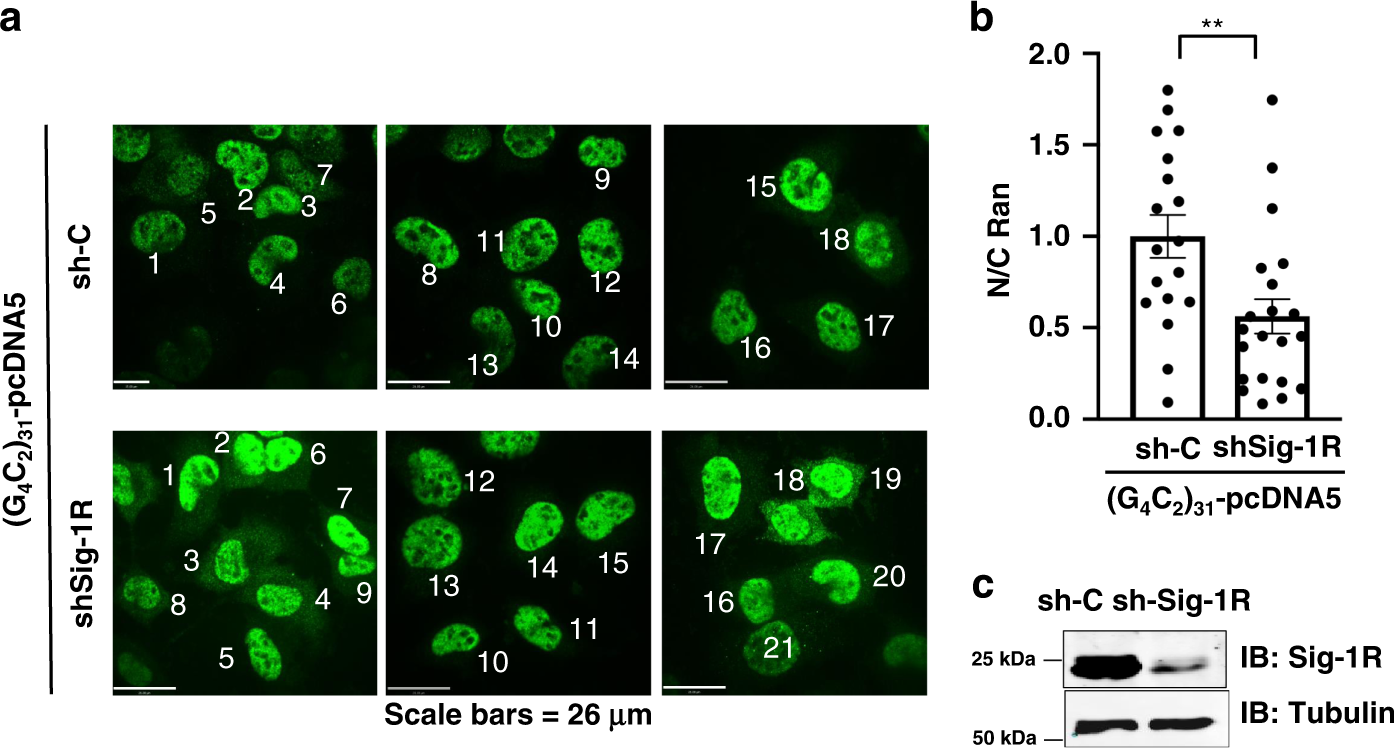
a The shRNA control (sh-C) or Sig-1R-shRNA (shSig-1R) vector was transiently transfected into HeLa cells for 24 h. The (G4C2)31-RNA vector was then transfected into HeLa cells to produce the (G4C2)31-RNA in the cell. Distribution of endogenous Ran (in green) was detected by immunostaining and confocal microscopy. b The semi-quantification of cytosolic or nuclear Ran was performed by using NIH Image J. (version 1.51b). Data are presented as means ± SEM; n = 18 for biologically independent control cells receiving scrambled shRNA plus (G4C2)31-RNA; n = 21 for biologically independent cells receiving shSig-1R plus (G4C2)31-RNA. Two-tailed unpaired Student’s t test, p = 0.0056. **p < 0.01. c The knockdown efficiency of Sig-1R protein in experiment above was detected by western blot. In this experiment, shSig-1R was transfected twice, i.e., 24 h after the first transfection the second transfection was carried out. Sig-1R, Sigma-1 receptor; Ran, ras-related nuclear protein. n = 3 (a) and n = 2 (b) independent experiments with similar results from biologically independent cells or cellular preparations.
Subcellular fractionation followed by western blotting was then carried out to quantitatively confirm the immunocytochemistry results. Specifically, the effect of Sig-1R knockout or overexpression on the N/C ratio of Ran was examined. Because the total tubulin level was the same in either wild type or knockout cells (Fig. 7a, lanes 1–4), the N/C ratio of protein of interest was calculated directly by the densitometric ratio from western blot. Results are presented in the rest of this section with the knockout results presented first. Sig-1Rs were knocked out in HeLa cells by the CRISPR technology.
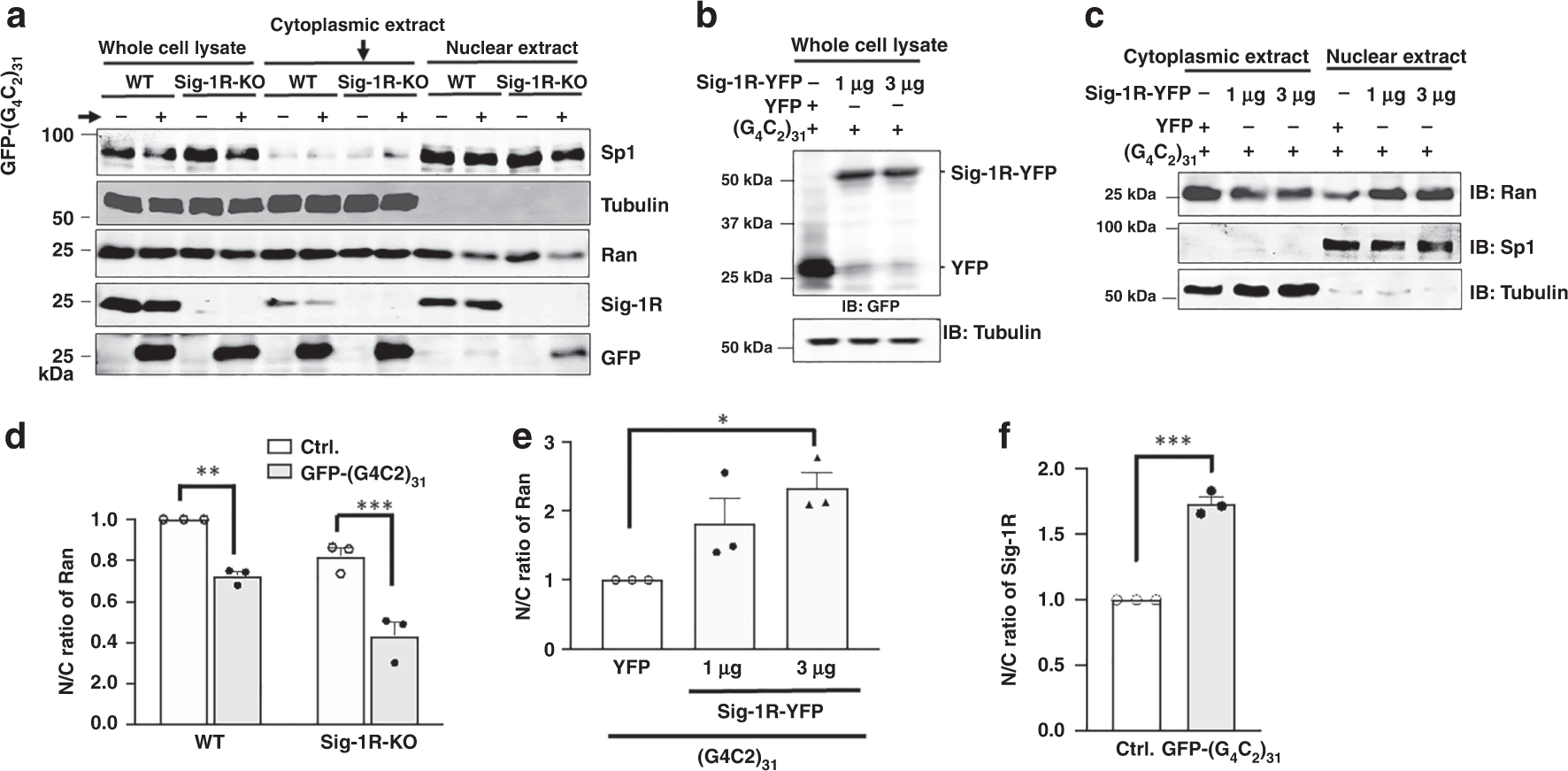
a Apparent decrease of nuclear Ran in Sig-1R knockout cells when compared to that seen in wild-type cells. Subcellular fractionation followed by western blot was used to examine the level of Ran in the cytoplasm or nucleus in HeLa cells in which Sig-1Rs were depleted by the CRISPR/Cas9 technique. GFP-(G4C2)31 vectors were transiently transfected for 24 h into either wild-type or CRISPR/Cas9 Sig1R-KO cells before subcellular fractionation. b Sig-1R successfully overexpressed in (G4C2)31-RNA-treated cells. c Overexpression of Sig-1R decreased Ran in the cytoplasm while concomitantly increased Ran in the nucleus. d Sig-1R knockout apparently exacerbates the N/C ratio of Ran when compared to that seen in wild type. See Results for explanations. Quantitative summary of results from three sets of independent experiments illustrated in a. Data are presented as means ± SEM; n = 3 for each group; two-way ANOVA followed by Sidak’s multiple comparisons test, p = 0.0027 for wild-type group, p = 0.0003 for Sig-1R-KO group, **p < 0.01; ***p < 0.001. eOverexpression of Sig–1Rs significantly rescues the aberrant N/C ratio of Ran caused by the (G4C2)31-RNA. Quantitative summary of results from three sets of independent experiments illustrated in c. Data are pμresented as means ± SEM; n = 3 for each group; one-way ANOVA followed by Sidak’s multiple comparisons test, p = 0.172 for YFP vs 1 μg, p = 0.0276 for YFP vs 3 μg, p = 0.4723 for 1 μg vs 3 μg, *p < 0.05. f Increase of nuclear Sig-1Rs in the (G4C2)31-RNA-treated HeLa cells. Quantitative summary of results from three sets of independent experiments illustrated in a. Data are presented as means ± SEM; n = 3 in each group. Two-tailed unpaired Student’s t test (p = 0.0001), ***p < 0.001. Note: the N/C ratio of the control in each independent experiment in d–f was taken as one. The N/C ratio from each experiment was normalized to respective control. Sig-1R: Sigma-1 receptor; Ran: ras-related nuclear protein. n = 3 (a), n = 3 (b), and n = 3 (c) independent experiments with similar results from biologically independent cellular preparations.
Visual examination on the representative western blot showed that in wild-type cells the cytoplasmic level of Ran apparently is not affected by the (G4C2)31-RNA treatment (Fig. 7a; lane 5 and lane 6) whereas the nuclear level of Ran is reduced by the same treatment (Fig. 7a, lane 9 and lane 10). In knockout cells, while the level of cytoplasmic Ran does not appear to differ between the control and the (G4C2)31-RNA-transfected cells (Fig. 7a; lane 7 and lane 8), the level of nuclear Ran apparently decreases to a large extent (Fig. 7a; lane 11 and lane 12). Interestingly, in wild-type cells, (G4C2)31-RNA causes a decrease of Sig-1R in the cytoplasmic extract (Fig. 7a; lane 5 vs lane 6) while concomitantly causes an apparently slight increase in the nuclear extract (Fig. 7a; lane 9 vs lane 10).
We examined next on the effect of the overexpression of Sig-1Rs on the cytoplasmic and nuclear levels of Ran in wild-type cells treated with (G4C2)31-RNA. Visual examination showed that, at 1 µg or 3 µg of the Sig-1R-YFP gene used for transfection, the expressed level of Sig-1R-YFP did not apparently differ in transfected cells (Fig. 7b; lane 2 and lane 3), suggesting a near maximum transfection at 1 µg of the vector employed. Results on the Ran level show that the cytoplasmic Ran decreases while the nuclear Ran increases with the overexpression of the Sig-1R-YFP (Fig. 7c).
The N/C ratio of Ran was then quantified by comparing results from three sets of independent western blotting experiments. Results are shown as follows.
In wild-type cells, the (G4C2)31-RNA-transfection significantly causes an increase of cytoplasmic Ran, thus a decrease in the N/C ratio (Fig. 7d). In Sig-1R-knockout cells, however, the N/C ratio of Ran apparently decreases in a greater magnitude when compared to that seen in wild-type cells (Fig. 7d). Those results suggest an even higher cytoplasmic accumulation of Ran in the presence of (G4C2)31-RNA when Sig-1Rs are reduced in the cell.
Overexpression of Sig-1Rs in wild-type cells causes an increase of the N/C ratio of Ran (Fig. 7e). This suggests that the overexpression of Sig-1Rs increases the nuclear Ran, counteracting the insult of (G4C2)31-RNA that causes the pathological accumulation of cytoplasmic Ran.
On a separate note, it is interesting to notice that in wild-type cells the (G4C2)31-RNA treatment causes an increase of N/C ratio of Sig-1Rs (Fig. 7f), suggesting a translocation of Sig-1Rs from cytoplasm into the nucleus after the treatment of (G4C2)31-RNA.
We extend our biochemical and cellular biological findings above to animals by using Drosophila as a model for ALS/FTD.
Drosophila studies
The fruit fly Drosophila melanogaster has proven to be a powerful model organism to study how HRE causes ALS and FTD neuropathologies51. Expression of expanded (G4C2)-RNA repeats in Drosophila leads to retinal degeneration, functional deterioration of motor neurons and locomotor defects52,53,54. Among pathogenic mechanisms, Drosophila genetics brought to light the importance of nucleocytoplasmic transport. Enhancing nuclear import was indeed found potent rescuer of HRE-induced toxicity in fly eyes12,54,55.
To evaluate in vivo whether or not human Sig-1R confers protection against expanded (G4C2)-RNA repeats, we used Drosophila (female) models expressing 3 or 30 RNA repeats of G4C2 ((G4C2)3 and (G4C2)30, respectively) under the regulation of UAS-GAL4 system52. We first confirmed that human Sig-1R is properly expressed in the presence of expanded (G4C2; Fig. 8a). Note that Drosophila has no detectable Sig-1R (Fig. 8a, left 2 lanes). Drosophila eyes are commonly used to evaluate toxicity of genes and in accordance to previous studies52, expression of (G4C2)30 into the retina progressively induces the formation of degenerative eyes with necrotic spots (Fig. 8b). Of interest, while this phenotype has an incomplete penetrance of 38%, the co-expression of Sig-1R significantly reduces the penetrance to 3% (Fig. 8c).
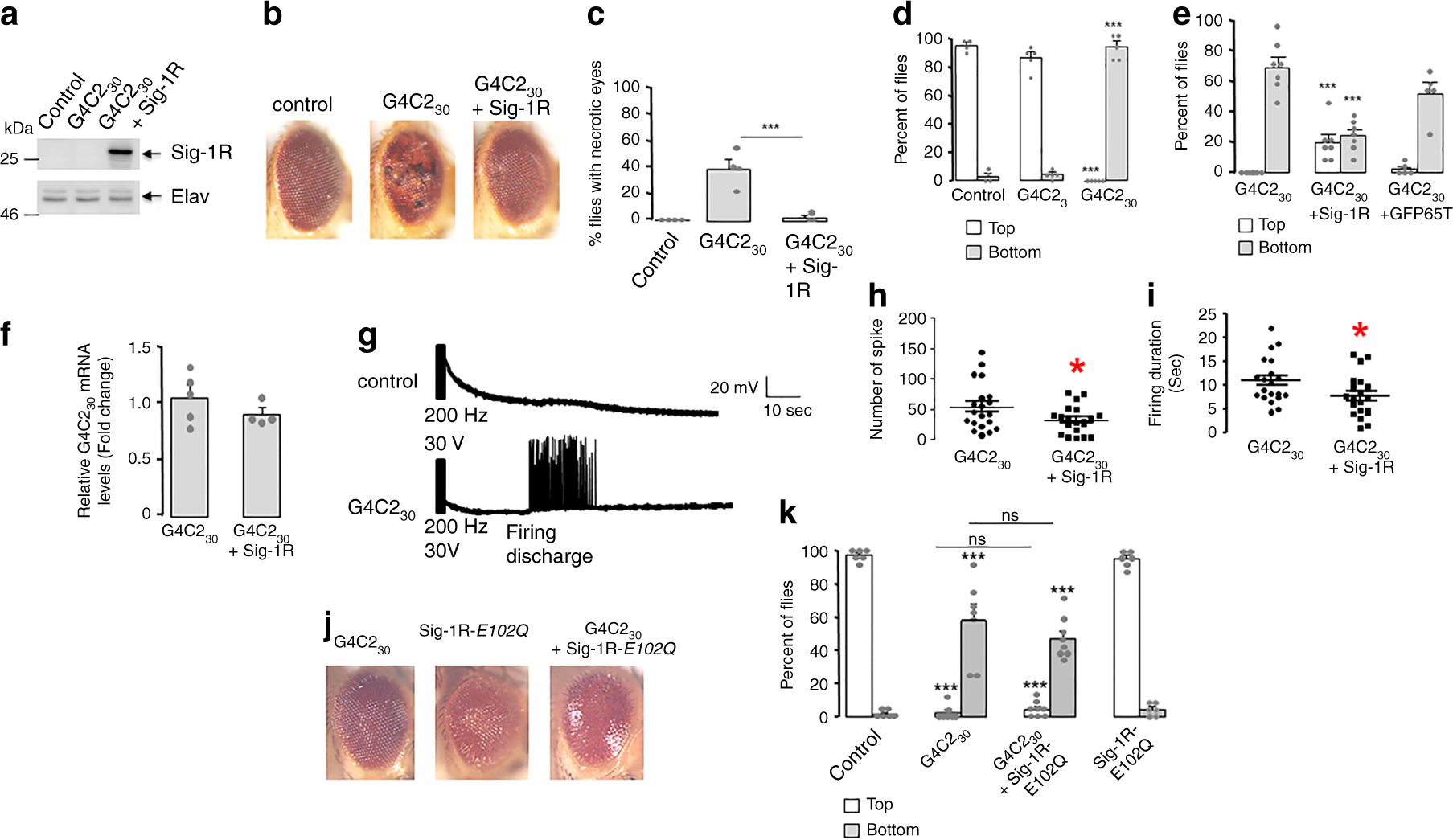
a Western blot of human Sig-1R (Sigma-1 receptor) in Drosophila; n = 3 independent experiments with similar results from biologically independent preparations. b Representative external eye morphology of flies (20 days after eclosion) expressing no transgene (control), 30 G4C2 repeats ((G4C2)30) alone or together with human Sig-1R. c Quantification of flies presenting necrotic spots in the eyes; n = 4 independent sets of studies from a total of 91 flies in control, 48 in (G4C2)30, and 68 in (G4C2)30 + Sig-1R group; statistics at the end. d Climbing performances (observation starts at 1 min) of 4-day-old flies expressing no transgene (control), 3 ((G4C2)3) or 30 G4C2 repeats ((G4C2)30). n = 8 flies/group; number of trials: control, 4; (G4C2)3, 5; (G4C2)30, 5. ***p < 0.001 versus control; statistics at the end. e Climbing performances of 4-day-old flies expressing (G4C2)30 alone or with Sig-1R or the green fluorescent protein GFP (GFP65T) in neurons. n = 8 flies/group; number of trials: (G4C2)30, 7; (G4C2)30 + Sig-1R, 7; (G4C2)30 + GFP65T, 5; statistics at the end. f Sig-1R does not modify G4C230 mRNA expression. mRNA levels of G4C230 (n = 5) or (G4C2)30 + Sig-1R (n = 4) were normalized to actin. Data are means ± S.E.M.; unpaired two-tailed t-test (p = 0.2950). g Representative traces of evoked responses after an electroconvulsive stimulation (30 V, 200 Hz) in flies expressing no transgene (control) or (G4C2)30. h Number of spikes in firing discharges induced by an electroconvulsive stimulation on flies (n = 20/group) expressing (G4C2)30 alone or together with Sig-1R. Data are means ± S.E.M; unpaired two-tailed t-test (*p = 0.0385). i Duration of firing discharges for flies (n = 20/group) expressing (G4C2)30 alone or together with Sig-1R. Data are means ± S.E.M.; unpaired two-tailed t-test (*p = 0.0375). j Expression of Sig-1R-E102Q in retina leads to rough eye phenotype. k Sig-1R-E102Q failed to ameliorate climbing performances of flies (4 days old) expressing expanded G4C2 repeats. Control: no transgene. n = 8 flies per group; number of trials: Control, 6; (G4C2)30, 7; (G4C2)30 + Sig-1R- E102Q, 8; Sig-1R- E102Q, 6. Statistics for c, d, e, k: data are means ± S.E.M., one-way ANOVA plus Tukey’s multiple-comparison tests, ***p < 0.001 versus control, ns not significant.
Flies expressing (G4C2)30 in neurons was also previously found to exhibit reduced locomotor activity52. We examined here the climbing response of flies after being tapped in the negative geotaxis test. This locomotor test uses the natural reflex of flies to walk against gravity and is a standard locomotor activity test in the field of Drosophila. Expression of (G4C2)30 but not (G4C2)3 in neurons, under the control of Elav-GAL4 driver, led to strong climbing response defects (Fig. 8d). While >80% of control or (G4C2)3-expressing flies attain the top of the column within 1 min, most of the (G4C2)30-expressing flies do not climb and none of them succeed to reach the top. Indeed, most of (G4C2)30-expressing flies suffered from a seizure-like episode after being startled. The bang-sensitive behavior was previously described as an abnormal response of the giant fiber escape circuit that controls motor neurons56,57. The presence of Sig-1R significantly ameliorates the climbing deficit of G4C230-expressing flies. In this case, 76% of flies climbed along the column and 19% of them reached the top within 1 min (Fig. 8e). As a control for potential UAS-GAL4 dilution effect, overexpressing the green fluorescent protein (GFP65T) fails to modify climbing performances of G4C230-expressing flies (Fig. 8e). Moreover Sig-1R does not modify G4C230 RNA expression (Fig. 8f), indicating that Sig-1R acts downstream of (G4C2)30 transcription.
Electrophysiological studies previously showed that the bang-sensitivity correlates to long firing discharge at the neuromuscular junction after a high-frequency electroconvulsive stimulation of the giant fiber pathway56,57. We thus set up electrophysiological recordings of flight muscles after stimulation (200 Hz for 2 s) of giant fiber neurons into the brain. As expected, an electroconvulsive stimulation of (G4C2)30-expressing flies resulted in delayed firing discharge in flight dorsal muscles (Fig. 8g). The presence of Sig-1R decreases the number of spike but also the duration of firing discharge (Fig. 8h, i). Thus, we demonstrate that the Sig-1R significantly reduces abnormal long firing of giant fibers and thereby ameliorates the startle-induced climbing response of flies expressing (G4C2)30.
We next examined whether the Sig-1R-E102Q mutant41,43 may have an impact on the (G4C2)30-induced phenotype. The Sig-1R-E102Q was either expressed in eyes only or in neurons, respectively, for examination of eye morphology or locomotion.
While flies expressing Sig-1R-E102Q in eyes only under the regulation of GMR-GAL4 driver shows a mild rough eye phenotype at 1 day of age, the presence of (G4C2)30 seems to worsen this phenotype (Fig. 8j). Unexpectedly, we also found that co-expression of (G4C2)30 with Sig-1R-E102Q in eyes is lethal for adult flies. They only survive a few days after eclosion (3–10 days), thus hampering a statistical analysis of the degenerative eye phenotype when they developed and aged.
We previously showed that mutant Sig-1R-E102Q has no deleterious locomotor effects in Drosophila notably when expressed at a moderate level in neurons such as in the line of Sig-1R-E102Q#1 flies43. Here we used the same Sig-1R-E102Q#1 flies for this portion of the study. Accordingly, Sig-1RE102Q#1 shows no climbing deficit when compared to control (Fig. 8; far right panels). When (G4C2)30 was expressed in neurons of Sig-1RE102Q#1 using the Elav-GAL4 driver, flies now present climbing defects seen in (G4C2)30-expressing flies at 4 days of age (Fig. 8k). Altogether, these data suggest that the mutant Sig-1R-E102Q confers no protection against (G4C2)30 toxicity.
Discussion
The nuclear pore complex (NPC) has been coined “The gate to neurodegenerative diseases”58. Here we report the existence of the first molecular chaperone at the NPC and show that this chaperone, the Sig-1R, counteracts the N/C ratio deficit of Ran induced by the (G4C2)-RNA repeats that underlies ~40% of the familial ALS cases. Although this study used cellular models in the first part of the study, the potential implications of results should not be lightly discounted for the following reasons. Firstly, we used a human cell line here in the present study. Secondly, we have shown in the past that results from cell lines perfectly mimic the results from rodent brain39,59. In addition, we have used Drosophila in this study which has been recognized as a suitable model for ALS and FTD and demonstrated that almost all biochemical and cellular biological observations in HeLa cells can be validated by the animal study.
Of course, more studies need to be done in the future to examine the clinical implication of the current report. Nevertheless, the direct implication of the current study is that by increasing Sig-1Rs in the ALS/FTD patients, suffering from the insult of the (G4C2)-RNA repeats, may attenuate the damage caused by the RNA repeats. In this regard, it is interesting to note that at least three drugs have been shown to increase Sig-1Rs in cell cultures and in rodents60,61,62,63. Follow-up studies in this line of thought may lead to potential therapeutic agents for treatment of this type of ALS/FTD patients.
Our study is not the first to relate Sig-1Rs to ALS/FTD but is the first to point out the NPC as the site of action of Sig-R in this regard. It is interesting to note that Sig1-Rs exist in diverse places in a cell including the nuclear envelope38,39 and nucleoplasm38. This may explain why Sig-1Rs may affect the stability of Nups that are on the cytosolic side but also other Nups that face the nucleoplasm such as Nup50. Whether Sig-1Rs may chaperone some other Nups not examined in this study is unknown. Since the crystal structure of NPC is known64,65,66, it would be interesting to know where and how this one-transmembrane Sig-1R25 fits into the structure of NPC. It is not totally clear at present how the chaperoning activity of Sig-1R may, at the molecular level, affect the N/C ratio of Ran. We surmise that the stabilized Nups, thus the NPC assembly, may facilitate the Ran entry from cytosol into nucleus.
The interaction between Sig-1R and (G4C2)10-RNA was discovered in this study out of the extension of the Sig-1R’s known ability in binding proteins as well as lipids49,50. The results of this study thus place the Sig-1R as an RNA-binding protein (i.e., ribonucleoprotein). Other ribonucleoproteins, including TDP-4367 and FUS68, are also known to involve in the neurodegenerative disease. Several studies have reported an increase of Sig-1Rs in the nucleus of neurons related to several neurodegenerative diseases (e.g. refs. 40,41,69,70). Here we also see an increase of Sig-1Rs in the nucleus of (G4C2)31-RNA-treated cells (Fig. 7f). We do not know at present why Sig-1Rs are increased in the nucleus of those “diseased” cells exactly opposite to that seen with dysfunctional TDP-43 or FUS. The relation, if any, between those three critical ribonuclear proteins in neurodegeneration remains to be cleared in the future.
We speculate that the Sig-1R’s rescue of the Ran N/C ratio may result from the Sig-1R ability to bind (G4C2)-RNA repeats as a molecular sponge and to reduce thus the effective concentration of the RNA repeats as they exert their insults on RanGAP12. It is interesting to note that the Sig-1R is in close proximity to RanGAP (Figs. 1a and 2a, b) and in fact can co-IP with RanGAP (Fig.1b). Thus, it can be imagined that Sig-1R-RanGAP-(G4C2)n-RNA may exist as a trimeric complex. If so, how does the Sig-1R help the RanGAP to get rid of the toxic (G4C2)n-RNA? More studies are certainly warranted to provide answer to this question.
Although we show here that the Sig-1R with a single amino acid mutation at 102 has a reduced ability to bind (G4C2)10-RNA (Fig. 4d), we do not know if this mutation of Sig-1Rs plays a role in the (G4C2)-RNA repeats-induced ALS/FTD. It rarely happens that a disease is caused by two mutations. Nevertheless, our result suggests a structural specificity of Sig-1R in its interaction with (G4C2)-RNA repeats. Whether the Sig-1R can interact with the HRE is unknown at present.
Questions as mentioned above notwithstanding, our results suggest the Sig-1R as a never-before reported target in understanding the NPC- and (G4C2)-RNA repeats-related neurodegeneration. The Sig-1R has been implicated as a beneficial factor in many types of neurodegenerative diseases in part due to the receptor’s ability to regulate the downstream targets at multiple loci of a cell35. Our current result indicates yet another locus whereby the Sig-1R plays a role against the neurodegenerative disease. In this case it is against the C9orf72 type of ALS/FTD at the NPC. Since the nuclear pore has been indicated to involve in many neurodegenerative diseases58, it is tempting to suggest that the Sig-1R action at the nuclear pore may serve as a common molecular target for those diseases.
Nevertheless, other potential mechanisms or loci may also be involved in the (G4C2)-RNA repeats-antagonizing action of Sig-1Rs. For example, the HRE-derived dipeptide repeats (DPR), can impede the maturation of mRNA in the nucleus or the biogenesis of ribosomal RNA in nucleoli, in model cells and even in patient’s cells, leading to defective nucleocytoplasmic transport71. Inasmuch as the Sig-1R exists in the nucleus, albeit with its nucleolar presence yet to be determined, it is tempting to speculate that Sig-1Rs may regulate the maturation of mRNAs or biogenesis of ribosomal RNA. Of course, whether Sig-1Rs may directly bind the DPR or the small RNA of the component of RNA spliceosome remains to be determined. The action of DPR was also reported to relate to the formation of stress granule assembly in the cytosol that plays a critical role in impeding the nucleocytoplasmic transport10. As Sig-1Rs exist at the reticular network of ER directly facing cytosol, they may participate in the formation of the granule assembly.
Existing evidence suggests a relation between the Sig-1R and TDP-43. For example, overexpression of TDP-43 causes a locomotor deficit as well as a reduced production of ATP in Drosophila, both of which nonetheless are rescued by overexpression of Sig-1R43. Those results suggest an action of TDP-43 at the mitochondria or perhaps at the IP3R of the MAM where the Sig-1R functions as a chaperone. TDP-43 pathology is recently shown to relate clinically to (G4C2)-RNA repeats72. Taken together, those results indirectly suggest a mitochondrial energy metabolism deficit caused by (G4C2)-RNA repeats, perhaps through TDP-43 which then is counteracted by the Sig-1R.
Lastly, it is interesting to see that the overexpression of Sig-1R-E102Q per se causes morphological deficit in the eye (Fig. 8j). In fact, this gain-of-toxicity of Sig-1R-E102Q may be related to its action on the IP3R at the MAM important for ATP production43. This explanation on the action of the Sig-1R-E102Q at IP3R at the MAM, together with current result showing the (G4C2)30 co-expression exacerbating the toxic effect of the mutant in the eye (Fig. 8j), render support to the above notion that (G4C2)-RNA repeats may lead to dysfunctional mitochondria and a reduced cellular bioenergetics. Further study is certainly required to confirm this speculation.