
Cellular interorganelle crosstalk in health and disease states: A glimpse on nephrology-related conditions

By Fateme Shamekhi Amiri
Excerpt from the article published in Archives of Clinical Nephrology 8(1): 007-027. DOI: 10.17352/acn.000059
Editor’s Highlights
- Mitochondria as a signalling organelle associates with the cellular signalling environment and mediates gene expression programs.
- Mitochondria are constantly interacting with other organelles through signalling pathways, and on some occasions even though physical contact sites.
- Sigma-1receptor (Sig-1R), a chaperone protein operates as an interorganelle signalling modulator.
- Sig-1Rs normally dwell in the ER to mitochondrion contact (MAM), where Sig-1Rs regulate ER to mitochondrion signaling and the ER to nucleus crosstalk.
- When cells are triggered by ligands or underwent prolonged stress, Sig-1Rs shift from the MAM to the ER reticular network and plasmalemma/plasma membrane to regulate a diversity of functional proteins, including ion channels, receptors, and kinases.
- Dysfunctional crosstalk between mitochondria with other organelles causes cancer, type 2 diabetes mellitus, metabolic syndrome/disorders, inflammation/infection, central nervous system disease, cardiovascular disease, renal disease, aging, and age-related disorders.
Abstract
Cellular interorganelle crosstalk in medical sciences is a new discussion of mechanisms and pathways of physiological functions and pathogenesis of diseases. The organelles (“mitochondria”, nucleus, lysosome, endoplasmic reticulum, Golgi apparatus) are members of such functional units that are needed to perform specific tasks. Mitochondria are essential metabolic organelles in cells, but they also contribute to iron and calcium homeostasis, as well as in the regulation of apoptosis, and they are increasingly recognized as key signaling platforms. In the kidney, crosstalk between mitochondria with endoplasmic reticulum is at mitochondria-associated endoplasmic reticulum membrane in regulating calcium homeostasis. Another crosstalk between these organelles is about autophagic mechanisms. Autophagy is triggered in the kidney in response to acute kidney injury and supports against kidney injury. High glucose-induced reactive oxygen species can be produced by both enzymatic and nonenzymatic pathways. The nucleotide-binding domain and leucine-rich repeat (NLR) family or nucleotide-binding and oligomerization domain (NOD)-like receptors family pyrin domain containing 3 (NLRP3) inflammasome plays a role in inducing infectious defenses via inflammatory cytokines. The NLRP3 inflammasome is activated by the mitochondria-associated endoplasmic reticulum membrane. It has a role in nephrocalcinosis-related chronic kidney disease. This review article is a summary of interorganelle crosstalk in health and disease states, especially in kidney and nephrology-related conditions.
Introduction
Isolated mitochondria are competent to produce energy during their work and are not particularly responsive to changes in their environment that mimic intracellular signals [1]. They are in a constant and dynamic interplay with their cellular environment providing adenosine triphosphate, buffering calcium (Ca2+), and fundamentally contributing to various signalling pathways. Mitochondria are constantly moving and undergoing fusion and fission processes, changing their shape and their interaction with another organelle. Moreover, the mitochondrial activity gets fine-tuned by intra- and interorganelle hydrogen (h+), potassium (K+), sodium (Na+), and Ca2+ signalling [2].
Functional communication and cross-talk of the mitochondria with other organelles in medical sciences
Description of studies
Mitochondria are essential metabolic organelles in cells, but they also take part in iron and calcium homeostasis, as well as in the regulation of apoptosis, and they are increasingly recognized as key signalling platforms. Like the rest of the organelles, mitochondrissa need to interact with other organelles to perform their functions, exchanging material and transmitting signals responsible for regulating metabolism, intracellular signalling, and cell maintenance. This communication can be established in different ways, such as vesicular transport (as initially revealed for organelles within the secretory pathway), the exchange of metabolites or signalling molecules by diffusion, or through direct physical contacts. Current enlargement of pharmacological, molecular, and genetic tools resulting to observe that these organelles have active intracellular as well as extracellular communications that are important for cells and organ homeostasis. Communication between organelles must happen to regulate the size, shape, and composition of individual organelles. For example, mitochondrial crosstalk is performed with the peripheral endoplasmic reticulum (ER) network at ER–plasma membrane (PM) contacts and this work helps in coupling Ca2+ influx to mitochondrial Ca2+ import [3]. In the following context, mitochondrial crosstalk with other organelles is described (Table 1).
Mitochondria-Nucleus crosstalk studies: The nucleus and mitochondria are the two principle genome-bearing organelles of the eukaryotic cell with central and orchestrating roles in cellular and organismal physiology. The coordination of nuclear and mitochondrial genomes plays a crucial role in the maintenance of mitochondrial biogenesis and functionality during stress and aging. Environmental and cellular stimulants signal to the nucleus and/ or mitochondria to trigger interorganelle compensatory responses. Loss of this tightly orchestrated coordination culminates in loss of cellular homeostasis and underlies different pathologies and age-related diseases. Several signalling cascades that govern interorganelle communication have been revealed so far, and have been classified as part of the anterograde (nucleus to mitochondria) or retrograde (mitochondria to the nucleus) response [4]. Many of these pathways depend on the double dispersion of nuclear or mitochondrial components under basal or stress conditions. These dually localized components are usually employed in peculiar tasks in their primary function of organelle, whilst upon cellular stimuli, they appear in other organelles where they engage in the same or various tasks and trigger a compensatory stress response [5]. For example, decreased nuclear-encoded mitochondrial genes (NEMG) expression, low mitochondrial number, and dysfunction are looked into in obesity and type 2 diabetes mellitus (T2DM). It can be hypothesized that the accumulation of medium and long-chain fatty acids due to a high-fat diet will induce incomplete beta-oxidation and could negatively regulate NEMG expression via retrograde communication with the nucleus [6]. Moreover, consideration of several gene transcription regulators within the nuclear compartment is being reported to be present in the mitochondria. Different findings demonstrate the importance of crosstalk between nuclear and mitochondrial genome, transcriptome, and proteome in the regulation of cellular functions. Both tumor suppressor p53 and estrogen receptor were originally clarified as nuclear transcription factors. In addition to their roles as regulators of various genes, these two proteins interact and resulting in major cellular consequences. Besides its nuclear role, p53 has been localized to the mitochondria where it executes various transcription-independent functions. Likewise, estrogen receptors are reported to be present in mitochondria; however, their functional roles remain to be clearly defined [7]. Peroxisome proliferator-activated receptor-ϒ coactivator1a (PGC-1a), a transcriptional coactivator of peroxisome proliferator-activated receptor-ϒ and other nuclear hormone receptors, is a major regulator of oxidative metabolism and mitochondrial biogenesis. PGC-1ɑ is expressed in organs and tissues with high energetic requirements, such as the heart, liver, skeletal muscle, brown adipose tissue, brain, and kidney. PGC -1a binds to and coactivates the transcriptional function of nuclear respiratory factor-1, which binds specifically to the mitochondrial transcription factor A (TFAM) promoter, a direct functionary of mitochondrial deoxyribonucleic acid (mtDNA) replication. The overexpression of PGC-1a results in a strong elevation in mitochondrial number, cellular respiration, and intracellular adenosine-5 triphosphate concentration in a variety of cell types. Two studies investigated crosstalk between nucleus and mitochondria herein that have been described clearly. In a study, “Yuan et al.” interrogated the role of PGC-1ɑ on reactive oxygen species (ROS) production, caspase activity, respiratory chain (RC) cytochrome c release, percentage of cells enduring apoptosis, urinary F2-isoprostanes, mitochondrial membrane potential (MMP), podocin and nephrin, malondialdehyde and adenosine triphosphatase (ATP) levels on podocyte damage. They demonstrated that aldosterone (Aldo) induced ROS production in two time- and dose-dependent manner. The time-dependent manner was noticeable initially at 15 minutes, maximal at 2 hours (h), and sustained to 24 hours, and a dose-dependent manner with a noticeable effect at 50 nanomoles per liter (nmol/l) and a maximal effect at 100 – 200 nmol/l. Mitochondrial morphology by transmission electron microscopy showed swollen mitochondria with disorganized and fragmented cristae in treated podocytes with aldosterone for 24 hours. Detection of JC-1 fluorescence by confocal microscopy showed a reduction in accumulated red fluorescence in the mitochondria and an increase in the distribution of green fluorescence in the cytoplasm of podocytes on treatment with aldosterone for 24 h suggested aldosterone as a declining inducer of MMP. Furthermore, this method confirmed flow cytometry usage. Alongside, aldosterone declined ATP production and mtDNA copy number in a dose-dependent manner. Moreover, aldosterone-induced podocyte apoptosis (percentage of cells undergoing apoptosis by 65%) in a dose-dependent manner using Hoechst 33258 staining and annexin V/flow cytometry detection. In this research, aldosterone-activated caspase-3 (70%) and – 9 (35%) but not caspase-8, which indicated that apoptosis in aldosterone-induced podocytes was mediated by the intrinsic mitochondrial pathway. Aldosterone is a dose-dependent manner inhibited nephrin (35%) and podocin (25%) expression significantly. In this relation, resveratrol (RSV) is a polyphenol that is enriched in the skins of red grapes and its beneficial effects of it have been characterized in the prevention of coronary heart disease and cancer. Two laboratory findings were assessed in the above-mentioned research: urinary F2-isoprostane levels and manoldialdehyde levels. Urinary F2-isoprostane levels, a marker of renal ROS production, were elevated in Aldo-infused mice but were improved in the RSV-treatment group. Manoldialdehyde levels, a marker of pro-oxidant status in kidneys, increased in parallel to F2-isoprostane levels, and treatment with RSV improved their levels. These findings showed that RSV treatment in Aldo-infused mice ameliorated the induction of ROS in the glomeruli. Another effect of aldosterone was on PGC-1ɑ that was expressed in the podocytes and PGC1ɑ levels were declined by aldosterone in the mentioned study. Therefore, these results suggest that RSV protects podocytes in Aldo-induced mice by regulating mitochondrial function. As mentioned above, endogenous PGC-1 is essential for the maintenance of podocytes in mitochondrial function under normal conditions. Furthermore, sirtuin-1(SIRT1) / PGC-1a axis activation protects against podocyte injury due to aldosterone induction likely by preventing mitochondrial dysfunction and RSV attenuated Aldo-induced mitochondrial dysfunction and podocyte injury both in vivo and in vitro [8, 9]. In my opinion, this research is crosstalk between the nucleus and mitochondria in podocytes as such PGC-1ɑ protects podocytes from aldosterone-induced renal injury. Another study by “Raharijaona et al.” described the regulation of several new pathways by the PGC-1 related coactivator (PRC). The authors showed that the PRC regulation of this process differed from that of PGC-1ɑ by focusing on oxidative phosphorylation (OXPHOS). The nuclear OXPHOS genes were tightly controlled by PGC-1ɑ but less by PRC. Even so, other genes such as subunit of 15 of respiratory chain complex IV (COX15) were detected to be more particularly regulated by PRC than by PGC-1ɑ. This precision control of mitochondrial energy metabolism should be located in the context of the complex regulation of the cell cycle. To end they concluded that the endothelial nitric oxide synthase (eNOS) / PRC signalling pathway can modulate the cell cycle by regulating the intracellular redox status. Therefore the action of PRC complementary to that of other PGC-1 factors should be further investigated, especially in the case of metabolic diseases [10]. In my belief, PRC regulates the nucleus to mitochondria crosstalk via eNOS in cells and it plays a cardinal role in metabolic functions in response to various stimuli. Collectively, crosstalk between two organelles of nucleus and mitochondria is regulated at genome, transcriptome, and proteome levels.
Mitochondria-endoplasmic reticulum crosstalk studies: Interorganelle signalling plays important role in many physiological functions and is crucial for cell life. Communication between organelles must happen to regulate the size, shape, and composition of individual organelles. Invitingly, the ER creates connections with several compartments in the cell (plasma membrane, Golgi compartments, endosomes, lysosomes, mitochondria, and peroxisomes). Direct ER-mitochondria communication is undertaken by the physical interaction of their membranes in specified structural domains, identified as mitochondrial-associated ER membranes (MAMs), which facilitate calcium, and lipid transfer between organelles and also act as platforms for signalling [11, 12]. Current research has made up and extended the functional roles of MAMs in a variety of cellular processes from lipid synthesis/transport, Ca2+ signalling, and ER stress, to mitochondrial shape and autophagy/mitophagy and inflammation and cell immunity [13]. For example, “as” elevation the number of ER-mitochondria contact sites intensifies Ca2+ transfer to the mitochondria, carrying organelle-extrinsic stress signals to activate adaptive responses or advance cell death. Based on the type and degree of cellular injury, the ER-mitochondria crosstalk coordinates the pertinent response: autophagy, Ca2+– driven apoptosis, or inflammation. In the case of the inflammatory response, nucleotide-binding domain and leucine-rich repeat (NLR) family of nucleotide-binding and oligomerization domain (NOD)-like receptors family pyrin domain containing 3 (NLRP3) and other inflammasome members propel to ER-mitochondria contact sites to quickly sense the level of danger and to align the suitable response. NLRP3 is a tripartite protein that consists of an amino-terminal pyrin domain (PYD), a central nucleotide-binding and oligomerization domain (NOD), and a C-terminal leucine-rich repeat (LRR) domain. Hence, MAMs not only depict a platform for the investigation of extracellular inputs but also render a structural platform that accommodates several regulators or effector proteins [14]. Therefore, the functions of crosstalk between mitochondria and ER can be subdivided into calcium homeostasis, signalling, and response to various stress, autophagy, cell metabolism, cell death, and cell control which are discussed in the following context.
Calcium homeostasis
The mitochondria-ER length indicator nanosensor (MERLIN), is a novel proximity sensor for the distances between the ER and mitochondria, which is susceptible to changes induced by genetic or pharmacologic treatments. Calcium is locally transmitted from ER to mitochondria at the connections and applies regulatory effects on many proteins. A frequent Ca2+ sensing mechanism is the EF-hand Ca2+ binding domain, many of which can be detected in proteins of the mitochondria, including Miro1& 2, mitochondrial calcium uniporter (MICU) 1-3, mitochondrial proton/calcium exchanger protein or Leucine zipper-EF-hand-containing transmembrane protein 1 (LETM1) and mitochondrial solute carriers. Currently, these proteins have induced much interest and were delineated in reports with branching conclusions [15]. Growing evidence robustly suggests that alterations of ER-mitochondria interactions are concerned in neurogenerative disorders, including amyotrophic lateral sclerosis (ALS), a destructive and quickly lethal motor neuron disease. Nonetheless, abnormally increased or unrelenting ER to mitochondria contact might culminate in enhanced calcium flux into mitochondria, evoking mitochondrial permeability transition and apoptosis. This assumption has been established in other pathologies, like obesity [16]. Vesicle-associated membrane potential-associated proteins (VAPA/B) are ER-resident membrane proteins comprising a major sperm protein (MSP) domain that interacts with proteins containing two phenylalanines FF in an acidic tract (FFAT) or FFAT-like motif. One such protein is PTPIP 51 a mitochondrial membrane protein that shows a reaction with VAPB to liaise mitochondria to ER associations, facilitating calcium exchange and governing autophagy. Calcium homeostasis and ROS are two main resultant functions from crosstalk between ER and mitochondria. In the discussion of topic-related to Ca2+ ions transfer, it must be said that even though the distance between the ER and the matrix side of the inner mitochondrial membrane is exceedingly low, less than 50 nanometers, the journey that Ca2+ ions must accomplish their mitochondrial terminus necessitate several regulatory steps and molecular checkpoints. Alterations in just one of these controlling pathways culminate in striking metabolic or apoptotic defects, indicating the importance of appropriate ER-mitochondria Ca2+ transfer to keep the physiologic status of the cell. Various diseases have been connected to the remodeling of MAMs functions, such as cancer and neurodegeneration [17]. The ER is the main site of calcium storage. Calcium from ER cisternae is flowing greatly through calcium release channels as inositol 1, 4, 5-triphosphate receptors (IP3R) and ANT [ryanodine receptor (RYR)].These channels are collected in MAMs, which associate with the mitochondrial outer membrane. Calcium ions from the cytoplasm come into the mitochondria via voltage-dependent anion channels (VDAC) or calcium uniporter. High levels of calcium trigger RC activity resulting in higher amounts of ROS. ROS can target ER-based calcium channels leading to the elevated release of calcium and further elevated ROS levels. Increased ROS and calcium load can open the mitochondrial permeability transition pore (mPTP) leading to the release of pro-apoptotic factors [18]. Another experimental study by “Basso et al.” explored parkin deficient cells and parkin mutant human fibroblasts that tether between ER and mitochondrion has declined. Furthermore, they recognized the site of parkin-dependent ubiquitination revealed that non-ubiquitinate mutant Mfn2 fails to renew the physical and functional interaction of ER to mitochondria. Furthermore, this study investigated the handling of ER to mitochondria tethering by stating an ER to mitochondria synthetic linker is enough to salvage the locomotor deficit related to an in vivo Drosophila model of parkinson’s disease [19]. Conclusively, physical ER to mitochondria crosstalk is regulated by parkin via Mfn2. Moreover, the transforming growth factor (TGF-β) gives rise uncoupling of mitochondria from the ER calcium release. Pacher et al in an experimental study explored whether TGF-β may affect the ER to mitochondrial communication pathway in preglomerular afferent arteriolar smooth muscle cells (PGASMC). TGF-β modifies cytosolic Ca2+ [Ca2+] c signals, which in occasional states may lead to the downregulation of IP3R Ca2+ channels. Ca2+ liberated by IP3R is effectively transmitted from ER to mitochondria to trigger ATP production and to permit feedback control of the Ca2+ mobilization. To assess the effect of TGF-β on the ER to mitochondria Ca2+ transfer, they first evaluated [Ca2+] c and mitochondrial matrix calcium [Ca2+] m signals in a single PGASMC. TGF-β pretreatment (24h) declined both the [Ca2+] c and [Ca2+] m responses stimulated by angiotensin II or endothelin. Remarkably, the [Ca2+] m signal was further declined and tarried than the [Ca2+] c signal. In permeabilized cells, TGF-β pretreatment weakened the rate but not the magnitude of the IP3-induced [Ca2+] c elevation, yet gives rise to bulky depression of the [Ca2+] m responses. ER Ca2+ depot and mitochondrial uptake of added Ca2+ were not influenced by TGF-β. In addition to it, TGF-β did not affect mitochondrial diversity, and the ER to mitochondria contacts were determined by two-photon nicotinamide adenine dinucleotide (phosphate) [NAD (P) H] imaging and electron microscopy. Downregulation of both IP3R1 and IP3R3 was detected in TGF-β treated PGASMC. Hence, TGF-β gives rise uncoupling of mitochondria due to ER Ca2+ release. The sole origin of this would be inhibition of the IP3R-mediated Ca 2+ efflux, showing that the ER to mitochondria Ca2+ transfer relies on the maximal rate of Ca2+ release. The disturbed ER to mitochondria coupling may play a role in the vascular pathophysiology associated with TGF-β [20]. In my opinion, TGF-β enrolls in uncoupling ER to mitochondria crosstalk through IP3R and calcium release and consequently disruption in this crosstalk is associated with vascular disease.
Signalling and response to stress
ER-mediated interorganelle signalling may permit crosstalk between defined organelles to fortify cellular responses to different stimuli. Therefore, the ER may work as a conduit for information transfer to integrate the functions of specialized organelles. Endoplasmic reticulum to mitochondria signalling impact on mitochondrial calcium homeostasis and cellular bioenergetics. ER to nucleus signalling weakens ER stress. ER to peroxisome signalling regulates cytosolic Ca2+ homeostasis and ER to mitochondria-plasma membrane signalling regulates hippocampal dendritic spine formation. Sigma-1receptor (Sig-1R), a chaperone protein operates as an interorganelle signalling modulator. Sig-1Rs normally dwell in the ER to mitochondrion contact (MAM), where Sig-1Rs regulate ER to mitochondrion signaling and the ER to nucleus crosstalk. When cells are triggered by ligands or underwent prolonged stress, Sig-1Rs shift from the MAM to the ER reticular network and plasmalemma/plasma membrane to regulate a diversity of functional proteins, including ion channels, receptors, and kinases [21]. Mitochondria are invariably communicating with the rest of the cell. Defects in mitochondria leads to severe pathologies, whose mechanisms remain poorly understood. It is becoming increasingly evident that mitochondrial malfunction comes across in other organelles, impairing their function and their biogenesis [22]. With this disturbance, chronic activation or decompensation of such crosstalks has been associated with a diversity of pathophysiological conditions such as metabolic disease, cardiovascular disease, and cancer. In this relation, several studies are discussed herein. In a study, “Bergeron et al.” explored the morphologic interrelationships between mitochondria, the ER, and other organelles in rat kidney cells by stereomicroscope of thick sections using either standard transmission or high voltage electron microscopy. Mitochondria lie in three various categories; 1) elongated cylinders seen in S 1, and S2 segments 2) irregular lamina in the cortical ascending limb or plates in the distal and convoluted segments 3) small spheres or short rods mostly in the intercalated and principle cells of the collecting tubule. The chondrioma residing an excess volume in all cells apart from in the thin limb and the principle cells of the collecting tubule. This volume resided by the chondrioma is likely to be related to metabolic functions but its polymorphic configuration could also be investigated by a passive adaptation of the mitochondria to the space left by the basilar membrane infoldings and the ER network, which was found to have an extensive three-dimensional organization which differs, as for the mitochondria, with the cell type. Actually, in the former nephron, mitochondria are circumvented by the ER and the plasma membrane seems to form a functional unit. Hence, organelle interrelationship is excessively altered during ontogeny or under hormonal influence. Disruption in organelle crosstalk or their motility could credibly have far-reaching results which would be further dangerous than the pathological lesions seen at the level of a single organelle [23]. In my viewpoint, crosstalk between mitochondria and ER in kidney cells was investigated in this research and showed each type of crosstalk disturbance influences on function of those. “Safiedeen et al.” in a cohort study investigated how microparticles (MPs) transmit their message to trigger endothelial dysfunction and they were decrypted crosstalk between ER and mitochondria in the regulation of oxidative stress. This study has shown that endothelial dysfunction provoked by MPs involves crosstalk between ER and mitochondria concerning spatial regulation of ROS through the neutral sphingomyelinase and interaction of MPs with Fas and/or low-density lipoprotein receptor. These results furnish a novel molecular view into the fashion that MPs mediate vascular dysfunction and permit to recognition of potential therapeutic targets to treat vascular complications associated with metabolic syndrome. Furthermore, temporary crosstalk between the endoplasmic reticulum and mitochondria mediates oxidative stress and regulates MP-induced endothelial dysfunction [24]. In this research, my opinion is expressed that mitochondria to ER crosstalk involves the control of oxidative stress via the transfer of new molecules and cellular pathways by MPs in the induction of endothelial dysfunction in vascular diseases. Mitochondrial dysfunction and ER stress due to disturbed autophagic organelle turnover in podocytes and the tubular epithelium is adequate to make many of the presentations of focal segmental glomerulosclerosis (FSGS) in mice.
Authophagy
Autophagy of a main intracellular lysosomal degradation system does homeostatic functions associated with metabolism and organelle turnover. Another study by “Kawakawi et al.” investigated the prevention of normal autophagic pathways in mice nephrons by mutating crucial autophagy genes, autophagy-related gene 5 (ATG5) or ATG7 during nephrogenesis. Mutant mice advanced mild podocyte and tubular dysfunction within 2 months, significant glomerular and tubular alterations bearing a strong similarity to human disease by four months. Ultrastructurally, podocytes, and tubular cells revealed vacuolization, abnormal mitochondria, and evidence of mitochondrial stress characteristics that comes before the appearance of histologic or clinical disease. The same alterations were seen in human idiopathic FSGS on kidney biopsy samples. Biochemical analysis of podocytes and tubules of 2-month-old mutant mice showed enhanced production of ROS and activation of ER stress pathways, phosphorylation of P38, and mitochondrial dysfunction. Moreover, cultured proximal tubule cells separated from mutant mice revealed mitochondrial dysfunction and increased mitochondrial superoxide scavenger. Disturbance of crosstalk between mitochondria and ER causes FSGS in mice with mutant autophagic genes in podocytes and tubules [25]. Furthermore, “MacVicar et al.” in an experimental study proposed that Ca2+ flux between ER and mitochondria at postulated ER to mitochondrial contact sites is required both for starvation-induced autophagy and for parkin-mediated mitophagy, more making up for the importance of interorganelle crosstalk for effective cellular homeostasis [26]. Another study by “Garofalo et al.” explored MAMs are subdomains of the ER that interact with mitochondria. They proposed the possible impact of MAM lipid rafts in autophagosome formation. This association appears to occur in MAM lipid microdomains during the early phases of the autophagic process. They proposed that the molecular interaction of GD3-AMBRA1 under autophagic stimulation and conversely the lack of interaction at the MAM level between key initiators of autophagy in the presence of ST8SIA1 siRNA, i.e., CANX-AMBRA1, could exhibit that MAM raft-like microdomains could be crucial in the basal organelle scrambling activity. This function consequently leads to the formation of an autophagosome. Moreover, this experimental condition results in the impairment of the ER to mitochondria crosstalk and the subsequent prevention of autophagosome nucleation [27]. In my viewpoint, this research analyzed ER to mitochondria crosstalk and involves autophagosome formation via lipid rafts at ER to mitochondria-associated membranes
Cell metabolism
Research has indicated that mitochondrial dysfunction and ER stress contribute to fundamental functions in the pathophysiology of obesity-related comorbidities, such as insulin resistance and type 2 diabetes mellitus [28]. Recently, mitochondria are identified to play a key role in the vascular remodeling in patients with pulmonary arterial hypertension. In these patients, smooth muscle cells aroused from pulmonary artery smooth muscle cells (PASMCs) incurred a metabolic shift, away from applying fatty acids and glucose oxidation to produce ATP, towards emphasizing cytoplasmic glycolysis as the principle origin of energy. Furthermore, ER stress is recognized to improve metabolic remodeling of mitochondria in pulmonary arteries during pulmonary arterial hypertension (PAH), due to the disruption of ER to mitochondria contacts. Mitochondria are central to energy metabolism as the origin of much of the cell’s ATP, also as a being a hub for cellular Ca2+ signalling. Mitochondrial Ca2+ is a positive effector of ATP synthesis, yet Ca2+overload can culminates in mitochondrial dysfunction and cell death. Furthermore, Ca2+ uptake by mitochondria is enrolled in shaping cellular Ca2+dynamics by mediating the concentrations of Ca2+ within microdomains between mitochondria and sarco/endoplasmic reticulum and plasma membrane Ca2+ transporters. ROS as a result of ATP generation in the mitochondria is valuable for cellular signalling. ROS regulates the activity of redox-sensitive enzymes and ion channels within the cell, which comprises Ca2+ channels. For both Ca2+ and ROS, a subtle equilibrium subsists between the beneficial and harmful effects on mitochondria [29]. Therefore, mitochondrial calcium is required for ATP production and free radicals of oxygen involved in calcium channels. Any disturbance in calcium load between mitochondria and ER will impair cell signaling. The functional roles of the stable contact sites between the ER and mitochondria (referred to as MAMs) have been discussed previously. Of these, Ca2+ released by the inositol 1, 4, 5-triphosphate receptor in the ER is taken up by mitochondria where it is needed for efficient bioenergetics functions. When this Ca2+ transfer is deficient, macroautophagy is activated to maintain cell survival. Interorganelle contacts also control mitochondrial biogenesis and dynamics. Mitochondrial fission is propelled by dynamin-related protein-1 (DRP1), a cytoplasmic protein invoked to the ER to mitochondria contact sites. Mitofusin 2, proceeding mitochondrial fusion, tethers contacts between mitochondria and the ER. Ultimately, live confocal microscopy has established that tethered ER and mitochondria coordinate and move along the cytoskeleton. The juxtaposition of the endoplasmic reticulum with mitochondria facilitates Ca2+fluxes between the organelles such a recent report has recognized a cardinal role for the dynamin-related mitofusins in the tethering mechanism, thereby guarantee the rapid and high accuracy of Ca2+ signalling between the organelles [30]. This research reveals that mitochondrial fission and fusion through DRP1 and Mitofusin 2 involve mitochondria to ER crosstalk. Changes in ER to mitochondria juxtaposition can endanger lipid metabolism, protein synthesis, folding, and functionality. The creation of metabolic syndrome is nearly linked with the deregulation of lipid metabolism. Nascent evidence has established that microRNAs (miRNA) are employed in lipid and lipoprotein metabolism by mediating genes engaged in the control of intracellular lipid synthesis, mitochondrial fatty acid oxidation, and lipoprotein assembly. Mitochondrial dysfunction triggered by changed miRNA expression has been suggested to be a contributing factor in the onset of metabolic diseases when at the same time, improper expression of distinct miRNAs is related to the induction of ER stress induced by nutrient surplus. These studies put miRNAs as a link between oxidative stress and ER stress, two cellular stress pathways that are deregulated in metabolic disease and are associated with very-low-density lipoprotein (VLDL) overproduction. Therefore, mitochondrial dysfunction occurs due to altered and improper miRNA expression that is associated with ER stress and thus causes metabolic syndrome. Moreover, there is a link between oxidative stress and inflammations in pathological states due to ER stress (Figure 1). Dyslipoproteinemia commonly attached to metabolic syndrome is begun largely by the overproduction of VLDL, and changed biogenesis of high-density lipoprotein (HDL) [31]. Cancer is determined by an uncontrolled cell proliferation rate even under low nutrient accessibility, which is maintained by metabolic reprogramming at present identified as a characteristic of cancer. Warburg first suggested a link between mitochondria and cancer by proposing that the source of cancer cells was a permanent lesion in mitochondrion as such increased the glycolytic rate as a compensatory mechanism. Gathering evidence indicates Ca2+ communication between the ER and the mitochondria as cardinal mitochondrial function and consequently for cancer progression [32]. Ca2+fluxes between ER and mitochondria impact several cancer hallmarks, comprising apoptosis, resistance, migration [33], and invasion. Oncogenes and tumor suppressors put at the MAMs operating part of their cellular function by changing ER to mitochondrial Ca2+ transfer, thereby advancing \ or hindering cancer cell survival [34]. Dependent on the cancer type and cancer stage, ER to mitochondrial Ca2+ transfer can either use anti-tumorigenic effects such as restoring apoptosis sensitivity or use pro-tumorigenic effectors like advancing metastatic behavior. Different chemotherapeutics depend on a Ca2+– signalling component to trigger cancer cell death. Ca2+ signalling modulation can re (sensitize) or raise the responsiveness of cancer cells toward chemotherapeutics [35]. In my belief, ER to mitochondria calcium transfer can involve in cancer. Dysfunction between ER and mitochondrial association gives rise to various diseases. Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis with associated frontotemporal dementia (ALS/FTD) are the main diseases for which there are no cures. Even though all are categorized as neurodegenerative diseases, they are clinically certain and cause damage to various neuronal populations. Furthermore, the damage is associated with the development of protein inclusions within affected neurons and the constituent proteins can be different for each disease. Despite these differences, all three diseases exhibit some common characteristics. Especially based on evidence they disturb several isolated cellular functions, including damage to mitochondria, Ca2+ homeostasis, lipid metabolism, axonal transport, and inflammatory responses. Currently, several studies have indicated that some insults associated with these neurodegenerative diseases disturb ER to mitochondria relationships and linked signalling. ER to mitochondria associations mediate many of the functions that are impaired in AD, PD, and ALS/FTD. Hence, damage to the ER to the mitochondria axis furnishes mechanisms that might create various diseases [36].
Cell death
Physical interaction between two organelles is conducted via calcium flux involving autophagic and mitophagic processes. The association between ER and mitochondria is due to the presence of tethers that associate both smooth and rough ER to the mitochondria. The length of the tethers exhibits some diversity, produce to different distances between ER and mitochondria. In response to apoptotic agents, the ER to mitochondria gap candles exhibit dynamic regulation of the interorganelle junction. In healthy cells, the ER to mitochondria tethering ascertains the circulation of inositol triphosphate receptor (IP3R)-linked Ca2+ signals to the mitochondria to align ATP with the stimulated state of the cell and to empower the mitochondrial Ca2+buffering. Even so, the gap between the organelles is adequately broad to separate mitochondria from the slow Ca+2 leakage from the ER. Relaxing the ER to mitochondria coupling inhibits the Ca2+ signal propagation to the mitochondria, laying at risk the Ca2+-dependent control of mitochondrial metabolism. In opposition to it, tightening of the coupling provokes mitochondria the manipulation of Ca2+ under resting states, sensitizing mitochondria to Ca2+ overloading and resulting in permeabilization and committing the cells to a cell death pathway. Tightening of the connections appears to be pertinent for several mechanisms of cell death. Hence these events indicate an abrupt dependence of cell function and survival on the maintenance of a suitable spacing between the ER and mitochondria [37]. The results of this research point to a proper gap of mitochondria to ER crosstalk in healthy cell function. Studies have detected that B-cell leukemia/lymphoma (Bcl-2) proteins are circulated in the mitochondrial outer membrane, ER, and nuclear membrane. Bcl-2 family proteins situated in the ER are identified to be involved in the mediation of endoplasmic reticulum signalling pathways. Therefore, the Bcl-2 family of proteins may also be involved in the mechanism of tumor chemotherapy resistance by influencing crosstalk between the ER and mitochondria [38, 39]. “Hacki et al.” in an experimental animal study explored how the disturbed ER communicates with the apoptotic machinery. They used two apoptotic stresses that make improper protein in the secretory system, brefeldin A (BFA), which inhibits retrograde conveyance of golgi-derived vesicles, culminating in a stop in secretion and the fusion of the golgi/ER compartments and tunicamycin which blocks the first reaction in the dolichol pathway of N-glycosylations of proteins in the ER lumen. Invitingly, both stresses evoke the release of cytochrome c before or at the time of effector caspase-3 activation suggesting crosstalk between the disturbed ER and mitochondria. This crosstalk does not involve caspase-8 mediated cleavage and/or mitochondrial translocation of Bid, but a yet unknown, caspase-independent mechanism. Furthermore, Bcl-2 can stop this crosstalk at the level of the ER and does not require being associated with mitochondria to complete its function. Damage in the ER compartment can be interpreted as an apoptotic response that involves the release of cytochrome c from mitochondria. One possibility is that the ER and mitochondria crosstalk via membranes as they can be in close contact and easily exchange lipids, calcium, and glycoproteins. A possible mediator may be the sphingomyelin hydrolysis in response to different apoptotic stimuli, comprising BFA, and has currently been indicated to directly operate on mitochondria to stimulate cytochrome c release. Bcl-2 supported cells from BFA/CHX- or tunicamycin-induced apoptosis at a step between the ER stress and caspase-3 activation. The results proposed that a cytochrome c release is a signalling event between the damaged ER and caspase-3 activation in BFA/CHX- and tunicamycin-induced apoptosis. In a comparison of wild-type Bcl-2, Bcl-2/cb5 was as effective in involving BFA/CHX- or tunicamycin-induced caspase-3 activation, cell shrinkage, and nuclear condensation/fragmentation and cell death. Bcl-2/cb5 also completely blocked BFA/CHX- or tunicamycin-induced cytochrome c release. Bcl-2 can operate on the ER to suppress an ER damage-induced mitochondrial event. Interestingly, the death protective effect of Bcl-2/cb5 was not restricted to ER stress agents but also to apoptotic stimuli like staurosporine that are identified to act via mitochondrial damage. Hence, neither BFA/CHX nor tunicamycin stimulated any major activation of caspase-8 or proteolysis/mitochondrial translocation of Bid over a time period of 24 h in R6 cells. Furthermore, cytochrome c release triggered by both BFA/CHX and tunicamycin was not inhibited in the presence of the general caspase inhibitor z-VAD.fmk exhibiting that none of the known caspases were interfered in carrying stress signals from the injured ER to mitochondria. Therefore, the ER to mitochondria communication in response to BFA/CHX or tunicamycin interferes with a novel, caspase-independent signalling pathway. Thus, apoptotic agents impairing ER functions trigger novel crosstalk between the ER and mitochondria that can be stopped by ER-based Bcl-2 [40]. The mitochondria to ER crosstalk cause apoptosis that is inhibited by Bcl-2 and this factor enrolls in both on mitochondrial membrane and ER. The crosstalk between ER and mitochondria is a cardinal characteristic of the spatial organization of cell signalling. This rigid link may be key to ensuring suitable mitochondrial responses (mitochondrial cardiomyocyte, sarcoplasmic reticulum) to calcium release but can also culminate in the intensification of cardiac myocyte death by an overplus of various stimuli [41]. “Gomez et al.” in an experimental study examined the role of glycogen synthase kinase-3β (GSK3β) in the Ca2+ transfer from the sarcoplasmic reticulum (SR) / ER to mitochondria at reperfusion time. They approved the involvement of GSK3β in the MAM protein complex and thereafter explored its association with CyD, VDAC, Grp75, and IP3R at the MAM interface. Both pharmacological and genetic blockade of GSK3β declined the interaction of GSK3β with IP3R, Grp75, VDAC, or CypD. SB21 substantially decreased the co-immunoprecipitation of IP3R with GSK3β, Grp75, VDAC, and CypD with IP3R. These findings propose that GSK3β modulates the IP3R-Ca2+ channeling complex at the MAM interface in the heart. They concluded that both pharmacological and genetic suppression of GSK3β declined protein interaction of IP3R with the Ca2+ channeling complex, disturbed SR/ER Ca2+ release, and declined the histamine-stimulated Ca2+ exchange between SR/ER and mitochondria in cardiomyocytes. During hypoxia/ reoxygenation, cell death is linked with an enhancement of GSK3β activity and IP3R phosphorylation, which culminates in enhanced transfer of Ca2+ from SR/ER to mitochondria. Blockade of GSK3β at reperfusion period declined both IP3R phosphorylation and SR/ER Ca2+ release, which finally declined both cytosolic and mitochondrial Ca2+ concentrations, as well as sensitivity to apoptosis. They demonstrated that blockade of GSK3β at reperfusion decreases Ca2+ leak from IP3R at MAMs in the heart, which restricts both cytosolic and mitochondrial Ca2+ overload and consequently cell death [42]. Conclusively, GSK3β is known as a valuable regulator of calcium transfer between SR/ER and mitochondria during IR injury. In exact, GSK3β acts as cytoprotective in permeability transition pore opening. GSK3β operates at the inner mitochondrial membrane in the heart and/or phosphorylates VDAC and cyclophilin D in cancer cells. Overexpression of antiapoptotic Bcl-2 or combined ablation of proapoptotic BAX and BAK diminishes ER calcium content and protects against cell demise in vitro. This hypothesis by “Diwan et al.” in an experimental animal study examined and demonstrated a new function for NIX as an integrator of transcriptional and calcium-mediated signals for programmed cell death. Transcriptional upregulation of the proapoptotic Bcl2 family protein of NIX restricts red blood cell formation and causes heart failure by triggering cell death. Endogenous cardiac NIX and recombinant NIX reside both in the mitochondria and the ER/SR. In genetic mouse models, cardiomyocyte ER/SR calcium depots are proportional to the level of expressed NIX. Because Nix ablation was protective in a mouse model of apoptotic cardiomyopathy, genetic correction of the declined SR calcium content of Nix-null mice restored sensitivity to cell death and reinstated cardiomyopathy. Nix mutants that are specific to ER/SR or mitochondria activated caspases and ER/SR-Nix caused loss of the mitochondrial membrane potential but were equally lethal [43]. The BH3-only protein NIX is expressed under physiological and pathological conditions in hematopoietic cells, cardiac myocytes, and other cells. In the heart, NIX is transcriptionally upregulated during pathologic cardiac hypertrophy in response to hemodynamic overload or neurohormonal excess. NIX-mediated apoptosis contributes to programmed loss of cardiac myocytes and consequently to heart failure. “Naon et al.” in an experimental study explored whether chronic or acute mitofusin 2 (Mfn) ablation affects ER to mitochondria proximity and Ca2+ transfer. The role of Mfn2 as a tether was approved in the heart, in pro-opiomelanocortin neurons, and the liver. They calculated the average distance between mitochondria and the ER put within 30 nm from the former. This study demonstrated Mfn2’s role in ER to mitochondria crosstalk. Electron microscopy and fluorescence-based probes of organelle proximity approved that ER to mitochondria juxtaposition was declined by constitutive or acute Mfn2 deletion. Indeed, mitochondrial uptake of Ca2+ released from the ER was diminished following acute Mfn2 ablation, as well as in Mfn2-/- cells overexpressing the mitochondrial calcium uniporter. Mitochondrial calcium uptake rate and extent were normal in isolated Mfn2-/- liver mitochondria, compatible with the finding that acute or chronic Mfn2 ablation or overexpression did not change mitochondrial calcium uniporter complex component levels. Mfn2 stands as a veritable ER to mitochondria tether whose ablation declined interorganelle juxtaposition and communication [44]. This research points to valuable results about Mfn2 operating as a linker between mitochondria and ER and tethering measurements perform by electron microscopy (EM) or confocal image analysis. Another study by “Bernard-Marissal et al.” in an experimental animal study explored that mutations in the MFN 2 gene encoding Mitofusin 2 culminate in the development of Charcot-Marie-Tooth type 2A syndrome (CMT2A), a predominant axonal form of peripheral neuropathy. The consequences of this study exhibited that changed interplay between ER and mitochondria share to the axonopathy in CMT2A Tg mice [45].
Homeostatic control of cell
Recent findings revealed proper communication between the ER and mitochondria is needed for proper maintenance of cellular iron levels. Loss of ERMES components activates an Aft-1-dependent iron deficiency response even in iron-replete conditions, leading to the accumulation of excess iron inside the cell. A mutation in the vacuolar protein sorting 13 (VPS13) gene that rescues the glycolytic phenotype of ERMES mutants suppresses the iron deficiency response and iron accumulation [46]. Taken together, two-way communications between the endoplasmic reticulum and mitochondria mediate both physiological processes in cells comprising mitochondrial energy metabolism, lipid metabolism, Ca2+ signalling pathway, and cell death. Furthermore, mitochondrial dysfunction and ER stress fulfill a cardinal role in the pathophysiology of various abnormalities.
Mitochondria-peroxisome crosstalk studies: With remembering the close association between peroxisomes and mitochondrial metabolism, it is completely obvious that the peroxisome to mitochondria partnership must have a main role in cellular metabolism and functions. Mitochondria and peroxisomes are jointly common in proteins, some metabolic functions, and link via vesicular transport. It has been identified that crosstalk between mitochondria and peroxisomes characterizes at the transcriptional level occasionally. Peroxisomes themselves are intimately linked physically with mitochondria and can be created from the ER. Moreover, there are implied, but also direct, observations of cross-talk and feedback systems that function to align peroxisome biogenesis and proliferation with the responses of other organelles [47]. There is strong evidence that multiple enzymes have resided from the mitochondrion to the peroxisome at different classes and evolutionary periods. Notably, retargeting of enzymes of alpha-proteobacterial origin into the peroxisome is old and interferes with one enzyme of the beta-oxidation pathway (Pox2p). Hence, this is not the enzyme generating oxygen radicals-the driving force hypothesized in both models. Furthermore, the enzyme that generates the damaging radicals, Pox1p (peroxisomal protein acyl-CoA oxidase-1) is involved in other pathways at the side of degradation of fatty acids via beta-oxidation, for example in the synthesis of polyunsaturated fatty acids. Given its much-defined phylogenetic origin, it is logical to imagine that Pox1p was not involved in the degradation of fatty acids via beta-oxidation until the cardinal enzyme-like Pox2p were retargeted from the mitochondrion. So a probable scenario is that Pox2p moved to an already existing peroxisome, initially originated to isolate the generation of oxygen radicals from the endoplasmic reticulum. The relocation of the multifunctional enzyme Pox2p from the mitochondrion, as protected by an alpha-proteobacterial source, into the formerly formed peroxisome would have empowered effective oxidation of very long fatty acids outside the mitochondrion by coupling to the former existing peroxisomal activity of Pox1p [48]. The nitric oxide-mediated increase of autophagy-related protein unc-51-like kinase (NO -ULK1) has been currently delineated as a novel pathway culminating in SIRT1 protein stabilization, although this appears to occur independently from autophagy in endothelial cells [49]. Impairment in peroxisomal metabolism can provoke redox-related signaling events that finally result in enhanced mitochondrial stress and the activation of mitochondrial stress pathways. Hence, the crosstalk pathways must be established herein. Potential mechanisms may consist of the diffusion of signaling molecules from one compartment to the other through the cytosol, the exchange of molecules through direct membrane contact sites or vesicular transport mechanisms, and retrograde signaling. Of course, the crosstalk pathway may vary depending on the identity, reactivity, and selectivity of the messenger. For instance, as hydrogen peroxide (H2O2) can pass on peroxisomes, it is very probable that this molecule can modulate the activity of extra-peroxisomal redox-sensitive proteins (e.g., transcription factors, kinases, and phosphatases) interfered in the (transcriptional) control of mitochondrial biogenesis and function. One such example may be v-Akt Murine Thymoma Viral Oncogene (AKT1), a serine-threonine protein kinase that confidently mediates the activity of cyclic adenosine monophosphate [(cAMP) response element-binding protein 1(CREB1)] and is broken by the ubiquitin-mediated proteasome pathway in conditions with enhanced H2O2. As the biological half-life of some ROS is exceedingly short (e.g., O2, 10-6s; OH, 10-9s), it is far that these molecules will be directly transmitted from one compartment to the other by diffusion or vesicular transport mechanisms. It is invitingly to note that the generation of excess oxygen (O2) inside peroxisomes gives rise to cellular lipid peroxidation and that this in turn provokes a complex network of signaling events ultimately resulting in elevated mitochondrial H2O2 generation. With notice presence of some evidence-based that the distribution of ROS signals from the ER to mitochondria is facilitated by membrane contact sites and such contact sites may also subsist between peroxisomes and mitochondria, these sites may be also interfered in the redox crosstalk between peroxisomes and mitochondria. Eventually, mitochondria can produce mitochondrial-derived vesicles (MDVs) that selectively transmit mitochondrial proteins to either peroxisomes or lysosomes as such vesicular transport pathways may also subsist for peroxisomes [50, 51]. Peroxisomes are related to other organelles for several of their metabolic functions. The interaction with mitochondria is particularly noteworthy for more metabolism of the end-products of beta-oxidation in peroxisomes including a declined form of NADH, acetyl-coenzyme A (CoA), propionyl-CoA, a variety of acyl-CoAs chain-shortened in peroxisomes. There is a direct interaction between peroxin 11 (Pex11), a membrane-bound Peroxin that interfered in peroxisome division and proliferation, and the endoplasmic reticulum-mitochondrial encounter structure (ERMES) complex of mitochondria. ERMES consists of four proteins: the mitochondrial outer membrane protein Mdm 10, the ER-resident Mmm 1 protein, and two peripheral membrane proteins, Mdm34 and Mdm12. All four proteins are needed for complex formation. When any one of the ERMES subunits is losing, other subunits fail to localize at the contact sites. Invitingly, Pex11 was detected to physically interact with Mdm34 to constitute the contact between peroxisomes and mitochondria. Tethering of both organelles is postulated to increase metabolism by declining the distance for efficient transmission of metabolites from the peroxisome to the mitochondrion. Loss of Pex11 function is related to declined peroxisome number and the formation of hypertrophied peroxisomes, while overexpression progresses peroxisome elongation and proliferation. Of the three Pex11 proteins known in humans, Pex 11β has currently been associated with the disease. Human Pex11 is a peroxisome-specific integral membrane protein with the N- and C-terminal motif encountered in the cytosol. Pex 11β forms homo-oligomers, interacts with Fis1 and Mff, and probably with membrane lipids to change the form and shape of the peroxisomal membrane; Moreover, the N-terminal motif comprises amphipathic helices needed for membrane elongation in vitro and in vivo as well as for dimerization [52]. In my belief, crosstalk between peroxisome and mitochondria performs through Peroxin 11 which participates in physical contact sites between these organelles. “Esposito et al.” in an experimental study approved that absence of Mdm10 or Mdm12 culminates in an increased number of mature peroxisomes. Moreover, they concluded that this is not merely due to respiratory function defects, mitochondrial DNA loss, or mitochondrial network alteration. Furthermore, they showed evidence that the contribution of ERMES subunits Mdm10 and Mdm12 to peroxisome numbers involves two various mechanisms [53]. Docosahexaenoic acid (DHA) is the most predominant n-3 polyunsaturated fatty acid in the adult mammalian brain and retina. DHA is synthesized from dietary linoleic acid in the endoplasmic reticulum through a series of elongation and desaturation reactions. This pathway needs that C22:5n-3 would be desaturated at position 4 by an acyl-CoA-dependent delta4-desaturase to construct C22:6 n-3. Several studies have presented that mammals do not have delta4-desaturase. Rather, a 24 carbon n-3 fatty acid is first synthesized which is then desaturated at position six to produce C24; 6 n-3 as the end-product [54]. Currently, DHA has been identified as a drug for the cure of aluminum phosphide poisoning. This drug has been first discovered at Shiraz University of Medical Sciences by persons who had been intoxicated with rice tablets in Iran country. Recently, there is abundant evidence at least in mammals that peroxisomes and mitochondria contribute to a redox-sensitive relationship. For instance, defects in catalase activity, peroxisomal β-oxidation, or peroxisome biogenesis have been depicted to trigger mitochondrial oxidative stress in different organs (e.g. liver), proximal tubules of the kidney, adrenal cortex, spinal cord, heart and brain, and cell types (e.g. fibroblast, mesangial cells, skeletal and smooth muscle cells, and hepatocytes). Invitingly, this increase in mitochondrial redox state succeeds the induction of peroxisomal function loss. In this case, peroxisome biogenesis is entirely inhibited. Such treatment also leads to other mitochondrial disturbances comprising structural changes of the inner mitochondrial membrane, a decrease in the activities of multiple respiratory chain complexes, declined mitochondrial DNA abundance, and an elevation in mitochondrial volume. On the other side, an elevation in catalase activity, peroxisomal β-oxidation, or peroxisomal number has been announced to eliminate mitochondrial fitness and support these organelles against oxidative insults [55]. In my opinion, peroxisome to mitochondria crosstalks exists in the context of redox states as such elevation or diminution of catalase activity, peroxisomal β-oxidation, and peroxisomal number influence on mitochondrial redox states. One of the most valuable common characteristics between mitochondria and peroxisome is the synergistic function for fatty acid oxidation. Fatty acids contain many paramount functions in energy generation, inflammation, resolution, etc. In mammalian cells, both peroxisomes and mitochondria comprise a beta-oxidative pathway. Beta-oxidation is a cardinal pathway for the breakthrough of fatty acids. These two pathways are common in many similarities, particularly in words of substrates and enzymatic reactions. Moreover, the metabolic implication and end-products are not alike in mitochondria and peroxisomes. The total pathways are of necessity like in both mitochondria and peroxisomes: fatty acids are initially activated as acyl-CoA and then the activated fatty acid is dehydrogenated. This displays the first step of beta-oxidation. Thereafter, hydration of the double bound happens and is succeeded by dehydrogenation and cleavage. This permits for the exit of 2 C from C: n acyl-CoA, culminating in the formation of C: n-2 acyl- CoA [56]. Peroxisomal beta-oxidation deficiencies most generally culminating in neurological abnormalities and/or hepatomegaly and disclose increased levels of very-long-chain fatty acids and/or bile acid intermediates along with a disturbance of peroxisomal beta-oxidation. Also, authors “Le Hir et al.” in an experimental animal study explored the localization of the peroxisomal and the mitochondrial pathways of beta-oxidation in the rat kidney. In this research fatty acyl- CoA oxidase and 3-hydroxy acyl-CoA dehydrogenase activities were weighed in glomeruli, in eight proximal and distal segments of the nephron. Morphologically defined segments were dissected and analyzed with microchemical assays. The peroxisomal fatty acyl-CoA is limited to the proximal tubule. The 3-hydroxy acyl-CoA dehydrogenase activity shows predominantly the mitochondrial pathway and is likely propagated in all cortical proximal and distal segments. It is much in glomeruli and collecting ducts. The distribution patterns of the two enzymes remain similar after 48 hr of fasting, even though the activity of fatty-acyl CoA oxidase enhances in glomeruli, proximal convoluted, and collecting ducts. The task for mitochondrial beta-oxidation of fatty acids is like in the proximal and the distal nephron. Moreover, the proximal tubule is a peroxisomal pathway for beta-oxidation with the capacity of a similar order of magnitude as in liver cells [57]. This above-mentioned research points to crosstalk between peroxisome and mitochondria about β-oxidation in glomeruli, proximal and distal segments of the nephron. Mitochondrial ROS interferes in diabetes complications consisting of diabetic nephropathy (DN). Plasma-free fatty acids (FFAs) plus glucose are enhanced in diabetes and it has formerly been established that organelles like peroxisomes and mitochondria enroll in FFA oxidation in an interconnected method. “Hwang et al.” in an experimental animal study explored if deficiency of catalase, a main peroxisomal antioxidant, accelerates diabetic nephropathy via peroxisomal dysfunction and abnormal metabolism of renal FFA. Diabetes was triggered by multiple infusions of low-dose streptozocin into catalase knock-out (CKO) and wild-type (WT) C57BL/6 mice. Murine mesangial cells (MMC) transfected with catalase small interfering RNA succeeded by catalase overexpression were utilized to clarify the role of endogenous catalase intricately. Regardless of hyperglycemia, tests of DN in line with markers of oxidative stress were more advanced in diabetic CKO mice than in diabetic WT mice up to 10 weeks of diabetes. CKO mice and MMCs represented disturbed peroxisomal/mitochondrial biogenesis and FFA oxidation. Catalase deficiency enhanced mitochondrial ROS and fibronectin expression in response to FFAs, which were effectively regenerated by catalase overexpression or N-acetylcysteine. These data furnish evidence that FFA-induced peroxisomal dysfunction aggravates DN and that endogenous catalase enzyme contains a most valuable function in supporting the kidneys from diabetic stress via preservation of peroxisomal and mitochondrial fitness [58]. These findings point to the status of catalase antioxidants in improving or accelerating diabetic nephropathy through peroxisomal organelle. PPARα is provoked by hypolipidemic drugs (fibrates) and by naturally occurring ligands of the arachidonic cascade, like leukotrienes. Several PPAR-α independent activators of peroxisomes like PGC-1a or SIRT1 have been announced (Figure 2). “Ivashchenko et al.” in an experimental study used a redox-sensitive variant of enhanced green fluorescent protein (roGFP2-PTS1) to follow the state of the peroxisomal matrix in mammalian cells. They presented that intraperoxisomal redox status is robustly impressed by environmental growth conditions. Moreover, impairments in peroxisomal redox balance, even though not necessarily correlated with the organelle, may provoke its breakdown. They also established that the mitochondrial redox balance is impaired in catalase deficient cells and upon generation of excess ROS within peroxisomes [59]. In my belief, crosstalk between peroxisome and mitochondria functionally interact probably via ROS generation, metabolites, and substances or perhaps through other yet-to-be-identified factors. The consequences of the association between mitochondria and peroxisomes are described as that mitochondria can crosstalk with peroxisomes through vesicular transport of MDVs. Cardinal fission components of mitochondrial fission 1 protein (FIS1), mitochondrial fission factor (MFF), and ganglioside-induced differentiation protein 1 (GDAP1) are contributed to both by peroxisomes and mitochondria, and they invoked dynamin-1-like protein (DNM1L) to the organelle degradation site to disturb organelles. The fatty acids ß oxidation can develop in mitochondria and peroxisomes but the lipid ß-oxidation in peroxisomes is not perfect, after metabolizing lipids to medium length, they will be co-transmitted with acetyl-CoA to the mitochondria for excess metabolism. Both mitochondria and peroxisomes can generate ROS, and they are also valuable organelles for extruding ROS and maintaining cells stability. Peroxisomes chiefly comprise catalase to degrade H2O2 and ROS are valuable signalling molecules for triggering cell apoptosis.
Mitochondria-lysosome crosstalk studies: Evidence has now emerged about crosstalk between mitochondria and lysosomes, as such the activity or stress status of one organelle may influence another organ. Mitochondrial dysfunction triggers lysosome impairment, disclosing a functional relationship between the mitochondria and lysosome [60, 61]. For instance, deletion of mitochondrial transcription factor has been used as a model of mitochondrial dysfunction, as TFAM is necessary for the replication and transcription of mitochondrial DNA (mtDNA). In the case of the absence of TFAM, there is less mtDNA, fewer transcripts encoding mt DNA genes, and disturbance of the electron transport chain. Upon deletion of TFAM in T cells, the impairment of mitochondria intensifies the number of lysosomes. Hence, lysosomal activity is strongly disturbed, triggering a deposition of sphingomyelin and autophagy intermediates, and provoking inflammatory responses. The elevated lysosomal mass was caused by the activation of the transcription factor EB (TFEB), a master transcription factor controlling lysosome biogenesis. The addition of a NAD + precursor for increasing the cellular levels of NAD+ improved lysosomal function in this model and this assay is evidence that the declined NAD+ levels in cells with impaired mitochondrial function cause lysosomal function. Moreover, the presence of mitochondrial molecules in exosomes discloses further indirect evidence of the crosstalk between mitochondria and the endolysosomal system. Physical contact between mitochondria and the endolysosomal compartment could synchronize processes that need to contribute to both organelles, like metabolic adaptation, phospholipid metabolism, regulation of calcium signalling, and the control of cellular homeostasis [62]. Mitochondrial and vacuolar biogenesis appears to influence each other. For example, vacuole morphology is affected by a loss of the cardiolipin synthase Crd1, while several endosomal and vacuolar mutants comprising all V-ATPase subunits, lead to a mitochondrial petite phenotype or affect mitochondrial functions in longevity [63, 64] (Figure 3). Another point that must be discussed herein is autophagy. In the year 1963, “Christian de Duve” was the first person that coined the term autophagy because he observed organelle degradation within lysosomes. Nearly 30 years later, the scientific world has begun to clarify the molecular events of autophagy. Overall, both acute and chronic kidney injuries increase oxidative stress and autophagic flux. The suggested mechanism is the contribution of oxidative stress in inflammation and even here sustained levels of ROS generation may cause cell death. This excessive oxidative stress, depending upon the intensity and/or duration, provokes cellular dysfunction and also contributes to the pathogenesis of acute kidney injury (AKI), CKD, and ultimately kidney failure / end-stage kidney disease (ESKD). Autophagy supports renal tissue by removing damaged organelles like mitochondria and thereby indirectly suppresses excessive ROS generation, inflammation, and cell death [65]. Furthermore, autophagy is a conserved multistep pathway that breaks down and recycles damaged organelles and macromolecules to maintain intracellular homeostasis. The autophagy pathway is upregulated under stress states comprising cell starvation, hypoxia, nutrient and growth factor deprivation, ER stress, and oxidant injury, most of which are interfered in the pathogenesis of AKI. Since lysosomes are involved in autophagy cargo clearance, it may be necessary to assess lysosomal dysfunction in the pathogenesis of AKI critically. Current studies have identified that autophagy is selective in degrading special targets consisting of damaged organelles [66]. The mTORC1kinase complex may mediate lysosomal stress response (UPRLy) and autophagy. Under conditions with high levels of amino acids, mTORC1 is recruited to the lysosomal membranes by the v-ATPase-Regulator complex, and activated by the small GTPase Rheb. Activated mTORC1 phosphorylates ribosomal protein S6 kinase (S6K) and eukaryotic initiation factor 4E-binding protein 1(4E-Ep1) which increases translation and cell growth. Phosphorylation of unc-51 like autophagy activating kinase 1(ULK1) and TFEB leads to inhibition of autophagy. These processes can be opposed upon starvation of amino acids culminating in expanded lysosomal compartments and activation of autophagy (Figure 4). Several studies have been performed about mitochondria to lysosome crosstalk that is discussed herein. “Fernandez-Mosquera et al.” in an experimental study confirmed that mitochondrial RC impairments provoke a stress signaling pathway that mediates lysosomal biogenesis through the microphthalmia transcription factor family. Invitingly, the effect of mitochondrial stress over lysosomal biogenesis relies upon the timeframe of the elicited stress; while RC inhibition with rotenone or uncoupling with carbonyl cyanide 3-chlorophenylhydrazone (CCCP) initially induces lysosomal biogenesis, the effect peaks after few hours and comes back to baseline. Long-term RC inhibition by long-term treatment with rotenone, or patients fibroblasts, mouse embryonic fibroblasts (MEFs) with genetic inhibition of the RC, and tissues from mice with mitochondrial RC defects result in repression of lysosomal biogenesis. Based on the short-term induction of lysosomal biogenesis, mitochondrial stress is dependent on TFEB and microphthalmia-associated transcription factor (MITF) both of these factors require AMP-associated protein kinase (AMPK) signalling. Furthermore, these factors are independent of calcineurin signalling. Conclusively, acute and chronic mitochondrial RC deficiency differentially mediate lysosomal biogenesis [67]. In my opinion, acute and chronic perturbations of the mitochondrial RC have opposite effects on lysosomal biogenesis. Acute mitochondrial RC stress triggers AMPK- and TFEB/MITF- dependent lysosomal biogenesis while chronic RC dysfunction results in repression of TFEB/MITF transcriptional activity and lysosomal biogenesis. “Ghavami et al.” in an experimental study explored underlying molecular mechanisms that interfere with both programmed cell death (PCD I, apoptosis) and PCD II (autophagy)-like death. Treatment of cells with S100A8/A9 caused the increase of Beclin-1 expression as well as Atg12-Atg 5 formation. S100A8/A9 – induced cell death was partially suppressed by the special PI3-kinase class III inhibitor, 3-methyladenine, and by the vacuole h+-ATPase inhibitor, bafilomycin-A1. S100A8/A9 triggered the translocation of BNIP3, a BH3-only pro-apoptotic Bcl2 family member, to mitochondria. Compatible with this result, ∆TM-BNIP3 overexpression partially suppressed S100A8/A9-induced cell death, declined reactive oxygen species production, and partially supported the decline in mitochondrial transmembrane potential in S100A8/A9-treated cells. Furthermore, either ∆TM-BNIP3 overexpression or N-acetyl-L-cysteine co-treatment decreased lysosomal activation in cells treated with S100A8/A9. Data of this study disclosed that S100A8/A9-promoted cell death happens via the crosstalk of mitochondria and lysosomes through ROS and this process involves BNIP3 [68]. This research shows crosstalk between mitochondria and lysosomes that autophagy besides apoptosis has a role in cell death due to S1008/A9. In other words, cell death-induced S100A8/A9 interferes with BNIP3, and ROS may have a role in cell death triggered by S100A8/A9. “Baixauli et al.” in an experimental study explored a mouse model with defective mitochondrial function in CD4+ T-lymphocytes by genetic deletion of the Tfam. Mitochondrial RC deficiency disturbs lysosome function, proceeding with p62 and sphingomyelin deposition. Other findings in this research consist of disruption of endolysosomal trafficking pathways and autophagy, thus linking a primary mitochondrial dysfunction to a lysosomal storage disorder. The disturbed lysosomal function in Tfam-deficient cells diverts T cell differentiation toward pro-inflammatory subsets and intensifies the in vivo inflammatory response. Maintenance of NAD+ levels relieves lysosome function and refines the inflammatory defects in Tfam-deficient T cells. This study furnishes evidence for a novel functional link between mitochondrial respiration and the endolysosomal system with implications for treating mitochondrial-related diseases. They demonstrated that mitochondrial respiration controls lysosomal function during inflammatory T cell responses [69]. In my viewpoint, cells that lack Tfam depict lysosomal defects similar to lysosomal storage disorders and these defects contain abnormal lipid trafficking, altered calcium mobilization, and defective autophagosome-lysosome fusion. In another study, “Brahimi-Horn et al.” performing an experimental study displayed that truncation is linked with elevated resistance to drug-induced apoptosis and is indicative of increased chemoresistance in patients. They depicted that silencing of the tumor suppressor TP53 declines truncation and elevates drug-induced apoptosis. In addition, they expressed that TP53 mediates truncation via induction of the mitochondrial protein Mieap. While they detected that truncation was independent of mitophagy, they depicted local microfusion between mitochondria and endolysosomal in hypoxic cells both in culture and patients’ tumor tissues. Because they detected that the endolysosomal asparagine endopeptidase was responsible for truncation, they suggested that it is a readout of mitochondrial-endolysosomal microfusion in hypoxia. The present study has confirmed the degradation of voltage-dependent anion channel 1(VDAC1) expressing a survival response as the readout of interaction between hyper-fused mitochondria with endolysosomes.They also depicted that this phenomenon exists not only in vitro but also in vivo in patients with lung adenocarcinoma. All of the above-mentioned results lead to the fact that this cross-talk between organelles is regulated by TP53-induced Mieap and binding to hypoxia-induced BNIP3 [70]. In my opinion, enlarged hypoxic mitochondria make fusional contact with late endolysosomes through TP53-induced Mieap in promoting cell survival. Taken together, mitochondria and lysosomal crosstalk perform by physical contacts for metabolic adaptation, phospholipid metabolism, regulation of Ca2+ signalling, and the control of cellular metabolism. Other functions of mitochondria to lysosome crosstalk in regulating cell function comprise autophagy, proliferation, and cell death. Of note, mitochondrial dysfunction has influenced lysosomal impairment via TFAM. Furthermore, mitochondrial RC deficiency mediates lysosomal biogenesis. Here another important point must be expressed that increasing cellular NAD+ precursor levels cause improving lysosomal function. Also, this point should be known that crosstalk between mitochondria and lysosome occurs through ROS by BNIP3. Ultimately this crosstalk plays a role in cancer by TP53-induced Mieap and binding to hypoxia-induced BNIP3.
Interorganelle Crosstalk in nephropathy-related conditions
In response to endothelial or epithelial cell stress, unfolded or misfolded proteins accumulate in the ER and provoke the unfolded protein response. Following acute ischemia and nephrotoxicity, ER stress is triggered in kidney epithelial cells both in vivo and “in vitro”. If the stress was too severe or prolonged, the maladaptive response is activated by the induction of CHOP, a transcription factor downstream from the PERK-ATF4 axis, responsible for apoptosis. Exactly, long-standing and overwhelming ER stress has influenced the development of renal fibrosis. Maladaptive repair following AKI culminates in CKD and ESRD (Figure 5). Hereby, targeting the UPR may represent a possible approach to prohibit or treat AKI. Peptide SS-31/cardiolipin supports mitochondrial structure and respiration during early reperfusion, hastening the recovery of AKI. Thus, agents that trigger mitochondrial biogenesis could use as biological therapies for AKI. The primary regulator of mitochondrial biogenesis is PGC-1a and PPAR-a. Agents that stimulate the expression and activation of PGC-1a comprise activators of AMPK like metformin and AICAR and as well as activators of SIRT1 which consist of isoflavones, resveratrol, and the small molecules SIRT1720. In sepsis-induced AKI without cell death, induction of PGC-1a proceeds renal recovery by improving mitochondrial respiration and maintaining the expression of mitochondrial electron transport genes. Formoterol, a β2-adrenergic agonist, maintains renal function and supports kidney tubules from injury and death by maintaining the expression and function of mitochondrial proteins, consisting of PGC – 1α, via the cAMP/PKA/CREB pathway [71]. Mediating mitochondrial dynamics is another therapeutic choice for the treatment or prevention of AKI. Pharmacological inhibition of Drp1— a fission protein that limits and degrades mitochondria, culminating in fragmentation — declines mitochondrial damage, cytochrome c release, apoptosis, and renal injury. Currently, SIRT3, a mitochondrial sirtuin, has been confirmed to recover mitochondrial dynamics by preventing Drp1-dependent fission, loss of mitochondrial membrane potential, and mitophagy. In addition, activation of the 5-hydroxytryptamine 1F receptor with selective, commercially available small molecules and cGMP-selective phosphodiesterase inhibitors proceeds mitochondrial biogenesis and recovery from AKI via elevated expression of PGC-1α and mitochondrial electron transport chain genes. Crosstalk between ER and mitochondria at MAM in line with Ca2+ release functionally associates ER stress to mitochondrial pathways due to activation with the cellular stress response. An important phenomenon in crosstalk between mitochondria and the UPR is autophagy, a catabolic process in which proteins, organelles, and cytoplasmic components are conveyed to lysosomes for degradation and recycling. Autophagy is triggered in the kidney in response to AKI and supports kidney injury [72]. High glucose-induced ROS can be produced by both enzymatic and nonenzymatic pathways. The enzymatic pathways consist of NADPH oxidase, uncoupling of NOS, CYTP450, COX, LOX, xanthin oxidase, and MPO. Nonenzymatic pathways comprise mitochondrial electron transport chain deficiencies, AGEs, glucose auto-oxidation, transition-metal catalyzed Fenton reactions, and polyol (sorbitol) pathways. In the diabetic kidney, NADPH (enzymatic) and mitochondrial electron transport chain (nonenzymatic) are the remarkable sources of ROS, which are opined to cause albuminuria and then progression to renal damage via podocyte depletion. Chronic hyperglycemia and resulting ROS generation can induce abnormal signal pathways interfering with various signalling mediators like transcription factors, inflammatory cytokines, chemokines, and vasoactive substances. Continuously, elevated expression and activation of these signalling molecules contribute to the irreversible functional and structural alterations in the kidney. This finding results in a critically declined glomerular filtration rate and then in final renal failure [73]. An important point that should be pointed out herein is the cellular expression of two apolipoprotein L1 renal risk variants (APOL1 RRVs). It has been demonstrated these variants are involved to induce cytotoxicity in several cell types for example in podocytes and contribute to necrosis, apoptosis, and pyroptosis. Mechanistically these toxicities were predicated on lysosomal swelling, K+depletion, mitochondrial dysfunction, autophagy blockade, protein kinase receptor activation, ubiquitin D degradation, and endoplasmic reticulum stress; notably, these effects were detected to be dose-dependent and happened only in overtly APOL1 RRV-expressing cells [74]. Mitochondrial cytopathies result due to disturbances in the process attributed to mitophagy or due to an imbalance between the process of fission and fusion. Renal mitochondrial cytopathies can characterize either as glomerular or tubular diseases or as renal cysts or neoplasia [75]. The NLRP3 inflammasome is activated by MAMs. The NLRP3 inflammasome plays a role in inducing infectious defenses via inflammatory cytokines. It has a role in nephrocalcinosis-related CKD. The NLRP3 inflammasome can react with metabolites such as urate crystal (caused by ingestion of excessive foods) and β-amyloid (caused by aging), thereby triggering excessive production of inflammatory cytokines. Eventually, these events can culminate in life-related diseases such as diabetes. Colchicine inhibits inflammation caused by urate crystals. Bardoxolone methyl activates nuclear factor (erythroid-derived 2)-like 2 (Nrf2) in diabetic kidney disease. The activation of Nrf2 protects tissues from inflammation by elevating intracellular antioxidant factors and inhibiting signaling pathways associated with inflammation, proposing the possibility that bardoxolone methyl impacts Nrf2Kelch-like ECH associated protein 1 (Keap1) in the MAM [76]. Novel therapeutic drugs such as sodium-glucose transporter 2 inhibitors (SGLT2i) and hypoxia-inducible factor (HIF)-prolyl hydroxylase (PH) inhibitor have useful renoprotective effects associated with mitochondrial metabolic reprogramming, suggesting the potent strategy for maintaining kidney function through mitochondrial metabolic homeostasis [77]. SGLT2 inhibitors are used to decrease blood pressure in patients with type 2 diabetes and HIF stabilizers increase endogenous erythropoietin (EPO) production and apply as novel therapeutic agents in the treatment of anemia in CKD [78] (Table 2). In my opinion, interorganelle crosstalk is a novel discovery in the scientific world especially in kidney science that can be utilized for both discoveries of unknowns and proceeding to detection of novel mechanisms for diseases.
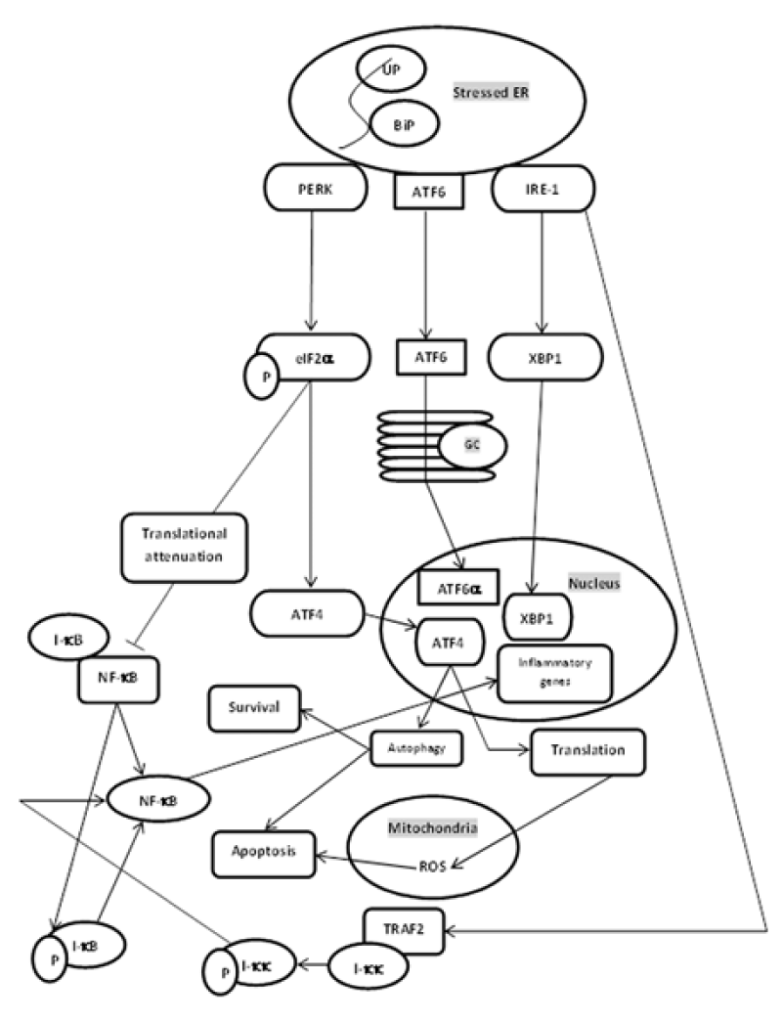
Axial arterial phase CT- ischemic area of cortical Right kidney (See arrow).
Coronal Recostruction arterial phase CT- ischemic area of cortical Right kidney (See arrow).
Results
Taken together, this review has integrated mitochondrial function into a complex but plenary network with other cellular organelles and processes [79]. The organelles (mitochondria, nucleus, lysosome, endoplasmic reticulum, Golgi apparatus) are members of such functional units that are needed to perform specific tasks. Those must be fully integrated and react to the activity of other subsystems. Mitochondrial crosstalk with other organelles has been summarized in Figure 6.
Conclusion
Mitochondria as a signalling organelle associates with the cellular signalling environment and mediates gene expression programs. Mitochondria are constantly interacting with other organelles through signalling pathways, and on some occasions even though physical contact sites. Therefore, the physiologic functions of both organelles are different and independent but their interaction is necessary for the optimal function of the cell. Dysfunctional crosstalk between mitochondria with other organelles causes cancer, type 2 diabetes mellitus, metabolic syndrome/disorders, inflammation/infection, central nervous system disease, cardiovascular disease, renal disease, aging, and age-related disorders. A better understanding of these interactions will help to explore new mechanisms and novel therapeutic modalities.