
Mitochondria in Huntington’s disease: implications in pathogenesis and mitochondrial-targeted therapeutic strategies
By Ana Maria Jurcau, and Carolina Maria Jurcau
Excerpt from the article published in Neural Regeneration Research 18(7):p 1472-1477, July 2023. | DOI: 10.4103/1673-5374.360289
Editor’s Highlights
- Neurons are cells with very high energy demands and which, unlike other cells in the body, have strict aerobic metabolism.
- ATP is generated in neurons mainly by the mitochondrial oxidative phosphorylation.
- Mitochondrial dysfunction in the pathogenesis of HD.
- The production of an abnormal protein, mutant huntingtin (mHtt), leads to widespread neuronal loss in the striatum, cortex, and hypothalamus.
- mHtt diminishes the expression of sigma-1 receptor (Sig-1R), an ER protein localized at MAMs which forms a chaperone complex with immunoglobulin protein/glucose-regulated protein 78. Upon ER Ca2+ depletion, Sig-1R dissociates from BiP and stabilizes IP3Rs, prolonging Ca2+ signalling.
Abstract
Huntington’s disease is a genetic disease caused by expanded CAG repeats on exon 1 of the huntingtin gene located on chromosome 4. Compelling evidence implicates impaired mitochondrial energetics, altered mitochondrial biogenesis and quality control, disturbed mitochondrial trafficking, oxidative stress and mitochondrial calcium dyshomeostasis in the pathogenesis of the disorder. Unfortunately, conventional mitochondrial-targeted molecules, such as cysteamine, creatine, coenzyme Q10, or triheptanoin, yielded negative or inconclusive results. However, future therapeutic strategies, aiming to restore mitochondrial biogenesis, improving the fission/fusion balance, and improving mitochondrial trafficking, could prove useful tools in improving the phenotype of Huntington’s disease and, used in combination with genome-editing methods, could lead to a cure for the disease.
Introduction
Huntington’s disease (HD) is a neurodegenerative disease with a global prevalence ranging between 0.02 and 17.27/100,000 inhabitants, which tends to increase in most regions due to increasing mutation rate, increasing life expectancy, greater availability of genetic testing, and access to symptomatic treatment (Rawlins et al., 2016). It is transmitted as an autosomal dominant disease with complete penetrance, the age of onset varying inversely proportional with the number of CAG repeats above 40 in exon 1 of the huntingtin gene located on chromosome 4 (Julayanont et al., 2020). The clinical picture consists of a variety of involuntary movements, such as chorea, dystonia, bruxism, variable degrees of bradykinesia and rigidity, psychiatric symptoms, and cognitive decline progressing to dementia (Zielonka, 2018).
Much research has tried to elucidate how the production of an abnormal protein, mutant huntingtin (mHtt), leads to widespread neuronal loss in the striatum, cortex, and hypothalamus. An intricate complex of vicious cascades involving excitotoxicity, impaired proteostasis, mitochondrial dysfunction, oxidative stress, transcriptional dysregulation, reduced bioavailability of growth factors, as well as astrocytic dysfunction and neuroinflammation, has been found to contribute to the different stages of the disease (Jurcau, 2022). The present review will focus on the involvement of mitochondrial dysfunction in the pathogenesis of HD and discuss some therapeutic targets.
Search Strategy and Selection Criteria
The references cited in this review have been obtained from the PubMed and Google Scholar databases. We referenced full-text review articles, randomized control trials, and meta-analyses published between 1995 and 2023.
Normal Functions of the Mitochondria
Neurons are cells with very high energy demands and which, unlike other cells in the body, have strict aerobic metabolism. The large amounts of energy are required for generation of action potentials and postsynaptic potentials as well as for membrane repolarization, neurotransmitter synthesis, vesicle recycling, axoplasmic transport, and synaptic plasticity. ATP is generated in neurons mainly by the mitochondrial oxidative phosphorylation (OXPHOS) (Deitmer et al., 2019), a process during which complexes I–IV located on the inner mitochondrial membrane (IMM) pump protons from the matrix to the intermembrane space using the electrons removed from the Krebs cycle by reduced nicotinamide adenine dinucleotide and flavin adenine dinucleotide (FADH2). This results in a potential gradient across the IMM (–150 to –180 mV), which is used in the final step of OXPHOS by complex V to synthesize ATP (Jurcau, 2021).
To meet the cellular energy requirements, mitochondria number, shape, and size must be tightly regulated, a process called mitochondrial dynamics.
Mitochondrial biogenesis is a complex process, involving transcription and translation of both mitochondrial DNA (mtDNA) and nuclear DNA-encoded proteins. The latter are synthesized in the cytosol as precursors, maintained in an unfolded conformation by forming complexes with chaperones, such as heat shock protein (HSP) 70 or HSP90, and imported to the mitochondria through cooperation of translocase of the outer mitochondrial membrane (TOM) and translocase of the inner mitochondrial membrane (TIM) 23 (Bausewein et al., 2017). A key role in this process is played by peroxisome proliferator-activated receptor (PPAR)γ-coactivator 1 α (PGC-1α), which regulates the expression of nuclear-encoded subunits of the electron transport chain complexes (ETC), of the mitochondrial transcription factor A (TFAM), of antioxidant defence proteins, and of nuclear respiratory factors 1 and 2 (Nrf1 Nrf2) (Panes et al., 2022).
For mitochondrial fission, a process that also allows for generation of new mitochondria, dynamin-related/-like protein 1 (Drp1) must be bound to the outer mitochondrial membrane (OMM) by adaptor proteins such as mitochondrial dynamics proteins 49 and 51 (MiD49 and MiD51) and mitochondrial fission factor, followed by recruitment of dynamin 2 (Dnm2), a GTPase which completes the process (Jurcau, 2021).
Fusion allows for repair of degraded organelles and sharing of essential components, and is regulated by Mfn1 and Mfn2, which tether the OMM of adjacent mitochondria, after which optic atrophy 1 (OPA1) mediates the fusion of the IMM (Adebayo et al., 2021).
Irreversibly damaged mitochondria are disposed through mitophagy, which involves the formation of an isolation membrane, followed by activation of the pre-initiation complex, containing Unc-51-like kinase 1, autophagy-related protein 13 and 101, and focal adhesion kinase family interacting partner 200 (FIP200; Swerdlow and Wilkins, 2020). Subsequent recruitment of phosphatidyl inositide 3-kinase (PI3K), autophagy-related protein 14, beclin1, autophagy and beclin 1 regulator 1, and vascular sorting protein 34 and 15 (Vsp 34 and 15) produce phosphatidyl inositol 3-phosphate, also called the initiation complex (Hurley and Young, 2017). Atg4 mediates cleavage of pro-light chain 3 (LC3) to LC3-I, which is further transformed by phosphatidylethanolamine to LC3-II, resulting in elongation and closure of the isolation membrane. The subsequent fusion of the autophagosome with lysosomes is mediated by Rab7 and lysosome-associated membrane protein 2. Recognition of the damaged mitochondria occurs through phosphatase and tensin homologue (PTEN)-induced putative kinase 1 (PINK1) and Parkin, an E3 ubiquitin ligase (Jurcau, 2021).
Due to the particular shape of the neurons, mitochondria must be trafficked along the neural outgrowths. The process is mediated by a protein complex consisting of the heavy chain of kinesin-1, Miro 1 and 2 (known in humans as mitochondrial Rho GTPase -RhoT1 and RhoT2), attached to the outer surface of the mitochondrion, milton (or TRAK1 and TRAK2 in humans), which links kinesin and Miro (Melkov and Abdu, 2018; Fenton et al., 2021), and dynein. While anterograde transport is mediated by kinesin-1, dynein is involved in retrograde trafficking. Calcium signals (transient elevations of cytosolic Ca2+), slow down or stop mitochondrial trafficking (Jurcau, 2021).
Mitochondria also contribute to maintenance of cellular calcium homeostasis. Normally, cytosolic calcium concentrations are nanomolar (50–300 nM), while extracellular calcium concentrations are milimolar (Jurcau, 2021). Elevated cytosolic Ca2+ concentration is rapidly restored through the activity of the Ca2+-ATPase, which hydrolyses ATP to pump Ca2+ against the concentration gradient, and of the Na+/Ca2+ exchanger, which extrudes calcium in exchange for sodium. Cytosolic Ca2+ also binds to Ca2+ -binding proteins, or is taken up by mitochondria or the endoplasmic reticulum (ER) through the ATP-dependent sarco-endoplasmic reticulum Ca2+-ATPase (Primeau et al., 2018). In mitochondria, Ca2+ is transferred into the intermembrane space by voltage-dependent anion-selective channels (VDACs), and into the mitochondrial matrix by the mitochondrial Ca2+ uniporter (MCU), where it activates ATP production by stimulating the activity of dehydrogenases in the Krebs cycle and the activity of complex I of the ETC (Naia et al., 2017). Once inside the mitochondria, calcium is either converted to insoluble salts, or extruded by the Na+/Ca2+ antiporter, powered by the Na+ gradient resulting from the activity of the Na+/H+ antiporter (MacAskill and Kittler, 2010). Mitochondria-associated membranes (MAMs) are microdomains enriched in inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs), ryanodine receptors (RyRs), and molecular chaperones, such as glucose-regulated protein 75, which generate functional complexes with voltage-dependent anion-selective channels to mediate Ca2+ transfer from the ER to mitochondria (Eysert et al., 2020). However, increases in mitochondrial Ca2+ concentrations cause mitochondrial depolarization and trigger the opening of the mitochondrial permeability transition pore (MPTP), resulting in release of cytochrome c and neuronal apoptosis (Jurcau and Ardelean, 2021).
Research has shown differences between synaptic and nonsynaptic mitochondria in their calcium-buffering ability. Synaptic mitochondria display faster rates of Ca2+ uptake and release to sustain neurotransmitter exocytosis (Naia et al., 2017). Furthermore, calcium influx evoked by N-methyl-D-aspartate (NMDA) receptor (NMDAR) activation, through the mitochondrial Rho-GTPase Miro acting as a calcium sensor, leads to postsynaptic translocation of mitochondria, calcium overload, mitochondrial dysfunction and excitotoxic cell death (Naia et al., 2017).
Mitochondria in Huntington’s Disease
Structural abnormalities in mitochondrial morphology were observed already in 1978 in post-mortem cortical tissue from HD patients (Zuccato et al., 2010), and evaluation of the cellular bioenergetics in HD-derived neurons showed an altered glycolytic capacity (The HD iPSC Consortium, 2020). In addition, one of the first HD models was obtained through administration of 3-nitropropionic acid, which irreversibly inhibits the mitochondrial complex II (Zuccato et al., 2010). These findings point toward a crucial role of mitochondria in HD pathogenesis.
Impaired mitochondrial energy production
Research has shown reduced glucose consumption, especially in the basal ganglia even in presymptomatic carriers of the HD mutation (Antonini et al., 1996), and increased lactate levels in the caudate and occipital cortex of HD patients (Jenkins et al., 1998). Due to the ubiquitous nature of huntingtin, the defective energy production affects peripheral tissues as well, such as lymphoblasts, muscle fibres, or adipose tissue, leading to weight loss in most patients with HD (Silva et al., 2013).
Normal mitochondrial ATP synthesis requires the mitochondrial transmembrane potential to be maintained at 80–90 % of its maximal value (–150 to –180 mV) (Carmo et al., 2018). The decrease in the activity of the mitochondrial complexes (mainly complexes II – succinate dehydrogenase, and III – ubiquinol-cytochrome c oxidoreductase), together with the interaction of the N-terminal fragment of mHtt with TIM23 and inhibition of mitochondrial protein import, synergistically decrease the mitochondrial transmembrane potential (Wright et al., 2017), leading to mitochondrial dysfunction which is directly correlated with the increased number of glutamine repeats. In addition, elongated CAG repeats in the mutated huntingtin gene promote age-dependent expansion of pathogenic mtDNA heteroplasmies (coexistence of wild-type and mutated mtDNA), which accelerate disease progression, as shown in a longitudinal study on HD lymphoblasts (Wang et al., 2021).
Furthermore, mHtt prevents the CREB/ TATA-binding protein-associated factor 4 (TAF4) complex from binding to the PGC-1α promoter and the transcription of PGC-1α, a key regulator of glucose metabolism and β-oxidation of fatty acids (Jesse et al., 2017). Pyruvate dehydrogenase, the enzyme that catalyses the oxidative decarboxylation of pyruvate to acetyl-CoA in the matrix, was decreased in the putamen and caudate of HD patients (Carmo et al., 2018), as were other enzymes of the Krebs cycle, such as aconitase (detailed further) or succinate dehydrogenase (Benchoua et al., 2006).
Glycolysis is also impaired, mainly due to the abnormal function of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), the enzyme mediating the sixth step of glycolysis. GAPDH preferentially interacts with cleaved polyglutamine (polyQ) domains, leading to enhanced nuclear translocation of mHtt via the ubiquitin-E3 ligase Siah1 and increasing its cytotoxicity, as well as to GAPDH sequestration in mHtt aggregates (Bae et al., 2006).
Increased oxidative stress
Mitochondria are one of the main generators of reactive oxygen species (ROS). About 1–2% of the consumed oxygen will generate superoxide anion by electrons leaking at the level of complexes I and III (Carmo et al., 2018) even in normal conditions. Despite neuronal and mitochondrial antioxidant defence systems (Mn-SOD, glutathione peroxidases, the cytosolic Cu,Zn-SOD or SOD1, catalase, and peroxiredoxins), the high rate of ROS production overwhelms these antioxidants and leads to oxidative stress, which impairs OXPHOS and energy production (Jurcau and Ardelean, 2021). Aconitase is particularly susceptible to ROS-induced impairment (Chen et al., 2017), its activity being reduced by 70–90% in the caudate and putamen of HD patients. Other ROS-producing enzymes, such as NADPH oxidase, also show augmented activity in cortical and striatal post-mortem samples from HD patients (Valencia et al., 2013).
The generated ROS oxidize proteins (including mitochondrial enzymes), membrane lipids (Manoharan et al., 2016), as well as nuclear and mitochondrial DNA (Askeland et al., 2018), having a crucial role in HD pathogenesis (Onyango et al., 2021). In addition, mHtt-induced transcriptional dysregulation interferes with the Nrf2/ARE pathway, which normally upregulates the expression of antioxidant enzymes (Moretti et al., 2021) in a vicious cascade.
Impaired mitochondrial quality control
The constant renewal of the mitochondrial network is modulated by a series of transcription factors, such as cAMP responsive element-binding protein (CREB) binding protein or TAF4. The interaction of mHtt with CREB binding protein or TAF4 and the indirect impairment of the expression of PGC-1α results in severely downregulated expression of nuclear-encoded subunits of the electron transport chain and of genes involved in antioxidant response (Johri et al., 2013; Jesse et al., 2017). In addition, PGC-1α regulates the expression of TFAM, the key regulator of mitochondrial DNA transcription (Palikaras and Tavernarakis, 2014), as well as the transcription of ATP synthase and manganese superoxide dismutase (Johri et al., 2013). In HD mice, there was a 6-fold decrease in PGC-1α in the medium spiny neurons of the caudate nucleus compared to wild-type mice, due to interactions of mHtt with the above-mentioned signalling pathways and to direct binding to the PGC-1α promoter (Cui et al., 2006). Overall, 72 genes involved in cellular metabolism, nervous system development, or apoptosis regulation have down-regulated microRNAs (miRNAs), while only 19 genes have up-regulated miRNAs (Dubois et al., 2021). Table 1 summarizes some of the down- and up-regulated miRNAs described by researchers in HD, with relation to mitochondrial dysfunction.
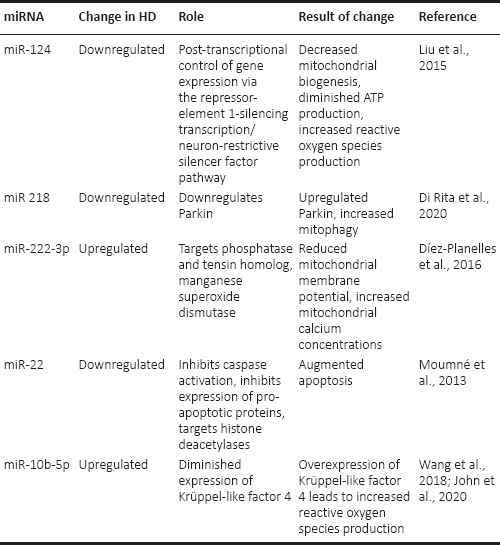
Changes in miRNAs described in Huntington’s disease and their effect on mitochondrial function
Mitochondrial morphology and dynamics are also significantly impaired by the direct interaction of the N-terminus fragment of mHtt with Mfn2, inhibiting its mitochondrial elongation-promoting action (Jurcau, 2022). Furthermore, the expression of Mfn1, Mfn2, and OPA1 were shown to decrease, paralleling the stage of HD (Machiela et al., 2021). As for mitochondrial fission, mHtt accumulates on mitochondria and increases the recruitment of Drp1. Activated calcineurin, together with mitogen-activated protein kinase 1, dephosphorylates Drp1, thereby potentiating Drp1 recruitment (Roe and Qi, 2018). Calcineurin also activates the striatal enriched tyrosine phosphatase (STEP), through which it leads to reduced synaptic NMDA receptor expression (Braithwaite et al., 2006). Inhibition of the calcineurin-Drp1 pathway proved neuroprotective against striatal degeneration, as did OPA1 overexpression (Machiela et al., 2021).
In depolarized mitochondria, PINK1 stabilizes at the OMM and is no longer imported to the IMM, favouring mitochondrial Parkin translocation (Kazlauskaite et al., 2015). Parkin molecules recruit ubiquitin chains to the OMM, recognized by autophagy adaptors such as p62. Fibroblasts from patients with juvenile HD showed increased Parkin levels and enhanced proteasomal degradation of Mfn1 (Aladdin et al., 2019), while PINK1 overexpression proved protective in HD fly models (Khalil et al., 2015). Wild-type Htt aids p62 to associate with LC-3. This scaffolding function is lost by mHtt, leading to impaired mitophagy (Rui et al., 2015). Htt, together with huntingtin-associated protein 1 (HAP1), have also an important role in controlling the dynamics of autophagosomes through regulation of kinesin and dynein.
The result of all these alterations is impaired mitochondrial quality control, with a reduced number and altered morphology shifting towards increased fragmentation of mitochondria due to increased levels of Drp1 and mitochondrial fission 1 protein (Fis 1) and decreased levels of Mfn1, Mfn2, and OPA1 (Sawant et al., 2021; Pantiya et al., 2020).
Impaired mitochondrial trafficking
Mitochondrial trafficking is critical in neurons, which need energy for regular bioenergetic homeostasis, as well as for synaptic transmission. As shown above, the process involves a complex machinery of motor proteins which attach to the organelles and transport them in an anterograde or retrograde direction (towards the cell soma). HAP1 interacts with both kinesin and dynein, regulating cargo transport on microtubules (Franco-Iborra et al., 2018), while Htt interacts with HAP1 and recruits GAPDH for producing the necessary amount of ATP. Further, the Htt–HAP1 complex serves as an adaptor protein, binding to both kinesin-1 and dynein as well as to the dynactin subunit p150glued (Twelvetrees et al., 2019). In HD, GADPH, HAP1 and dynamin are sequestered into mHtt aggregates (Qin et al., 2004). Further, huntingtin-interacting protein 1 (HIP1) contributes to the assembly of cytoskeletal structures, a function which is disturbed in HD due to a weaker interaction of HIP1 with mHtt. In addition, PINK1 and Parkin accumulate on the OMM and phosphorylate the GTPases RhoT1 and RhoT2, leading to a detachment of kinesin from mitochondria and arrest of the organelle (Jurcau, 2021). The impaired mitochondrial trafficking results in accumulation of fragmented mitochondria in the cell bodies, further augmenting the energetic disturbances and leading to synaptic loss and neuronal degeneration.
Impaired mitochondrial calcium handling
The soluble N-terminal fragment of mHtt interacts directly with the OMM and, via TIM23, with the IMM, disturbing protein import and leading to mitochondrial depolarization (Schrank et al., 2020). In addition, polyQ tracts are able to form proton-selective ion channels in the mitochondrial membrane, further diminishing the electrochemical proton gradient across the IMM (Monoi et al., 2020). These abnormalities interfere with the mitochondrial ability to buffer excess cytosolic Ca2+. The consequence of mitochondrial depolarization is opening of the MPTP at lower Ca2+ concentrations, leading to cytochrome c release and mitochondrial swelling (Jurcau, 2022).
The carboxy-terminus of mHtt also interacts with the IP3 receptors (IP3Rs) in a complex with HAP1, increasing their responsiveness and causing ER Ca2+ release at lower IP3 concentrations (Mackay et al., 2018). In addition, mHtt diminishes the expression of sigma-1 receptor (Sig-1R), an ER protein localized at MAMs which forms a chaperone complex with immunoglobulin protein/glucose-regulated protein 78. Upon ER Ca2+ depletion, Sig-1R dissociates from BiP and stabilizes IP3Rs, prolonging Ca2+ signalling (Delprat et al., 2020). The RyRs have been less extensively studied in HD, but the finding that inhibition of RyRs with dantrolene protects cortical and striatal neurons expressing polyQ stretches from degeneration clearly implicate these receptors as well in the dysfunction of MAMs and calcium leakage from the ER (Sun and Wei, 2021). Figure 1 summarizes the alterations in cellular calcium homeostasis in HD.
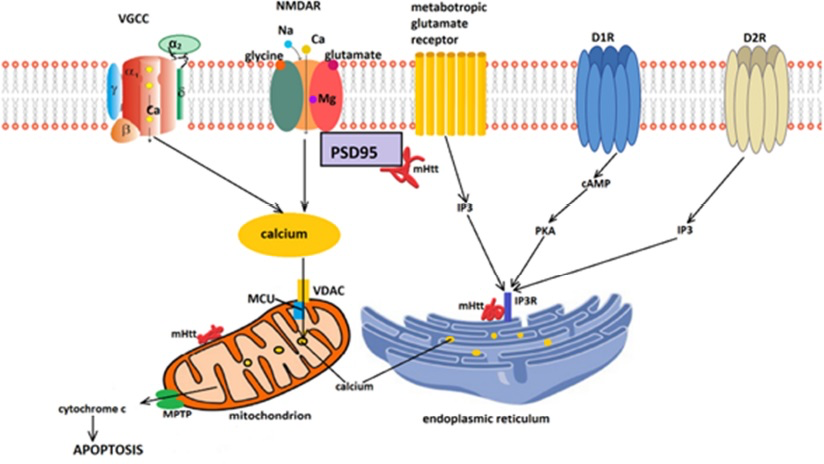
Impaired calcium homeostasis in Huntington’s disease leads to apoptosis.
Mutant Htt enhances the function of NMDA receptors (NMDAR) most likely through decreased interaction with postsynaptic density 95 (PDS95). The result is increased calcium influx. Dopamine released by nigral neurons acts on D1 and D2 receptors (D1R and D2R) and directly (for D2Rs) or indirectly (via activation of adenyl cyclase and phosphokinase A – for D1Rs) stimulate inositol 1,4,5-phosphate receptors (IP3R), leading to further increases of cytosolic Ca2+concentrations by stimulating Ca2+release from the endoplasmic reticulum (ER). The excess cytosolic calcium is buffered by mitochondria via the mitochondrial voltage-gated anion channel (VDAC) and mitochondrial calcium uniporter. However, mitochondrial Ca2+overload will cause opening of the mitochondrial permeability transition pore (MPTP). This outcome is enhanced by the direct association of mHtt with mitochondrial membrane proteins. VGCC: L-type voltage-gated calcium channel.
Mitochondria-dependent apoptosis
Following the opening of the MPTP and cytosolic release of cytochrome c, the latter forms with deoxy-ATP, Apaf-1 and caspase 9 the apoptosome that activates downstream targets such as caspases 3, 6, and 7, initiating caspase-dependent apoptosis (Jurcau and Ardelean, 2021). Other mitochondrial proteins, such as apoptosis-inducing factor or second mitochondrion-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI (SMAC/DIABLO) are released as well. Apoptosis-inducing factor translocates to the nucleus and causes DNA fragmentation and poly ADP ribose polymerase inhibition, accelerating cellular damage and destruction. SMAC/DIABLO binds to X chromosome-linked inhibitor-of-apoptosis protein and triggers apoptosis by suppressing the anti-apoptotic activity of X chromosome-linked inhibitor-of-apoptosis protein (Jurcau and Ardelean, 2022). Caspase-2 can cleave mHtt and generate toxic N-terminal fragments which cause degeneration of neurites, while caspase-7, preferentially expressed in MSNs, interacts with mHtt and triggers the activity of other caspases, accelerating apoptosis (Carmo et al, 2018). Increased cytosolic Ca2+ activates calpains and caspase 8, leading to cleavage and activation of Bcl-2 (B-cell lymphoma 2) interacting domain, which in turn induces conformational changes in other proapoptotic proteins, such as Bax, Bad, Bcl-XS, and inactivate anti-apoptotic proteins like Bcl-2 or Bcl-XL (Stevens and Oltean, 2019), thereby inducing caspase-independent apoptosis. In addition, mHtt binds more efficiently to p53, a major transcription factor, leading to upregulation of nuclear p53 and increasing the levels of target genes such as Bax and p53 upregulated modulator of apoptosis (Schapira et al., 2014). Furthermore, with increasing length of the polyQ tracts, the interaction of mHtt with HIP1 decreases, allowing HIP1 to interact with apoptotic proteins such as procaspase 8 and potentiating the extrinsic apoptotic pathway (Choi et al., 2006).
Therapeutic Strategies Targeting Mitochondrial Dysfunction
Although the most logical approach in a disease caused by transcription of an abnormal protein would be to stop the transcription of that protein, clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9) or RNA interference approaches are still in their infancy, require usually invasive modes of administration, and target the transcription of mHtt only in cerebral tissue (Jurcau and Jurcau, 2022). Given the widespread presence of mitochondrial dysfunction, and the fact that diagnosis is often established late, in symptomatic patients, the search for means to efficiently counterbalance these impairments is worth pursuing. Many molecules have been tested in cell lines or animal models of HD, but few of them have advanced to clinical testing.
Conventional mitochondria-targeted therapies
Melatonin, a hormone produced by the pineal gland, inhibited mHtt-induced mitochondrial depolarization and the release of pro-apoptotic factors in vitro (Wang et al., 2011). It also exhibits anti-inflammatory effects by inhibiting the binding of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) to nuclear DNA and the production of pro-inflammatory cytokines, up-regulated Nrf2 and sirtuin 1 (SIRT1), upregulated γ-aminobutyric acid activity and prevented excitotoxicity (Cardinali, 2019). A pilot clinical trial (NCT04421339) is currently recruiting HD gene carriers to evaluate efficacy in improving sleep and quality of life (https://www. clinicaltrials.gov).
A series of natural compounds act as antioxidants and improve mitochondrial function by interfering with various pathways. The dietary flavonoid quercetin acts as a free radical scavenger and induces the Nrf2/ARE pathway, upregulating SOD, glutathione transferase, glutathione peroxidase, catalase and thioredoxin. It showed potent antioxidant effect in a rat chemical model of HD (Grewal et al., 2021). Resveratrol, a dietary polyphenol, activates SIRT1 and enhances the PI3K/protein kinase B pathway and the nuclear translocation of Nrf2, exhibiting also anti-inflammatory effects through inhibition of the NF-κB and MAPK pathways (Hannan et al., 2020). In YAC128 mice, resveratrol improved motor function by increasing the expression of mitochondrial ETC complexes and of mitochondrial-related transcription factors such as TFAM and PGC-1α (Naia et al., 2017b). Another natural compound, β-lapachone, also increased SIRT 1, PGC-1α deacetylation and CREB levels, reducing the formation of ROS and leading to phenotypic improvement in R6/2 mice (Lee et al., 2018). Curcumin interferes with the NF-κB, signal transducer and activator of transcription 3 and Nrf2 pathways, acting as an antioxidant and anti-inflammatory molecule, being also able to suppress protein aggregation (Labanca et al., 2021). It showed beneficial effects in R6/2 mice (Elifani et al, 2019). Unfortunately, dietary antioxidant molecules have poor blood brain barrier penetrance, but nanotechnology-based delivery methods, such as nanomaterials, nanozymes and drug-loaded nanosystems, could help overcome this drawback (González et al., 2021).
Other antioxidants have also been shown to improve mitochondrial function. Nicotinamide mononucleotide adenylyl transferase reduced mHtt aggregation and restored ATP levels in a fly model of HD (Zhu et al., 2019), N-acetylcysteine restored the activities of complexes II, IV, and V in 3-nitropropionic acid-treated mice (Sandhir et al., 2012), while insulin-like growth factor 1 restored mitochondrial function by upregulating the PI3K/protein kinase B signalling pathway (Ribeiro et al., 2014). Bezafibrate is an activator of PGC-1α expression, which in R6/2 and full-length mHtt mouse models of HD reduced striatal atrophy and improved the disease phenotype (Johri et al., 2012). Alpha-lipoic acid and acetyl-L-carnitine, two other antioxidants, improved the lipid composition and structure of mitochondria, leading to improvement of memory impairments in chemical HD models (Mehrotra et al., 2015).
However, a series of clinical trials with antioxidants failed or yielded minor improvements. Cysteamine, shown to improve motor dysfunctions and increase life expectancy in R6/2 HD mice, failed in a phase 2 clinical trial (NCT02101957) (Verny et al., 2017). Coenzyme Q10, shown in preclinical settings to diminish ROS production and improve mitochondrial bioenergetics, also yielded negative results in a phase 3 clinical trial on 609 patients with early-stage HD (NCT00608881, 2CARE) (McGarry et al., 2017). Similar results were obtained for creatine, shown to stimulate mitochondrial respiration, in the CREST-E study (NCT00712426) (Hersch et al., 2017) and ethyl-eicosapentaenoic acid, shown to bind to mitochondrial PPAR, inhibit caspases, and downregulate the c-Jun N-terminal kinase pathway, in the TREND-HD trial (NCT00146211) (Huntington Study Group TREND-HD Investigators, 2008). The effect of resveratrol was evaluated in a phase 3 clinical trial (REVHD, NCT02336633), but the results have not been published so far (https://clinicaltrials.gov).
Potential targets proved to be the Sig-1 receptors. Pridopidine, initially used as a dopamine stabilizer, was subsequently shown to interact with Sig-1 receptors and improve neuronal calcium dyshomeostasis (Grachev et al., 2021). Pridopidine showed modest beneficial effects in clinical trials (NCT02006472, Open-PRIDE-HD-NCT02494778). In addition, through restoring calcium levels in the ER and preventing excessive Ca2+ release, pridopidine may also be synaptoprotective by stabilizing dendritic spines (Wu et al., 2018). Another molecule, known as PRE-084, improves the expression of Sig-1 receptors and of antioxidants such as Cu, Zn-SOD, Mn-SOD, or thioredoxins, diminishes caspase-3 cleavage and activates the NF-κB pathway (Motawe et al., 2020) in preclinical research.
Olesoxime is a small cholesterol-like molecule able to accumulate in mitochondria and target to the voltage dependent anion channels. It showed beneficial effects in a BACHD rat model of HD by improving mitochondrial bioenergetics, enhancing the expression of OMM transport proteins, and inhibiting the MPTP opening (Clemens et al., 2015).
Future potential therapeutic strategies
A series of novel molecules, which are able to more efficiently restore normal mitochondrial functions, are in research and development.
The mitochondrial fission/fusion process can be regulated with fission inhibitors. Currently, three molecules are evaluated (Kalra, 2023). Mdivi-1 has been explored in rodent models for epilepsy and shown to inhibit mitochondrial fission, also acting as a ROS scavenger (Kim et al., 2016). Dynasore acts as a dynamin GTPase inhibitor (Kalra, 2023), while P110 appears to block mitochondrial fission through inhibiting the enzymatic activity of Drp1 (Qi et al., 2013).
Conjugating conventional mitochondrial-targeted antioxidants, such as coenzyme Q10 or vitamin E, with the lipophilic triphenylphosphonium cation significantly increases the mitochondrial availability of these antioxidants, delivered as MitoQ or MitoVitE (Kalra, 2023). MitoQ (mitoquinone mesylate) was shown to penetrate to the IMM and improve healthy mitochondrial biogenesis in striatal progenitor neural cells by increasing the expression of PGC-1α and TFAM (Yin et al., 2016).
Szeto-Schiller peptides (SS peptides) are small cell-permeable tetrapeptides which penetrate to the IMM and prevent ROS formation and ROS-mediated damage (Ding et al., 2021), being also able to inhibit mitochondrial depolarization and MPTP opening. SS-31 (also known as Elamipretide, MTP-131, or Bendavia) increased ATP production by upregulating the mRNA levels of complex I, IV, and V, as well as the mRNA levels of mitochondrial biogenesis regulators (PGC-1α, TFAM). In addition, it improved the mitochondrial fission/fusion balance by upregulating Mfn1, Mfn2, and OPA1 and downregulating Drp1 and Fis1 in striatal HD progenitor neurons (Yin et al., 2016).
Mitochondrial trafficking, impaired by tubulin acetylation in HD and Alzheimer’s disease, was shown to improve after histone deacetylase 6 inhibition with tubastatin A, which also increased mitochondrial fusion in striatal neurons (Guedes-Dias et al., 2015).
Another emerging strategy for various diseases in which mitochondrial dysfunction plays a key role is mitochondrial transplantation (or mitochondrial replacement therapy). Although still in development, research has shown that systemically delivered mitochondria can improve neurotoxin-induced Parkinson’s disease in mice (Shi et al., 2017). Increased availability of mitochondria can be obtained through peptide- mediated delivery or using synaptosomes as natural vehicles (Picone et al., 2021). However, isolation of mitochondria can prove difficult, needing to be completed quickly and at low temperature, and methods for long-term storage of the organelles are not yet available (Park et al., 2021).
Conclusions
Despite compelling evidence for the contribution of mitochondrial dysfunction in the pathogenesis of HD, therapeutic attempts aiming to restore normal mitochondrial function have so far yielded disappointing results in clinical trials, as opposed to encouraging results in animal models. Various reasons may contribute to this discrepancy.
First, a variety of natural compounds, such as resveratrol, quercetin, curcumin, can improve mitochondrial dysfunction by acting on multiple pathways, but they have poor bioavailability. Nanotechnology-based delivery methods could significantly improve our ability to use these compounds and more efficiently rescue mitochondrial function.
Second, a fine balance between mitochondrial fission and fusion is difficult to achieve, while mitochondrial trafficking may be impaired by the disturbed function of the motor proteins and the mutant huntingtin protein (mHtt) aggregates. In addition, in many cases the diagnosis is made in the symptomatic stage, when the vicious cascades involved in HD pathogenesis have already led to significant neuronal loss and synaptic dysfunction, making therapeutic success difficult to achieve.
Nonetheless, in our opinion, the future holds promise. Widespread genetic testing in the families of affected patients could improve diagnosis in pre-symptomatic stages. The further refinement of genetic editing tools could silence the expression of the defect gene and stop the transcription of the mutant protein while preserving transcription of the wild-type allele. A multimodal approach, enhancing the clearance of mHtt and administering drugs to improve mitochondrial dysfunction, could improve neuronal survival; in extreme cases mitochondrial transplantation could be offered, while stem cell therapies could replace lost cells.