
Novel compound inhibits glycolysis, proteotoxicity, inflammation, and impairments in animal models of Alzheimer’s, Huntington’s, and stroke: Aging as a consequence of glycolysis

By Rachel Litke, Kun-Hyung Roh, Yonejung Yoon, Sindhu Vangeti, Irene Ramos-lopez, Husnu Kaniskan, Jian Jin, Christopher Kellner, and Charles Mobbs
Excerpt from the article published in bioRxiv 2023.06.12, 544352; doi: https://doi.org/10.1101/2023.06.12.544352
Editor’s Highlights
- Glycolysis plays a major role in driving age-related diseases and mortality, and reduction of glycolysis mediates protective effects of dietary restriction and other manipulation on these diseases and mortality.
- The age-related increase in adiposity, and subsequent pancreatic decompensation leading to diabetes, is plausibly a consequence of age-related increase in beta cell glycolysis.
- Activating the Sigma-1 receptor, whose protective mechanisms in turn also entail inhibiting glycolysis.
- The Sigma-1 receptor robustly inhibits inflammation whereas ablation of the Sigma-1 receptor produces extreme vulnerability to mortality due to excess inflammation.
- The Sigma-1 receptor also reduces aerobic glycolysis and increases mitochondrial metabolism.
Abstract
Alzheimer’s Disease (AD) and other age-related diseases are increasingly among the most devastating strains on public health systems as the population ages, yet few treatments produce clinically significant protection. Although it is widely agreed that proteotoxicity drives impairments in AD and other neurological diseases, many preclinical and case-report studies indicate that increased microglial production of pro-inflammatory cytokines such as TNF-a mediate proteotoxicity in AD and other neurological conditions. The critical importance of inflammation, especially TNF-a, in driving age-related diseases is indicated by the fact that Humira, simply a monoclonal antibody to TNF-a, has become the top-selling drug in history, even though it does not cross the blood-brain barrier. Since target-based strategies to discover drugs to treat these diseases have largely failed, we developed parallel high-throughput phenotypic screens to discover small molecules which inhibit age-related proteotoxicity in a C. elegans model of AD, AND microglia inflammation (LPS-induced TNF-a). In the initial screen of 2560 compounds to delay Abeta proteotoxicity in C. elegans, the most protective compounds were, in order, phenylbutyrate (HDAC inhibitor), methicillin (beta lactam antibiotic), and quetiapine (tricyclic antipsychotic). These classes of compounds are already robustly implicated as potentially protective in AD and other neurodegenerative diseases. In addition to quetiapine, other tricyclic antipsychotic drugs also delayed age-related Abeta proteotoxicity and microglial TNF-a. Based on these results we carried out extensive structure-activity relationship studies, leading to the synthesis of a novel congener of quetiapine, #310, which inhibits a wide range of pro-inflammatory cytokines in mouse and human myeloid cells, and delays impairments in animal models of AD, Huntington’s, and stroke. #310 is highly concentrated in brain after oral delivery with no apparent toxicity, increases lifespan, and produces molecular responses highly similar to those produced by dietary restriction. Among these molecular responses are induction of CBP and inhibition of CtBP and CSPR1, and inhibition of glycolysis, reversing gene expression profiles and elevated glycolysis associated with AD. Several lines of investigation strongly supported that the protective effects of #310 are mediated by activating the Sigma-1 receptor, whose protective mechanisms in turn also entail inhibiting glycolysis. Reduced glycolysis has also been implicated in the generally protective effects of dietary restriction, rapamycin, reduced IFG-1 activity, and ketones during aging, suggesting that aging is at least in large part a consequence of glycolysis. In particular, the age-related increase in adiposity, and subsequent pancreatic decompensation leading to diabetes, is plausibly a consequence of age-related increase in beta cell glycolysis. Consistent with these observations, the glycolytic inhibitor 2-DG inhibited microglial TNF-a and other markers of inflammation, delayed Abeta proteotoxicity, and increased lifespan. To our knowledge no other molecule exhibits all these protective effects which makes #310 a uniquely promising candidate to treat AD and other age-related diseases. Thus it is plausible that #310 or possibly even more effective congeners could displace Humira as a widely used therapy for age-related diseases. Furthermore, these studies suggest that the efficacy of tricyclic compounds to treat psychosis and depression could be due to their anti-inflammatory effects mediated by the Sigma-1 receptor, rather than the D2 receptor, and that better drugs to treat these conditions, as well as addiction, with fewer metabolic side effects could be developed by focusing on the Sigma-1 receptor rather than the D2 receptor. These results strongly support the value of phenotypic screens to discover drugs to treat AD and other age-related diseases and to elucidate mechanisms driving those diseases.
Introduction
Glucose, as the salient product of photosynthesis, is the quintessential molecule around which life is organized. In the context of antagonist pleiotropy, however, most obviously reflected in the epidemic of diabetes, the threat of glucose used not wisely but too well, is vastly underappreciated. The present report seeks to remedy that paradox.
Life expectancy has increased from about 40 years in 1900 to about 80 years today, even excluding infant mortality without, unfortunately, a concomitant increase in healthspan [1]. Today morbidity and mortality are largely due to age-related chronic diseases, including dementias caused by Alzheimer’s, Parkinson’s, Huntington’s, and neurovascular (stroke) diseases, which generally entail, and are plausibly driven by, both age-related increased microglia inflammation (innate immunity), [2] [3] and age-related proteotoxicity [4, 5]. The age-related increase in chronic inflammation, sometimes referred to as inflammaging, “is considered the major risk factor for age-related diseases [6].” Furthermore, dietary restriction (DR), which generally delays age-related diseases and increases lifespan in a wide range of species and diseases, inhibits inflammation in humans and other species [7], and evidence supports that inhibition of inflammation mediates protective effects of DR [46] [8]. The functional significance of inflammation during aging is also supported by the fact that Humira, an antibody to the pro-inflammatory cytokine TNF-a, is the best-selling drug in the world by revenue, even though it does not cross the blood-brain barrier [9]. Furthermore, both inflammation [10] [11] and proteotoxicity [12], like cancer [13], entail and are arguably driven by increased glycolysis (the Warburg effect), and considerable evidence suggests that protective effects of DR are mediated by reduced glycolysis, reversing the Warburg effect in cancer as well as in inflammatory diseases [14–18]
Alzheimer’s Disease (AD) and other forms of dementia arguably produce the greatest strain of any condition on world-wide health systems, up to $1 trillion in 2018, affecting more than 50 million people [19]. The emotional toll on caregivers is also substantial [20] and there is generally agreement that dementia substantially reduces quality of life of both patients and caregivers [21]. Until recently four individual drugs and one combination drug have been approved for use in patients with AD [22], but the effect sizes of these drugs are relatively modest and the clinical significance of these effects, especially whether the benefits outweigh the harm, have been widely questioned [23]. For example, Eliti [24] simply states that “the efficacy of these is widely debated.” The limited efficacy of the approved drugs is probably because these treatments address symptomatic relief, rather than addressing causes of dementia. Clinical trials of other therapies, primarily targeting Abeta, the main constituent of disease-defining amyloid plaques, have generally failed [25, 26]. More recently the FDA approved an antibody against Abeta aggregates, but this was a highly controversial approval and scientific consensus is that this antibody is not clinically effective [27, 28].Thus development of pharmaceutical treatments for AD and other forms of dementia, which produce clinically significant amelioration of the condition, thus elucidating the causes of dementia, is of the utmost priority.
AD is clinically defined according to post-mortem analysis which quantifies the density of silver-stained plaques (consisting primarily of aggregates of the Abeta peptide) and tangles (consisting primarily of hyperphosphorylated Tau). There is general consensus that proteotoxicity involving both Abeta and Tau and possibly other proteins is largely responsible for driving AD pathophysiology [29]. Since neurodegeneration is the hallmark feature of AD, the neuron has understandably been the focus of AD research since the discovery of the disease by Alois Alzheimer [30]. Nevertheless, recent genetic studies have clearly implicated microglial activity, which serve as the primary driver of innate immunity in the brain, as a key driver of the disease [31]. This activity is plausibly driven at least somewhat independent of Abeta [32], although Abeta1-42 peptide does produce microglial release of TNF-a and IL-6 [33]. Of particular significance, numerous studies have demonstrated that inhibiting TNF-a ameliorates impairments in rodent models of AD [34] [35, 36] and even, according to case reports, in humans [37] [32, 34–55]. Particularly compelling, chemical ablation of microglia improves outcome in the 3XTg mouse model of AD [56], strongly implying that microglial hyperactivity (likely including increased secretion of TNF-α) plays a major role in driving the pathophysiology of AD.
Since target-based screens to discover compounds to treat AD and other age-related diseases have largely failed [26], whereas phenotypic screens allow the discovery of potentially novel mechanisms [57–60], we developed parallel high-throughput phenotypic screens to discover small molecules which delay impairments in animal models of AD, Huntington’s, and stroke, associated with reduced proteotoxicity, inflammation, and glycolysis, increase lifespan, and cross the blood-brain barrier after oral delivery with minimum toxicity. The effects of these small molecules were used to further elucidate mechanisms driving the pathophysiology of dementia with implications for further development of small molecules protecting against dementia.
Results
Initial screen for compounds which delay Abeta proteotoxicity in a C. elegans model of AD
Our initial screen used the Microsource Spectrum Collection (2560 compounds) in a C. elegans model (CL2006) of transgenic human Abeta1-42 proteotoxicity, which we [61] and others [62] have used to probe mechanisms mediating protective effects of DR on proteotoxicity in the context of the whole animal, since such effects may be cell non-autonomous [5]. This screen entailed administering each compound at 8 uM (lower concentration than in similar C. elegans screens [63]) in liquid medium cultivated with C. elegans strain CL2006 beginning at Adult Day 1. Abeta-induced proteotoxicity was quantified as paralysis at Adult Days 10, 11, 12, at which time about 97% of the CL2006 strain are paralyzed (wild-type C. elegans never exhibit this phenotype). Remarkably, the most protective compound (exhibiting only 12% paralysis compared to 97% controls) was phenylbutyrate (PBA), an HDAC inhibitor approved as a therapy for urea cycling disorder, an orphan disease, and which is being developed as a possible therapy for a wide range of neurological conditions [64] including AD [65–71]. An early indication of the potential for HDAC inhibitors to treat AD was our previous report that induction of CBP, a histone acetyltransferase (HAT), is required for the protective effects of dietary restriction and inhibited insulin-like signaling in a C. elegans model for AD, as well as lifespan, whereas the HDAC inhibitor sodium butyrate mimics those protective effects dependent on CBP [61]. These observations have been extensively corroborated and extended [72], including that the protective effects of CBP during dietary restriction are mediated by GABAergic neurons [73]. We further demonstrated that the protective effects of drugs which reduce glucose toxicity and proteotoxicity in the C. elegans model for AD and increase lifespan are also dependent on CBP as are the protective effects of dietary restriction and inhibition of glycolysis to delay proteotoxicity in a C. elegans model of Huntington’s Disease [74]
Since butyrate and phenylbutyrate inhibit several HDACs, we carried out an RNAi screen to determine which HDAC was most implicated in driving impairments in the C. elegans model of Alzheimer’s Disease. These studies indicated that RNAi inhibition of HDAC2 was most protective in delaying impairments in this model (Fig. 1A). We then screened several small molecules that were relatively specific to specific HDACs; the most protective of these HDAC inhibitors was Kevetrin, a relatively specific inhibitor of HDAC2 and HDAC6 (Fig. 1B).
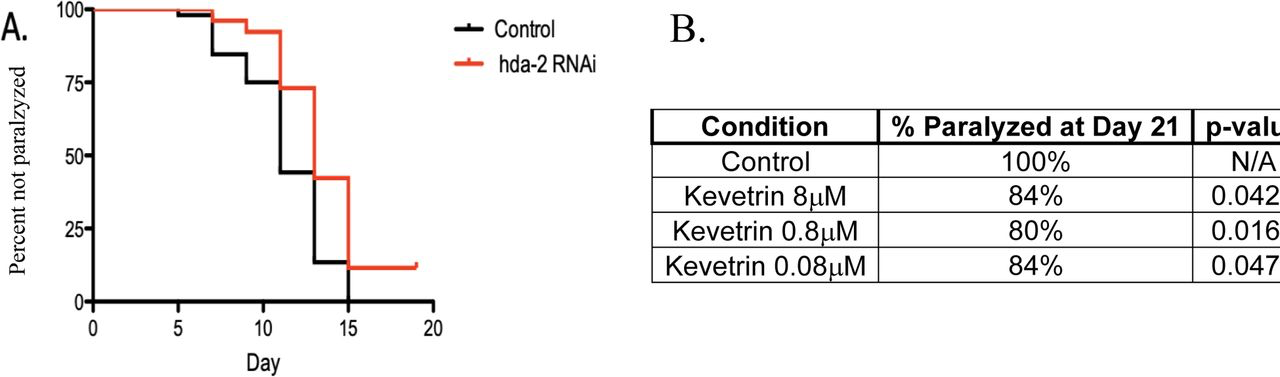
Genetic and pharmacologic HDAC2 inhibition delays impairments in a C. elegans muscle model of Alzheimer’s disease. (A) RNAi inhibition of HDAC2, but not RNAi of other HDAC isoforms, significantly (p<0.001) delayed impairments in a C. elegans model of human Abeta 1-42 (CL2006). (B) Kevetrin, a relatively specific HDAC2 inhibitor, but not other inhibitors relatively specific to other HDAC isoform, significantly (p-values shown in Table) delayed impairments across a range of concentrations.
The second-most protective drug to delay Abeta proteotoxicity in the C. elegans CL2006 model was the beta lactam antibiotic methicillin (17% paralyzed vs. 97% paralyzed controls). Beta lactam antibiotics are protective in a wide range of neurological conditions in animal models, including in animal models of AD [75–78] [75–77], although the mechanisms mediating these protective effects are not fully elucidated. We therefore assessed if other beta lactam antibiotics would also delay Abeta protoxicity in the C. elegans CL2006 model. These studies indicated that several other beta lactam antibiotics, including Cefproxil, Ampicillin, and Amoxicillin, also delayed Abeta proteotoxicity (Fig. 2), suggesting that this is a general property of beta lactam antibiotics.
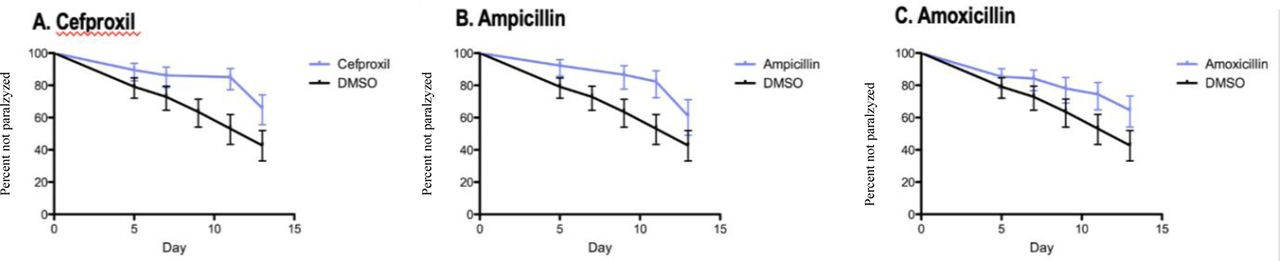
Beta lactam antibiotics significantly (p<0.001) delay Abeta proteotoxicity (% non-paralyzed/% survival) in C. elegans CL2006 model.
Finally, the third-most protective drug was quetiapine, a tricyclic atypical antipsychotic sometimes also used to treat depression (18% paralyzed vs. 97% paralyzed controls). As with the first 2 protective drugs, quetiapine delayed cognitive impairments and several indicators of pathology in a mouse model of AD [79].
As described above, microglial gene expression and in particular microglial secretion of inflammatory cytokines such as TNF-a, are increasingly implicated in driving the pathophysiology of AD [34–45, 80–82] and stroke [83–86]. A different cytokine, IL-6, has also been implicated in driving the neuropathology of both AD and stroke [87], suggesting that dementia in both conditions might be driven by a general increase in inflammatory cytokines. NFkappaB, the key transcription factor which generally induces pro-inflammatory cytokines, has also been directly implicated in the pathophysiology of AD [88] and stroke [89].
We therefore re-screened the 40 compounds which produced the most robust inhibition of Abeta-induced proteotoxicity in the C. elegans studies described above (from 12% to 33%, paralyzed, compared to 97% paralyzed in vehicle-treated controls) for efficacy to inhibit BV2 microglial secretion of TNF-a. BV2 cells were exposed to 5ug/ml of LPS for 24 hours for 24 hours, then 8 uM of the test compound which in the first screen inhibited Abeta-induced proteotoxicity, then supernatant was removed 24 hours later to measure cytokine release; cell viability was assessed using the MTT assay [90]. In our hands stimulating inflammation by adding LPS (1.25-5ug/mL) was a more reliable stimulus of inflammation than hAbeta1-42 [91]. 8 drugs were toxic as demonstrated by MTT, and were precluded from the analysis. As shown in Figure 3, all of the 32 compounds which inhibited Abeta toxicity in the C. elegansmodel of AD, without cytotoxicity in BV2 cells, also quantitatively inhibited TNF-a secretion (normalized to MTT), although inhibition was only significant in 23/32 compounds. This far from random overlap greatly supports that inhibition of the innate immune system (inflammation) mediates the protective effects of these compounds to reduce proteotoxicity. Further supporting this hypothesis, the two most inhibitory compounds (#3 and #77), and the fifth most inhibitory compound (#4) were cortisol, flumethasone, and dexamethasone, classic glucocorticoids which are best known for, and whose main clinical use is for, their robust inhibition of innate immunity. It is perhaps surprising that these compounds were also highly effective to inhibit proteotoxicity in C. elegans, but cortisol exerts protective effects in this species, at least in part by activating the longevity-enhancing gene Daf-16 [92], which turn is known to inhibit proteotoxicity in a C. elegans model of AD [93]. The other two most inhibitory compounds, #61 (Berbamine) and #18 (Emodin) have already been demonstrated to protect in mouse models of AD [94] [95] further validating the predictive power of our parallel phenotypic screening protocol. The other compounds which inhibit Abeta proteotoxicity and TNF-a secretion represent several classes of drugs approved to treat a wide range of different health conditions (see Discussion).
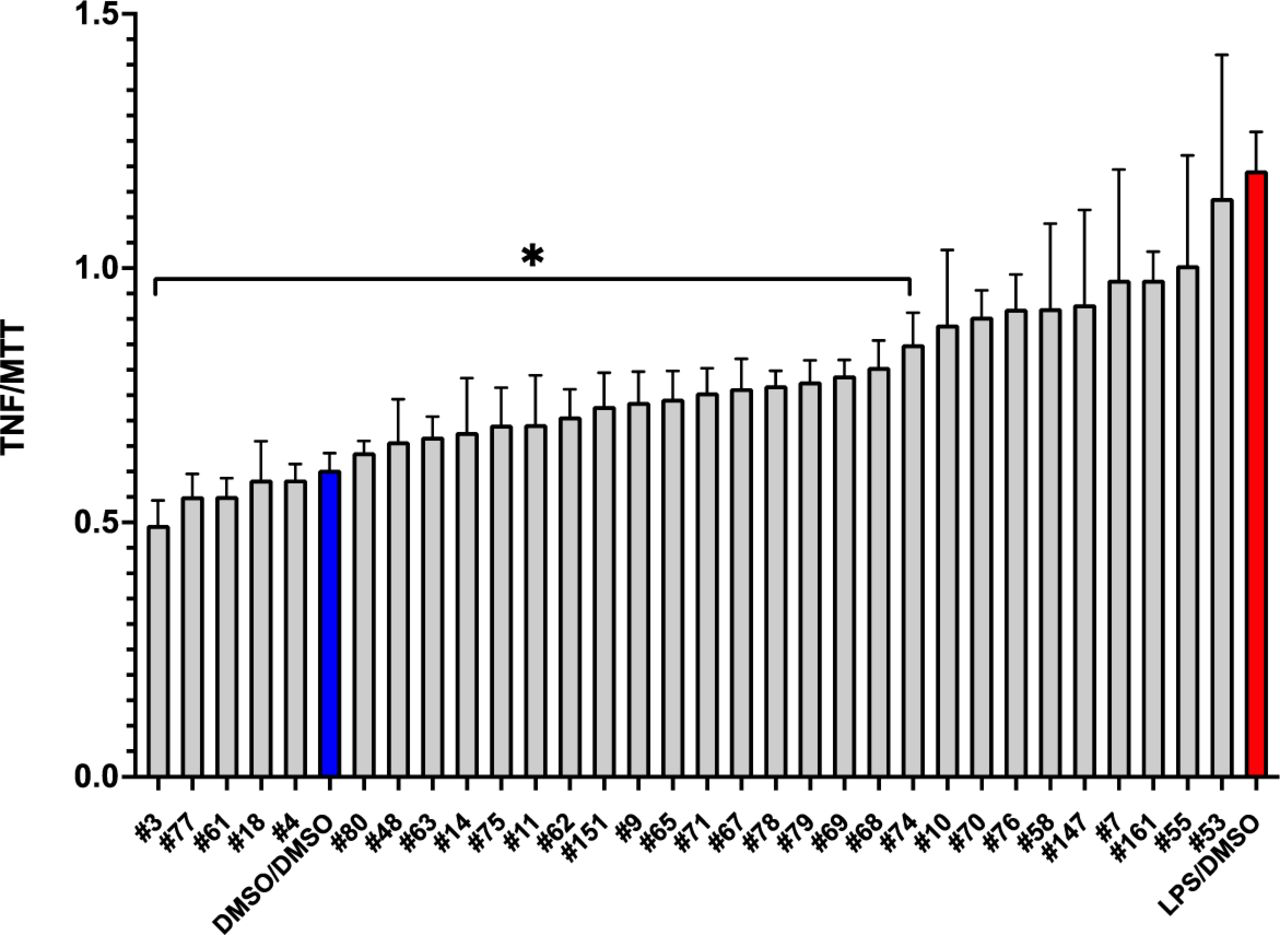
Inhibition of secreted TNF-a in LPS-stimulated microglia treated with most protective compounds. Secreted TNFa was evaluated in BV2 microglia stimulated with 5ug/mL LPS for 24 hours followed by 8μM drug for 24 hours using ELISA (A.). Selected compounds were the top protective compounds from pretreatment studies. *P < 0.05 vs LPS condition. (Only compounds which were not toxic were included in the analysis).
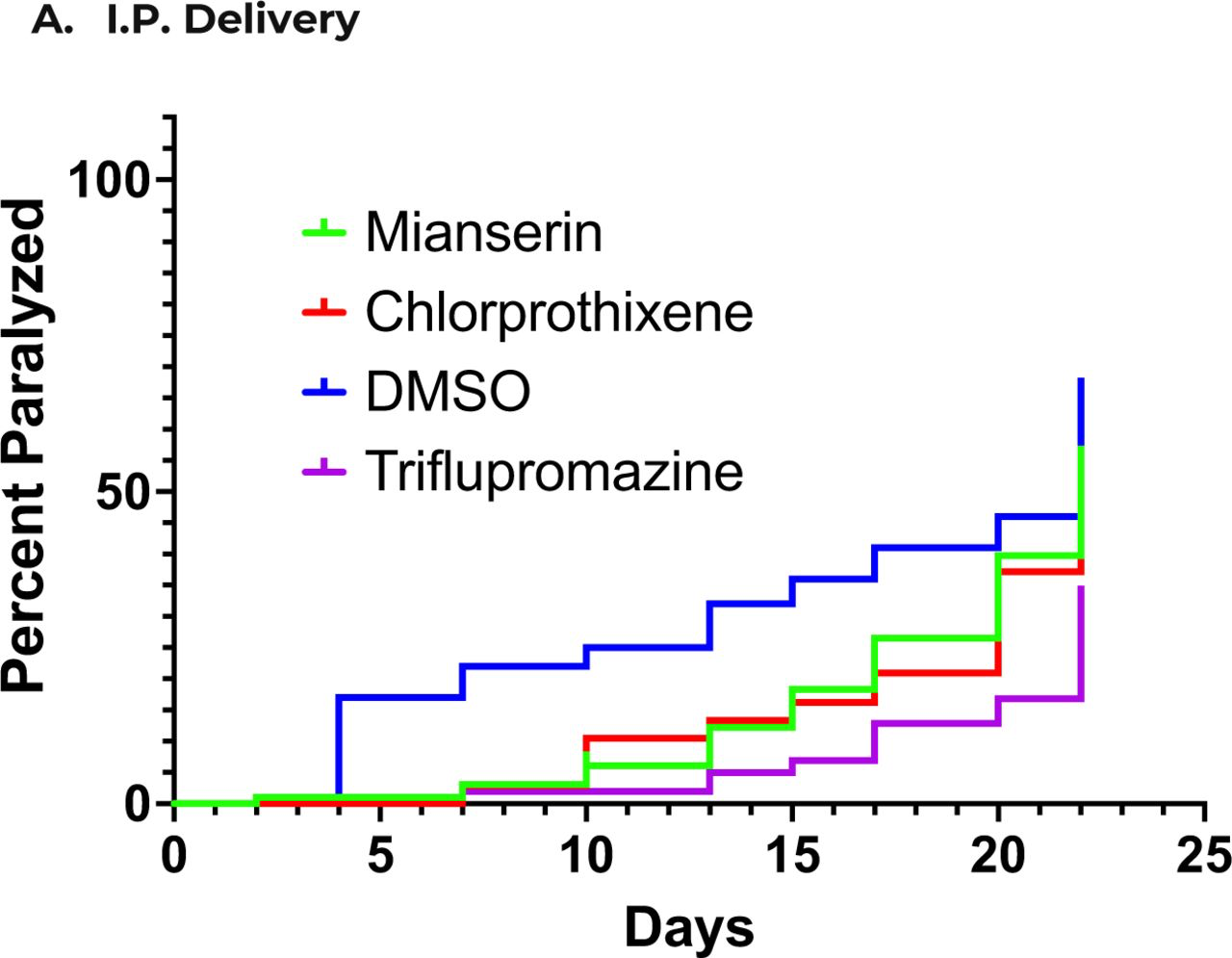
A key property of all potential treatments for treatment of AD is the requirement to cross the blood-brain barrier, preferably after oral administration to be effective in psychiatric conditions, they are plausible candidates to treat dementia, offering a much more effective (and certainly less expensive) alternative to currently approved anti-inflammatory treatments such as Humira. Tricyclic antipsychotic and antidepression drugs are obviously effective after oral delivery, and quetiapine reduces microglial inflammation [96]. We also observed that other tricyclic compounds, in addition to quetiapine, like chlorprothixene (#45) and triflupromazine (#75), were highly effective to protect against proteotoxicity and even to reduce TNF-a secretion from BV2 microglial cells (Figure 3). To develop novel compounds which might be even more effective and would be likely to cross the blood-brain barrier, and even potentially elucidate mechanisms driving age-related neurodegenerative as well cognitive impairments and neuropsychological conditions associated with AD, we carried out structure-activity relationship studies using congeners to quetiapine which we obtained from the NCI compound library [97, 98] to assess efficacy to reduce TNF-a secretion. Remarkably, as with the broader screen depicted in Figure 3, all of the non-toxic tricyclic congeners (as indicated by the MTT assay), also quantitatively reduced TNF-a secretion from BV2 microglia without producing toxicity although the results were significant for only about half of these compounds (Figure 4).
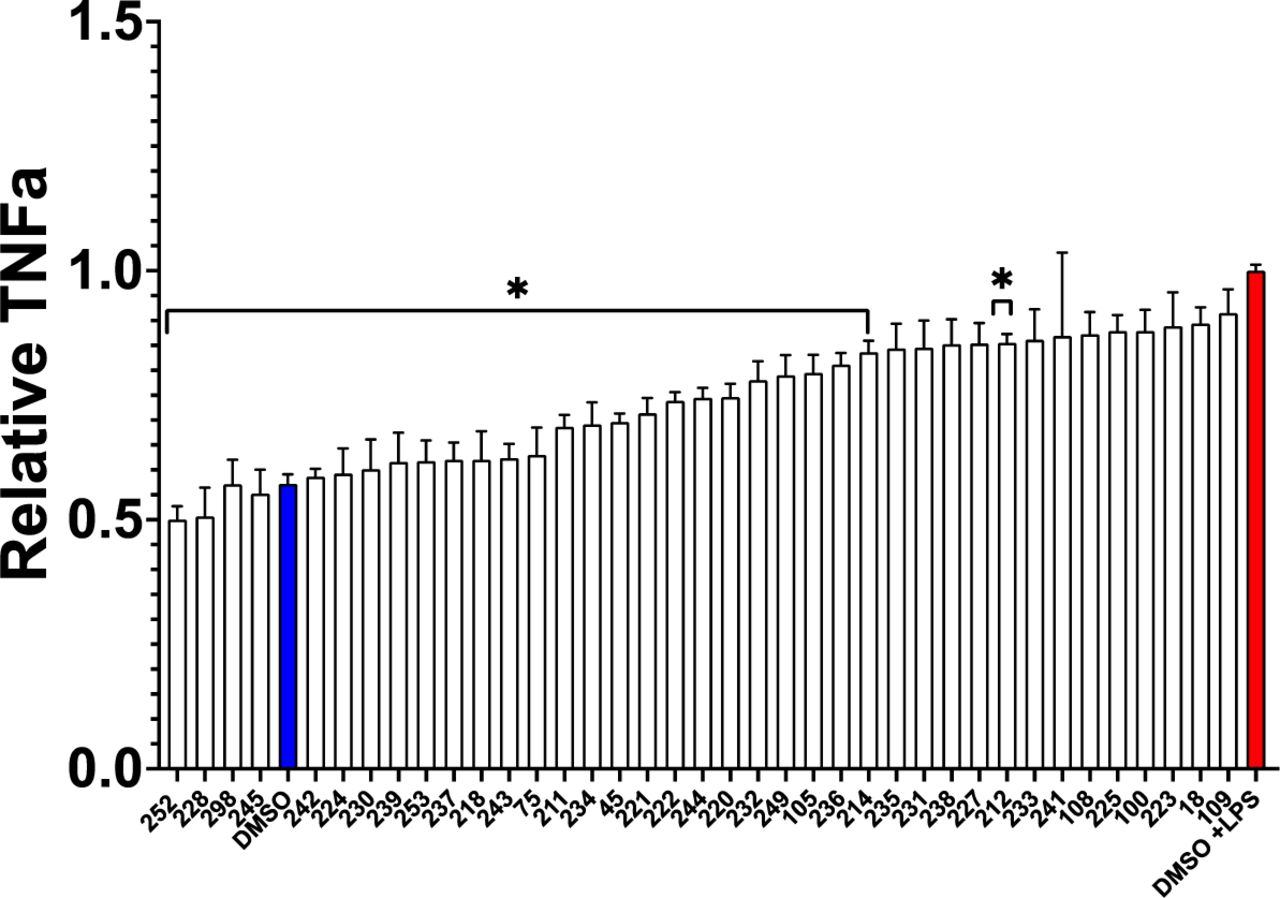
Inhibition of secreted TNF-a in LPS stimulated microglia treated with tricyclic congeners of quetiapine from the NCI library of compounds. Secreted TNFa was evaluated in BV2 microglia stimulated with 5μg/mL LPS for 24 hours followed by 8μM drug for 24 hours using ELISA (A.). None of the drugs affected viability as measured by MTT.
To further probe protective effects of tricyclic antipsychotic compounds, we assessed efficacy of several other tricyclic drugs approved by the FDA to treat psychosis to delay Abeta proteotoxicity in the C. elegans CL2006 model of AD. Remarkably, every tricyclic assessed in this assay significantly delayed Abeta proteotoxicity in this C. elegans model of AD (Figure 5), confirming and extending the observation of the protective effects of quetiapine in the same model.
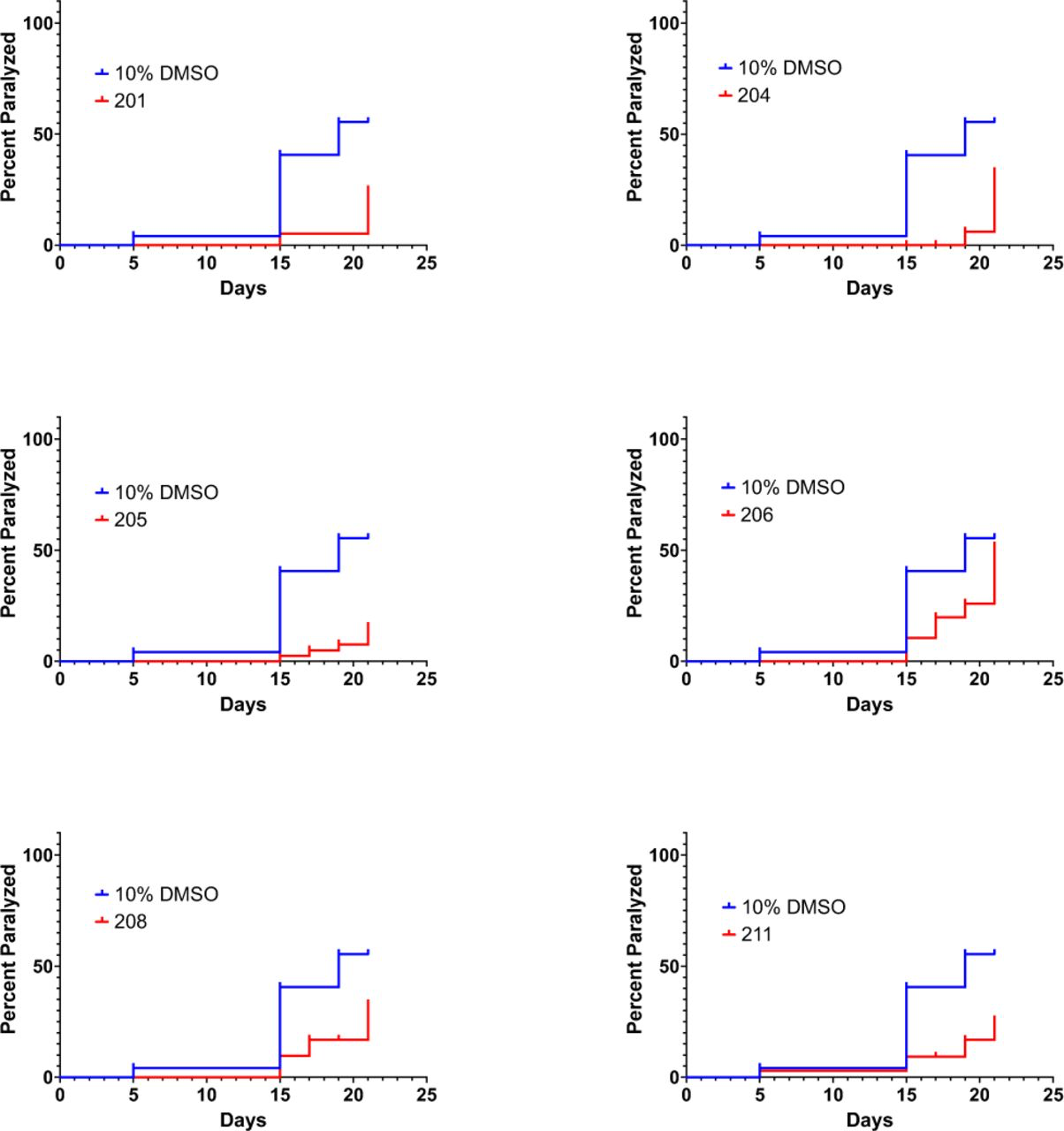
FDA-approved tricyclic antipsychotic drugs delay proteotoxicity in a C. elegans model of AD. All results are statistically significant (p<0.05).
To develop novel compounds even more protective with minimum toxicity, we used the results from these structure-activity relationships to predict novel tricyclic compounds with such protective properties, which were then synthesized and assessed for their protective properties. As shown in Figure 6, novel tricyclic compound #310 produced the greatest inhibition of TNF-a secretion without producing toxicity, as indicated by the MTT assay.
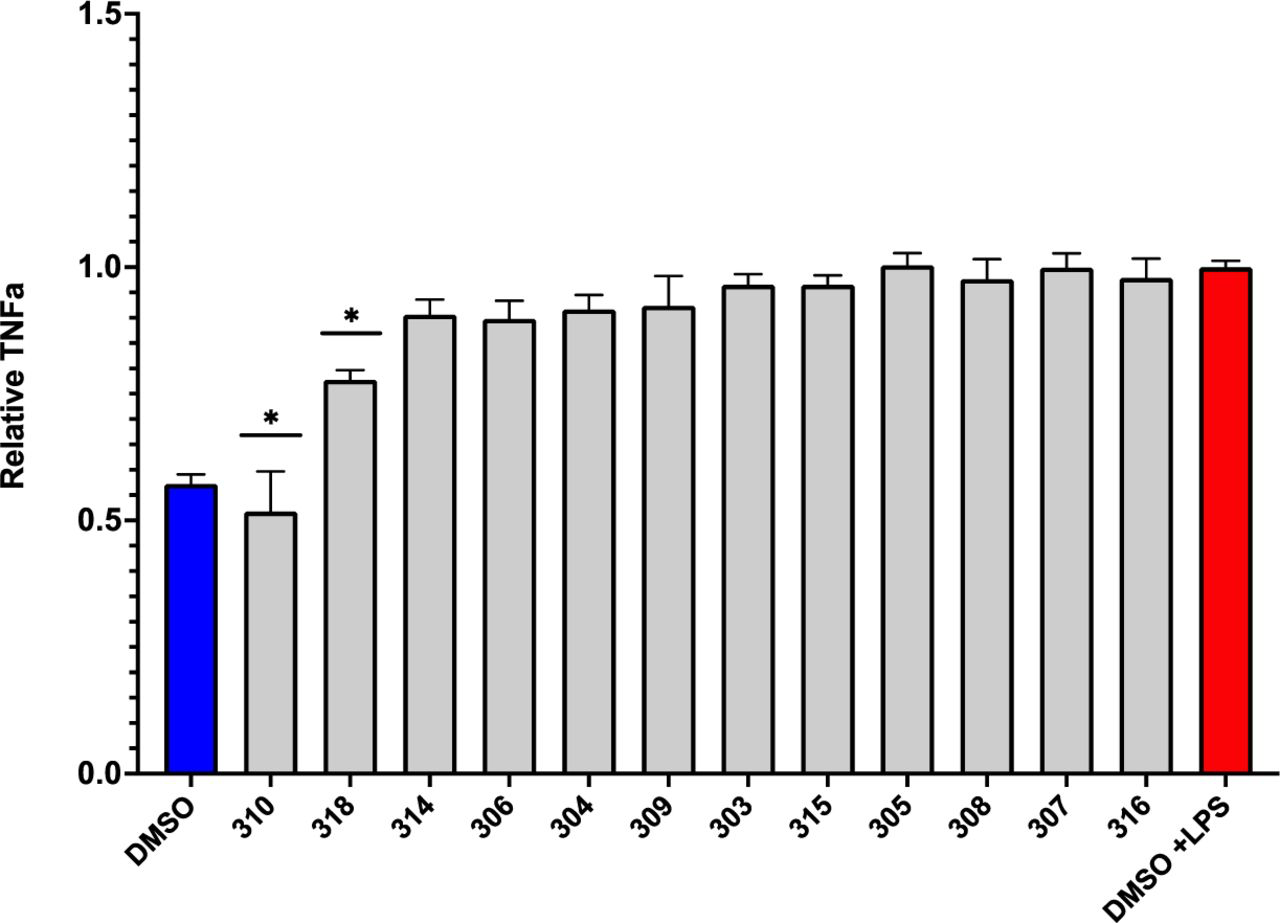
Of the novel tricyclic congeners to FDA-approved tricyclic drugs which inhibit LPS-induced microglial TNFa secretion, only #310 and #318 significantly reduced LPS-induced microglial TNFa (only compounds which were not toxic were included in the analysis).
We then further assessed the protective effects of these novel tricyclic compounds to delay Abeta proteotoxicity in the C. elegans model of AD. Compounds #45 (As shown in Figure 7, all 9 of of these novel compounds quantitatively delayed proteotoxicity at Day 15 in the C. elegans model of AD, although inhibition was only significant in 5/9 compounds. but only #310 also significantly inhibited TNF-a secretion. We therefore carried out a more extensive dose-response analysis of the effects of #310 over the entire lifespan of the CL2006 strain assessed every day until Day 24 of the adult phase. As shown in Figure 8, #310 produced a robust dose-dependent delay in paralysis in this C. elegans model of AD, although at the highest concentration the compound was no longer protective. We have observed such behavior in most of the dose-response studies we have carried out in many different assays, and have interpreted such results as indicating some toxicity at the highest doses. This interpretation is consistent with the FDA requirement that dose-response studies be carried for a sufficiently wide range of doses to be able to calculate not only ED50 but also LD50.
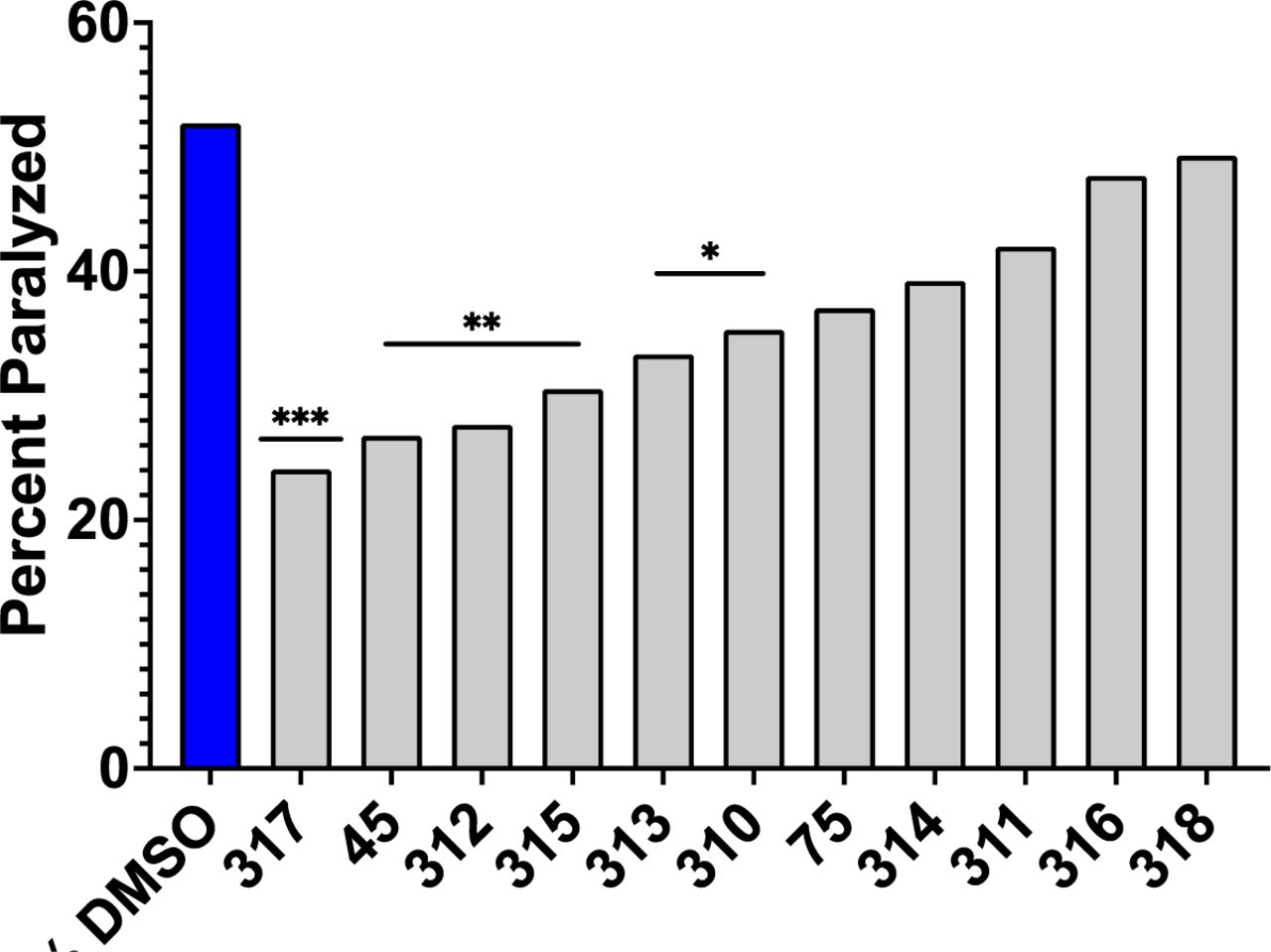
Of the novel tricyclic congeners, #317, 312, 315, 313, and 310 significantly inhibited Abeta proteotoxicity at day 15 in the C. elegans model of AD (*p<0.05,Chi-squared). Only #310 inhibited both microglial TNFa secretion AND Abeta proteotoxicity.
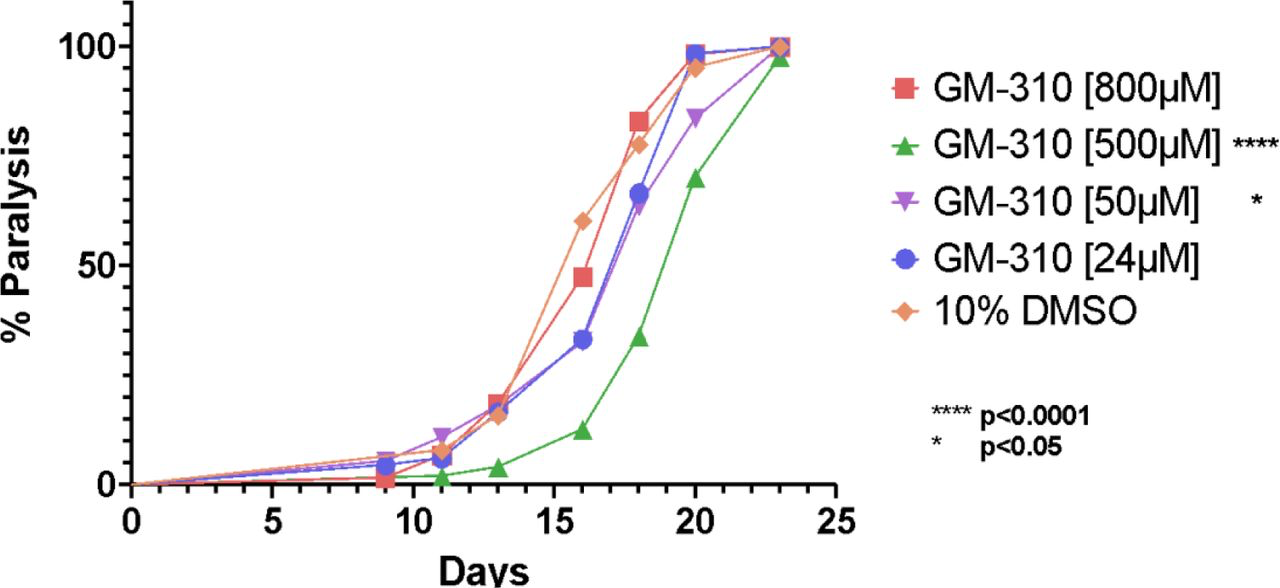
Novel tricyclic compound #310 produces dose-dependent delay in proteotoxicity the C. elegans model of AD.
Since AD entails at least two forms of proteotoxicity (Abeta and Tau), and likely other proteins as well, we have focused on the functional impairments (proteotoxicity) produced by transgenic expression of the human Abeta 1-42 peptide, rather than the levels of Abeta or Abeta aggregates. However, it would still be possible that the protective effects of the tricyclic compounds could be due to reduction of some aspect of Abeta which we did not measure. To address this issue, we assessed the protective effects of several tricyclic compounds in a completely different model of proteotoxicity produced by the disease-causing allele of the human Huntingtin protein, which we have used to extend our mechanistic studies of Abeta proteotoxicity [99]. As shown in Figure 9, several tricyclic compounds which inhibited BV2 cytokine secretion and protected in the C. elegans model for AD, were also protective in the C. elegans model for Huntingtin proteotoxicity. These studies indicate that the tricyclic compounds are generally protective against common pathophysiological processes that drive proteotoxicity.
Tricyclic compounds which reduce microglial secretion of TNFa and delay proteotoxicity in a C. elegans model of AD also delay proteotoxicity in a C. elegans model of Huntington’s Disease.
Although the tricyclic compounds, including #310, protect against proteotoxicity and inhibit microglial secretion of TNFa, a concern with small molecules is that they may nevertheless produce other off-target toxic effects which have not been assessed but could preclude clinical application. To address this concern, we examined effects of #310 on lifespan in the wild-type (N2) strain. As shown in Figure 10, #310 significantly increased lifespan in wild-type C. elegans. This result indicates that even if this compound produces some off-target deleterious effect we simply have not yet detected, it produces protective effects that more than compensate, thus increasing lifespan.
Novel tricyclic compound #310 also increases lifespan in wild-type (N2) C. elegans.
One of the major, yet often surprisingly overlooked, obstacles in developing drugs to treat neurological and psychiatric conditions, including dementia, is that “more than 98% of small molecule drugs and almost 100% of large molecule drugs are precluded from drug delivery to brain” [100, 101]. A major feature of the tricyclic compounds that have been approved to treat psychiatric conditions such as psychosis and depression is that in general and almost by definition these compounds cross the blood-brain barrier after oral administration [102]. We therefore assessed if compound GM310 would be concentrated in the brain after i.p. or oral delivery. As shown in Figure 11, #310 is indeed highly concentrated in the brain after either i.p. or oral delivery.
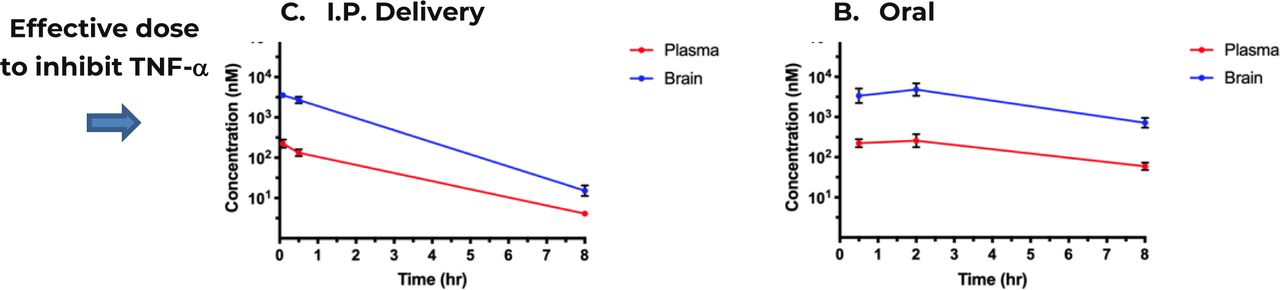
Plasma and brain concentrations of GM310 over 8 hours following a single IV injection (A) at 1 mg/kg or oral administration (B) at 10 mg/kg in male C57Bl/6J mice. Plasma and brain (cortical) concentrations at each time-point represent the mean +/− SD from 3 mice. Arrow indicates effective dose of GM310 to block impairments in a mouse model of stroke (Fig. 10).
To assess the potential efficacy of #310 to treat dementia, we assessed efficacy of the compound to ameliorate behavioral impairments, including memory, in a mouse model of intracerebral hemorrhage (ICH) [103]. Although dementia due to AD is more common than vascular dementia, post-stroke cognitive impairment and dementia (PSCID) is a major source of morbidity and mortality after stroke, including ICH [104]. Furthermore, vascular dementia and dementia due to AD appear to be driven by similar pathophysiological mechanisms, including a role for ApoE4 [105, 106] which entails inflammation, a mechanism common to these forms of dementia [107] and it has been concluded that interventions effective for one would likely be effective for the other [108, 109]. More specifically, ApoE4 promotes the secretion of microglial TNFa which in turn reduces neuronal viability, compared to other ApoE alleles [81]. This observation is particularly pertinent since ApoE is the major genetic risk factor for AD, and many studies have demonstrated that increased TNF-a is a major contributor to the pathophysiology of both AD [34–45, 80–82] and stroke [83–86]. A different cytokine, IL-6, has also been implicated in driving the neuropathology of both AD and stroke [87], suggesting that dementia in both conditions might be driven by a general increase in inflammatory cytokines. NFkappaB, the key transcription factor which generally induces pro-inflammatory cytokines, has also been directly implicated in the pathophysiology of AD [88] and stroke [89], suggesting that an even wider range of cytokines might similarly promote neurotoxicity in both conditions. As stated by Zhou et al., “These results confirm that AD and ICH may have common pathogenesis and share preventive treatment measures” [110]. Furthermore, assessing efficacy of interventions in mouse models of AD requires significant investment in time and funds, yet these models continue to be highly controversial [111]. In contrast mouse models of ICH are generally accepted as likely predictive of effects of interventions for ICH in humans [103, 112, 113], and such studies require far less investment in time and funds than for mouse models of AD, so more suitable to assess feasibility.
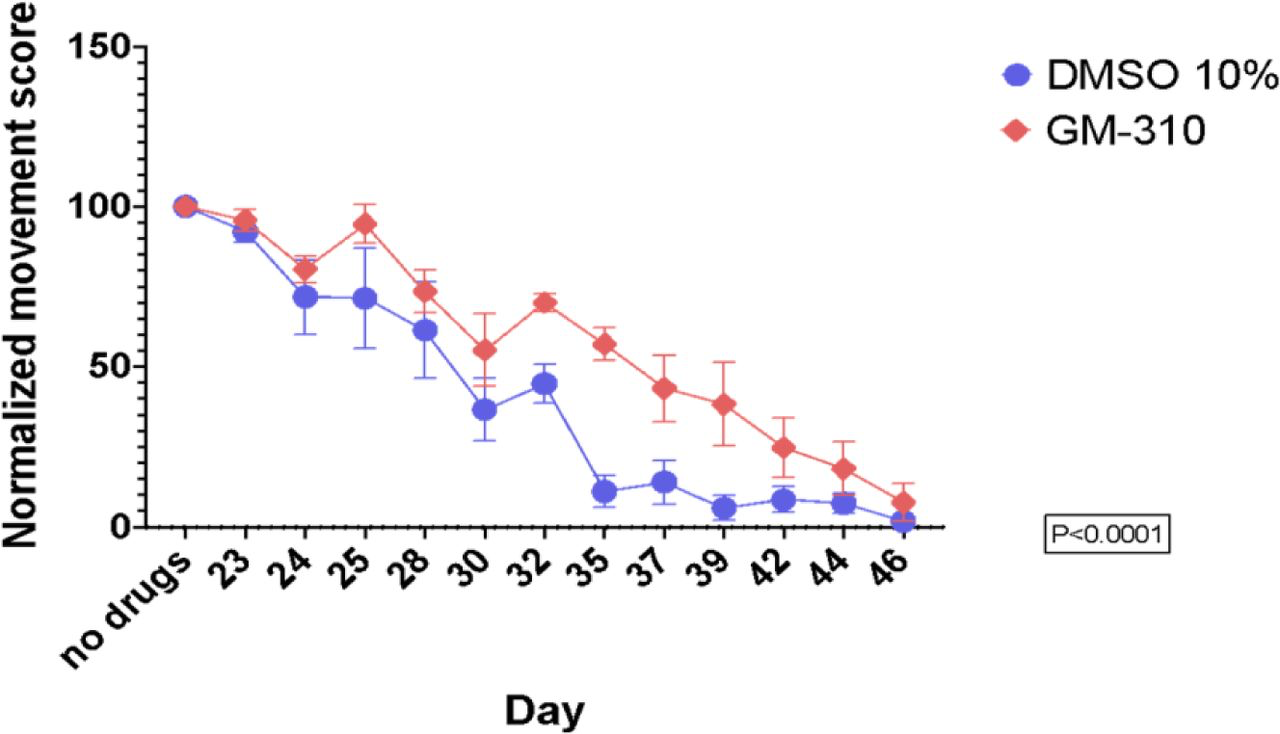
We therefore assessed efficacy of #310 to ameliorate impairments, including memory impairments, in a mouse model of ICH [103]. In this study ICH was produced by autologous infusion of whole blood into the mouse striatum, followed immediately by infusion of 10 nM #310 into the same area. As shown in Figure 12, infusion of 10 nM #310 completely prevented behavioral impairments and inflammation (increased TNF-a) after ICH.
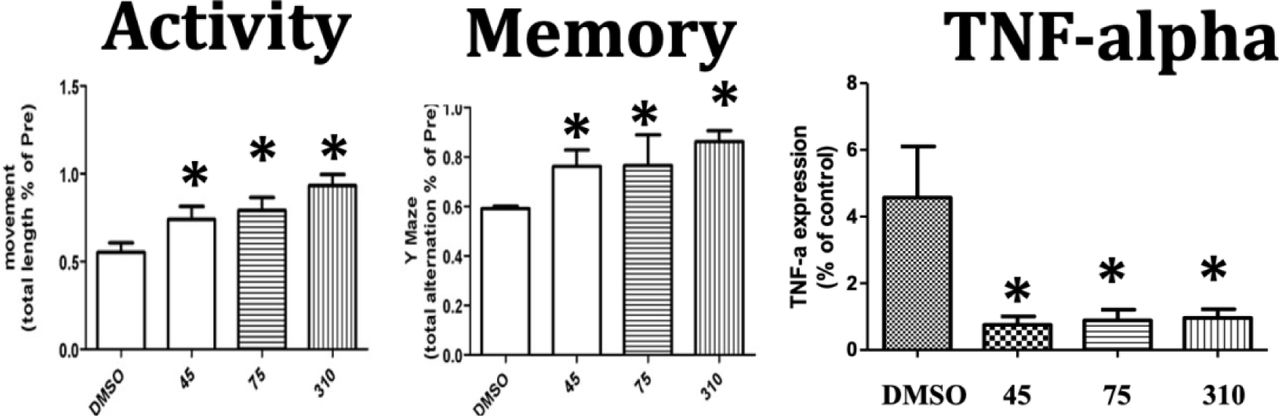
Infusion of 10 GM310 into site of ICH completely protects against impairments in activity and memory and increased TNFa concentrations at the site of the ICH (*p<0.05).
As described above, TNF-a and inflammation in general have been implicated as driving pathophysiology of AD [34–45, 80–82] and stroke [83–86]. A different cytokine, IL-6, has also been implicated in driving the neuropathology of both AD [42] and stroke [87]. We therefore assessed the extent to which #310 generally inhibits the microglial (BV2) secretion of pro-inflammatory cytokines, assessed by the Luminex Cytokine Array multiplex assay. As shown in Figure 13, #310 inhibited microglial secretion of almost all proinflammatory cytokines in this assay, corroborating and extending previous studies focused on TNF-a and IL-6, suggesting that #310 might be clinically useful to treat a wide variety of conditions in which inflammation has been implicated, including dementia due to AD and vascular dementia (see above). The wide range of cytokines inhibited by #310 is consistent with the hypothesis that #310, like other tricyclic compounds, inhibits NFKappaB expression by inhibiting the transcription complex that stimulates NFKappaB expression [97]. A plausible mechanism mediating these effects is that #310 activates the Sigma-1 receptor, which in turn inhibits the production of NFKappaB [114] (see below).
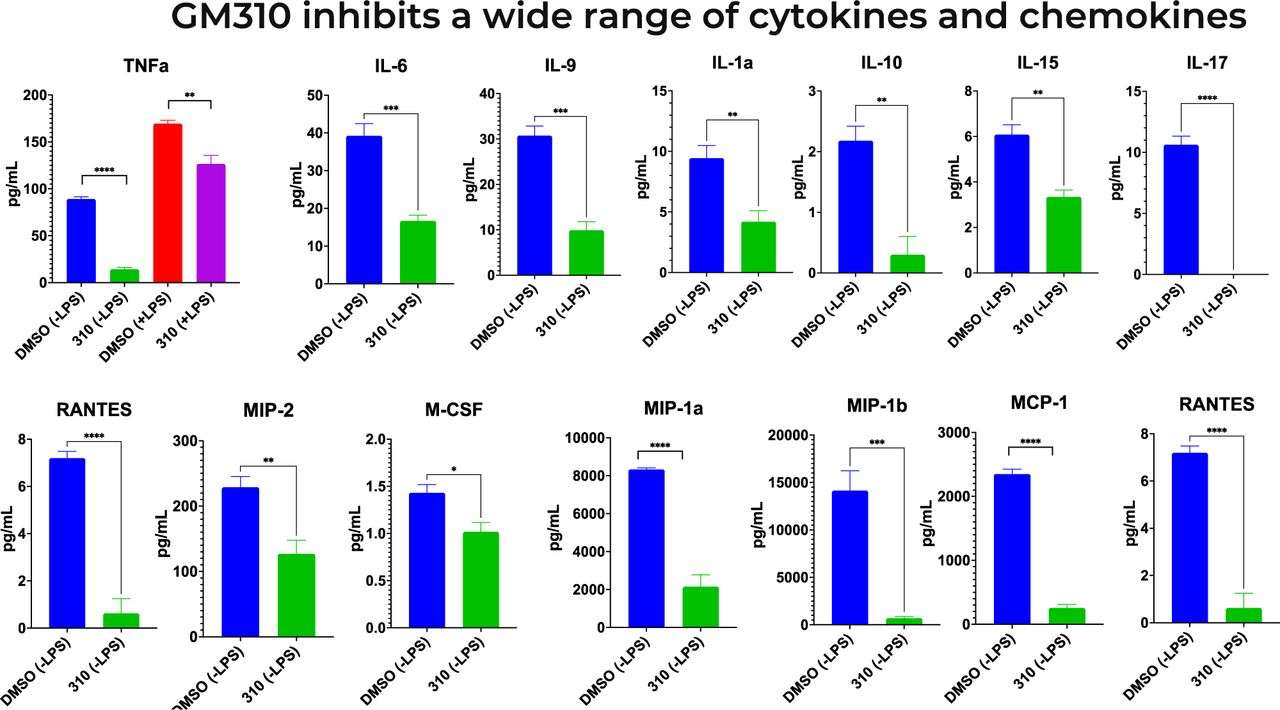
Novel tricyclic compound GM310 inhibits BV2 microglial secretion of cytokines. Supernatant was taken 48 hours after addition of 8uM drug and 24 hours of LPS stimulation using Luminex Cytokine Array multiplex assay. *** P < 0.0001, treated condition vs. LPS control.
Although it is now widely agreed that GWAS studies clearly implicate microglia gene expression as driving major aspects of the pathophysiology of AD, and as described above the evidence is overwhelming that enhanced microglial inflammation similarly drives major aspects of the pathophysiology of many forms of dementia [34–45, 80–82] and stroke [83–86], it is plausible that impaired microglial repair mechanisms, especially phagocytosis, could also contribute to these impairments [115]. Thus it is possible that if a compound reduces microglial inflammation but also reduces phagocytosis, the net effect could be that these processes in effect cancel each other out, or even worse, if impaired phagocytosis is quantitatively more important than enhanced inflammation, such a compound could conceivably worsen impairments in dementia. We therefore assessed the effect of the compounds which inhibited both Abeta proteotoxicity AND microglial cytokine secretion on microglial phagocytosis. Two of the tricyclic compounds, #228 and #246, did reduced phagocytosis, so would not be good candidates to develop to treat dementia (Fig. 14). The only tricyclic compound which inhibited microglia inflammation and also increased microglial phagocytosis is #310 (Fig. 14), which further supports that this novel compound is the most plausible lead so far to treat AD and other forms of dementia.
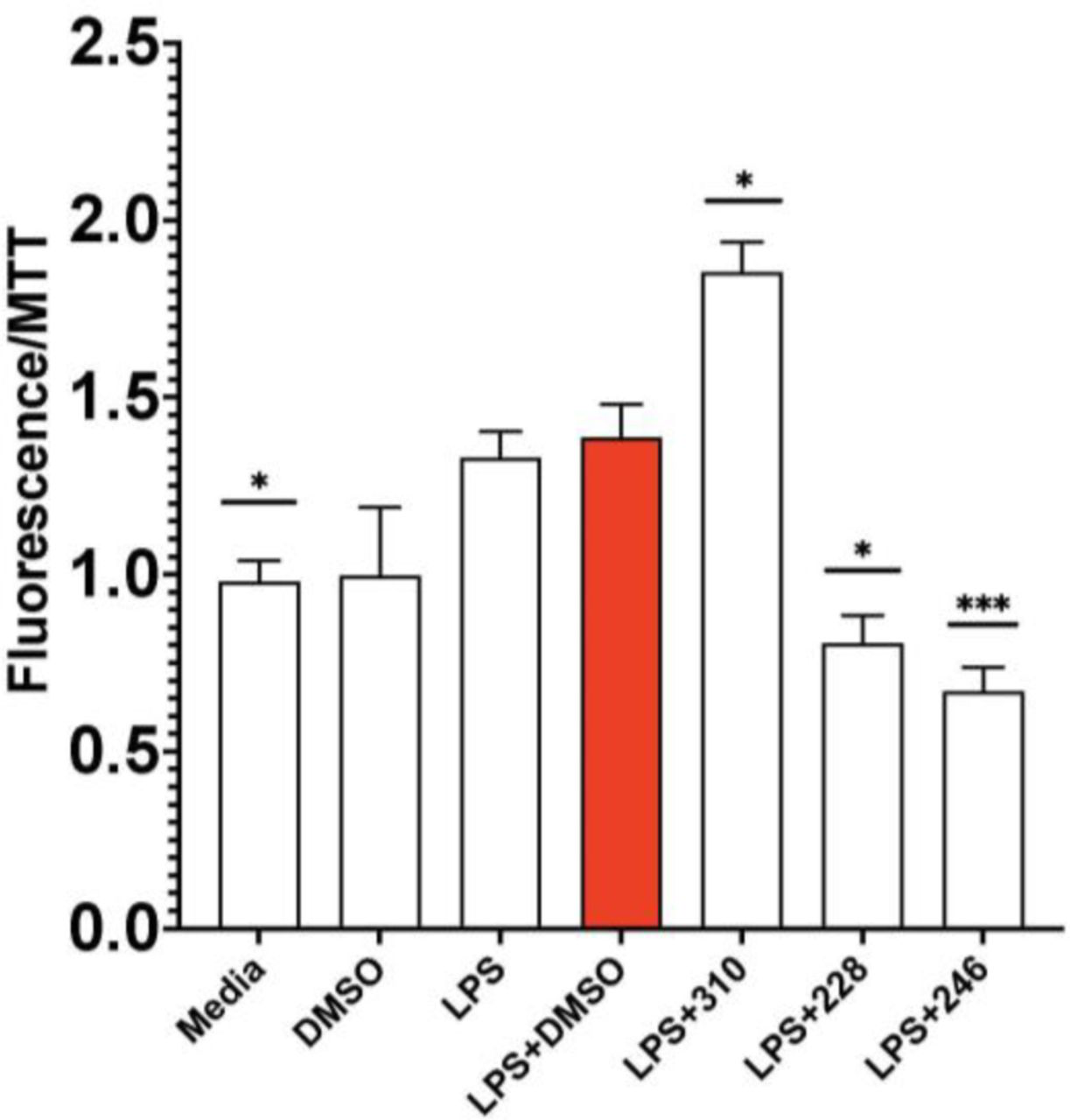
GM-310 is only compound tested which decreased inflammation and increased phagocytosis in microglia.
To assess potential efficacy of #310 and its congeners to treat dementia in human patients, we assessed if these compounds would also inhibit LPS-induced TNF-a secretion in human primary monocytes (Figure 15) and human PBMCs (Figure 16). As shown in Figure 15, almost all of the test compounds significantly inhibited TNF-a secretion from human monocytes. Similar results were obtained using PBMCs isolated directly from blood of patient volunteers (Figure 16). Of particular interest, #310 was the most effective compound to reduce TNF-a in human PBMCs.
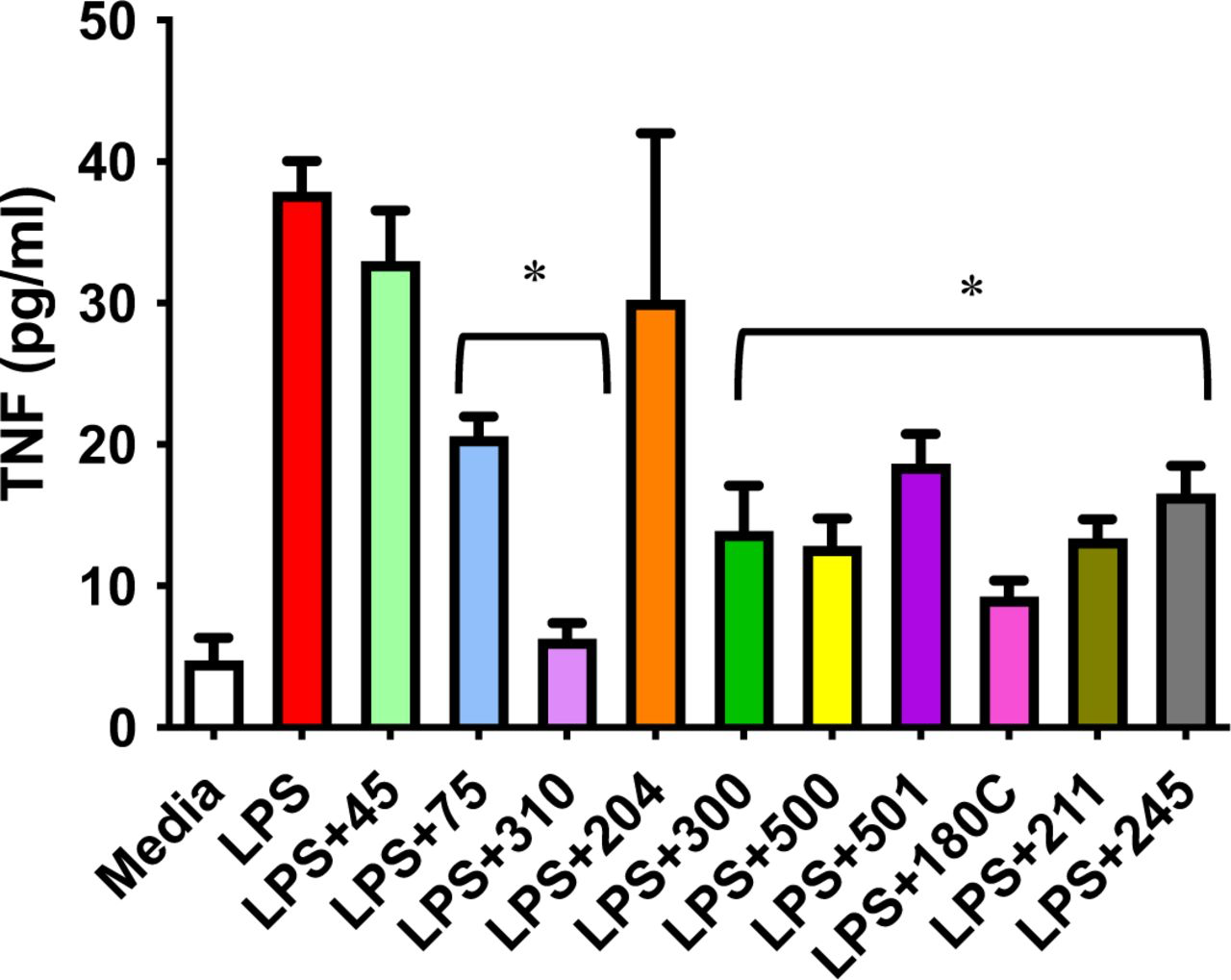
GM310 and congeners inhibit LPS-induced secretion of TNFa in human primary monocytes (effects of compounds 75, 310, 300, 500, 501, 180C, 211, and 245 all p<0.01).
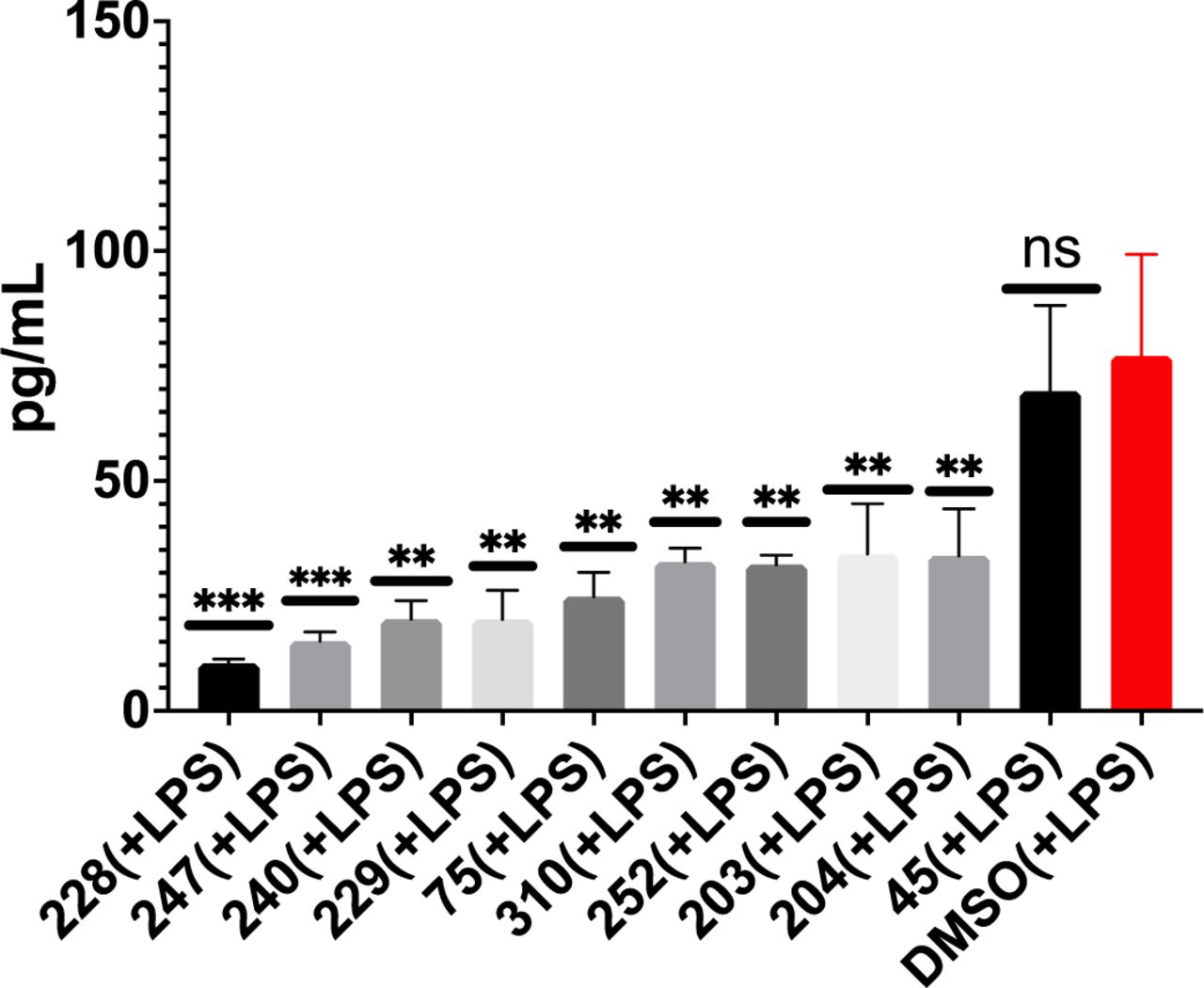
GM-310 and congeners inhibit LPS-induced secretion of TNFa in human PBMCs.
Further examination of the effects of these FDA-approved tricyclic drugs on gene expression in human cells using the CMAP/L1000 database [116] indicated that among the most robust responses to these genes were inhibition of transcriptional co-inhibitor related genes CtBP1 and CtBP2. Since at least at the allosteric level CtBP activity is stimulated by glycolysis-derived NADH is of interest to us because its transcriptional co-inhibitor activity is mediated at least in part by inhibition of CBP activity [117], which we have shown mediates protective effects of dietary restriction. Furthermore, CtBP is allosterically activated by glucose metabolism, specifically via NADH [118], which we have argued is the metabolite most responsible for deleterious effects of glucose metabolism during aging and diabetes [14]. Thus it is of particular interest that inhibition of CtBP increases lifespan by enhancing lipid metabolism at the expense of glucose metabolism [119], precisely the metabolic profile produced by dietary restriction and mediated by CBP [118]. Remarkably, this life-extending effect is mediated by CtBP expressed in neurons [120], as are the life-extending effects of CBP. Of particular interest to the present study, activation of CtBP by NADH promotes inflammation by enhancing transcriptional activity of NFKappaB, apparently by inhibiting the opposite effects of CBP [121]. by allosteric inhibits the transcriptional co-activator CBP, whose induction we showed mediates the protective effects of dietary restriction on lifespan and proteotoxicity in the CL2006 C. elegans model of Abeta proteotoxicity [61], We also showed that hypothalamic expression of CtBP inversely correlates with lifespan, opposite the relationship between CBP and lifespan [61] consistent with reports that neuronal CtBP shortens lifespan [120], the opposite of CBP [73]. As a co-transcriptional inhibitor, CtBP also recruits HDACs to the chromatin to inhibit transcription, and as described above, we [73] and others have demonstrated that HDAC inhibition is highly protective in a wide range of models relevant to dementia [70, 122–125]. It is therefore of great interest that CtBP2 gene expression is significantly elevated in almost every brain region examined in patients with AD, compared to controls, according to the AGORA database [126].
Another gene robustly inhibited by tricyclic congeners of quetiapine, as indicated in the CMAP database, cysteine-rich protein 1 (CSRP1), which is conversely highly induced in all brain cell types, including microglia, in brains of Alzheimer’s patients, as indicated by the scREAD database [127] and has been nominated as a potential target for AD by the agora.ampadportal.org [128]. Of particular interest, the reversal of age-related human cardiovascular impairments by FOX03, the human analog of daf-16, appears to be mediated by inhibition of CSRP1 [129].
To further assess potential mechanisms mediating anti-inflammatory effects of #310 and other tricyclic compounds in microglia, we assessed gene expression in BV2 microglia 24 hours +/LPS, +/− 8 uM #310, using RNAseq. Remarkably, the gene expression profile produced by LPS alone was, as expected, characteristic of increased glycolysis and expression of several cytokines, including TNF-a, whereas #310 produced a gene expression profile largely reversing the LPS-induced profile, and highly similar to that which we had shown is produced by dietary restriction in mouse hypothalamus, including induction of Creb-Binding Protein (CBP, p<10-5), Catalase (Cat, p<10-14), and Snf2-related CREBBP activator protein (Srcap, p<10-3) with inhibition of genes which oppose Cbp activity such as HDACs, inhibited, and a robust shift in gene expression indicating a shift toward metabolism of alternative substrates relative to glycolysis (e.g., increased CPT1) (Table 1).
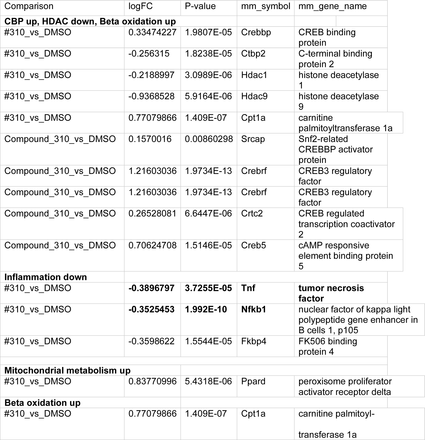
of GM-310 on microglial gene expression is similar to effects of dietary restriction on hypothalamic gene expression: induction of Cbp, inhibition of opposing HDAC gene expression and CtBP2 expression, and a gene profile promoting metabolic pathway genes, such as beta oxidation and mitochondrial metabolism relative to glycolysis. Consistent with the metabolic profile, TNF-alpha and pro-inflammatory gene expression are inhibited. Sign of logFC indicates induction vs. inhibition.
To functionally assess if this change in gene profile also functionally produced a reduction in glycolysis, we assessed these functions in BV2 microglia using flux analysis (Seahorse Fluxometer) 24 hours +/− LPS, +/− tricyclic compounds including GM310. As demonstrated in Figure 17, LPS induced glycolysis, as expected, and GM310, inhibited glycolysis with or without LPS.
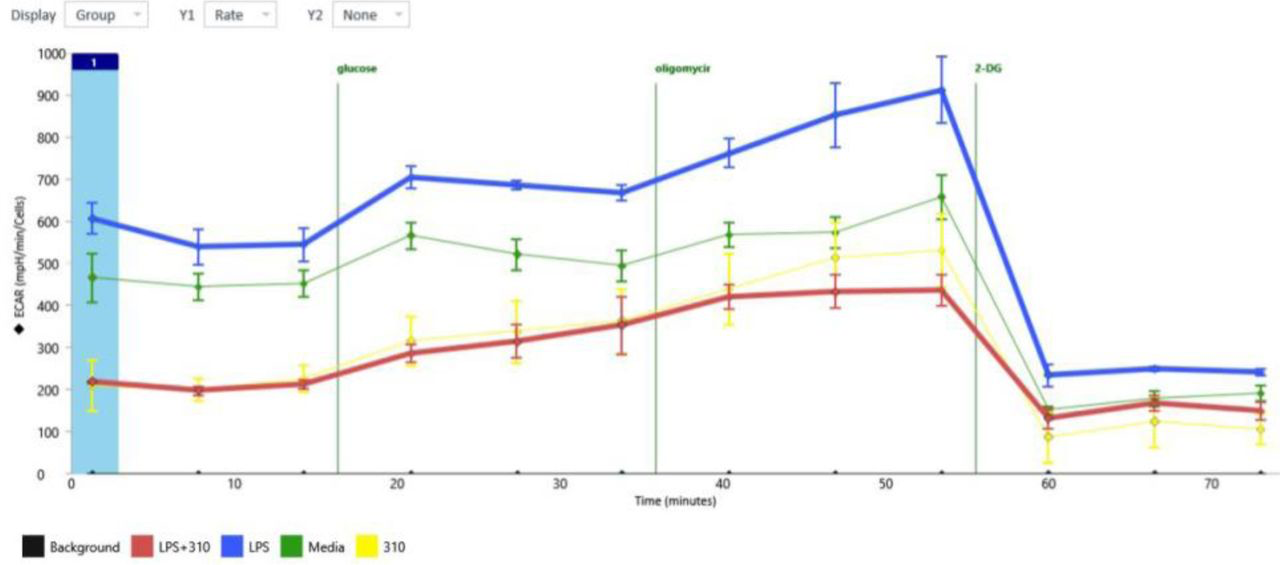
LPS induced glycolysis in BV2 cells, and compound GM310 reduces glycolysis with or without LPS.
To more fully assess the role of glycolysis in driving Abeta proteotoxicity, we assessed the effect of reducing glycolysis by the hexokinase inhibitor 2-deoxglucose (2-DG) on paralysis in the CL2006 C. elegans model. Similar to results we obtained in a C. elegans model of Huntington’s Disease [99], 2-DG significantly delayed Abeta proteotoxicity and increased lifespan in wild-type C. elegans (Figure 18).
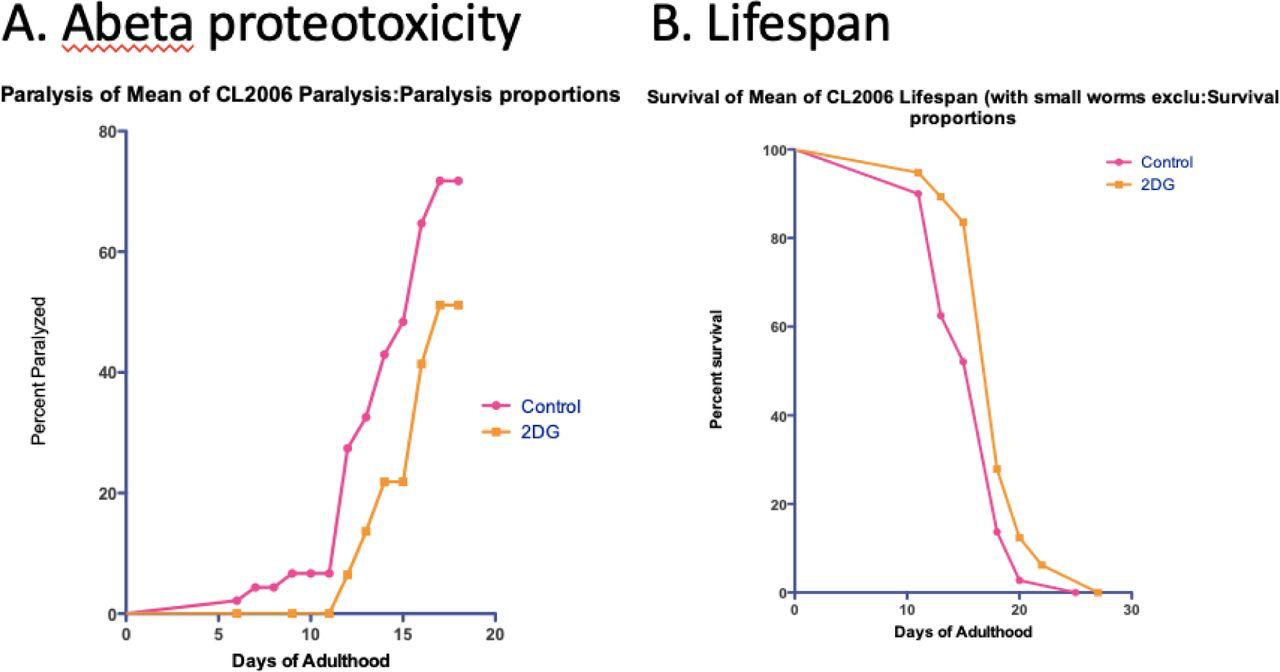
Glycolytic inhibitor 2-deoxyglucose (2-DG) significantly delays Abeta proteotoxicity in the CL2006 strain and increases lifespan in wild-type C. elegans.
To address the likely direct target by which #310 produces these many protective effects as a potential treatment for AD and other age-related diseases, we entered the structure of #310, as well as the other less protective tricyclic compounds including quetiapine, into the SwissTargetPrediction database [130]. First, we validated the algorithm by assessing predicted targets of several very well-established small molecules such as cortisol, and the algorithm clearly predicted the known targets, with other potential targets far less likely. Continuing the analysis, as would be expected, the most likely target for quetiapine was the Dopamine D2 receptor, but other neurotransmitter receptors were also likely targets, in descending order: 5-HT1a, Muscarinic M2, Muscarinic M1, 5-HT2c, Histamine H1, Dopamine D1, and finally Sigma-1, which was about as likely to be a target by this analysis as the D1 receptor. Similarly, Chlorprothixene (our compound #45) was predicted to be equally likely to bind to all of these receptors, although still predicted, though less likely, to bind to the Sigma-1 receptor. Interestingly, Triflupromazine (our compound #75), like chlorprothixene a classic FDA-approved antipsychotic [131], was predicted to bind equally to the Sigma-1 receptor and the D2 receptors, and less likely to bind any other receptors. Assessing the likely target of tricyclic antipsychotics generally indicated that these drugs (e.g., promethazine, methiomeprazine, flupenthixol, etc.) were generally as likely to bind the Sigma-1 receptor as dopamine or any other neurotransmitter receptors, and #310 was similarly predicted to bind the Sigma-1 receptor as other neurotransmitter receptors.
To further assess the potential roles of neurotransmitter receptors in mediating the anti-inflammatory effects of #310, we assessed if neurotransmitters such as dopamine, GABA, serotonin, acetylcholine, and epinephrine reduced LPS-induced TNF-a secretion from BV2 microglia, but none of these neurotransmitters significantly reduced TNF-a secretion (not shown). In fact, to the extent that dopamine has an effect on metabolism, it is to increase glycolysis in glioma initiating cells [132]. Since the medium in which these studies were carried out were devoid of such neurotransmitters, the anti-inflammatory effects of #310 could not be due to antagonism of these receptors. In contrast activation of the Sigma-1 receptor robustly inhibits inflammation whereas ablation of the Sigma-1 receptor produces extreme vulnerability to mortality due to excess inflammation [133]. The Sigma-1 receptor also reduces aerobic glycolysis and increases mitochondrial metabolism [134], thus mimicking protective effects of #310 and dietary restriction (see below). In fact, the Sigma-1 receptor is of increasing therapeutic interest because activation of this receptor elicits “ potent neuroprotective effects and promote neuronal survival via multiple mechanisms… decreasing oxidative stress and regulating neuroimmnological functions” [135]. Thus we conclude that the anti-inflammatory effects of #310 are due to activation of the Sigma-1 receptor, which is consistent with the known activities of this receptor (see below).
DISCUSSION
The purpose of the studies reported here was to discover small molecules bioavailable in brain after oral deliver which could mimic the protective effects of dietary restriction to generally delay AD and other age-related impairments [136]. Since target-based screens to discover compounds to treat AD and other age-related diseases have largely failed [26], we developed parallel phenotypic assays which could plausibly also allow the discovery of potentially novel mechanisms, which could facilitate further drug discoveries [57–60]. Ideally such compounds could constitute the equivalent, for age-related diseases, of the remarkably successful “magic bullet” paradigm leading to generally effective antibiotics, which, while highly controversial when first proposed, led (along with the allied concept of vaccination) to arguably the greatest improvement in human health, and thus human welfare, in history [137]. Successful prosecution of such a program requires appropriate choices of phenotype that reflect the general goal (e.g., general delay of age-related diseases in humans) while allowing high-throughput screening. A far more common strategy to develop such therapies has been to focus on specific targets hypothesized to be a critical driver of a disease (e.g., Abeta peptide for AD), and develop compounds acting on that target. However, the history of drug discovery clearly indicates that first-in-class effective drugs (penicillin, anti-psychotics, etc.) have almost never been discovered, almost by definition, using target-based strategies [26], whereas phenotypic screens allow the discovery of potentially novel targets and mechanisms [57–60].
An alternative plausible approach to discover drugs that are broadly effective against age-related diseases has been to carry out high-throughput screens to discover compounds [63] or genes whose inhibition increase lifespan [138–140]. The model organism C. elegansconstitutes the only plausible platform for carrying out such high-throughput screens in whole animals, so the extent to which mechanisms and interventions which delay age-related impairments in C. elegans may apply to humans is an issue of the greatest importance. To date results have been encouraging-the remarkable discovery of the role of insulin-like signaling in determining lifespan in C. elegans [141, 142] has been shown to be equally important in flies [143], mice [144, 145], and even humans [146, 147]. However, we have observed that only a minority of molecular and pharmaceutical interventions which increase lifespan in C. elegans also delay impairments associated with models of age-related diseases such as the CL2006 model for Abeta proteotoxicity/Alzheimer’s Diseases (Gonzalez et. al, unpublished). We therefore argue that discovering drugs to treat age-related diseases should use models of the processes implicated in driving age-related diseases, rather than lifespan, an approach we have taken in these studies.
One such process plausibly involves inflammation-for neurological diseases, microglial inflammation. GWAS studies have convincingly demonstrated that microglial activity is a major factor in the pathophysiology of AD [32] [51]. Microglia are the resident myeloid cells in the brain, and like myeloid cells (macrophages) in the periphery, when activated they undergo a process entailing destructive inflammation (the M1 phase entailing secretion of cytokines and reactive oxygen species) followed by repair (the M2 phase entailing phagocytosis). Many lines of evidence strongly implicate microglial inflammation in the pathophysiology of AD [36, 38, 47, 50, 148–151]. Of particular interest, several reports have concluded that inhibitors of TNF-a would plausibly be protective in AD [34, 35, 37, 43–45, 80, 82]. However, these inhibitors are generally biologicals (antibodies and protein binders of TNF-alpha) so have limited capacity to cross the blood-brain barrier, suggesting that small molecule inhibitors of TNF-a secretion could be far more effective. Almost as extensive are the many studies implicating IL-6 in driving pathophysiology of AD [36, 38, 42, 46, 48, 49], although evidence that inhibiting IL-6, in contrast to TNF-a, would ameliorate impairments in AD is lacking. Of course proteotoxicity, particularly driven by Abeta and Tau, also contribute to the pathophysiology of AD, but a strong case can be made that this proteotoxicity is mediated at least in part by microglial inflammation [54]. It is therefore of great interest that pro-inflammatory cytokines, especially IL-6 and TNF-a, have been increasingly implicated in the development of neuropsychiatric conditions [152], including depression [153] and psychosis [154, 155], and likely it is not a coincidence that these conditions are highly co-morbid with AD.
The three compounds most effective to reduce proteotoxicity the C. elegans model of AD, out of 2560, were, in decreasing order of efficacy, phenylbutyrate, methicillin, and quetiapine, members of classes of compounds already extensively implicated as promising to treat a wide range of age-related neurological conditions, including AD [65, 66, 68–70, 75, 78, 79, 156]. Taken together these results strongly support that the phenotypes and models chosen for the present screen are indeed likely to predict protective effects in human age-related, especially neurological, diseases, including AD.
The compound most effective to reduce proteotoxicity, out of 2560 compounds, was the HDAC inhibitor phenylbutyrate (PBA) (12% paralyzed vs. 97% paralyzed controls), corroborating our previous report of similar protective effects produced by a similar HDAC inhibitor, sodium butyrate, in the CL2006 model for AD and to extend lifespan in wild-type C. elegans [61]. Corroborating the highly protective effects of PBA, the compound increases lifespan in Drosophila [157] and is highly protective in a wide range of animal models for AD, in some cases actually reversing cognitive impairments in these models [65–71, 156, 158], as well as Parkinson’ Disease [159], Huntington’s Disease [160], and stroke [161]. Studies in preparation for clinical trials for patients with Huntington’s Disease have already begun [162]. Of particular interest, phenylbutyrate combined with the bile acid taurursodiol has now been approved to treat ALS [163, 164] and clinical trials using the same combination to treat AD are now under way with promising results indicated by surrogate endpoints (https://n.neurology.org/content/98/18_Supplement/131).
We further assessed which HDAC was likely involved in the protective effect of phenylbutyrate, and both genetic and pharmacological evidence suggested the importance of HDAC2. This result was of particular interest because brain HDAC2 is preferentially induced with age, an effect blocked by dietary restriction [165], and is preferentially induced in patients with AD and in animal models of AD [166]. Similarly genetic ablation of HDAC2 specifically in adult microglia delays impairments in the 5XFAD mouse model of Alzheimer’s Disease [167]. Furthermore, genetic ablation of HDAC2 in microglia, either genetically or pharmacologically, promotes the transition from the proinflammatory state to the M2 phagocyotic state and is highly protective in a mouse model of stroke [168, 169]. Inhibition of HDAC2 was also specifically associated with protection by dietary restriction in mouse and C. elegans models of Huntington’s Disease [99, 170]
The second-most effective compound to reduce proteotoxicity was the beta lactam antibiotic methicillin (17% paralyzed vs. 97% paralyzed controls). As with the HDAC inhibitors, this class of compounds has also been extensively demonstrated to protect in a wide range of models for neurological diseases, including stroke [77], traumatic brain injury [171], and AD [75, 78].
The third-most effective compound to reduce proteotoxicity was the tricyclic antipsychotic quetiapine (18% paralyzed vs. 97% paralyzed controls), which, like phenylbutyrate and methicillin, has already been reported to improve symptoms in mouse models of AD and other forms of dementia [79, 172, 173]. Tricyclic compounds constitute the major class of drugs to treat psychosis and depression [174]. The combination of two tricylic antipsychotic phenothiazines, chlorpromazine and promethazine, is reported to be neuroprotective in a rat model of stroke associated with reduction in neuroinflammation [175, 176]. Two other tricyclic compounds, chlorprothixene (#45), second-most prescribed drug for psychosis [177], and also effective in treating depression [178], and triflupromazine (#75) also approved to treat psychosis [179], were also highly effective to reduce proteotoxicity in this initial screen.
In a secondary screen to assess efficacy of the drugs which inhibited proteotoxicity to also inhibit microglial inflammation, again tricyclic antipsychotic drugs (e.g., #75, triflupromazine) were prominently effective (Figure 3). Since a major challenge in developing drugs to treat neurological diseases is the requirement to cross the blood-brain barrier, preferably after oral delivery, the relative prominence of this class of compounds to prevent proteotoxicity AND to reduce microglial inflammation, coupled with the fact that these compounds are highly concentrated in the brain after oral delivery, led us to carry out a structure-activity relationship study between tricyclic congeners of quetiapine (including chlorprothixene and triflupromazine), obtained from the NCI library, and inhibition of microglial secretion of TNF-a. As shown in Figure 4, most tricyclic congeners significantly reduce TNF-a secretion, including chlorprothixene (#45) and triflupromazine (#75).
Remarkably, after these analyses were completed, a subsequent independent and novel bioinformatic analysis concluded that “antipsychotic drugs with circadian effects, such as Quetiapine … will have efficacious effects in AD patients” [180]. Together with the present results, this observation is consistent with the high prevalence of both psychosis [181], which actually predicts the progression of cognitive impairments [182], and depression [183] [184–187] in patients with AD. However, in clinical trials atypical antipsychotic drugs actually *accelerated* cognitive impairments [188]. This observation would appear to rule out treating AD with currently available atypical antipsychotic drugs. However, these results suggest that compounds with similar bioactivities, but possibly fewer or at least different iatrogenic effects might still prove to be effective in treating AD and other neurological diseases. Furthermore, the high degree of co-morbidity of neuropsychiatric diseases and AD raise the interesting possibility these conditions may share some common pathophysiological mechanisms.
Based on the relationship between structure and anti-inflammatory activity (Figure 4) and proteotoxicity (Figure 5) observed in these studies, we hypothesized novel congeners that might be even more effective at lower doses and less toxic. After these novel compounds were synthesized, we assessed their efficacy and found that only two of the twelve novel compounds (#310 and #318, internal lab designation) significantly reduced TNF-a, with #310 being the most protective, completely blocking the effect of LPS to induce TNF-a (Figure 6). When we assessed if these compounds would significantly reduce proteotoxicity, 5 of the novel congeners were protective, but only #310 was significantly protective in both phenotypes (Figure 7). We therefore carried out a detailed dose-response study and observed that there was a clear relationship between dose and protection against proteotoxicity except at the highest dose protection was no longer observed (Figure 8). This is a very common phenomenon observed during drug screening, which is why the FDA requires assessment of both ED50 and LD50. These studies thus provide the basis for predicting the optimum dose for clinical trials.
Likely efficacy of #310 in treating human dementia was further supported by the observation that this compound increases lifespan (Figure 10), suggesting that if the compound does produce deleterious side effects, they are more than compensated by protective effects, likely mediated by reduction in glycolysis (see below). Furthermore, after oral delivery the concentration of #310 was more than 10X higher in brain than in blood, a characteristic of this class of drugs and a major factor in the efficacy of these drugs to treat psychosis and depression. In fact after oral delivery, for at least 8 hours the concentration in the brain (Figure 11) was higher than necessary to produce complete protection including memory (Figure 12), in a mouse model of vascular dementia caused by intracerebral hemorrhage (ICH) [103]. Vascular cognitive impairment (VCI) is the second most common type of dementia after Alzheimer’s disease (AD) [189]. Post-stroke cognitive impairment and dementia (PSCID) is a major source of morbidity and mortality after stroke, including ICH [104, 110, 190, 191].
To further clarify mechanisms mediating these protective effects, we also observed that #310 reduced microglial secretion of almost every major pro-inflammatory cytokine (Figure 13), thus demonstrating the profoundly inhibitory effect of #310 on microglial M1 inflammatory activity. Since failure of microglial M2 repair activity (phagocytosis) might also contribute to AD, it was of particular interest that #310 was the only compound examined which not only reduced microglial inflammation but also increased microglial phagocytosis (Figure 14). #310 and several congeners were also highly effective to reduce TNF-a from human primary monocytes (Figure 15) and PBMCs (Figure 16), further supporting its possible use in humans.
To clarify potential molecular mechanisms mediating the protective effects of #310 and tricyclic congeners, we used the CMAP database to discover genes expressed in human cells responsive to FDA-approved tricyclic drugs. Among the most robust effects of these tricyclic drugs was the inhibition of CtBP1, whose inhibitory effects on gene expression is mediated at least in part by inhibition of CBP [117], whose induction we showed mediates protective effects of dietary restriction on impairments in the CL2006 model of AD [61]. Of particular interest, CtBP activity is stimulated by glycolysis-derived NADH [118, 192–194], inhibition of which appears to mediate protective effects of 2-DG [192] and dietary restriction [119]. Conversely, CtBP activity promotes inflammation [121]. This hypothesis was further supported by our observation that, conversely to CBP, expression of CtBP1 correlates inversely with lifespan (Poplawksi et al., unpublished). Using RNAseq we corroborated that #310 inhibits CtBP gene expression in BV2 cells (Table 1), presumably due to inhibition of glycolysis (Figure 17). Furthermore, the effects of #310 on gene expression in BV2 cells is highly similar to that produced by dietary restriction in the brain, including induction of CBP and related co-factors, inhibition of HDAC gene expression, and induction of genes which promote beta oxidation and inhibit glycolysis (Table 1). These results were corroborated by direct measurement of glycolysis using fluxometery, which demonstrated that LPS induced glycolysis, necessary to support inflammation [11, 195], particularly microglia in AD [10] and #310 inhibited both baseline and LPS-induced glycolysis (Figure 17). The critical role of glycolysis in driving age-associated proteolysis and mortality was further supported by the delay of proteolysis by 2-DG, which inhibits glycolysis, in this model of AD (Figure 18A) as well as in a similar model for Huntington’s Disease [99], as well as lifespan in the wild-type strain (Figure 18B).
These results further support that glycolysis plays a major role in driving age-related diseases and mortality, and reduction of glycolysis mediates protective effects of dietary restriction and other manipulation on these diseases and mortality, as we have previously argued [14, 17, 196, 197]. While it is widely accepted that metabolic processes drive the aging process, as reflected by the rate of living [198] and the “free radical” [199] theories of aging, and bolstered by the generally protective effects of dietary restriction to delay age-related diseases and mortality, the general gist of those paradigms is that age-related damage is due to free radicals produced by oxidative metabolism. However, a stronger case can be made that metabolism specifically driven by glycolysis, rather than oxidative metabolism generally, is the key process driving age-related disease, impairments, and mortality [14, 196]. Obvious support for this hypothesis is that elevated glucose reduces lifespan [200, 201] and increases the risk of many age-related diseases, including AD [202–204]. Toxicity of elevated glucose is most likely due to increased oxidative stress as a result of increased glycolysis [205, 206], since glucose follows the general biochemical principle that substrates induce their own metabolism and inhibit metabolism of alternative metabolites. This principle is famously exemplified by the lac operon, through which lactose induces its own metabolism while inhibiting glucose metabolism [14], and also exemplified by the activation of Ppar transcription factors by free fatty acids, released during fasting, which promote the transcription of genes which enhance metabolism of free fatty acids and inhibit metabolism of glucose, the metabolic response which apparently mediates protective effects of dietary restriction [14, 99, 170, 196] In fact, dietary restriction actually *increases* oxidative phosphorylation [207] and this increase is required for the protective effects of dietary restriction [208, 209], while decreasing glycolysis [14, 196]. The specific deleterious effect of glycolysis on cellular function arises in part from the fact that ATP generation from glycolysis relies primarily on the production of NADH, whose donation of an electron to the mitochondrial ETC Complex 1, followed by transfer to Complex III then IV, is efficient to produce ATP but for that very reason also generates reactive oxygen species [14, 196]. Production of ATP from lipid (beta) oxidation entails transfer of an electron to ETC complex II via NADH2, which is somewhat less efficient but does not produce as much oxidative stress [14, 196]. This phenomenon explains the otherwise puzzling observation that inhibition of Complexes I, III, IV, and V increase lifespan, whereas inhibition of complex II increases lifespan [140]. Also as indicated above NADH drives the activity of CtBP transcriptional inhibitors, whose activities are deleterious at least in part by inhibiting CBP activity, and also increasing NADH reduces NAD+ concentrations, which are protective at least in part by activating Sirtuin-related transcription factors [208]. Related mechanisms of toxicity driven by glucose metabolism are that glycolysis competes for glucose metabolism with the pentose pathway, such that glycolysis produces toxic NADH at the expense of protective NADPH produced by the pentose pathway, the source of reducing power of glutathione and other anti-oxidative defenses [14, 196].
A compelling example of the importance of glycolysis in driving AD is that the ApoE4 allele, the best-established genetic risk factor for AD, is associated with increased aerobic glycolysis in both humans in in mouse models of AD, long before the development of symptoms [210]. The protective effects of rapamycin, the only pharmaceutical intervention so far shown to robustly increase lifespan in mammals, also appear to be due to reduction in glycolysis [211]. Similarly, protective effects of taurine, whose production decreases with age and whose supplementation can ameliorate inflammaging, also appear to be mediated by reduced glycolysis [212]. In addition, as discussed extensive above, age-related increase in inflammation, which drives many or perhaps most, age-related impairments including AD, appears to be driven increased glycolysis [10, 11, 91, 195, 213]. Increased glycolysis is necessary for inflammation to produce the required metabolic state for the production of reactive oxygen species that mediate the toxic effects of inflammation [214]. The protective effects of ketones to increase lifespan and delay age-related impairments in C. elegans [215] and mice [216], and reverse diabetic complications [217], also appear to be due to reduced glycolysis, which was the motivation for carrying out that latter study. And finally, cancer of course is also driven by and requires increased glucose metabolism, as per the Warburg effect [13, 218, 219], probably to support macromolecular synthesis of nucleotides and lipids required for cell division [14, 196]. In fact, it is increasingly clear that simply reducing exposure to glucose mimics protective effects of dietary restriction in yeast [208] and in the senescence of human cells in which, as predicted by the glucose hysteresis model, glycolysis increases with senescence [220].
To the extent that not just glycolysis, but an age-related increase in glycolysis, may account for age-related increases in inflammation and other toxic processes, what explains this age-related increase in glycolysis? Similarly, the reduction of microglial inflammation by the combination of the two tricylic antipsychotic drugs chlorpromazine and promethazine appear to be mediated by a reduction not just in baseline glycolysis but in “hyperglycolysis” [221]. We have proposed that cumulative exposure to normal glucose leads to persistently-induced glycolysis in a phenomenon observed in diabetic patients and referred to as “metabolic memory” [16, 206] [16, 222–229], leading to persistently elevated oxidative stress, inflammation, and risk of cancer [17]. A similar form of “metabolic memory” is also exhibited by the lac operon [14, 230, 231]. We demonstrated this phenomenon in vitro and demonstrated that it could be prevented with agonists to Ppar-alpha and Ppar-gamma, which produces the opposite metabolic profile of increased lipid oxidation and decreased glycolysis [16]. This phenomenon may thus explain why inflammation increases with age [2], in turn leading to the many age-related conditions driven by increased inflammation, including neurodegenerative diseases like AD (see above). Thus use of phenotypic screens to discover and develop drugs to delay age-related diseases not only leads to promising therapies, but also substantially elucidate relevant mechanisms, leading to discovery of even more effective drugs, etc., in a virtuous cycle of discovery.
Specifically, the effective interventions, especially of the novel tricyclic compound #310, produced similar molecular responses as produced by dietary restriction, particularly the induction of CBP and inhibition of glycolysis, whose functional significance was corroborated by the demonstration that inhibition of glycolysis by 2-DG similarly delayed Abeta proteotoxicity and reduced microglial secretion of TNF-a. We had previously reported that inhibition of glycolysis by 2-DG was highly protective in a model of Huntington’s Disease, also dependent on NHR-49, the C. elegans homolog of Ppar-alpha [99]. These results strongly support the functional significance of these molecular responses in mediating protective effects of dietary restriction, as we have previously argued [14, 61]. In turn these observations suggest the likely success of further screens to discover drugs that act through these mechanisms.
Although #310 and of course the tricyclic drugs can bind dopamine and other neurotransmitter receptors under some circumstances, the protective effects of #310 are apparently mediated by activation of the Sigma-1 receptor. As indicated above, according to the SwissTargetPrediction algorithm [130], #310 and many of the FDA-approved tricyclic drugs are about as likely to bind to the Sigma-1 receptor as to dopamine or other neurotransmitter receptors. However, in contrast to #310 and some FDA-approved tricyclic antipsychotic drugs, neither dopamine or any other neurotransmitters had any effect on TNF-a secretion. In fact, to the extent that dopamine has an effect on metabolism, it is to increase glycolysis in glioma initiating cells [132], the opposite effect of #310. Furthermore, there were no neurotransmitters in the medium so #310 could not be acting as an antagonist to those receptors either. In contrast activation of the Sigma-1 receptor robustly inhibits inflammation whereas ablation of the Sigma-1 receptor produces extreme vulnerability to mortality due to excess inflammation [133]. The Sigma-1 receptor also reduces aerobic glycolysis and increases mitochondrial metabolism [134], thus mimicking protective effects of #310 and dietary restriction. In fact, the Sigma-1 receptor is of increasing therapeutic interest because activation of this receptor elicits “potent neuroprotective effects and promote neuronal survival via multiple mechanisms… decreasing oxidative stress and regulating neuroimmunological functions” [135]. Specifically Sigma-1 activators are being actively investigated to treat Alzheimer’s Disease as well as stroke [232, 233]. Sigma-1 receptor activators are also being actively investigated to treat depression and psychosis as well [234]. Considering that the sigma-1 receptor was originally identified as a target of opioids, it is not surprising that this receptor is showing great promise as a target to treat drug addiction [235, 236]. Of possibly even greater clinical signi Therefore it is plausible that #310 or congers may be safe and effective to treat several neurodegenerative and psychiatric pathophysiological processes in AD and other neurodegenerative diseases by reducing microglial inflammation. Thus the present studies strongly support the utility of phenotypic screens to discover better pharmaceutical therapies to treat age-related diseases as well as to elucidate mechanisms driving these diseases.