
N, N-Dimethyltryptamine, a natural hallucinogen, ameliorates Alzheimer’s disease by restoring neuronal Sigma-1 receptor-mediated endoplasmic reticulum-mitochondria crosstalk
By Dan Cheng, Zhuo-Gui Lei, Kin Chu, Oi Jin Honey Lam, Chun Yuan Chiang, and Zhang-Jin Zhang
Excerpt from the article published in Alzheimer’s Research & Therapy volume 16, 95, 01 May 2024, DOI: https://doi.org/10.1186/s13195-024-01462-3
Abstract
Background
Aberrant neuronal Sigma-1 receptor (Sig-1r)-mediated endoplasmic reticulum (ER)- mitochondria signaling plays a key role in the neuronal cytopathology of Alzheimer’s disease (AD). The natural psychedelic N, N-dimethyltryptamine (DMT) is a Sig-1r agonist that may have the anti-AD potential through protecting neuronal ER-mitochondrial interplay.
Methods
3×TG-AD transgenic mice were administered with chronic DMT (2 mg/kg) for 3 weeks and then performed water maze test. The Aβ accumulation in the mice brain were determined. The Sig-1r level upon DMT treatment was tested. The effect of DMT on the ER-mitochondrial contacts site and multiple mitochondria-associated membrane (MAM)-associated proteins were examined. The effect of DMT on calcium transport between ER and mitochondria and the mitochondrial function were also evaluated.
Results
Chronic DMT (2 mg/kg) markedly alleviated cognitive impairment of 3×TG-AD mice. In parallel, it largely diminished Aβ accumulation in the hippocampus and prefrontal cortex. DMT restored the decreased Sig-1r levels of 3×TG-AD transgenic mice. The hallucinogen reinstated the expression of multiple MAM-associated proteins in the brain of 3×TG-AD mice. DMT also prevented physical contact and calcium dynamic between the two organelles in in vitro and in vivo pathological circumstances. DMT modulated oxidative phosphorylation (OXPHOS) and ATP synthase in the in vitro model of AD.
Conclusion
The anti-AD effects of DMT are associated with its protection of neuronal ER-mitochondria crosstalk via the activation of Sig-1r. DMT has the potential to serve as a novel preventive and therapeutic agent against AD.
Introduction
The escalating incidence of Alzheimer’s disease (AD) has posed a great challenge to the individuals, families, and societies [1]. Although researchers have made numerous attempts in the development of pharmacological drugs for AD, the clinical outcomes have been unsatisfactory. This is largely due to the fact that most approved drugs merely targeted the downstream pathways that restore acetylcholine (ACh) transmission and eliminate pathological products, mainly amyloid β (Aβ) and tau [2]. There have been various subcellular pathological alterations occurred in neuronal organelles that are closely associated with the upstream pathways of Aβ and tau accumulations [3]. The most apparent is aberrant signalling between the endoplasmic reticulum (ER) and mitochondria [3]. ER-mitochondria interplay plays an essential role in various neuronal functions, e.g., lipid metabolism, calcium homeostasis, autophagy, ER stress responses, axonal transport, and synaptic plasticity [4]. It is heavily involved in the pathogenesis of AD [4, 5]. This has led to the assumption that agents capable of restoring neuronal ER-mitochondria crosstalk may have anti-AD potentials.
ER-mitochondrial interface involves in specialized subdomains of the ER, known as mitochondria-associated membranes (MAMs) that are reversibly tethered to mitochondria [6]. The signalling between the two organelles is primarily achieved by releasing calcium from inositol 1,4,5-trisphosphate receptors (IP3Rs) of the ER to mitochondria, where calcium is taken into via voltage dependent anion channel 1 (VDAC1) and mitochondrial calcium uniporter (MCU), which are located in the outer mitochondrial membrane and inner mitochondrial membrane, respectively [4]. On the other hand, an ER protein, Sigma-1 receptor (Sig-1r), acts as a chaperone for IP3Rs to regulate calcium release from the ER to mitochondria, bioenergetics, ER stress, and cell differentiation [7,8,9,10]. Sig-1r is widely expressed throughout the central nervous system, with a high density in brain regions associated with learning and memory, such as the hippocampus and cortex [11,12,13]. Sig-1r activation inhibited Aβ deposition [14], whereas Sig-1r deactivation resulted in the development of AD [14, 15]. Several Sig-1r agonists are suggested to have benefits in alleviating neurodegenerative diseases in cultured cells and animal models [16]. These findings indicate that Sig-1r agonists may have the anti-AD potentials.
N, N-dimethyltryptamine (DMT) is a natural hallucinogenic alkaloid richly existing in Ayahuasca, an entheogenic beverage traditionally used in South Americans [17, 18]. While DMT acts at 5-HT2A receptors as agonist, it is also a Sig-1r agonist and has been proven to have potent protective effects against hypoxia and cerebral ischemia by activating Sig-1r [17, 19, 20]. DMT enhanced spatial learning and memory tasks by promoting adult neurogenesis via Sig-1r, but not 5-HT1A and 5-HT2A receptors [21]. Intravenous injection of DMT produced antidepressant efficacy in people with treatment-resistant depression [22]. These studies imply that the psychotropic effects of DMT may be related to the activation of Sig-1r, which restores ER-mitochondria signalling. Unlike most hallucinogenic drugs, DMT did not cause or caused less tolerance, dependence, and withdrawal symptoms [23]. It thus deserves to be further developed as a novel anti-AD agent.
The working hypothesis of this study was that DMT exerted anti-AD effects by restoring neuronal ER-mitochondria signaling via the Sig-1r activation. To test this hypothesis, we firstly examined the effects of DMT chronic treatment on cognitive impairment and Aβ deposits in transgenic 3×TG-AD mice, one of animal models of AD. We then evaluated the effects of DMT on the level of Sig-1r and MAM-associated proteins, physical contact, calcium dynamic between ER and mitochondria in in vitro and in vivo paradigms, and oxidative phosphorylation (OXPHOS) and ATP synthase with an additional utilization of Sig-1r knockdown mice and the Sig-1r antagonist BD1063.
..
Results
DMT treatment improved cognitive impairment and diminished Aβ pathology in 3×TG-AD mice
The spatial learning and memory of mice was evaluated in water maze test. In the training trials, the latency to find the platform in the 3×TG-AD mice injected with vehicle significantly prolonged on the 2nd day relative to the WT mice. While 3×TG-AD mice injected with DMT had a significant reduction in the latency at Day 2, 3, and 5 compared to the vehicle-treated animals (Fig. 1C). The performance of mice in the probe trail showed the behavioral variables, time spent in platform, target quadrant, and entries into platform largely decreased in the 3×TG-AD mice relative to WT mice (Fig. 1D-F). DMT treatment markedly increased time and entries in the platform in the model mice (Fig. 1D-F). No significantly statistical differences were observed in the DMT improvement on these two parameters between male and female mice (Fig. S4). The three groups did not differ in other parameters (time and entries in target quadrant, distance in target quadrant, and total travel distance) (Fig. 1G-I). These results suggested that chronic DMT treatment was sufficient to suppress cognitive decline of AD.
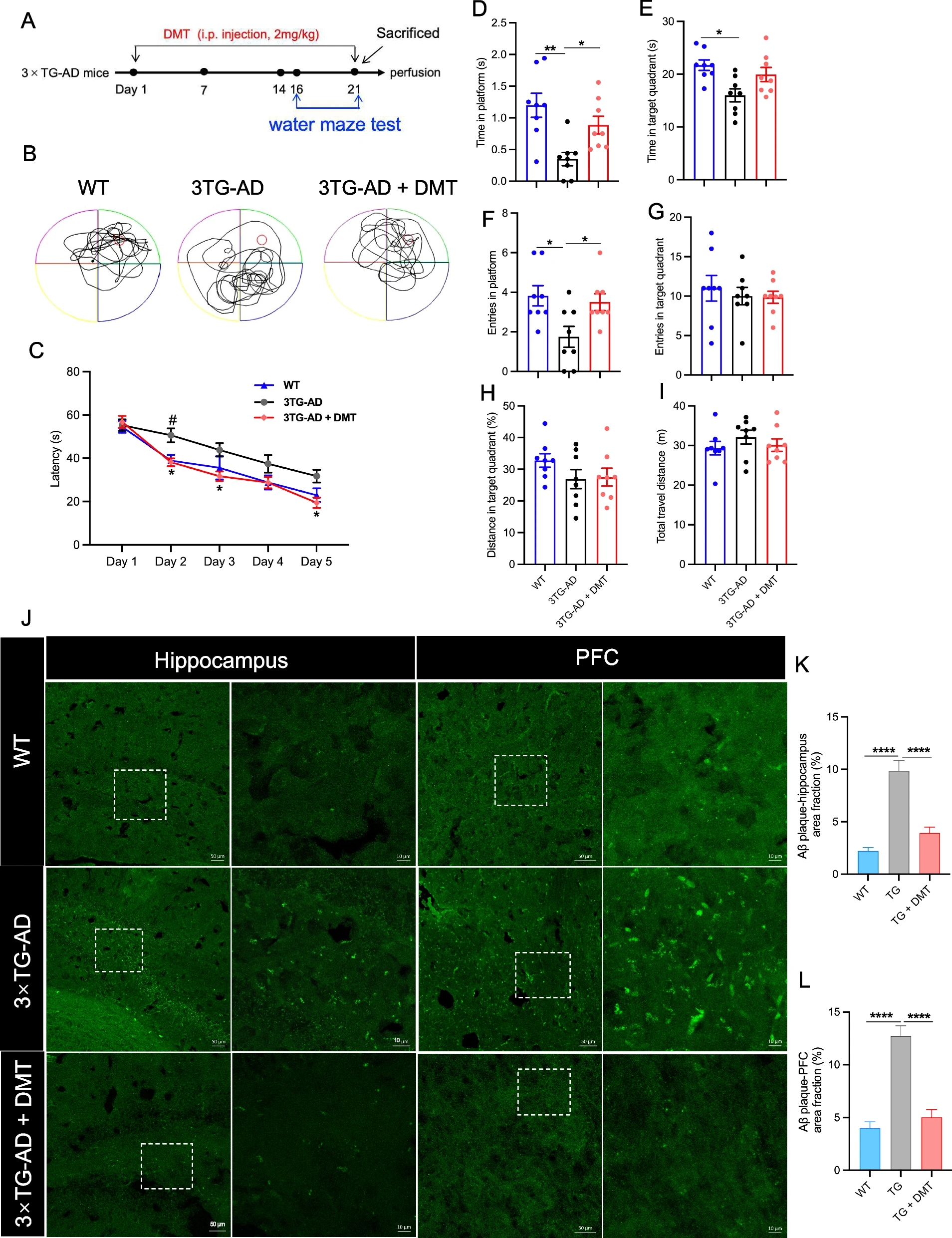
DMT improves cognitive impairment and diminishes Aβ pathology in the 3×TG-AD mice.
AExperimental timeline. B The representative swim paths of mice. C Latency to platform of mice during the training trial. D Time in platform, E Time in target quadrant, F Entries in platform, G Entries in target quadrant, H Distance in target quadrant, I Total travel distance of mice in the probe test. (n = 8 in each group). J The representative images of Aβ plaque in the hippocampus and prefrontal cortex (PFC) of 3×TG-AD mice. The images on the right are enlarged views of the image on the left in each panel. Scale bar: 50 μm (left), 10 μm (right). K, L The quantification of the Aβ accumulation in the hippocampus and PFC, respectively. The results were presented as the mean area fraction of the positive particle. In the training trials of water maze test, data were analyzed using two-way ANOVA test; in the probe test, data were analyzed using one-way ANOVA. Data analysis of Aβ plaque was performed on average 5 randomly selected sections from each mouse (n = 3 per group) using one-way ANOVA. *P < 0.05, **P < 0.01, ****P < 0.0001. TG, 3×TG-AD
Next, we investigated whether Aβ plaques in the mice brain were diminished with DMT treatment by using Thioflavin-S staining (Fig. 1J-L). The transgenic mice had a strikingly higher density of Aβ plaques in the hippocampus and prefrontal cortex (PFC) regions than WT mice. DMT treatment significantly facilitated the degradation of Aβ plaques in these two regions.
Effects of DMT on Sig-1r level in mice and cultured neurons
We found that compared with age-matched WT mice, the expression level of Sig-1r in the hippocampus of 3×TG-AD mice significantly decreased with the progression of AD (Fig. S3). This result was consistent with previous studies [5], and Sig-1r levels were also found to be reduced in the brains of AD patients [5, 40]. However, whether the reduced expression of Sig-1r was a result or one of the causes of AD remains for further investigation [41].
The effects of DMT on Sig-1r level was examined in the in vivo paradigms. The expression of Sig-1r level was determined in Sig-1r knockdown mice and 3×TG-AD mice using immunofluorescent staining. The Sig-1r knockdown mice exhibited a marked decrease in the Sig-1r expression in the mice hippocampus relative to the control (Fig. 2A-B). The mRNA level of Sig-1r was further confirmed the decreased Sig-1r level in the Sig-1r knockdown mice (Fig.2C). The Sig-1r fluorescence intensity in the hippocampus of 3×TG-AD mice obviously decreased compared to WT mice. Chronic DMT administration completely reversed the expression of Sig-1r to the level in the control mice (Fig. 2D-E). Our findings indicated that low-dose chronic treatment with DMT increased Sig-1r levels in in vivo models of AD. These results were consistent with other previous studies, which found that the agonist of Sig-1r increased the expression of Sig-1r protein and mRNA [42, 43].
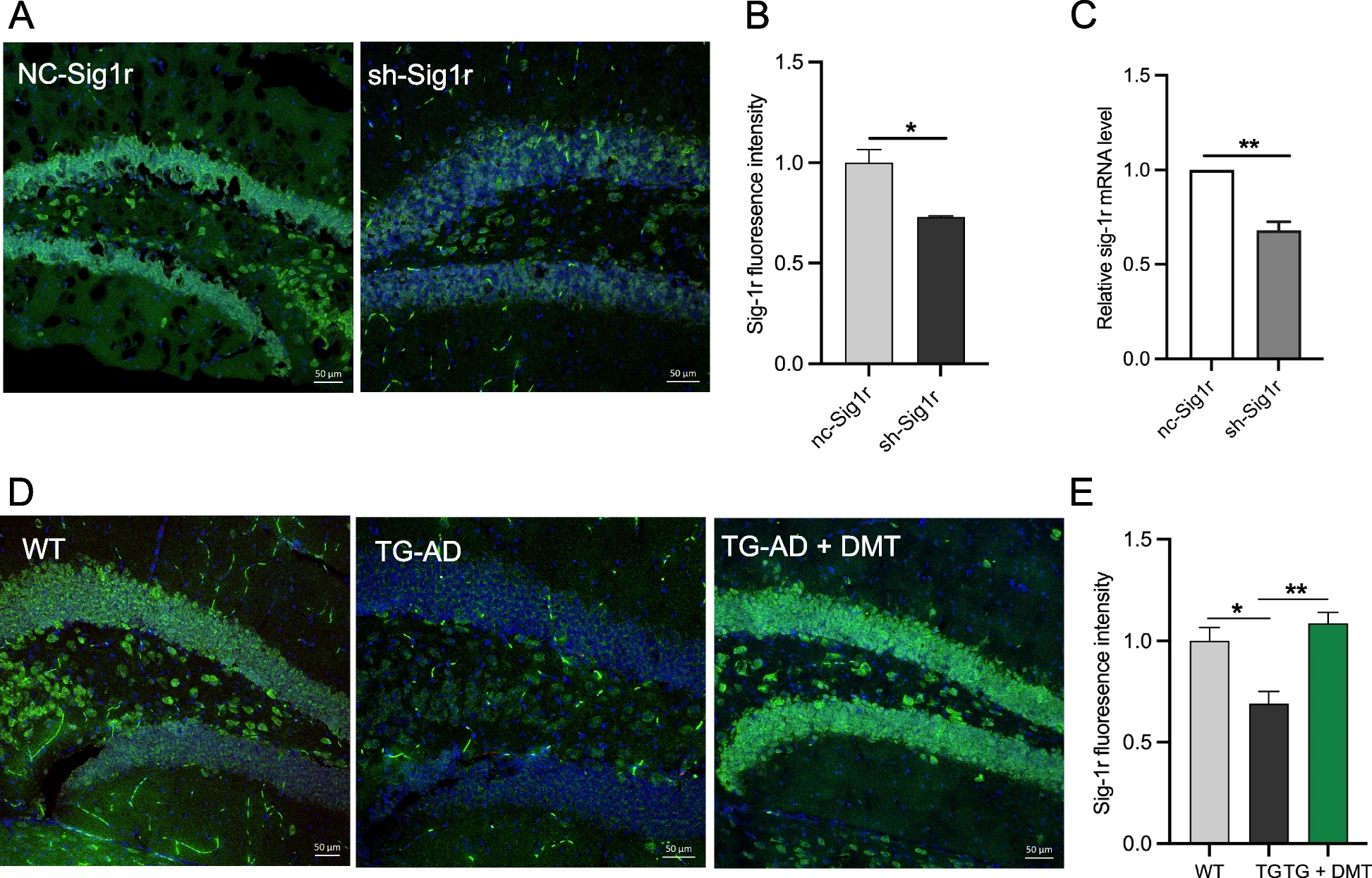
DMT improves the Sig-1r levels in the mice hippocampus and primary hippocampal neurons.
A, B Representatives and the quantification of the Sig-1r level in the hippocampus of sh-Sig1r knockdown mice. C The mRNA level of Sig-1r in the hippocampus of sh-Sig1r knockdown mice. D, E Representative images and the quantification of the Sig-1r level in the hippocampus of 3×TG-AD mice. In the in vivo study, data was obtained from average 5 randomly selected sections from each mouse (n = 3 per group). Data was analyzed using one-way ANOVA. *P < 0.05, **P < 0.01. TG, 3×TG-AD
Effects of DMT on neuronal ER-mitochondria distance in neuronal tissues
ER-mitochondrial contact sites are known to orchestrate many important cellular functions, including calcium signaling, mitochondrial function, and ER stress, which are known to be dysregulated in AD [31]. The expression of Sig-1r was known to be critical for ER-mitochondrial contacts since the Sig-1r activation regulate calcium transport from ER to mitochondria and indirectly regulate TCA cycles [44, 45]. Herein, the effects of DMT on the ER-mitochondria distance was examined in hippocampal neurons of 3×TG-AD and Sig-1r knockdown mice. Quantitative transmission electron microscope analysis displayed a significant increase in the ER-mitochondrial distance of 3×TG-AD mice relative to WT mice, whereas chronic DMT shortened the ER-mitochondrial distance to the value of WT mice (Fig. 3A, B). Sig-1r knockdown mice had an approximately 40% wider distance than controlled mice. DMT completely rescued the distance between the two organelles (Fig. 3C, D).
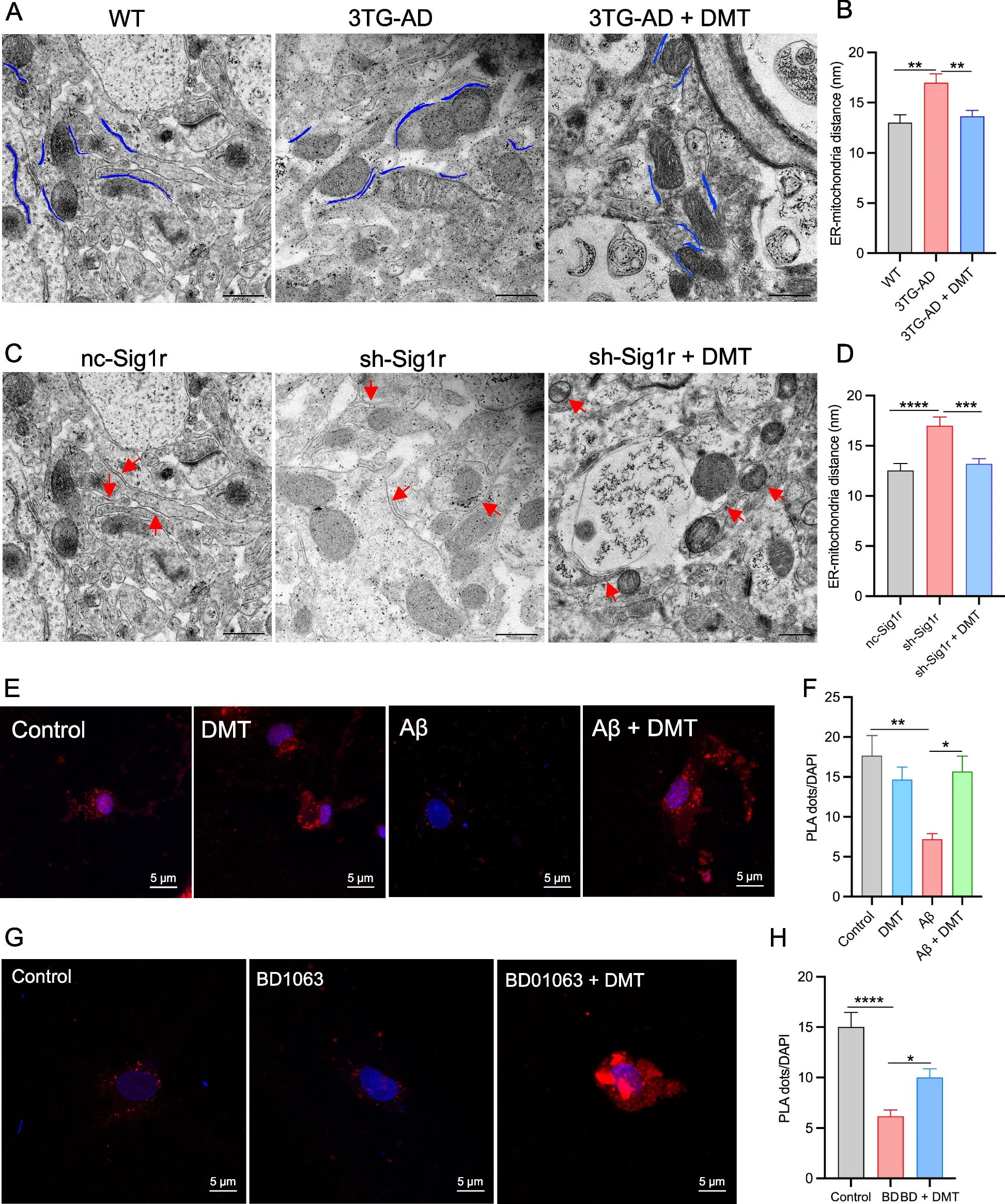
DMT modulates endoplasmic reticulum (ER) – mitochondrial distance and contacts.
Two different animal models, 3×TG-AD mice and sh-Sig1r knockdown mice were used. A, B Representatives and quantification of ER-mitochondrial distance in the hippocampus of 3×TG-AD mice. C, D Representatives and quantitative analysis of ER-mitochondrial distance in the hippocampus of sh-Sig1r knock down mice. Scale bar: 500 nm. Data was obtained from 3 mice per group with 10 images in each mouse. E, F Representatives and quantification of the PLA signal in the cells exposed to Aβ with or without DMT treatment. G, H Representatives and quantification of the PLA signal in the cells exposed to BD1063 with or without DMT treatment. Scale bar: 5μm. Data was representative of 3 independent experiments and analyzed using one-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
To further detect effects of DMT on ER-mitochondrial physical contact, cultured neurons were treated with 20 μM Aβ25-35 plaque and 1 μM BD1063 with or without DMT treatment for 24 h. The contact points between the ER and mitochondria were verified using PLA technology. The PLA dots was used to measure the proximity of IP3R3-VDAC1 pair, indicating the contact points between the ER and mitochondrial, as the general distance between the two organelles is 20-40 nm and PLA can detect protein pairs with less than 40 nm away from each other [46]. A striking decrease in PLA dots was observed in the Aβ-exposed cells relative to the normal cells. The presence of DMT greatly increased the number of PLA dots in the cells exposed to Aβ, but did not show great effects in the control cells (Fig. 3E, F). The number of PLA dots markedly decreased in the cells exposed to BD1063. BD1063 co-treatment with DMT increased the number of PLA dots (Fig. 3G, H). These results suggested that DMT improved the ER-mitochondrial crosstalk under the pathological conditions.
DMT modulated the expression of MAM-associated proteins in mice brain
In addition to IP3R and VDAC1 regulating calcium transfer from ER to mitochondria, other MAM-associated proteins, such as PACS2, PSS1, MFN2, Sig-1r, VAPB, and CHOP, also directly mediate ER-mitochondrial contact and calcium homeostasis [30,31,32, 47]. Give that DMT significantly modulated ER-mitochondrial contact in neuronal tissues, we further evaluated the effects of DMT on the expression of these MAM-associated proteins in the whole brain of mice using Western blot (Fig. 4).
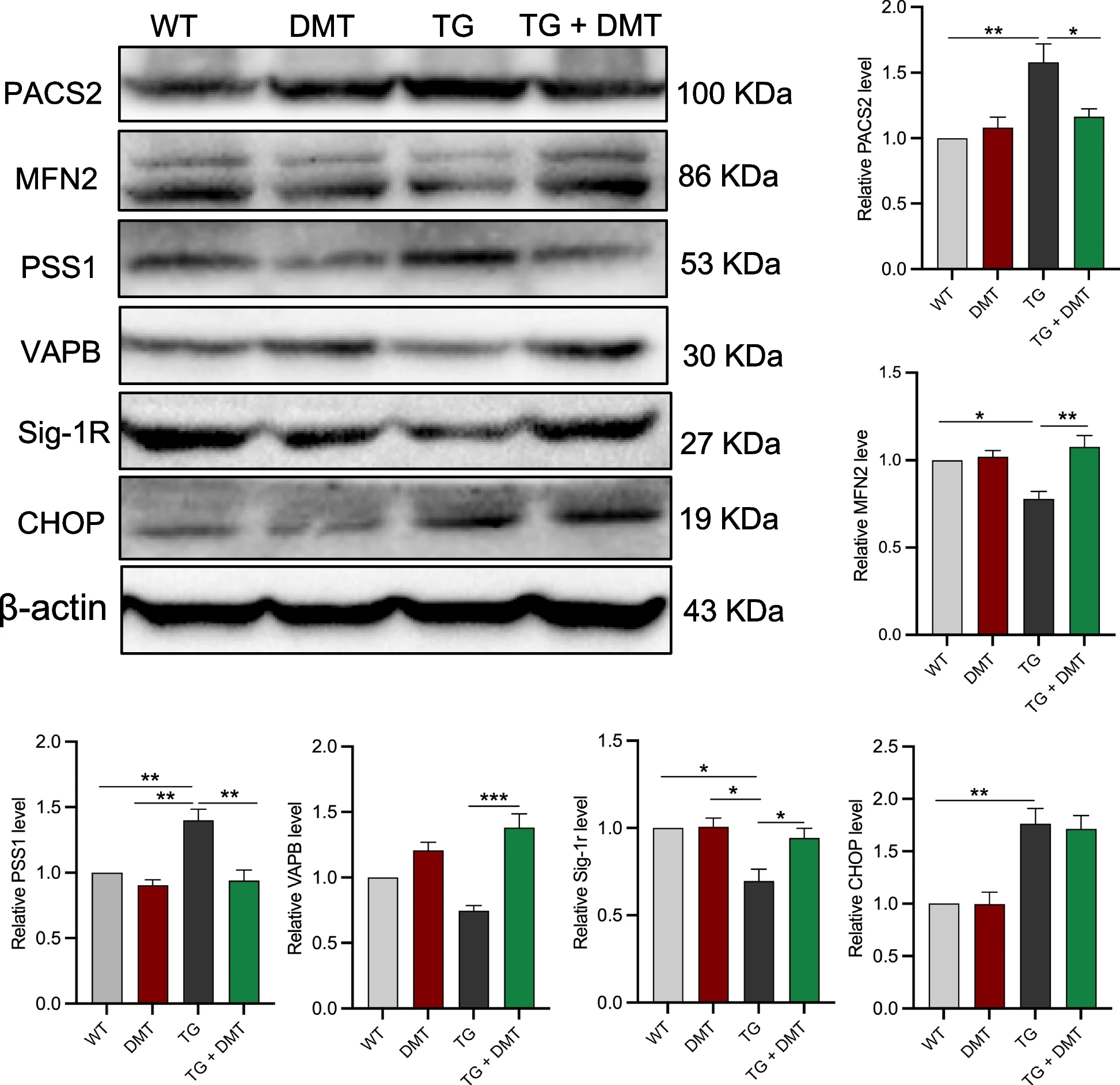
DMT modulates the expression of MAM-associated proteins in 3×TG-AD mice brains.
Western blot was used to determine the expression of MAM-associated proteins. Data were analyzed using one-way ANOVA (n = 3 per group). *P < 0.05, **P < 0.01, ***P < 0.001
The expression levels of PACS2, PSS1, and CHOP in the 3×TG-AD mice were much higher than those of WT mice, DMT markedly suppressed the higher levels of PACS2 and PSS1 (P ≤ 0.0339), but did not show significant effects on CHOP expression. The expression of the remaining three proteins, MFN2, Sig-1r, and VAPB, markedly decreased in 3×TG-AD mice relative to WT mice (P ≤ 0.0390). DMT remarkably restored the levels of the three proteins (P ≤ 0.0216). The level of these 6 MAM-associated proteins did not show significant changes in the WT mice treated with DMT.
DMT modulated Ca2+ dynamic between ER and mitochondria
After evaluating the effect of DMT on the main factors impacting on the Ca2+ transport, ER-mitochondrial contacts and MAM-associated proteins, we further determined effects of DMT on the Ca2+ transfer efficiency between ER and mitochondria. Ca2+ uptake in the organelles was assessed in the presence and absence of DMT with and without co-treatment with BD1063 and Aβ in primary hippocampal neurons. Following 24 hours of treatment, DMT had no effects on mitochondrial and cytosolic Ca2+ response to ATP stimulation (Fig. 5A-D). The Aβ exposure evoked an approximate 15% decrease and 24% increase of Ca2+ amplitude in mitochondria and cytosol, respectively. This result was consistent with previous studies, in which the decrease in mitochondrial Ca2+ in the Aβ -treated cells was considered to be the loss of MMP that significantly reduced mitochondrial Ca2+ uptake capacity [48]. Co-administration with DMT almost entirely reversed the Aβ-evoked alteration of Ca2+ amplitude in the two subcellular components (Fig. 5A-D). BD1063 markedly also lowered mitochondrial Ca2+ level. The cytosolic Ca2+ amplitude of the BD1063-treated cells was only 59.1% of the controlled cells. Co-treatment with DMT restored mitochondrial Ca2+ amplitude to the control value, but did not change the cytosolic level (Fig. 5E-H).
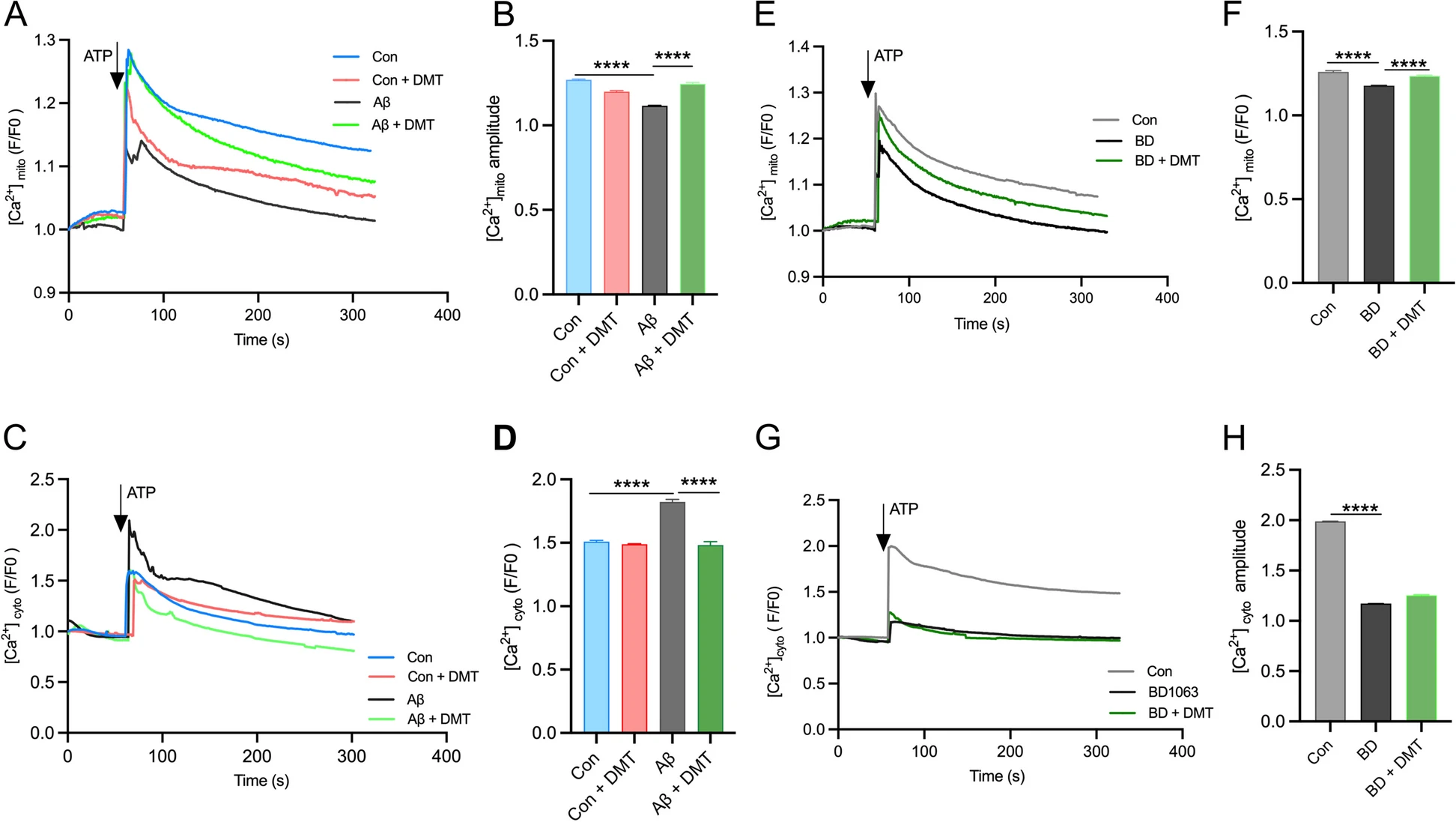
DMT modulates the calcium transfer between ER and mitochondria.
Cytosolic and mitochondrial Ca2+ were measured in the primary hippocampus by using live cell calcium imaging. A, B and C, D show the mitochondrial and cytosolic calcium, respectively, in the cells exposed to Aβ with or without DMT treatment. E, F and G, H show the mitochondrial and cytosolic calcium, respectively, in the cells exposed to BD1063 with or without DMT treatment. A, C, E, G show the representative figure of Ca2+ peaks after ATP treatment. B, D, F, H show the the amplitude peak of calcium levels after ATP treatment. Data were obtained from 3 independent experiments and analyzed using two-way ANOVA. [Ca2+]mito, mitochondrial calcium; [Ca2+]cyto, cytosolic calcium. BD, BD1063. ****P < 0.0001
DMT protected mitochondrial function in the primary hippocampal neurons
Having established that DMT was sufficient to maintain mitochondrial Ca2+ homeostasis, we next considered the effect of DMT on the mitochondrial Ca2+ -linked cellular process, including oxidative phosphorylation (OXPHOS) and ATP synthase, in the in vitro model of AD. Aβ-treated cells were monitored for changes in OXPHOS by measuring mitochondrial oxygen consumption rates (OCR) in a seahorse assay. The Aβ-exposed cells also had strikingly lower OCR values on basal respiration, proton leak, maximal respiratory capacity, and ATP-linked respiration (Fig.6A-E). Correspondingly, MMP and cellular ATP levels also decreased (Fig. 6F-G). This result was expected because it has been known that maintaining the TCA cycle requires sufficient flux of Ca2+ from the ER to mitochondria [49]. Therefore, the loss of mitochondrial Ca2+ in the cells exposed to Aβ may impair OXPHOS activity, leading to mitochondrial damage, collapse of MMP, and the decrease of ATP synthase. The aforesaid parameters, maximal respiratory capacity and ATP-linked respiration, were rescued post DMT treatment (Fig.6D, E). DMT treatment in control cells had no effect on OCR. DMT and Aβ co-incubation markedly reversed intracellular ATP levels and MMP compared to incubation with Aβ alone (P < 0.050) (Fig. 6F, G).
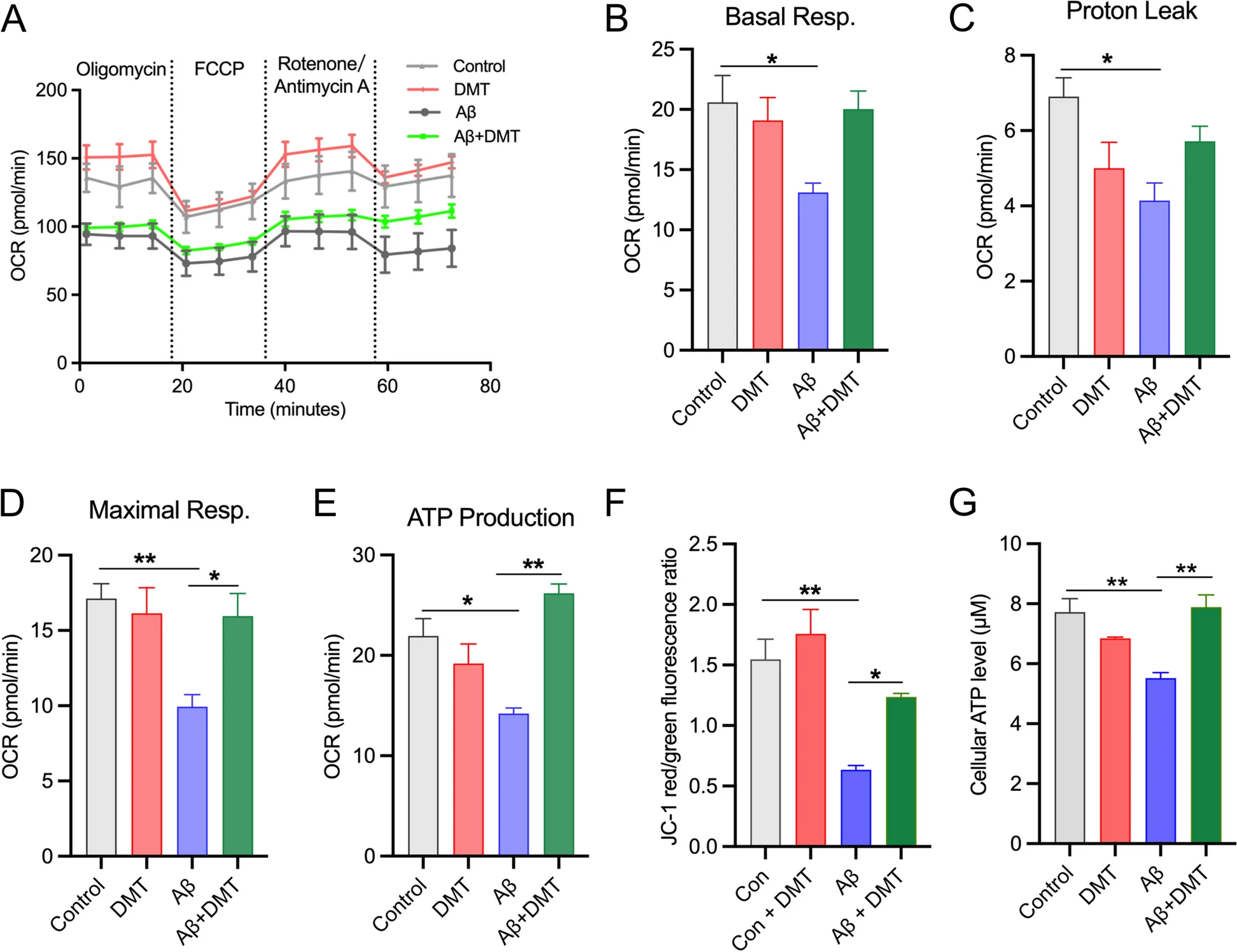
DMT rescues mitochondrial dysfunction in the primary hippocampal neurons.
A Oxygen consumption rate (OCR) at baseline and following: oligomycin, FCCP, and rotenone + antimycin A. B, C, D, E show the quantification of basal respiration, proton leak, maximal respiration, and ATP production. F shows the quantification of the mitochondrial membrane potential (MMP). G shows the quantification of the cellular ATP level. Data were obtained from 3 independent experiments and analyzed using two-way ANOVA. *P < 0.05, **P < 0.01
Discussion
The current study represents a systematic investigation on the anti-AD effects of DMT and its association with the modulation of neuronal Sig-1r-mediated ER-mitochondria signaling in the 3×TG-AD transgenic mouse model of AD and Aβ-exposed primary hippocampal neurons with additional utilization of Sig-1r knockdown mice and the Sig-1r antagonist BD1063.
This study revealed robust effects of DMT in mitigating spatial memory impairment and Aβ deposits in the transgenic AD model mice. In the water maze test, following 3 weeks of the treatment, DMT markedly shortened the latency to the platform in multiple days of training phase, and increased time and number of entries into the platform in the probe trial of the model mice. In parallel, chronic DMT largely reduced Aβ plaques in the hippocampus and PFC, the two brain regions closely associated with encoding, retrieval, and consolidation of memories [50]. One previous study has shown that chronic DMT (2 mg/kg for 21 days) also improved learning and memory in water maze and novel object recognition test in intact mice; the combination with ritanserin, a 5-HT2 receptor antagonist, however, did not abolish the behavioral effects of DMT [21]. DMT promoted adult neurogenesis of the hippocampus via Sig-1r, but not 5-HT1A and 5-HT2A receptors [21]. These results suggest that the anti-AD effects of DMT observed in this study is more closely associated with its modulation of Sig-1r than 5-HT receptor systems.
Subsequently, we further examined the role of Sig-1r in DMT’s effects. While a marked decreased level of Sig-1r in the hippocampus of 3×TG-AD and Sig-1r knockdown mice was observed using immunofluorescent detection, chronic DMT consistently and entirely returned the expression of Sig-1r to the control levels in the 3×TG-AD mice. These results proved that DMT as agonist could protect brain Sig-1r activity from AD pathological insults. It also indicated an involvement of the decreased Sig-1r function in the neuropathology of AD, which is consistent with previous studies, demonstrating a large decrease and loss of Sig-1r in brain regions of individuals in early stage of AD [5, 51]. In other studies, it was found that another Sig-1r agonist, PRE084, improved mitochondrial respiratory impairment and alleviated blood-brain barrier disruption in the Aβ-injected animals [14, 52]. Both PRE084 and DMT significantly reduced Aβ-induced neuroinflammatio n[42]. Compared with DMT, PRE084 significantly improved hippocampal neurogenesis in the Aβ-injected mice, which may be due to PRE084’s higher affinity for Sig-1r relative to DMT [42]. However, whether there exit differences between PRE084 and DMT in improving others AD pathology remains for further evaluation, as DMT is an agonist acting at multiple receptors such as Sig-1r, 5-HT2, and TAAR, while PRE084 is a highly selective Sig-1r agonist [23, 42].
Given that ER-mitochondria interface with MAMs is a key site where Sig-1r mediates calcium signaling between the two organelles [6], we hence examined the effects of DMT on the integrity of ER-mitochondria physical contacts. Both transgenic and Sig-1r knockdown mice displayed markedly wider gaps at ER and mitochondria contact points as compared to wild type mice. Similarly, following the exposure of cultured hippocampal neurons to Aβ oligomers and BD1063 for 24 hours, number of PLA-labeled dots indicative of ≤ 40 nm contact clefts between the two organelles strikingly decreased, proving the assertion that the impaired ER-mitochondria integrity is a universal and probably early event occurred in the pathogenesis of AD [4]. Co-administration with DMT, however, largely and even completely recuperated the two organelles’ contact gaps in both in vivo and intro paradigms. It appeared that the protective effects of DMT against the AD-related impairment of the subcellular integrity were mainly derived from its activation of Sig-1r.
This study further revealed that the model mice had apparently abnormal expression of the six MAM-associated proteins examined, manifesting as upregulated expression of PACS2, PSS1 and CHOP, and downregulated expression of MFN2, VAPB and Sig-1r. The first three proteins are widely involved in free radical production and ER stress-induced apoptosis, while they are key components of MAMs in regulating calcium transfer [3]. The last three proteins play a critical role in the maintenance and operation of mitochondrial network in particular ER-mitochondrial communication [3]. One previous study also has shown the upregulated expression of PACS2 and PSS1 in multiple brain regions of AD mouse model and in cortical tissues of AD patients; and the upregulation occurred prior to the appearance of Aβ plaques [5]. It is suggested that the dysregulation of MAM-associated proteins may represent an early aberrant event of ER-mitochondrial interplay that perhaps triggers AD pathological cascades. However, chronic DMT partially and even entirely reversed the expression of all the MAM-associated proteins to the levels of WT control mice, except for CHOP, indicating an association of DMT’s effects in preventing ER-mitochondria interplay with its modulation of MAM-associated proteins. The increased CHOP has been suggested in the progression of AD by sensitizing neuronal cells to apoptosis [53]. Other two MAM-associated proteins, PACS-2 and PSS1, are involved in modulating lipid synthesis of the ER. PACS-2 also could integrate ER-mitochondria communication and apoptosis [54, 55]. This study revealed that DMT had significant effects in suppressing the increased expression of PACS2 and PSS1, but not CHOP. It seems that DMT may particularly restore lipid synthesis of the ER, but had less effects in modulating cell apoptosis.
Dysregulation of MAM-mediated calcium homeostasis has been implicated in the neuropathology of AD and other neurodegenerative diseases [56]. In this study, we examined the effects of DMT on neuronal calcium dynamics by measuring mitochondrial and cytosolic calcium amplitude under DMT co-treatment with Aβ and BD1063. While the Aβ exposure for 24 hours induced a decrease of mitochondrial and an elevation of cytosolic calcium amplitude, BD1063 reduced both compartment calcium amplitudes. It is likely that the neurotoxin and the Sig-1r antagonist may target subcellular compartments differentially, i.e., Aβ mainly impaired mitochondrial calcium dynamics by inhibiting the uptake, in turn, causing the accumulation of the ion in the cytosol. It might be plausible as mitochondria among organelles is the most vulnerable to oxidative and neurotoxic insults [57]. Whereas BD1063 probably suppressed calcium influx and storage via the blockade of ER’s Sig-1r. While DMT co-treatment overturned mitochondrial calcium uptake in the two types of pathological insults and Aβ-evoked decrease of cytosolic calcium level, it did not affect the BD1063-induced suppression of cytosolic calcium level. Similar results were also observed in a previous study, revealing that mitochondrial, but not cytosolic, calcium uptake was markedly increased following SA4503, an agonist of Sig-1r, co-treatment with Aβ or tunicamycin, a neurotoxin, in SH-SY5Y cells, a cell model of neurodegenerative disorders [5]. It thus appeared that the effects of DMT in protecting neuronal calcium homeostasis may be principally achieved by restoring Sig-1r-mediated MAM calcium mechanisms.
We also noted that DMT improved the expression of Sig-1r in the AD models. It is possible that DMT helps to compensate for the loss of signalling pathways and other receptors. The Sig-1r itself has been found to have pleiotropic neuroprotective effects, such as shuttling calcium from the ER to mitochondria to prevent mitochondrial stress, prevent apoptosis, and induce cell proliferation and growth [41]. Targeting Sig-1r in disease states may therefore have multiple neuroprotective benefits. Herein, our results indicated that DMT regulated calcium transfer between and mitochondria to restore mitochondrial function via Sig-1r activation. Also, DMT effectively repaired mitochondrial function manifesting as the improvement in the mitochondrial OXPHOS capacity and ATP synthesis in the AD models.
DMT has been found to enhance cognitive performance in adult mice probably via Sig-1r modulation of neurogenesis [21], confirming, once again, the crucial function of Sig-1r in the DMT’s nootropic effects. It also could, at least in part, explain DMT as an entheogenic beverage often used to enhance perception, mood, consciousness, cognition, and behavior in ritual and recreational activities. Furthermore, as the endogenous ligand, DMT could exert the behavioural effect by binding to Sig-1r, which is abundant in the brain [19]. This study elucidated the underlying intracellular molecular mechanism that DMT improved cognitive dysfunction in AD via activation of Sig-1r. Additionally, DMT is also the agonist of TAAR and 5-HT receptors, which are regarded as targets for the treatment of AD [58, 59]. Therefore, DMT may exert anti-AD effect through multiple receptors-mediated signalling. Previous studies have discussed the importance of targeting multiple pathways in combating the neurodegenerative disease AD, particularly using drugs that target multiple receptors [60]. Therefore, DMT is a potentially important drug for the treatment of AD.
There are multiple limitations that should be taken into consideration. Firstly, this study only delineated the neuronal mechanisms, but did not detect the role of glia in the nootropic effects of DMT. Sig-1r is also expressed in microglia and it is involved in the protective effects of DMT against brain ischemia [61]. Whether and to what extent microglial Sig-1r is involved in the anti-AD effects of DMT deserve for further estimation. Secondly, although the previous study has revealed that the nootropic effects of DMT were less associated with 5-HT receptor systems than Sig-1r [21], whether there exist synchronic, additive, or even synergistic effects of the two receptor systems should be an intriguing question for the future study of DMT. Finally, although, unlike most hallucinogenic drugs, DMT did not cause or caused less tolerance, dependence, and withdrawal symptoms for acute and short-term use [23], the potential side effects of the long-term use should be carefully examined. Additionally, DMT should have acceptable safety parameters for use due to its high vulnerability to abuse. It also should be pay attention to whether long-term effects of DMT use is associated with Hallucinogen Persistence Perception Disorder.
Conclusion
Taken together, the current study demonstrated that the anti-AD effects of DMT are associated with its restoration of neuronal ER-mitochondria crosstalk via the Sig-1r activation (Fig. 7). Sig-1r was identified as a target for maintenance of mitochondrial calcium homeostasis and restoration of mitochondrial function, particularly mitochondrial calcium uptake. It then improved mitochondrial OXPHOS capacity and ATP production. DMT could serve a novel preventive and therapeutic agent against AD.

The putative mechanism of DMT improving the spatial learning and memory in animal models of AD.
The current study demonstrated that the anti-AD effects of DMT are associated with its restoration of neuronal ER-mitochondria crosstalk via the Sig-1r activation. DMT modulates the mitochondrial calcium uptake and ER-mitochondrial contacts in the in vivo and in vitro models, facilitates the TCA cycle, and protects against mitochondrial dysfunction in Alzheimer’ disease