
Sigma-1 receptor agonist PRE-084 confers protection against TAR DNA-binding protein-43 toxicity through NRF2 signalling
By Christelle Lasbleiz, Amandine Peyrel, Pauline Tarot, Jérôme Sarniguet, Lucie Crouzier, Nicolas Cubedo, Benjamin Delprat, Mireille Rossel, Tangui Maurice, Jean-Charles Liévens
Excerpt from the article published in Redox Biology, 17 November 2022, 102542, ISSN 2213-2317,
DOI: https://doi.org/10.1016/j.redox.2022.102542.
Editor’s Highlights
- Amyotrophic lateral sclerosis (ALS) familial and sporadic cases are associated with mutations in genes encoding the copper/zinc superoxide dismutase 1 (SOD1), TAR DNA-binding protein of43 kDa (TDP43), fused in sarcoma (FUS) or the chromosome 9 open reading frame 72 (C9orf72) proteins, among others.
- Of importance, TDP43 is ubiquitously expressed and localizes predominantly in the nucleus.
- The sigma-1 receptor (S1R) fulfils a key role in activation of defense against oxidative stress.
- S1R exerts neuroprotection in TDP43 proteinopathy.
- This study confirmed the high therapeutic value of S1R agonists in ALS.
Autors’ Highlights
- PRE-084 restored motor performances and mitochondrial respiration of TDP43 zebrafish.
- Antioxidant ER stress response and NRF2 signalling were boosted by PRE-084.
- Increasing NRF2 levels conferred protection against TDP43 toxicity.
Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease affecting upper and lower motor neurons. As a consequence, ALS patients display a locomotor disorder related to muscle weakness and progressive paralysis. Pathological mechanisms that participate in ALS involve deficient unfolded protein response, mitochondrial dysfunction and oxidative stress, among others. Finding a therapeutic target to break the vicious circle is particularly challenging. Sigma-1 receptor (S1R) is an endoplasmic reticulum (ER) chaperone that may be one of those targets. We here address and decipher the efficiency of S1R activation on a key ALS gene, TDP43, in zebrafish vertebrate model. While expression of mutant TDP43 (TDP43G348C) led to locomotor defects, treatment with the reference S1R agonist PRE-084 rescued motor performances in a zebrafish model. Treatment with the agonist ameliorated maximal mitochondrial respiration in the TDP43 context. We observed that TDP43G348C exacerbated ER stress induced by tunicamycin, resulting in increased levels of ER stress chaperone BiP and pro-apoptotic factor CHOP. Importantly, PRE-084 treatment in the same condition further heightened BiP levels but also EIF2α/ATF4 and NRF2 signalling cascades, both known to promote antioxidant protection during ER stress. Moreover, we showed that increasing NRF2 levels directly or by sulforaphane treatment rescued locomotor defects of TDP43G348C zebrafish. For the first time, we here provide the proof of concept that PRE-084 prevents mutant TDP43 toxicity by boosting ER stress response and antioxidant cascade through NRF2 signalling.
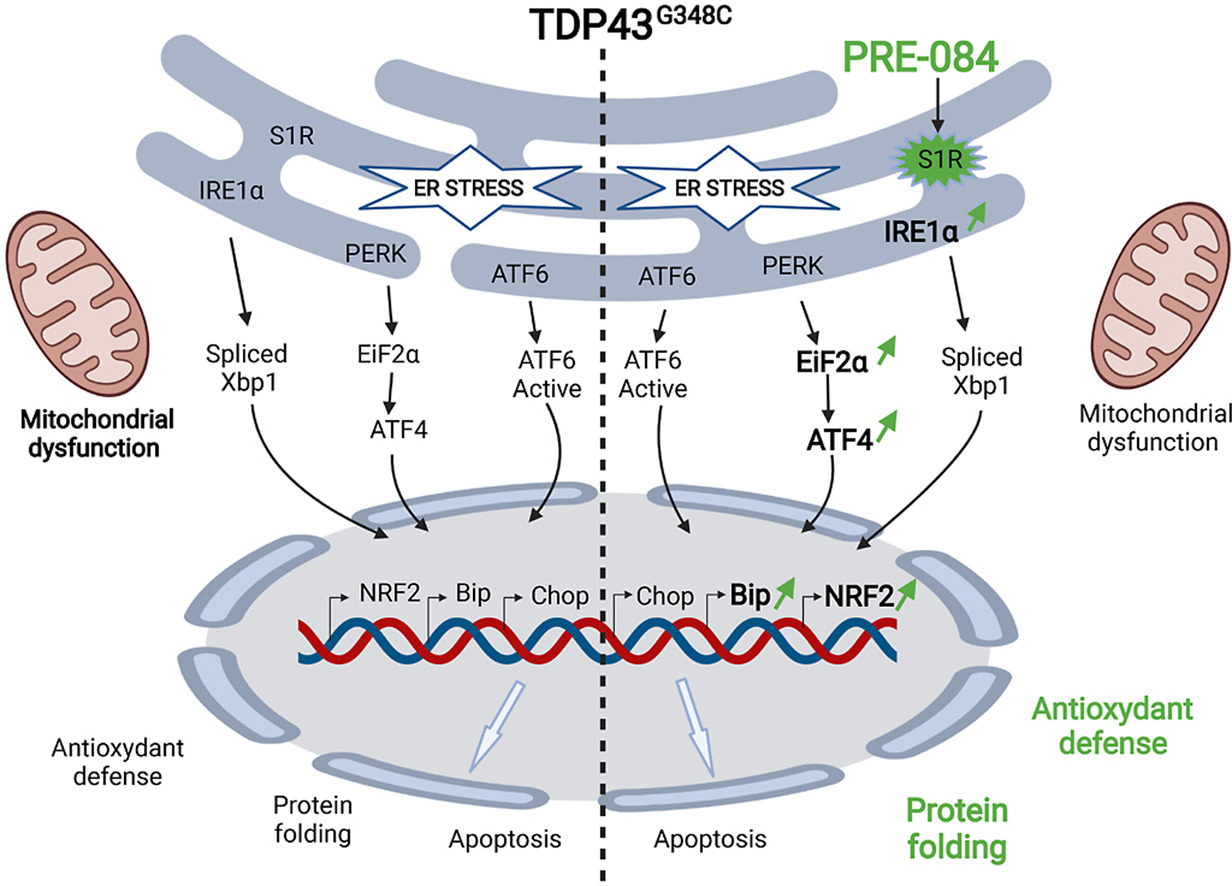
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a very disabling disorder characterized by muscle weakness and progressive paralysis. The disease is due to a degeneration of motor neurons in the spinal cord, brain stem and motor cerebral cortex. Ultimately, the disease rapidly evolves with respiratory distress and death within 5 years after the first motor symptoms in most cases. Currently, there is no cure for ALS and no effective treatment to halt or reverse the progression of the disease. The only available treatments with Riluzole and Edaravone have limited benefits [1,2].
ALS patients are divided into sporadic cases with no family history, accounting 90% of the total cases, and those with a clear genetic inheritance. ALS results from a complex interaction between both genetic and environmental factors. Advances in the understanding of ALS pathogenesis come from the identification of genes linked to ALS. Familial and sporadic cases are associated with mutations in genes encoding the copper/zinc superoxide dismutase 1 (SOD1), TAR DNA-binding protein of43 kDa (TDP43), fused in sarcoma (FUS) or the chromosome 9 open reading frame 72 (C9orf72) proteins, among others [3,4]. Of importance, TDP43 is ubiquitously expressed and localizes predominantly in the nucleus. However, it has been found that in the majority of ALS patients TDP43 abnormally accumulates in the cytoplasm and forms aggregates, suggesting that deregulation and mislocalization of wild-type TDP43 mediates both sporadic and familial ALS [5]. TDP43 is also found mislocalized in 45% of patients with frontotemporal lobar degeneration (FTLD). TDP43 plays a critical role in several steps of mRNA metabolism by regulating transcription, mRNA transport and stabilization into the cytoplasm, translation and microRNA processing [6]. TDP43 dysregulation leads to many cellular abnormalities including abnormal accumulation of unfolded protein, excitotoxicity, nucleocytoplasmic transport defects, mitochondrial dysfunction and oxidative damage. Finding a therapeutic target to alleviate all those defects is particularly challenging.
The sigma-1 receptor (S1R) is considered to promote cell survival in living systems. S1R is a cellular chaperone, still named receptor, as it can be activated/inactivated by small molecules, thus considered as S1R agonists or antagonists [7]. S1R is mostly located in the endoplasmic reticulum (ER), and more particularly at the mitochondria-associated ER membrane, where it modulates the inositol triphosphate (IP3) receptor type 3 to ensure proper Ca2+ signalling from the ER into mitochondria [[8], [9], [10], [11]]. In case of accumulation of misfolded proteins, S1R can participate to the unfolded protein response, by acting on the inositol-requiring enzyme 1 (IRE1)/X-box binding protein 1 (XBP1) or activating transcription factor 4 (ATF4) pathways [[12], [13], [14]]. This leads to the subsequent expression of antioxidant proteins and chaperone proteins. Very interestingly, it has been proposed that during oxidative stress conditions S1R may also enhance nuclear production of antioxidant proteins by enhancing nuclear factor erythroid-2 related factor 2 (NRF2) signalling [[15], [16], [17]]. Moreover, upon over-expression or stimulation, S1R can translocate at the plasma membrane to modulate ion channels, receptors and kinases [12,18] or at the nuclear envelope to regulate nucleocytoplasmic trafficking and gene transcription [19,20].
In the last decade, S1R has been the focus of many studies on ALS. Not only, mutations in S1R were found to be the cause of juvenile cases of ALS [21,22], but also S1R knock-down exacerbated ALS-linked mutant SOD1 pathology in mouse models [22,23]. Of importance, S1R is a druggable target and its activation by agonists alleviated deficits of mutant SOD1 murine models [[24], [25], [26], [27]]. Since mutations in SOD1 are linked to 15–20% of familial cases, observations from mice expressing this mutant protein do not allow extrapolating to the rest of ALS conditions. Further analyses are required to provide evidence that S1R is a valuable target to prevent ALS pathology. Of interest genetic approaches in Drosophilashowed that overexpression of human S1R rescued locomotor defects of ALS flies expressing either TDP43 or the C9ORF72 mutation [20,28]. In the present study, we provide the proof of concept that S1R activators are efficient therapeutic strategies to reduce TDP43-induced damages in a vertebrate model.
3. Results
3.1. PRE-084 rescued locomotor and motor neuron defects of TDP43G348Czebrafish larvae
Locomotor behavior tests were first used to determine locomotor deficits induced by TDP43G348C overexpression. The touch-escape assay was used to quantify the locomotor performances consecutively to a mechanical stimulation at 5 dpf. While larvae expressing a control mCherry protein swam a similar distance in 5 s as compared to controls, larvae expressing TDP43G348C showed a significant reduced motor response (Fig. 1A). To provide a direct proof that a neuroprotective strategy could be based on S1R, a transgenic zebrafish overexpressing human S1R was generated (Suppl. Fig. 1C). In transgenic S1R larvae, TDP43G348C did not affect swimming escape response compared to controls (Fig. 1B). Overexpressing S1R alone had also no impact on the touch-escape response (Fig. 1B). Next, we examined the effect of the reference S1R agonist, PRE-084. Two concentrations of PRE-084, 5 and 10 μM; the drug being added to the bath water for 24 h between 4 and 5 dpf. Interestingly, the swimming distance of TDP43G348C-expressing zebrafish was rescued after the treatment with PRE-084 at the two doses (Fig. 1C). To confirm the specificity of the protective effect of PRE-084, larvae were treated with PRE-084 alone or with the S1R antagonist, NE-100, at 5 μM each. Treatment with NE-100 significantly abolished the protective effect of PRE-084 (Fig. 1D). As control experiments, we observed that PRE-084 by itself had no impact on the locomotion of control larvae and NE-100 alone did not modify the locomotor behavior of TDP43G348C larvae (Suppl. Fig. 2). Another S1R agonist, SA4503, was tested in TDP43G348C larvae and again this treatment rescued the locomotor escape response (Fig. 1E).
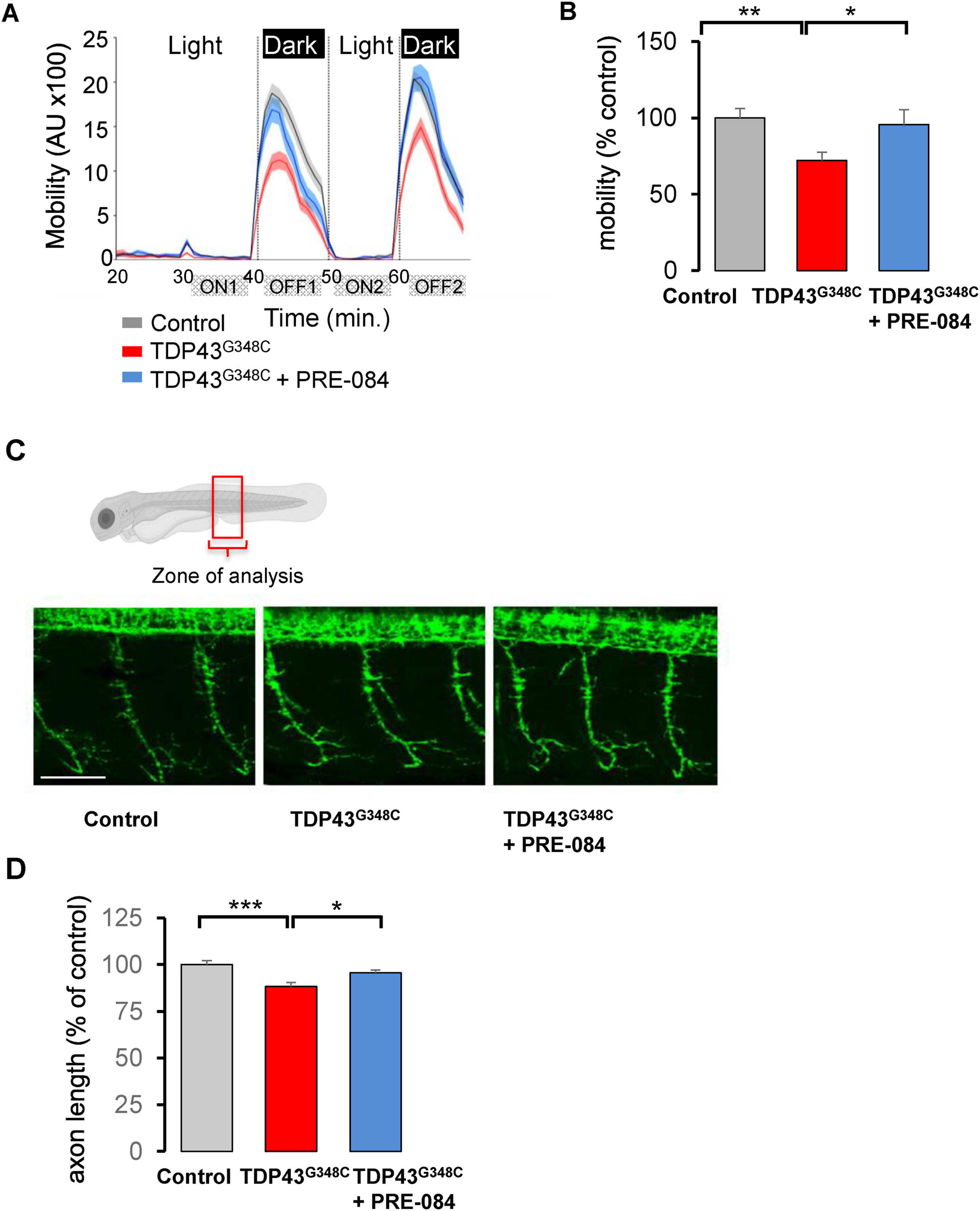
PRE-084 ameliorated TDP43G348C-larvae mobility and axonal projection.
(A) Mobility of control larvae, larvae expressing TDP43G348C alone or treated with 5 μM of PRE-084 during the light/dark phases in the VMR test. The mobility was measured during 1 min each. AU: arbitrary unit. (B) Quantitative analyses from (A) showing the relative mobility of controls, larvae expressing TDP43G348C alone or treated with PRE-084. Data were the mean of the mobility during the two OFF periods minus ON periods (OFF1-ON1; OFF2-ON2). Relative mobility from >100 larvae was expressed as percent of control and presented as mean ± SEM. Statistical analysis was performed using ANOVA followed by Tukey’s test (**p < 0.01; ***p < 0.001). (C) Representative photomicrographs showing three axonal projections in the zone of analysis. Scale bar: 50 μm. (D) Quantitative analyses of axonal projections (as shown in (C)) in control larvae or larvae expressing TDP43G348C alone or treated with PRE-084. Data from 42 to 54 motor neurons were averaged and presented as mean ± SEM. Statistical analysis was performed using ANOVA followed by Tukey’s test (*p < 0.05; ***p < 0.001).
We also performed the locomotor response to light intensity changes, using the visual motor response assay (VMR). Larvae are placed in a 96-well plate in a zebrabox and after 30 min, two consecutive periods of light onset (ON) and light offset (OFF) for 10 min each. Larvae showed increased locomotion during dark periods (Fig. 2A). The expression of TDP43G348C reduced the mobility of larvae during dark periods (Fig. 2B). The mean mobility significantly decreased by 28% in the presence of TDP43G348C. More importantly, treatment with PRE-084 at 5 μM rescued the response of TDP43G348C-expressing larvae in the VMR test (Fig. 2B).
Expression of TDP43G348C was reported to shorten motor neuron extension [35]. We therefore examined the axonal length of motor neurons on 2 dpf larvae. While in TDP43G348C-expressing larvae the mean axon length was significantly reduced by 12%, PRE-084 treatment rescued this alteration (Fig. 2C and D). Altogether our results emphasized the beneficial effects of S1R activation in a zebrafish vertebrate model of ALS.
3.2. PRE-084 ameliorated ATP production during high energy demand
Since S1R is a chaperone protein, we first investigated whether or not S1R activation by PRE-084 affected TDP43 levels. Western blot analyses revealed that PRE-084 did not affect expression of human TDP43 levels (Suppl. Fig. 3). Among its functions, S1R regulates energy production by mitochondria. Mitochondrial respiration activity was then evaluated by using a 24 plate-Seahorse XF analyzer (Agilent™). This system allows the measure on the whole zebrafish larvae of the basal mitochondrial activity to produce ATP, the maximal capacity of mitochondria to function and the non-mitochondrial respiration. The presence of TDP43G348Csignificantly reduced the maximal mitochondrial respiration, suggesting a mitochondrial dysfunction during high energy demand (Fig. 3A and B). Of importance, PRE-084 treatment enhanced maximal mitochondrial respiration in TDP43G348C context but not in control condition (Fig. 3B).
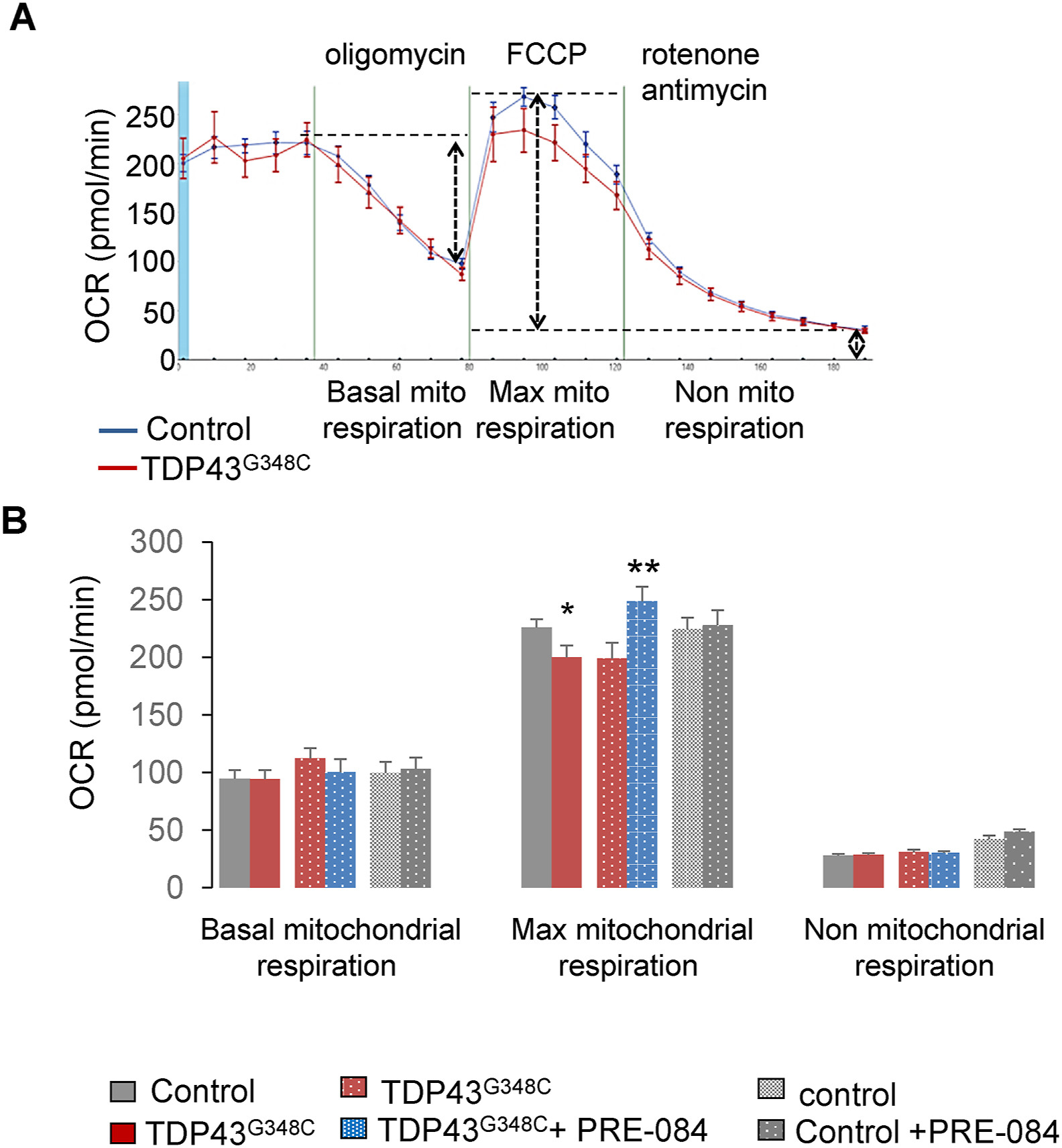
PRE-84 boosted maximal mitochondrial respiration of TDP43G348C-expressing larvae.
(A) Oxygen consumption rate (OCR) was measured using the Seahorse system on control and TDP43G348C larvae. (B) Basal mitochondrial respiration, maximal capacity of mitochondrial respiration and non-mitochondrial respiration were measured on control and TDP43G348Clarvae treated or not with PRE-084. Data from n = 18–33 larvae were presented as mean ± SEM. Statistical analysis was performed by the Student t-test (*p < 0.05; **p < 0.01).
3.3. TDP43G348C exacerbated tunicamycin-induced ER stress sensitivity
Abnormal accumulation of unfolded proteins in the ER, leads to an ER stress response, also termed the UPR. During unstressed conditions, the chaperone protein, GRP78/BiP, is associated to ER stress sensors (IRE1, PERK, ATF6), keeping them inactive. Upon ER stress, GRP78/BiP dissociates from these sensors resulting to the activation of the three UPR arms. As a consequence, this leads to the UPR characterized by increased levels of chaperone proteins like GRP78/BiP itself. However, under persistent ER stress, the normal functions of the ER fail to recover and results to apoptosis, as characterized by overexpression of the transcription factor C/EBP homologous protein (CHOP). Impact of TDP43 on ER stress has been poorly studied in vivo.
We have investigated levels of ER stress response genes by using RT-qPCR on 5 dpf zebrafish larvae. TDP43G348C expression did not modify mRNA levels of the three canonical UPR pathways: i) IRE1 and spliced (active) or unspliced forms of Xbp1, ii) PERK/EIF2α and ATF4 isoforms (ATF4a/ATF4b) and iii) ATF6 (Fig. 4A). Next, we used tunicamycin at 2 μg/ml for 24 h to induce ER stress and subsequently, the overexpression of UPR genes as shown in Fig. 4A. Likewise, levels of downstream UPR genes, BiP and CHOP, were stimulated by tunicamycin (Fig. 4B). Noteworthy one canonical UPR pathway was significantly overwhelmed by the presence of TDP43G348C during ER stress: the IRE1/Xbp1 cascade (Fig. 4A). The unspliced (inactive)/spliced (active) ratio of Xbp1 was found significantly increased (+33%, p < 0.05) in TDP43G348C-expressing larvae. In parallel, TDP43G348C further boosted BiP and CHOP upregulation in the presence of tunicamycin (Fig. 4B). Thus, mutant TDP43 increased the vulnerability to ER stress in vivo. At the protein level, while TDP43G348C did not modify BiP protein expression alone (Suppl. Fig. 4), it further enhanced BiP protein expression in the presence of tunicamycin (Fig. 4D and E and Suppl. Fig. 5).
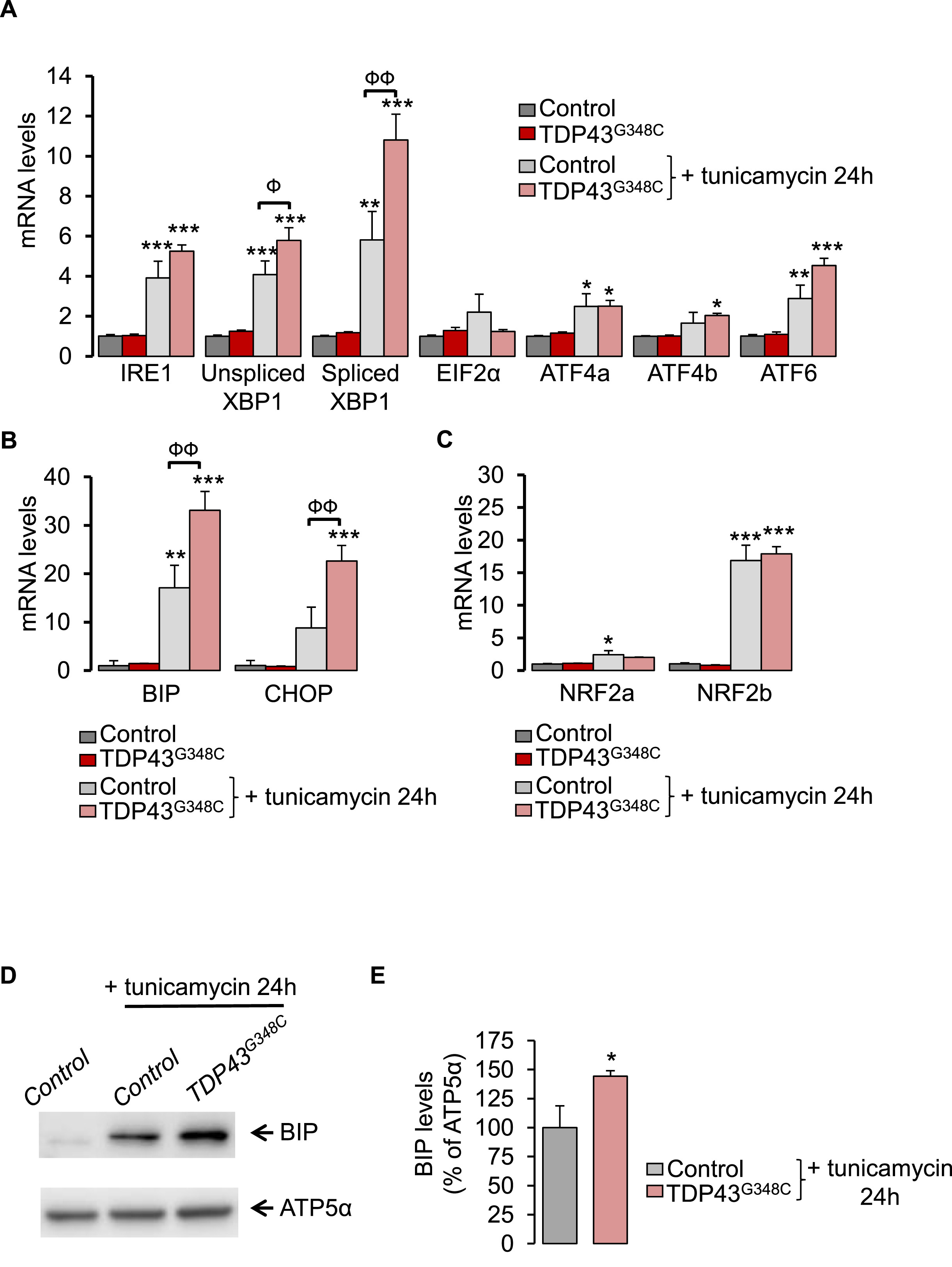
TDP43G348C increased sensitivity to ER stress.
(A) Transcript levels were evaluated from control or TDP43G348C larvae, treated or not with the ER stress inducer, tunicamycin, for 24 h. Data from n = 5 were presented as mean ± SEM. Statistical analysis was performed by using ANOVA followed by Tukey’s test (*p < 0.05, **p < 0.01 and ***p < 0.001 versus control condition; Фp<0.05, ФФp<0.01). (B) As in (A) for UPR gene targets: BiP and CHOP (**p < 0.01 and ***p < 0.001 versus control condition; ФФp<0.01). (C) As in (A) for NRF2 isoforms: NRF2aand NRF2b (*p < 0.05 and ***p < 0.001 versus control condition). (D) Western blot showing BiP and ATP5α protein levels from control or TDP43G348C larvae treated or not with tunicamycin for 24 h. (E) Densitometric analysis of BiP protein levels in zebrafish larvae from (D). ATP5α was used as a reference to normalize BiP levels. Data from 5 independent samples were averaged and presented as mean ± SEM. Statistical analysis was performed using a Student’s t-test (*p < 0.05).
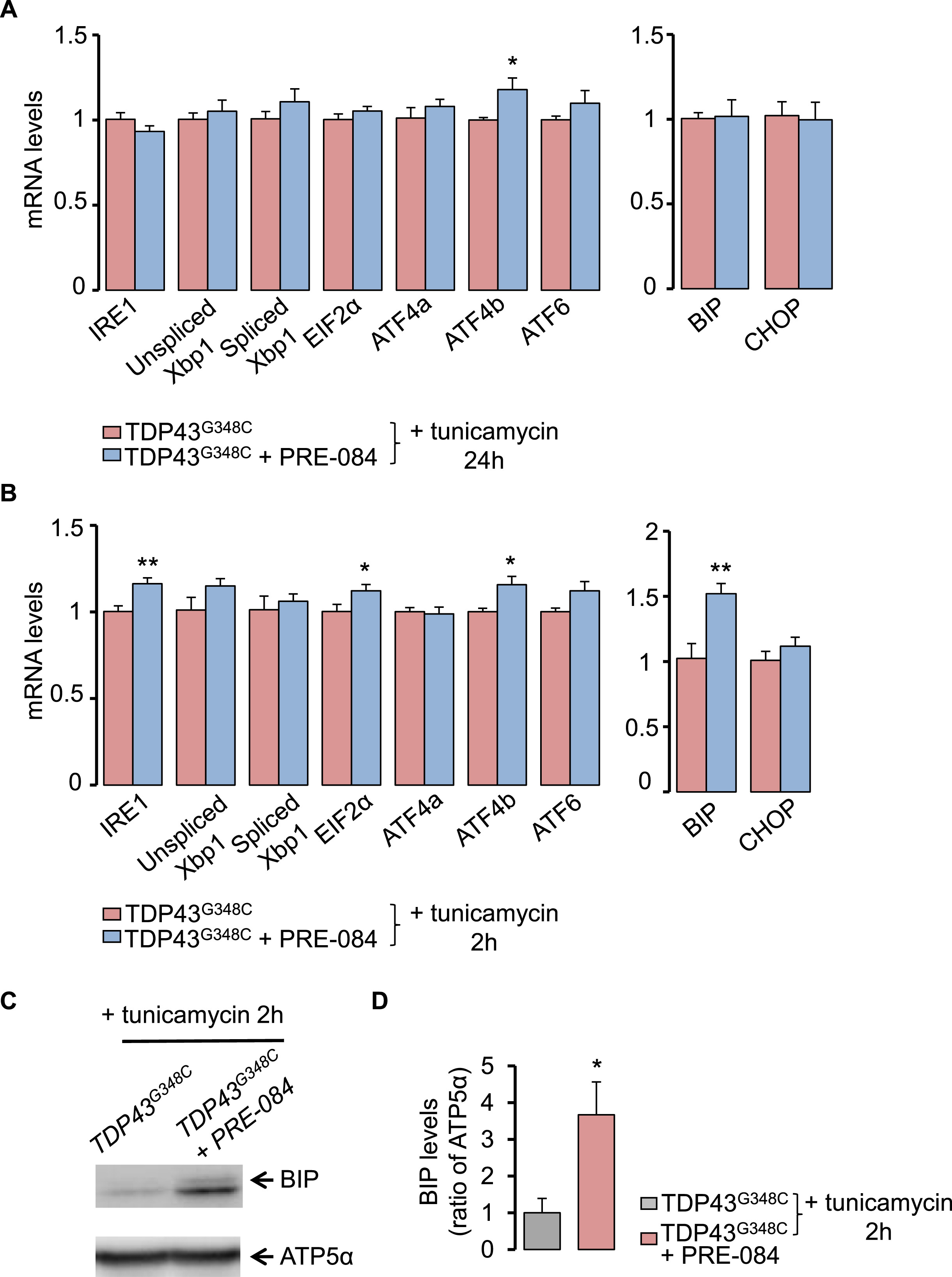
PRE-084 enhanced ER stress response in TDP43G348C-zebrafish.
(A) Transcript levels of UPR genes in TDP43G348C larvae, treated or not with PRE-084 after an ER stress induction with tunicamycin for 24 h. Data from n = 8–9 were presented as mean ± SEM. Statistical analysis was performed by using Student t-test (*p < 0.05). (B) As in (A) but after an ER stress induction for 2 h. Data from n = 6–7 were presented as mean ± SEM. Statistical analysis was performed by using Student t-test (*p < 0.05; **p < 0.01). (C) Western blot showing BiP and ATP5α protein levels larvae expressing TDP43G348C treated or not with PRE-084 after 2 h with tunicamycin condition. (D) Quantitative analysis of BiP protein levels in zebrafish larvae from (C). ATP5α was used as a reference to normalize BiP levels. Data from 8 to 9 independent samples were averaged and presented as mean ± SEM Statistical analysis was performed by using Student t-test (*p < 0.05).
Antioxidant NRF2 signalling pathway and ER stress response are closely related. In particular, PERK/EIF2α not only phosphorylates and activates NRF2 but also ATF4 was shown to promote NRF2 transcription [[36], [37], [38]]. We thus examined mRNA levels of the two NRF2 isoforms: NRF2a and NRF2b. Treatment of 5 dpf wild-type larvae with tunicamycin for 24 h led to a 17-fold increase in NRF2b but had no effect on NRF2a transcript levels (Fig. 4C). TDP43G348C did not affect levels of NRF2 levels in the presence or not of tunicamycin as compared to control.
3.4. PRE-084 stimulated ER stress response in TDP43G348C larvae
To better understand how S1R activation may prevent TDP43 toxicity, we studied the impact of PRE-084 on ER stress response after exposure of larvae with tunicamycin. Larvae were treated with 5 μM PRE-084 at 4 dpf for 24 h and were analyzed for RNA levels of canonical UPR genes. After treatment with tunicamycin for 24 h, PRE-084 only increased levels of ATF4b transcript in TDP43G348C-expressing larvae (Fig. 5A). Expression of UPR downstream genes, BiP and CHOP, remained unchanged (Fig. 5A). To determine if changes also occur and are more pronounced at shorter term during ER stress, a treatment of 2 h was performed with tunicamycin (Fig. 5B). In this condition, PRE-084 revealed significant increase in mRNA encoding ATF4b but also IRE1, EIF2α and BiP (Fig. 5B). Altogether this data indicates that PRE-084 may have beneficial effects by boosting UPR signalling pathway. Analyses at protein levels further showed that PRE-084 increased by 3.6-fold BiP levels in the TDP43 context (Fig. 5C and D).
3.5. PRE-084 activated antioxidant NRF2 pathway in TDP43G348C larvae
Next, we examined the effect of PRE-084 on NRF2 signalling pathway that controls antioxidant pathway. Transcript levels of NRF2a/NRF2b were found increased in the presence of PRE-084 with tunicamycin for 2 h or 24 h (Fig. 6A). NRF2 is known to activate expression of antioxidant key genes in glutathione-based system (GCLC, GCLM), thioredoxin-based system (PRDX6), NADPH-generating enzyme 6-phosphogluconate dehydrogenase (PGD), as well as on its inhibitor KEAP1. Consecutively to a 24 h treatment with tunicamycin, PRE-084 elevated mRNA levels of GCLC, PDG and KEAP1a isoform but to reduce PRDX6 levels (Fig. 6A). At a shorter term of 2 h, PRE-084 enhanced expression of GCLC and GCLM but reduced that of PRDX6 (Fig. 6B). The impact of PRE-084 on NRF2 levels was further studied in physiological condition. Interestingly, treatment of control larvae with PRE-084 for 2 h and 24 h increased levels of NRF2a/NRF2b and NRF2b transcripts, respectively (Suppl. Fig. 6). Altogether these observations strongly suggest a role of NRF2 signalling in the beneficial impact of PRE-084.
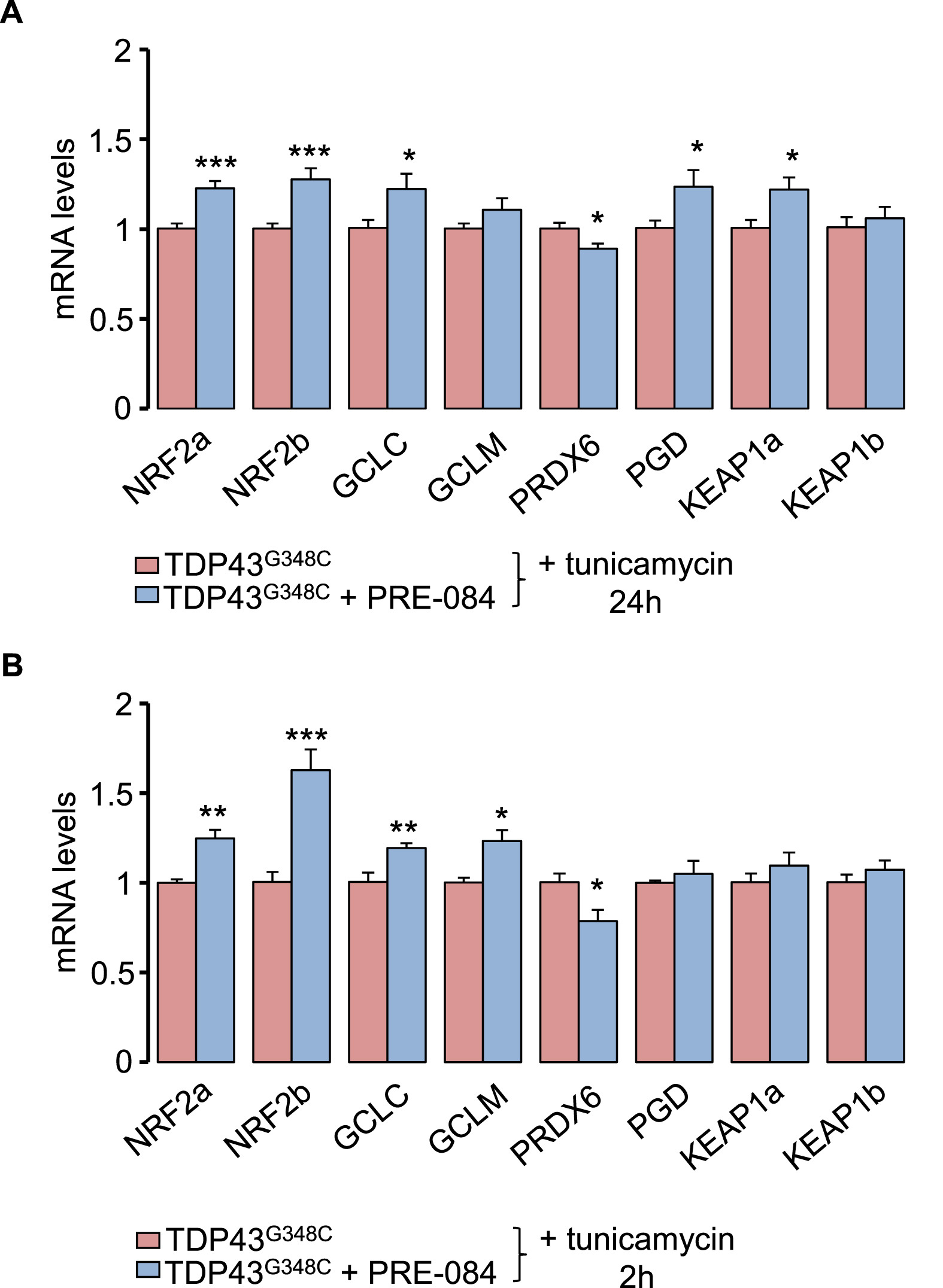
PRE-084 boosted NRF2 signalling cascade and alleviates TDP43G348C toxicity.
(A and B) RNA expression levels of NRF2 and its targets: GCLC, GCLM, PRDX6 and PGD in TDP43G348Clarvae treated or not with PRE-084 after an ER stress induction with tunicamycin for 24 h (A) or 2 h (B). Data from n = 8–9 (24 h) and n = 6–7 (2 h) were presented as mean ± SEM. Statistical analysis was performed by the Student t-test (*p < 0.05; **p < 0.01; ***p < 0.001).
3.6. Enhancing NRF2 expression ameliorated locomotor defects of TDP43G348C larvae
To further demonstrate that increasing NRF2 levels confers protection against TDP43 toxicity, we treated our ALS zebrafish model with sulforaphane, that was previously used to activate NRF2 signalling in zebrafish [39,40]. A treatment with 40 μM sulforaphane was sufficient to enhance mRNA expression of NRF2b as well as its targets GCLC, GCLM, PDG and PRDX6 (Fig. 7A and B). Surprisingly, NRF2amRNA levels were found decreased. Noteworthy, sulforaphane rescued the locomotor response of TDP43G348C– expressing zebrafish in the touch-escape test (Fig. 7C). Finally, we directly tested the impact of injected NRF2 zebrafish transcript together with TDP43G348C mRNA (Fig. 7D). We found a rescue of the locomotor escape response of TDP43G348C-expressing zebrafish (Fig. 7D).
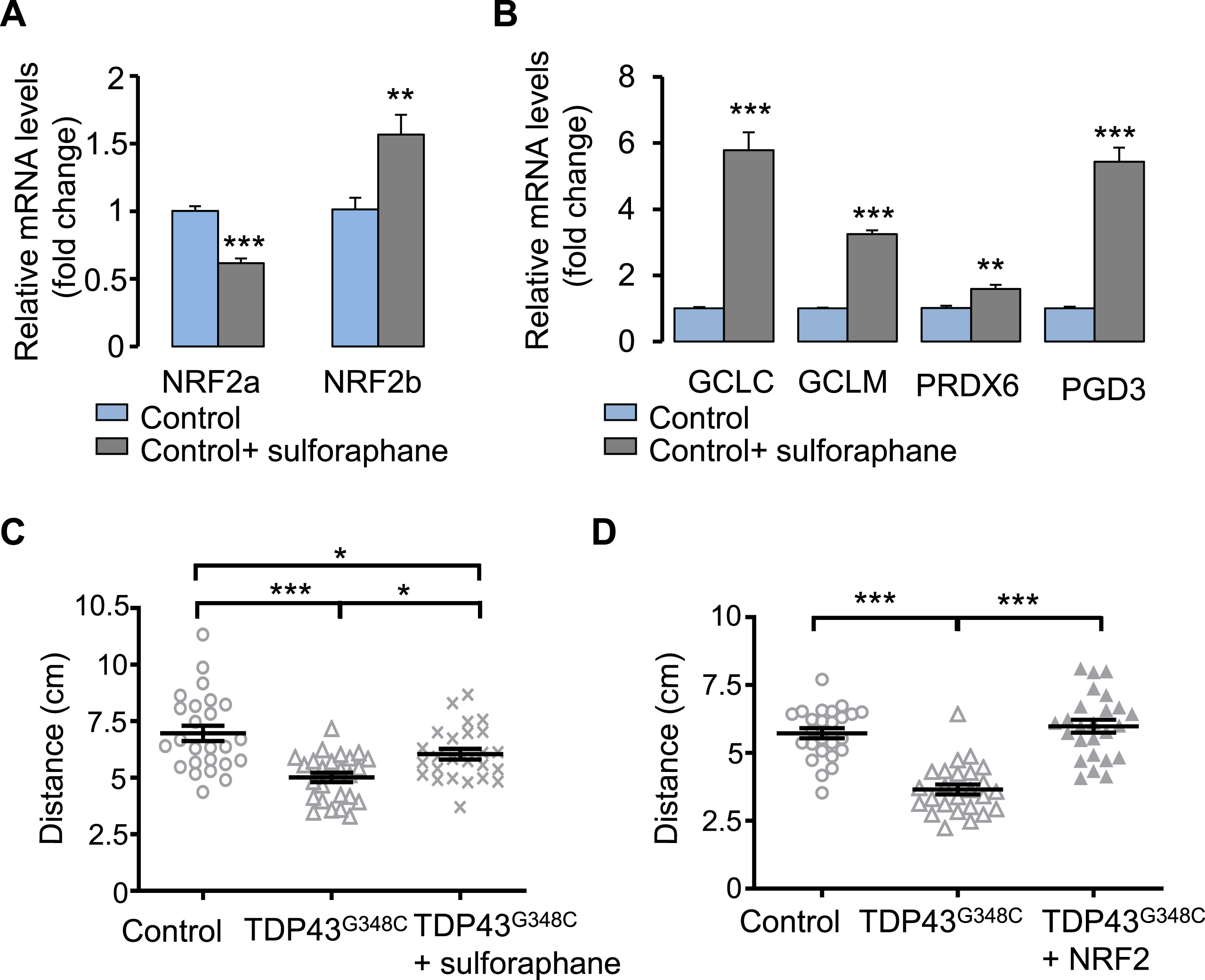
Increasing NRF2 levels reduced TDP43 toxicity.
(A) RNA expression levels of NRF2 in larvae treated or not with sulforaphane. Data from n = 6 were presented as mean ± SEM. Statistical analysis was performed by the Student t-test (**p < 0.01; ***p < 0.001). (B) RNA expression levels of NRF2 targets: GCLC, GCLM, PRDX6 and PGD in larvae treated or not with sulforaphane. Data from n ≥ 5 were presented as mean ± SEM. Statistical analysis was performed by the Student t-test (**p < 0.01; ***p < 0.001). (C) Touch-escape response of control larvae or larvae expressing TDP43G348C and treated or not with sulforaphane. Data from 25 were averaged and presented as mean ± SEM. Statistical analysis was performed by Tukey’s test (*p < 0.05; ***p < 0.001). (D) Touch-escape response of control larvae or larvae expressing TDP43G348C alone or with NRF2. Data from 25 were averaged and presented as mean ± SEM. Statistical analysis was performed using ANOVA followed by Tukey’s test (***p < 0.001).
4. Discussion
The interest of S1R in ALS is fostered by the fact that mutations in the gene result in juvenile cases of ALS [21,22]. Moreover, in the SOD1G93A mouse model of ALS, several S1R agonists were found to slow the progression of neuronal alterations or behavioral phenotype [[24], [25], [26], [27]]. Altogether these data made S1R agonists a promisingly potent therapeutic tool to counteract ALS pathology. However, it is now imperative to further validate the therapeutic concept in other ALS contexts and to ascertain their mode of action. By using genetic approaches in Drosophila, we previously demonstrated that overexpressing S1R reversed climbing deficits of TDP43-expressing flies [28]. We here showed in a vertebrate model that locomotion of zebrafish overexpressing human S1R are less affected by mutant TDP43 than controls. The purpose of the present study was to provide the proof of concept that the S1R activation by the agonist, PRE-084, is efficient to counteract TDP43 toxicity. While expression of mutant TDP43 leads to locomotor defects, PRE-084 treatment rescues performances not only in the touch-escape test but also the VMR. Moreover, we dissected some of the mechanisms underlying PRE-084 protective effects. Treatment with the agonist ameliorates maximal mitochondrial respiration in the TDP43 context. One the other hand, we demonstrated that TDP43G348C exacerbates ER stress by enhancing response to tunicamycin, resulting in increased levels of ER stress chaperone BiP and pro-apoptotic factor CHOP. Interestingly, PRE-084 treatment in the same condition further heightens BiP mRNA levels but not CHOP. Moreover PRE-084 stimulates EIF2α/ATF4 and NRF2 signalling, both known to promote antioxidant protection during ER stress. To further validate the protective effect of NRF2 in TDP43 context, we showed that overexpressing NRF2 alleviates locomotor defects to mechanical stimulation of TDP43G348C zebrafish. Additionally, we looked for potential benefits of sulforaphane, well known to enhance NRF2 signalling pathway, and again we found an amelioration of the touch-escape response of zebrafish expressing TDP43G348C. Altogether, these findings highlight the beneficial impact of activating S1R in ALS pathogenesis, boosting ER stress response and antioxidant cascade.
The preferential localization of S1R at the ER-mitochondria interface supports its action on mitochondria. At this domain its interaction with IP3R type 3 adapts calcium transfer from ER to mitochondria and thereby modulates production of NADH cofactor by intra-mitochondrial dehydrogenases [41]. The activation of these enzymes by calcium is considered important to stimulate mitochondrial respiration and hence matches ATP supply with increased energetic needs [42]. We showed that PRE-084 stimulates maximal oxidative mitochondrial respiration that was significantly reduced by the presence of TDP43G348C. In contrast, basal ATP-dependent respiration remained unchanged in the presence of TDP43G348C and/or PRE-084. It was previously reported that TDP43 may have a direct impact on mitochondria functioning, since cytoplasmic TDP43 could accumulate on the inner mitochondrial membrane and reduce complex I assembly thereby impairing ATP production [43]. Moreover, S1R agonists, including PRE-084, were found to directly potentiate complex I activity and mitochondrial respiration in physiological and pathological conditions [44]. Thus, S1R activation may confer protection by restoring mitochondrial respiratory capacity to fit with high energetic demand in TDP43 context. However, TDP43 toxicity results from a plethora of altered cellular processes, suggesting that rescuer effect of S1R may not solely be due to increased mitochondrial respiration.
In stress condition, S1R can activate the unfolded protein response, by acting on two UPR cascades: IRE1/X-box binding protein 1 (XBP1) and activating transcription factor 4 (ATF4) pathways [[12], [13], [14]]. Whether wild-type or mutant TDP43 triggers ER stress is still under debate [45]. Using NSC34 cells, it was found that TDP43 transfection did not activate IRE1/XBP1, ATF6 or PERK/ATF4 pathways while CHOP levels were increased [46]. Moreover, in HEK293 and primary motor neurons, mutant TDP43 failed to modify levels of spliced XBP1 and BiP [47], and even in rat forebrain neurons the authors noticed a decreased levels of unspliced/spliced XBP1 [48]. In contrast other studies in Neuro2a cells showed upregulation of XBP1, ATF6 and CHOP [49] while in SH-SY5Y cells they described higher levels of ATF4 and the ER stress effectors, BiP and CHOP [50,51]. All these discrepancies may be due to the different mutations and cell models that have been used and even likely the time-points of the observation. We showed here that induction of ER stress by tunicamycin increased transcript levels of all UPR genes but, more importantly, led to the higher exacerbation rate of IRE1/XBP1 cascade in TDP43G348C condition. This was accompanied by enhanced overexpression of BiPand CHOP, suggesting a higher vulnerability to ER stress in the presence of TDP43G348C. BiP promotes survival by alleviating accumulation of misfolded proteins while CHOP activates apoptosis when ER stress is persisting (For reviews see [52,53]). CHOP expression can be activated by both ATF4 and ATF6 ER stress arms [53]. In ER stress conditions, we showed that PRE-084 treatment further enhanced BiP but not CHOP levels and upstream significantly increased mRNA levels of IRE1, EIF2α and ATF4b. In previous work in a S1R deficient zebrafish line, transcript levels of IRE1 and PERK-related effectors like ATF4 were found decreased [54]. Also, ATF4 gene expression decreased after S1R silencing in transduced siRNA neurons [14]. We here provide evidence that, conversely, S1R activation can further increase UPR genes, IRE1 and ATF4, as well as the pro-survival effector BiP.
Besides ER stress, our data indicate that S1R fulfils a key role in activation of defense against oxidative stress. In physiological conditions, Keap1 binds NRF2 and then targets Nrf2 for ubiquitination and degradation by the ubiquitin proteasome system. In contrast during oxidative stress NRF2-Keap1 interaction is interrupted and NRF2 translocates to nucleus to activate expression of antioxidant genes. So far it has been reported that i) silencing S1R decreased NRF2 protein levels and led to oxidative stress in retinal cone cells, ii) S1R activation by ligands increased levels of NRF2 and NRF2-regulated antioxidant targets in retinal photoreceptors and Müller glial cells and iii) S1R colocalizes with NRF2 in photoreceptor cells, suggesting that NRF2 may be a direct target of S1R [[15], [16], [17],55]. To date, analyses were performed mostly in retina and it is of interest to determine if the effect of S1R on NRF2 is a common mechanism. We are here demonstrating that PRE-084 enhances NRF2 gene expression in physiological condition and in tunicamycin-induced stress. The impact on NRF2 targets seems to depend on the exposure time to tunicamycin, first affecting glutathione metabolism then NADPH levels with PGD and KEAP1 as negative feedback. The downregulation of PRDX6 mRNA is in accordance with a direct gene regulation of this peroxiredoxin by S1R since silencing S1R was previously reported to upregulate PRDX6 transcripts, although how remains to be established [14]. Impact of S1R on NRF2 may be one of the most significant mechanism in the multifaceted neuroprotective action of S1R against TDP43 toxicity. Accordingly, increasing NRF2 levels in zebrafish by sulforaphane and more directly by NRF2 RNA injection both rescue locomotor defects induced by mutant TDP43. It is noteworthy that while sulforaphane and PRE-084 increase similarly NRF2b mRNA levels, the impact on transcript levels of NRF2-regulated targets is much more pronounced after sulforaphane treatment. Both drugs may impact NRF2 signalling pathway through different mechanisms and not solely on transcript expression, among which phosphorylation by kinases and nuclear translocation of NRF2 protein are critical. Of interest, NRF2 was found to colocalize with S1R in photoreceptor cells, suggesting that NRF2 may be a direct target of S1R [56]. Future investigations should address the significance of this interaction in physiological condition or after activation by PRE-084. Moreover, we previously reported that S1R overexpression rescued the nucleocytoplasmic transport deficit caused by the ALS mutation in C9orf72 by stabilizing nuclear pore proteins and restoring the Ran gradient across nuclear membrane, that is essential for the nuclear import of RNA and proteins [20]. Since mutant TDP43 was also reported to alter nucleocytoplasmic transport [57], another alternative to explore is that PRE-084 may ameliorate nucleocytoplasmic transport of mRNA encoding NRF2 targets.
Altogether our data highlight the importance of NRF2 but also ATF4 in the beneficial impact of S1R activation. In general, ATF4 and NRF2 bind to their respective response elements, C/EBP-ATF response element (CARE) sequences and antioxidant responsive element (ARE), respectively, in response to distinct cellular stress. However, ATF4 is also described as part of the integrated stress response modulating crucial biological processes such as obviously ER stress but also antioxidant response and mitochondrial quality control [37,58,59]. Gene expressions of ATF4 and NRF2 is even intimately related, regulating each other [37,38,60,61]. Moreover, NRF2 form homodimers or heterodimers with other factors to stimulate targets. Several lines of evidence now indicate that ATF4 and NRF2 cooperatively stimulate genes controlling antioxidative defence, notably by forming heterodimers [36,62,63]. In future studies it would be very interesting to use morpholinos to downregulate NRF2 and ATF4, alone or together, to provide direct evidence of the synergic role of NRF2 and ATF4 in PRE-084-induced neuroprotection. While more work is needed to clarify all the mechanisms underlying S1R neuroprotection in TDP43 proteinopathy, our study confirmed the high therapeutic value of S1R agonists in ALS. An important issue in future investigations would be to understand the potential impact of S1R on the nucleocytoplasmic transport of NRF2 and its targets that has not been yet investigated in the TDP43 context.