
Bispecific sigma-1 receptor antagonism and mu-opioid receptor partial agonism: WLB-73502, an analgesic with improved efficacy and safety profile compared to strong opioids
By Alba Vidal-Torres, Begoña Fernández-Pastor, Mónica García, Eva Ayet, Anna Cabot, Javier Burgueño, Xavier Monroy, Bertrand Aubel, Xavier Codony, Luz Romero, Rosalía Pascual, Maria Teresa Serafini, Gregorio Encina, Carmen Almansa, Daniel Zamanillo, Manuel Merlos, and José Miguel Vela
Excerpt from the article published in Acta Pharmaceutica Sinica B, September 25, 2022, ISSN 2211-3835, DOI: https://doi.org/10.1016/j.apsb.2022.09.018
Editor’s Highlights
- WLB-73502 is a bispecific compound that binds sigma-1 (S1R) and mu-opioid (MOR) receptors, currently developed as ADV502
- WLB-73502 has an analgesia comparable (nociceptive and mixed inflammatory and OA pain) or superior (neuropathic pain) to full MOR agonists, but it does not induce tolerance and causes no/less constipation, respiratory depression, and nausea/vomiting than strong opioids.
- WLB-73502 is a promising alternative for treating chronic refractory pain, potentially neuropathic cancer pain, where regular stand-alone opioids do not achieve satisfactory outcomes and are limited by drug tolerance and adverse effects.
Abstract
Opioids are the most effective painkillers, but their benefit-risk balance often hinder their therapeutic use. WLB-73502 is a dual, bispecific compound that binds sigma-1 (S1R) and mu-opioid (MOR) receptors. WLB-73502 is an antagonist at the S1R. It behaved as a partial MOR agonist at the G-protein pathway and produced no/unsignificant β-arrestin-2 recruitment, thus demonstrating low intrinsic efficacy on MOR at both signalling pathways. Despite its partial MOR agonism, WLB-73502 exerted full antinociceptive efficacy, with potency superior to morphine and similar to oxycodone against nociceptive, inflammatory and osteoarthritis pain, and superior to both morphine and oxycodone against neuropathic pain. WLB-73502 crosses the blood–brain barrier and binds brain S1R and MOR to an extent consistent with its antinociceptive effect. Contrary to morphine and oxycodone, tolerance to its antinociceptive effect did not develop after repeated 4-week administration. Also, contrary to opioid comparators, WLB-73502 did not inhibit gastrointestinal transit or respiratory function in rats at doses inducing full efficacy, and it was devoid of proemetic effect (retching and vomiting) in ferrets at potentially effective doses. WLB-73502 benefits from its bivalent S1R antagonist and partial MOR agonist nature to provide an improved antinociceptive and safety profile respect to strong opioid therapy.
Graphical abstract
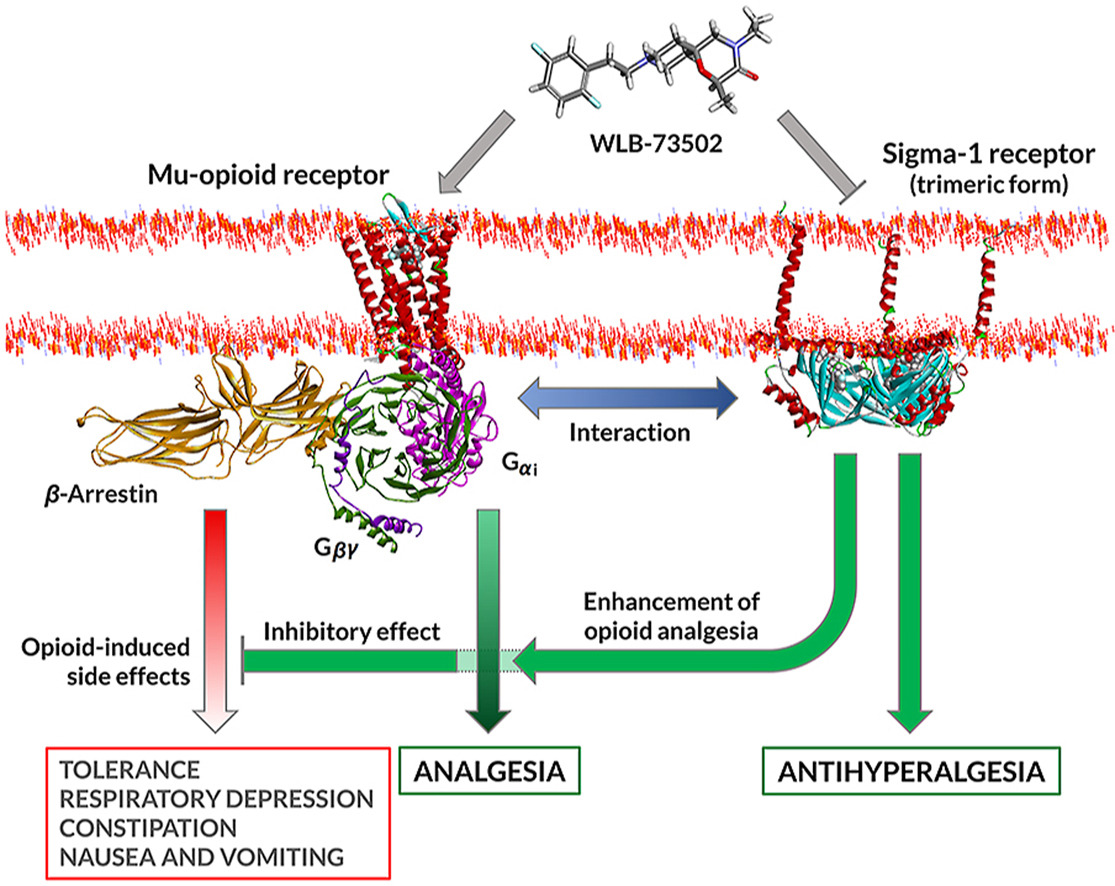
1. Introduction
Chronic pain is one of the most common health problems worldwide1. Despite its large prevalence, chronic pain is still poorly understood and difficult to treat. Pharmacological activation of opioid receptors, particularly the μ-opioid receptor (MOR), is one of the main treatment options. However, the use of opioids is associated with a wide range of side effects including constipation, nausea, vomiting, respiratory depression and dysphoria or euphoria2. In addition, under repeated administration, tolerance develops that results in a diminished effectiveness against chronic pain conditions3. Analgesic tolerance leads to the use of higher drug doses that increase overdose risk. Due to their reinforcement properties, opioids have a high potential for causing addiction, as proven by the opioid abuse/misuse epidemics (opioid crisis) affecting the United States4. Because of the negative aspects of their use, new opioids drug discovery focuses on the design of compounds having greater safety/efficacy ratio5.
One approach consists in the development of MOR agonists with low intrinsic activity as safer alternatives6. However, the reduced side effects often run parallel to limited analgesia and potential advantages of partial opioid agonists are not clearly borne out in clinical practice7. In the case of buprenorphine, a non-selective schedule III partial MOR agonist (also binds to kappa and delta opioid receptors), analgesia is remarkable and some responder and safety analyses8, and benefit-risk assessments9 support its use over full MOR agonists for chronic pain treatment.
Alternative approaches include biased ligands that promote preferential signalling through one of the MOR transducers10. This concept generated considerable excitement as the Gi/o protein pathway was proposed to mediate analgesia while the β-arrestin pathway the opioid-related adverse effects11. Oliceridine (TRV130, Olinvyk®, Trevena, Inc., King of Prussia, PA, USA), a G-protein-biased MOR agonist indicated for intravenous treatment of acute pain was reported to exhibit a favourable safety profile compared to morphine12. However, oliceridine produces typical opioid abuse-related effects in rodents and humans13. Confirmation of its improved benefit-risk profile would probably require specific trials examining its safety versus conventional opioids14.
The benefit-risk of opioid analgesics may also be improved by multimodal mechanisms, combining opioid and non-opioid pathways to enhance efficacy and/or minimize adverse effects15,16. Innovation in multimodal analgesia involving opioid mechanisms goes from i) free combinations (2 tablets, 2 active ingredients; polypharmacy, frequent in clinical practice); ii) to fixed-dose combination formulations (1 tablet, 2 active ingredients; e.g., tramadol/acetaminophen)17; iii) to drug–drug co-crystals (1 tablet, 2 active ingredients non-covalently bonded in a co-crystal lattice; e.g., Seglentis®, a co-crystal of tramadol and celecoxib)18,19; and finally iv) to multimodal drugs (1 tablet, 1 active ingredient with at least 2 activities; e.g., Nucynta®, tapentadol, a MOR agonist and norepinephrine reuptake inhibitor)20. The discovery of single compounds that bind to two distinct targets, also known as dual, bivalent, bifunctional or bispecific ligands, is challenging, but such drugs offer advantages respect to drug combinations besides improved efficacy and/or safety. These include superior patient compliance with medication (one instead of various pills at potentially different dosage frequencies), lower risk of drug–drug interactions, simpler (single) pharmacokinetics and synchronic/overlapping engagement of targets, and less variability among patients, both in drug exposure and response to treatment21. The non-opioid mechanism should ideally have an independent effect and synergistic analgesic activity when used with opioids, but it should not enhance or preferably counteract opioid-related side effects. This is the case of the sigma-1 receptor (S1R, σ1R) antagonism mechanism. Based on drug combination studies, S1R antagonists increase the therapeutic index of opioids by enhancing analgesia22,23 and adding a unique analgesic action in “opioid-resistant pain”, particularly neuropathic pain24. Besides inhibition of pain hypersensitivity per se, S1R antagonists “release the brake” enabling opioids to exert enhanced antinociceptive effects25,26 as S1R is a tonically active system limiting opioid analgesia22. Potentiation of opioid analgesia by S1R blockade results in an opioid-sparing effect at equianalgesia, which translates into improved safety of the combination compared to opioid monotherapy. Contrary to analgesia, opioid side effects are not potentiated and some of them are counteracted22,23,26,27, which further contributes to a better safety profile of the combination.
A drug discovery program was undertaken to identify a multimodal, bispecific drug with S1R and MOR activities using a pharmacophore merging approach, which led to the discovery of WLB-7350228. Here we disclose the pharmacological in vitro and in vivo efficacy and safety profile of WLB-73502 in comparison with strong opioids (morphine and oxycodone). Results are discussed according to their potential as a replacement alternative to strong opioid monotherapy.
…
3. Results
3.1. In vitro functional profile
3.1.1. MOR functionality
WLB-73502 induces negative coupling of MOR to adenylyl cyclase and thus behaves as MOR agonist at the cAMP-dependent pathway, but accurate determination of its functional nature (i.e., full vs. partial agonism) cannot be established in cell lines overexpressing MOR, with a high receptor reserve37,38. Accordingly, the efficacy of WLB-73502 was evaluated by a cAMP determination assay on CHO-K1 cells stably expressing the human MOR, following partial receptor inactivation in presence of increasing concentrations of the irreversible MOR antagonist β-FNX (Fig. 1). MOR agonist inhibition of forskolin-stimulated cAMP production in presence of β-FNX revealed a partial agonist activity of WLB-73502 (∼30% maximal efficacy in presence of the highest concentrations of β-FNX). The maximal efficacy of the full MOR agonist oxycodone was much less affected by β-FNX. To accurately quantify and compare, concentration–response curves were subjected to operational analysis and τ values calculated (Supporting Information Table S1). This analysis revealed a statistically significant partial agonist activity for WLB-73502 (lgτ = 1.58) as compared to classical full opioid agonists such as morphine (lgτ = 2.11), oxycodone (lgτ = 2.18) or fentanyl (lgτ = 2.23).
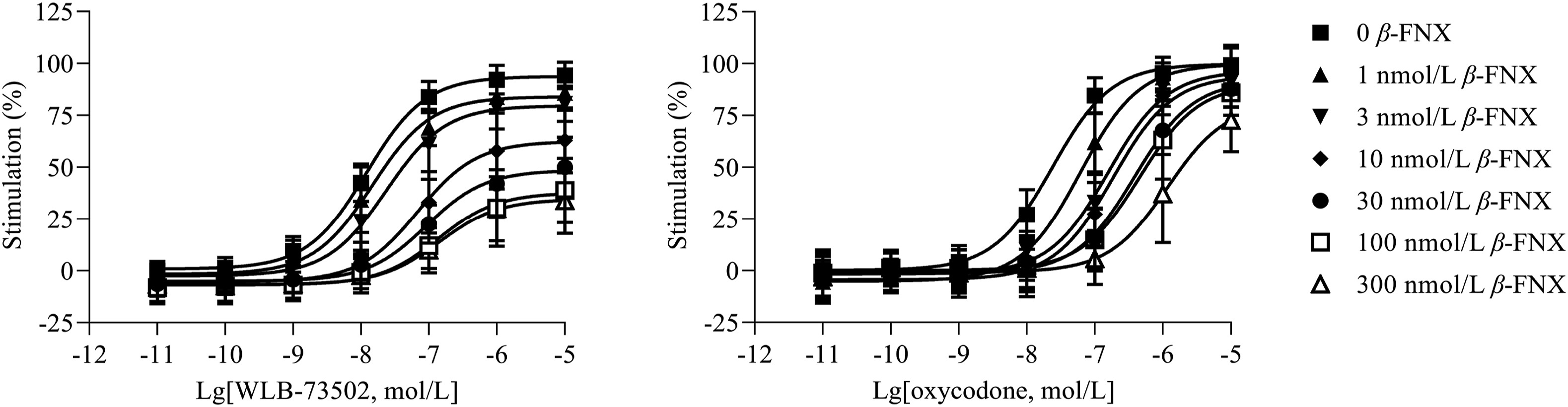
MOR agonist-mediated inhibition of forskolin-stimulated cAMP production.
The concentration–response curves of WLB-73502 and oxycodone were determined relative to DAMGO (1 μmol/L) as reference full agonist in this assay, in the absence (0) and presence (1, 3, 10, 30, 100, 300 nmol/L) of the irreversible MOR antagonist β-FNX. n = 5 independent experiments. In each experiment, data points (mean ± SEM) were obtained from duplicates.
A complementation assay was used to assess β-arrestin-2 recruitment elicited by MOR agonist ligands. Concentration–response curves (Fig. 2A) were subjected to operational analysis and τ values calculated (Table S1). WLB-73502 had no effect in this signalling pathway and thus the operational lgτ parameter could not be determined. Traditional opioids, like morphine (lgτ = −0.19), oxycodone (lgτ = −0.28) or fentanyl (lgτ = 0.31) recruited the β-arrestin signalling pathway to some extent, as published previously37. Accordingly, the maximal effect (Emax) for β-arrestin-2 recruitment elicited of WLB-73502 was significantly different from the Emax of the other MOR agonists assayed (morphine, oxycodone and fentanyl) (Table S1).
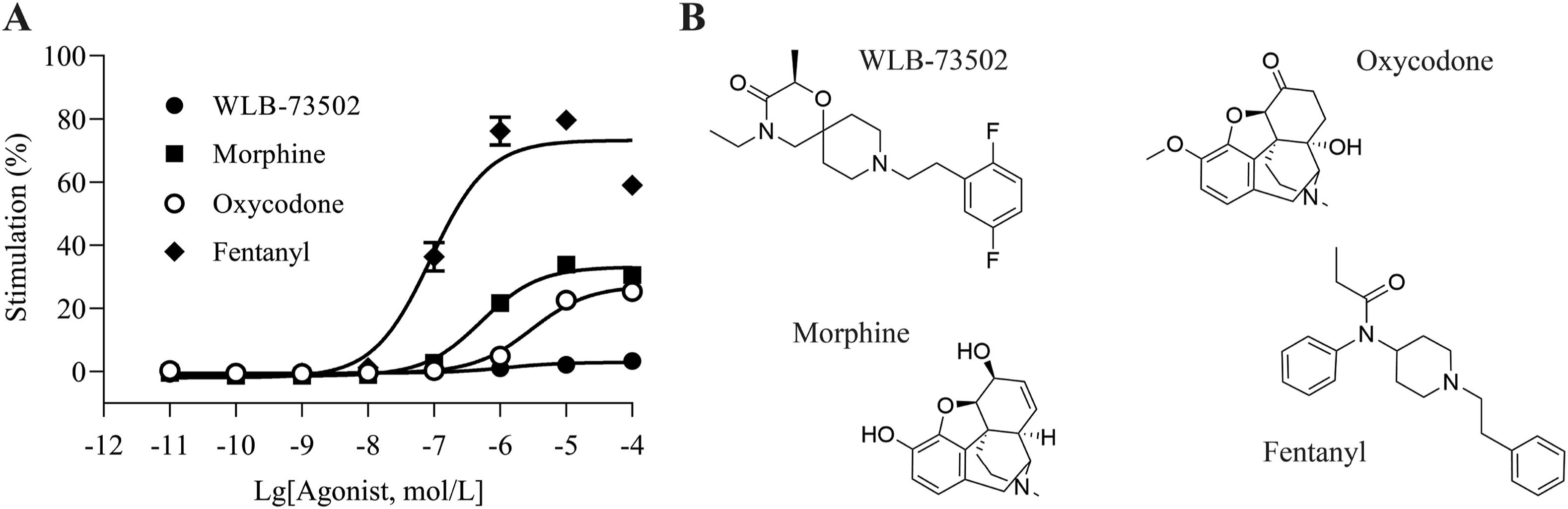
MOR agonist-mediated β-arrestin recruitment (A) and molecular structure of assayed ligands (B).
The concentration–response curves of WLB-73502, morphine, oxycodone and fentanyl were determined relative to the concentration–response curve of DAMGO as reference full agonist in this assay. n = 5 except for fentanyl (n = 3) independent experiments. In each experiment, data points (mean ± SEM) were obtained from quadruplicates.
3.1.2. S1R functionality
Phenytoin (diphenylhydantoin, DPH) is a positive allosteric S1R modulator that shifts S1R agonists to significant higher binding affinities (Ki ratios without DPH vs.with DPH > 1), while no shift or a very little shift to lower affinity values (Ki ratios without DPH vs. with DPH ≤ 1) occurs with antagonists29. WLB-73502 produced a small, non-significant shift to lower affinity values when incubated with DPH (Kiwithout DPH/Ki with DPH = 0.9), which indicated antagonist properties at the S1R (Fig. 3). A similar small non-significant shift to the right was also found for the reference S1R antagonist haloperidol, whereas DPH significantly shifted the curve of the reference S1R agonist dextromethorphan to higher affinity (Ki without DPH/Ki with DPH = 9.0).
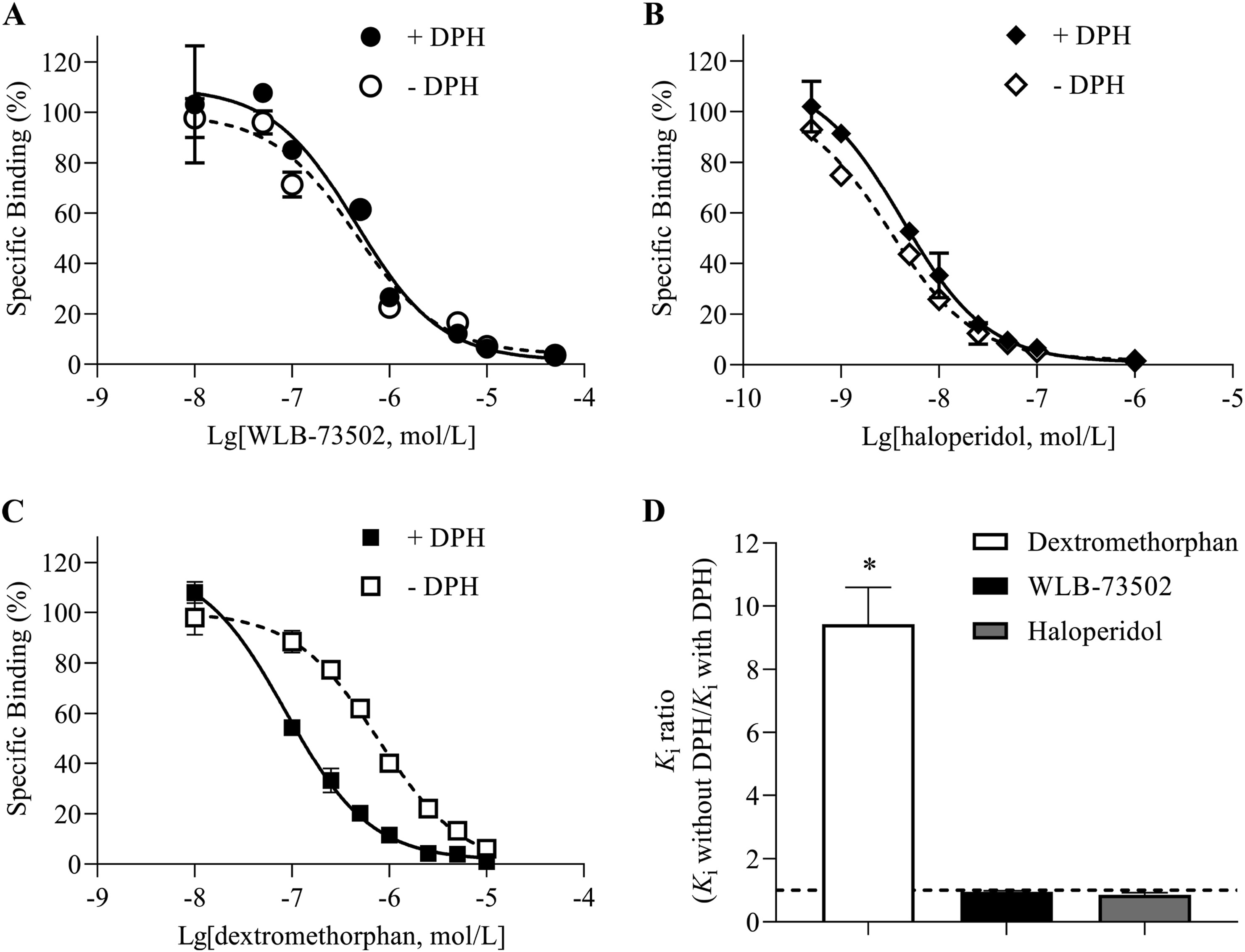
DPH modulation of S1R binding. Inhibition by WLB-73502 (A), haloperidol (B) and dextromethorphan (C) of [3H](+)-pentazocine binding to guinea pig brain membranes in the absence (open symbols) or presence (closed symbols) of 1 mmol/L DPH. Ratios of the Ki value in the absence and presence of 1 mmol/L DPH for WLB73502, haloperidol and dextromethorphan (D). n = 5 independent experiments. In each experiment, data points (mean ± SEM) were obtained from duplicates. ∗P < 0.05 vs. ratio = 1 (one-way ANOVA followed by Bonferroni’s post hoc test).
3.2. Antinociceptive effect after single administration in rat models of pain
The effect of single i.p. administration of WLB-73502, morphine and oxycodone on mechanical (paw pressure test) and thermal (tail-flick test) sensitivity, and on mechanical hypersensitivity (mechanical allodynia) following somatic inflammatory (carrageenan paw injection), osteoarthritis (MIA knee injection) and neuropathic (SNI) pain induction was evaluated (Fig. 4 and Table 1).
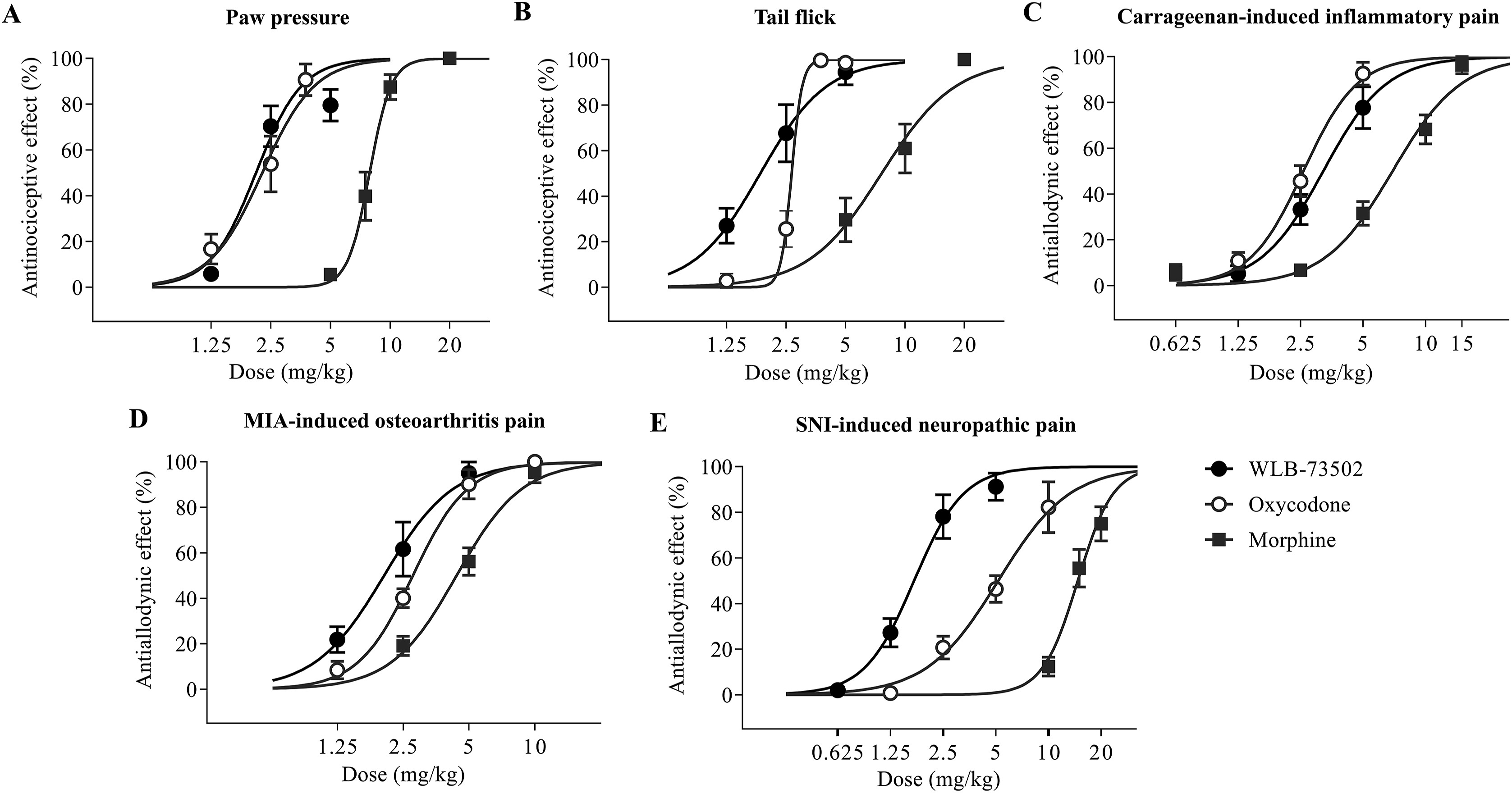
Acute antinociceptive/antiallodynic effect. Dose–response curves of the effect of single administration of WLB-73502, morphine or oxycodone on mechanical nociception in the paw pressure test (A), on thermal nociception in the tail flick test (B), and on mechanical allodynia in carrageenan-induced inflammatory (C), MIA-induced OA (D) and SNI-induced neuropathic (E) pain models in male Wistar rats. n = 7 (paw pressure and tail flick), n = 9–10 (carrageenan and MIA) and n = 8 (SNI) per treatment group. Values are expressed as percentage of effect. Points and vertical lines represent the mean ± SEM.
| ED50 (mg/kg)a | |||
|---|---|---|---|
| Animal pain model | WLB-73502 | Oxycodone | Morphine |
| Mechanical nociception | |||
| Paw pressure | 2.11 ± 0.18 | 2.27 ± 0.22 | 7.88 ± 0.24∗ |
| Thermal nociception | |||
| Tail-flick | 1.85 ± 0.22 | 2.67 ± 0.46 | 7.63 ± 0.96∗ |
| Inflammatory pain – mechanical allodynia | |||
| Carrageenan-induced inflammation | 3.11 ± 0.34 | 2.68 ± 0.15 | 6.94 ± 0.42∗ |
| Osteoarthritis pain – mechanical allodynia | |||
| MIA-induced osteoarthritis | 2.05 ± 0.20 | 2.77 ± 0.13 | 4.39 ± 0.28∗ |
| Neuropathic pain – mechanical allodynia | |||
| SNI-induced neuropathy | 1.71 ± 0.14 | 5.17 ± 0.44∗ | 14.72 ± 0.73∗ |
ED50 values of the antinociceptive/antiallodynic effect of WLB-73502, oxycodone and morphine on acute mechanical (paw pressure) and thermal (tail-flick) nociception, and on mechanical allodynia in pain models of different aetiology following single i.p. administration in male Wistar rats.
Data are mean ± SEM; n = 7 (paw pressure and tail flick), n = 9–10 (carrageenan and MIA) and n = 8 (SNI) per treatment group. ∗P < 0.05 vs. WLB-73502 (one-way ANOVA followed by Bonferroni’s post hoc test).
3.2.1. Paw pressure test
Single administration by i.p. route of WLB-73502 (1.25, 2.5, and 5 mg/kg), morphine (5, 7.5, 10, and 20 mg/kg) or oxycodone (1.25, 2.5, and 3.75 mg/kg) dose-dependently inhibited paw pressure-induced mechanical nociception (Fig. 4A, Supporting Information Fig. S1A and S1B), reaching a maximal efficacy ≥80%. WLB-73502 was 3-fold more potent than morphine (P < 0.05) and had a potency similar to oxycodone (Table 1).
3.2.2. Tail flick test
Single systemic i.p. administration of WLB-73502 (1.25, 2.5, and 5 mg/kg), morphine (5, 10, and 20 mg/kg) and oxycodone (1.25, 2.5, 3.75, and 5 mg/kg) dose-dependently inhibited acute thermal nociception (Fig. 4B, Fig. S1C and S1D), with efficacies reaching a maximal effect ≥95%. WLB-73502 showed 4-fold more potency than morphine (P < 0.05) and similar potency to oxycodone (Table 1).
3.2.3. Carrageenan-induced model of acute inflammatory pain
A significant decrease in the withdrawal thresholds to von Frey stimulation (i.e., mechanical allodynia) was observed in the ipsilateral paw 4 h after intraplantar carrageenan injection. Single i.p. administration of WLB-73502 (1.25, 2.5, and 5 mg/kg), morphine (0.625, 2.5, 5, 10, and 15 mg/kg) or oxycodone (1.25, 2.5, and 5 mg/kg) dose-dependently reversed carrageenan-induced mechanical allodynia in the ipsilateral paw (Fig. 4C and Supporting Information Fig. S2). Maximal effect was ≥80% for all treatments. WLB-73502 was 2-fold more potent than morphine (P < 0.05) and had similar potency to oxycodone (Table 1). Mechanical allodynia did not develop in the contralateral paw and treatments did not exert significant effects on the contralateral paw (Fig. S2).
3.2.4. MIA-induced model of OA pain
A significant decrease in the withdrawal thresholds to von Frey stimulation was observed in the ipsilateral paw 14 days after MIA injection into the knee joint. WLB-73502 (1.25, 2.5, and 5 mg/kg), morphine (2.5, 5, and 10 mg/kg) or oxycodone (1.25, 2.5, 5, and 10 mg/kg) dose-dependently reversed mechanical allodynia in the ipsilateral paw reaching a maximal effect ≥95% (Fig. 4D and Supporting Information Fig. S3). WLB-73502 showed 2-fold more potency than morphine (P < 0.05) and similar potency to oxycodone (Table 1). Mechanical allodynia did not develop in the contralateral paw and treatments did not exert significant effects on the contralateral paw (Fig. S3).
3.2.5. SNI model of neuropathic pain
A significant decrease in the withdrawal thresholds to von Frey stimulation was observed in the ipsilateral paw 14 days after SNI surgery. Administration of WLB-73502 (0.625, 1.25, 2.5, and 5 mg/kg), morphine (10, 15, and 20 mg/kg) or oxycodone (1.25, 2.5, 5, and 10 mg/kg) dose-dependently reversed mechanical allodynia in the ipsilateral paw reaching a maximal effect of 91.2 ± 5.9%, 82.2 ± 11.1% and 74.9 ± 7.5% respectively (Fig. 4E and Supporting Information Fig. S4). WLB-73502 was 8- and 3-fold more potent than morphine and oxycodone, respectively (P < 0.05) (Table 1). It is worth noting that morphine and oxycodone required higher doses to exert antinociception in the SNI-induced neuropathic pain model than in the other pain models, whereas WLB-73502 showed a similar potency in all of them (Table 1). Mechanical allodynia did not develop in the contralateral paw and treatments did not exert significant effects on the contralateral paw (Fig. S4).
3.3. Brain penetration, receptor occupancy and PK/PD relationship after single administration
3.3.1. Plasma and brain pharmacokinetics
Drug levels were analysed by LC–MS/MS in plasma and brain samples following single i.p. administration of WLB-73502 at 3 mg/kg (a dose that produces 45%–85% antinociceptive effect, depending on the test) in male Wistar rats. Standard PK parameters were determined by non-compartmental analysis of the concentration–time curves (Table 2). Plasma Cmax following i.p. administration was reached very quickly (Tmax = 5 min; first sampling time) and the compound was rapidly eliminated (t1/2 = 20 min). The compound crossed the blood–brain barrier (BBB). Indeed, brain kinetics was parallel to plasma kinetics, with high brain penetration: both the peak (Cmax) and extent (AUC) of WLB-73502 exposure were higher in brain than in plasma (brain-to-plasma partition coefficient = 2.6).
| Compound | Matrix | t1/2(h) | Tmax(h) | Cmaxa | AUC0–∞a | Brain-to-plasma ratio (AUC brain/AUC plasma) |
|---|---|---|---|---|---|---|
| WLB-73502 | Plasma | 0.30 | 0.08 | 441 ± 73 (ng/mL) | 136 ± 15 (ng·h/mL) | 2.57 |
| Brain | 0.34 | 0.08 | 982 ± 191 (ng/g) | 350 ± 40 (ng·h/g) |
PK parameters of WLB-73502 in plasma and brain following i.p. administration of 3 mg/kg to adult male Wistar rats.
Data are mean ± SEM; n = 6 (8 sampling points, 6 animals per sampling point).
3.3.2. Brain receptor occupancy
The occupancy of MOR and S1R following i.p. drug administration was studied by ex vivo autoradiography in brain sections from male Wistar rat (Supporting Information Fig. S5 and Supporting Information Table S2).
Robust binding of [3H]-DAMGO (MOR radioligand) and [3H]-(+)-pentazocine (S1R radioligand) on brain sections was found in cortex and striatum of vehicle-treated rats, according to the reported distribution of MOR39 and S1R40 in rat brain (Fig. S5). Both cortex and striatum exhibited high levels of specific binding, as defined by incubations in presence of 50 μmol/L naloxone and 10 μmol/L haloperidol.
Administration of WLB-73502 at 3 mg/kg i.p. significantly inhibited [3H]-DAMGO specific binding in rat brain cortex (51% occupancy) and striatum (50% occupancy) 15 min after dosing when compared to vehicle-treated controls. Similarly, WLB-73502 at 3 mg/kg i.p. significantly inhibited [3H]-(+)-pentazocine specific binding in cortex (57% occupancy) and striatum (63% occupancy) 15 min after dosing when compared to vehicle-treated controls (Table S2).
A significant inhibition of [3H]-DAMGO specific binding by morphine at 10 mg/kg i.p. was observed in rat cortex (51% occupancy) and striatum (52% occupancy) 30 min after dosing when compared to vehicle-treated controls. Oxycodone at 3 mg/kg i.p. also significantly inhibited [3H]-DAMGO specific binding in rat cortex (40% occupancy) and striatum (44% occupancy) when compared to vehicle-treated controls. In contrast, neither morphine nor oxycodone had a significant effect on [3H]-(+)-pentazocine specific binding in rat cortex and striatum (Table S2).
3.3.3. PK/PD relationship
A preliminary PK/PD relationship was established from the brain exposure and the antinociceptive response at a single point following single i.p. administration of WLB-73502 at 3 mg/kg, the dose and route of administration used in brain receptor occupancy studies.
Antinociceptive efficacy, calculated from the dose–response curves (Fig. 4) in the different models 20 min after administration of WLB-73502 at 3 mg/kg i.p., corresponds to 45%–85% (average 75%), depending on the test. Experimental processing for receptor occupancy was initiated 15 min after compound administration at 3 mg/kg i.p. and occupancies were 50%–51% for MOR and 57%–63% for S1R, depending on the brain region (Table S2). Finally, experimental brain concentrations quantified at 15 and 30 min after i.p. administration of WLB-73502 at 3 mg/kg (PK parameters summarized in Table 2) were 127.6–236.5 ng/g, that correspond to a brain concentration of 362.0–671.1 nmol/kg. Considering the brain tissue binding of WLB-73502 in the rat (49.9%)41, the free, unbound fraction corresponds to 181.4–336.2 nmol/kg. As a reference, the binding to human MOR and S1R is 64 and 118 nmol/L, respectively28. Table 3 summarizes the above in vitropharmacological, ex vivo autoradiography and in vivo PK and PD data. There was a consistency between brain exposure, MOR and S1R affinities, brain MOR and S1R occupancy, and antinociceptive efficacy, although efficacy was a bit higher than expected based on a direct translation of receptor occupancies.
| Antinociceptive efficacy (range in the different pain model) | Brain receptor occupancy (cortex-striatum) | Total brain concentration (30–15 min) | Unbound brain concentration (30–15 min) | In vitrobinding affinity (Ki) |
|---|---|---|---|---|
| 45%–85% | 50%–51% (MOR) | 362.0–671.1 nmol/kg | 181.4–336.2 nmol/kg | 64 nmol/L (MOR) |
| 57%–63% (S1R) | 118 nmol/L (S1R) |
PK/PD relationship after single i.p. administration of WLB-73502 at 3 mg/kg to male Wistar rats.
Antinociceptive efficacy obtained from dose–responses curves in the different pain models (paw pressure, tail-flick, and carrageenan-, MIA- and SNI-induced pain) 20 min after compound administration. Experimental processing for brain MOR and S1R occupancy was initiated 15 min after compound administration. Brain concentrations obtained from experimental quantifications at 15 and 30 min after compound administration. Unbound, free fraction was calculated subtracting the bound fraction (rat brain tissue binding: 49.9%)41. Binding affinities (Ki) in vitro correspond to human MOR and S1R28. n = 7 (paw pressure and tail flick), n = 9–10 (carrageenan and MIA) and n = 8 (SNI) for antinociceptive efficacy; n = 5 for receptor occupancy; and n = 6 for brain concentration studies.
3.4. Tolerance to the antinociceptive effect
3.4.1. MIA-induced OA model (pharmacodynamics)
WLB-73502 (0.5, 1.5, and 3 mg/kg), oxycodone (2.5 mg/kg) or morphine (5 mg/kg) were administered during 28 (WLB-73502) or 21 days (oxycodone and morphine) by s.c. instead of i.p. route to diminish first-pass liver metabolism and b.i.d. to favour around-the-clock drug exposure. Drug treatments started 14 days after intraarticular MIA injection (when mechanical allodynia had fully developed) and the effect on MIA-induced mechanical allodynia was evaluated every 3–4 days, 30 min after daily administrations. Systemic b.i.d. administration of WLB-73502 induced a dose-dependent reduction of MIA-induced mechanical hypersensitivity in the ipsilateral paw. The effect of WLB-73502 remained unchanged throughout the treatment period, with a tendency to increase efficacy overtime (significant by the end of treatment when compared with Day 1 at 0.5 and 1.5 mg/kg doses exerting submaximal effects), thus suggesting absence of tolerance to its analgesic effect after 4 weeks of repeated administration (Fig. 5A and Supporting Information Fig. S6). Oxycodone (2.5 mg/kg) and morphine (5 mg/kg) also reduced mechanical hypersensitivity, but their antinociceptive effect progressively diminished throughout the treatment period (i.e., pharmacodynamic tolerance developed) (P < 0.05), the effect being almost completely lost after 3 weeks of treatment (Fig. 5A and Fig. S6). Mechanical allodynia did not develop in the contralateral paw and treatments did not exert significant effects on the contralateral paw. No effect was observed either before daily drug administration (pre-treatment) or following vehicle-treatment, supporting that mechanical allodynia remained stable throughout the treatment period and that effects were indeed attributable to drug treatments (Fig. S6).
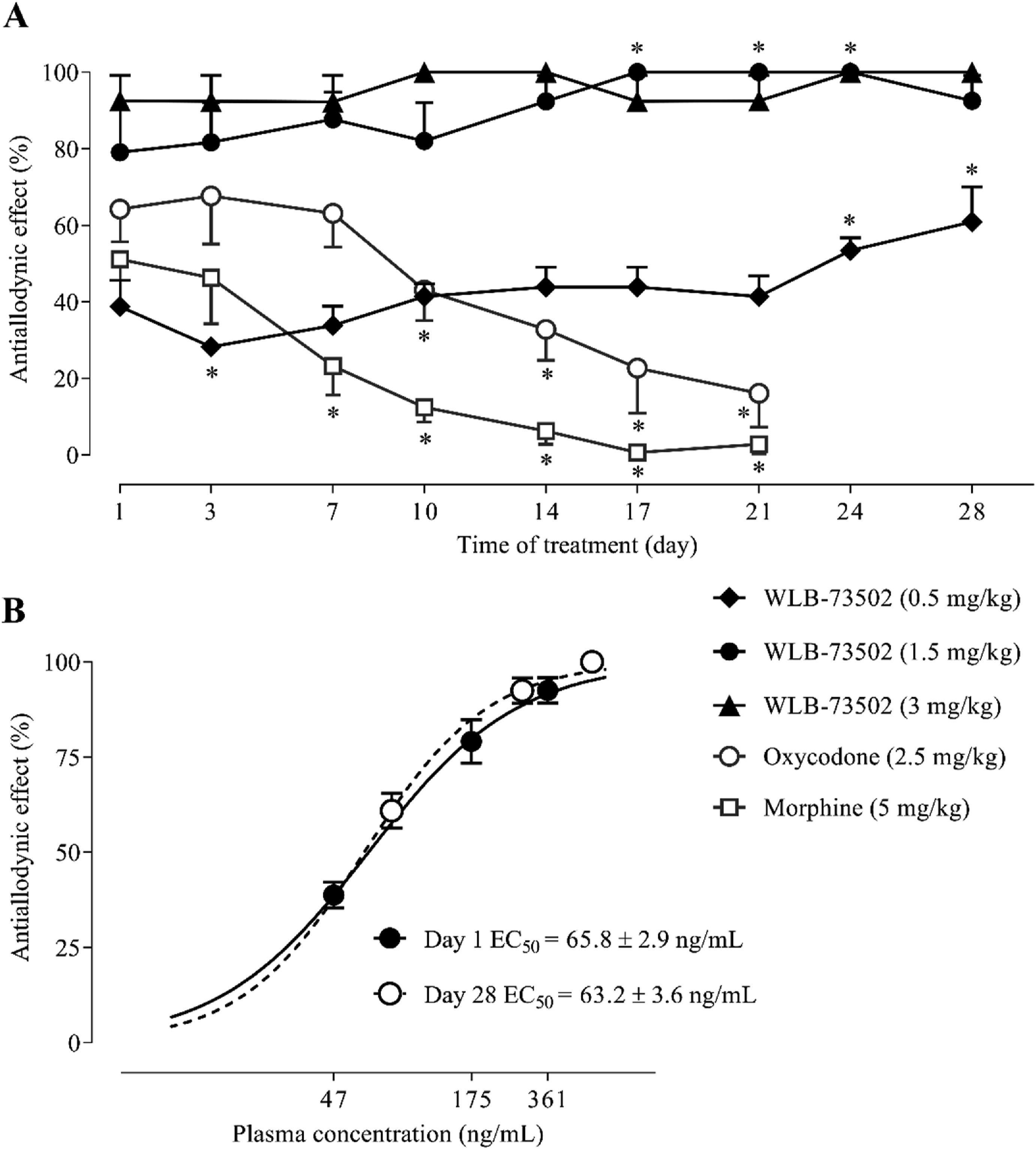
Antiallodynic effect on MIA-induced mechanical allodynia following subchronic administration. Time–course effect of WLB-73502, morphine or oxycodone on mechanical allodynia after repeated b.i.d. s.c. administration starting 14 days after intraarticular MIA injection in male Wistar rats (A). Plasma concentration–response curves on Day 1 (single administration) and after 28 days of b.i.d. s.c. administration of WLB-73502 (B). Points and vertical lines represent the mean ± SEM. n = 7–9 (A) and n = 5 (B, from animals used in A) per treatment group. ∗P < 0.05 vs. corresponding Day 1 (one-way ANOVA followed by Bonferroni’s post hoc test).
.4.2. MIA-induced OA model (pharmacokinetics)
Plasma samples from rats treated with WLB-73502 were obtained on Days 1 (first day of administration) and 28 (last day of administration) immediately after the PD evaluation (i.e., 35–40 min after drug administration) in the MIA-induced model of OA knee pain (same animals used in the PD study) to explore the possibility that progressive drug accumulation could mask the development of tolerance. The mean plasma concentration of WLB-73502 was 361 ± 24.7 ng/mL on Day 1 and 550.6 ± 33.8 ng/mL on Day 28 when the compound was administered at 3 mg/kg. When administered at the dose of 0.5 mg/kg the values ranged 47.3 ± 3.8 ng/mL on Day 1– 82 ± 3.2 ng/mL on Day 28; and 174.9 ± 7.4 ng/mL on Day 1–283.7 ± 10.4 ng/mL on Day 28 when administered at 1.5 mg/kg (Supporting Information Table S3). Accordingly, there was drug accumulation (factor 1.5–1.7) following repeated b.i.d. s.c. treatment for 28 days based on the comparison of levels attained at a single point after PD evaluation the first and last day of administration. Efficacy on MIA-induced mechanical allodynia was also superior on Day 28 compared to Day 1 (factor 1.2–1.6), particularly at the lower doses (0.5 and 1.5 mg/kg) (P < 0.05) exerting submaximal effects (Fig. 5A and Fig. S6). To find out if tolerance really did develop, plasma concentration–effect curves on Day 1 and Day 28 were done and mean concentrations providing half of maximum analgesia (EC50s) calculated (Fig. 5B). There were no differences between Days 1 and 28 (Day 1 EC50 = 65.8 ± 3 ng/mL; Day 28 EC50 = 63.2 ± 4 ng/mL), which demonstrated that efficacy of WLB-73502 was maintained over time, with neither increase nor decrease, and that the absence of pharmacodynamic tolerance was not due to a pharmacokinetic effect.
3.5. Gastrointestinal transit (constipation)
The effect of WLB-73502 (2.5, 5, 10, and 20 mg/kg) and oxycodone (2.5, 5, and 10 mg/kg) on gastrointestinal motility was evaluated in male Wistar rats. Drug was i.p. administered 30 min before p.o. administration of the charcoal suspension, and passage of charcoal through the intestine was measured 30 min later. Treatments induced a significant dose-dependent intestinal transit inhibition as shown by a reduced percentage of the distance travelled by the charcoal meal vs. the total length of the small intestine (Fig. 6A). WLB-73502 did not significantly inhibit the intestinal transit at 2.5 and 5 mg/kg, and induced partial intestinal transit inhibition at 10 mg/kg (19%) and 20 mg/kg (59%). Oxycodone had no effect at 2.5 mg/kg but it reduced intestinal transit by 72% at 5 mg/kg and produced close to full blockade of intestinal transit (93% of inhibition) at 10 mg/kg. It is worth noting that, contrary to oxycodone, WLB-73502 elicited remarkable/maximal antinociceptive effect at doses devoid of significant effect on intestinal transit (Fig. 6A).
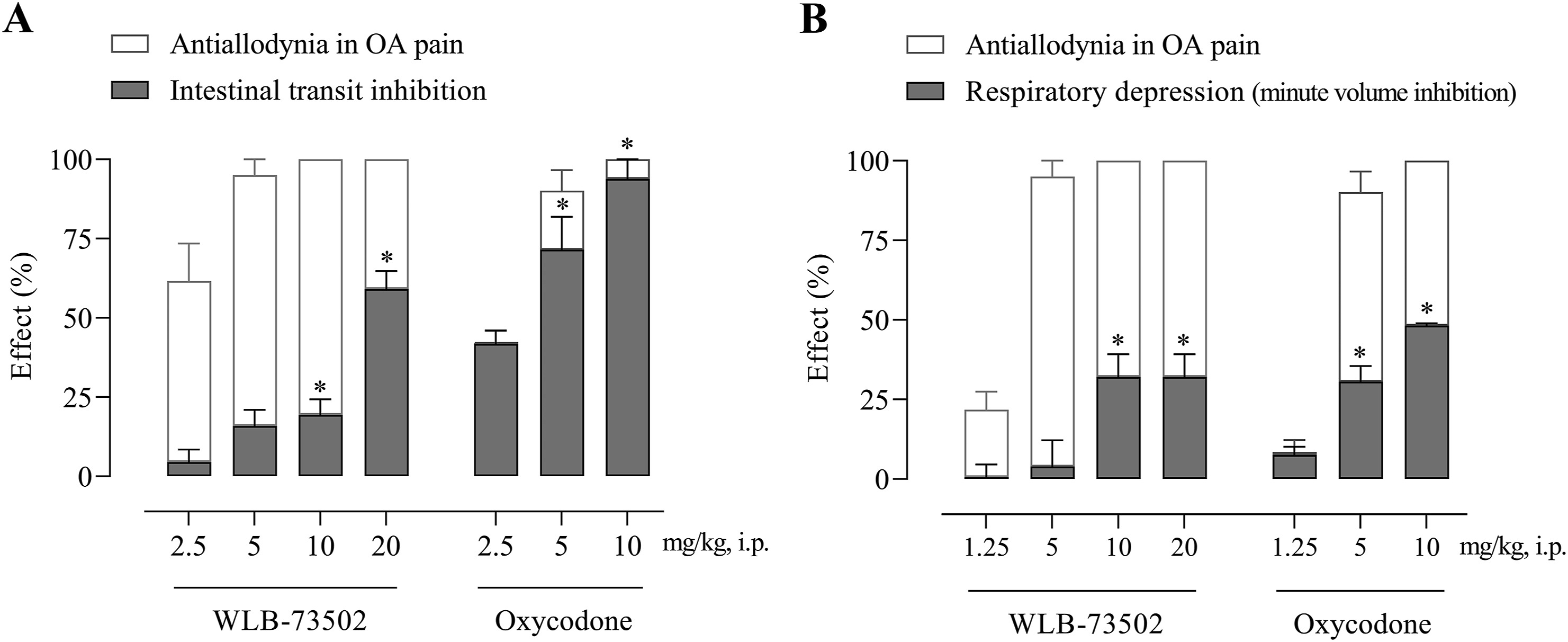
Dose–response effect of single administration of WLB-73502 and oxycodone on intestinal transit measured in the charcoal test (A) and respiratory function measured as minute volume (B) in male Wistar rats. Values in grey bars are mean ± SEM percentage of effect (A, intestinal transit inhibition; B, respiratory depression) vs. vehicle. For comparison, the percentage of antiallodynic effect on OA pain (white bars) achieved at the same doses is also shown. n = 8 per treatment group. ∗P < 0.05 vs. vehicle (one-way ANOVA followed by Bonferroni’s post hoc test).
3.6. Whole body plethysmography (respiratory depression)
The effect of WLB-73502 (1.25, 5, 10, and 20 mg/kg) and oxycodone (1.25, 5, and 10 mg/kg) on respiratory function in male Wistar rats was evaluated by whole-body plethysmography. Basal respiratory parameters were recorded continuously for 15 min and the effect of drugs was then evaluated along 30 min, starting immediately after i.p. compound administration when animals returned to the plethysmography chambers. WLB-73502 did not significantly change MV when administered in the analgesic dose range (1.25 and 5 mg/kg), but it reduced MV at 10 and 20 mg/kg. The inhibitory effect reached a plateau of ∼30% reduction, with no differences between the 10 and 20 mg/kg doses. Oxycodone did not significantly modify MV when administered at 1.25 mg/kg, but it inhibited MV by 30% and 50% when administered at 5 and 10 mg/kg, respectively. Note that, contrary to oxycodone, WLB-73502 elicited antinociceptive effect at doses devoid of significant effect on respiration (Fig. 6B).
3.7. Emesis (nausea and vomiting)
3.7.1. Pharmacodynamics
The study was done in ferrets as rats lack the emetic reflex and predictive value of pica behaviour in rodents remains questionable42. Male ferrets received a single i.p. administration of WLB-73502 (0.5 and 1 mg/kg, i.p.) or morphine (0.5 mg/kg, s.c.) and were then observed continuously for 4 h for emesis and nausea-like behaviours quantification. Emetic responses (retching and vomiting) and other typical nausea-like-related behaviours were summed up to get a nausea-like global score (Fig. 7). Control animals administered with vehicle did not show any emetic response, neither retching nor vomiting episodes, and the nausea-like global score was ∼2. WLB-73502 did not induce any emetic response at 0.5 mg/kg or 1 mg/kg i.p., and scored ∼2 and ∼4, respectively. In contrast, morphine (0.5 mg/kg, s.c.) induced emetic responses during the 4 h period after its administration (2.17 ± 0.60 retches and 1.17 ± 0.65 vomits) and scored ∼12 (Fig. 7).
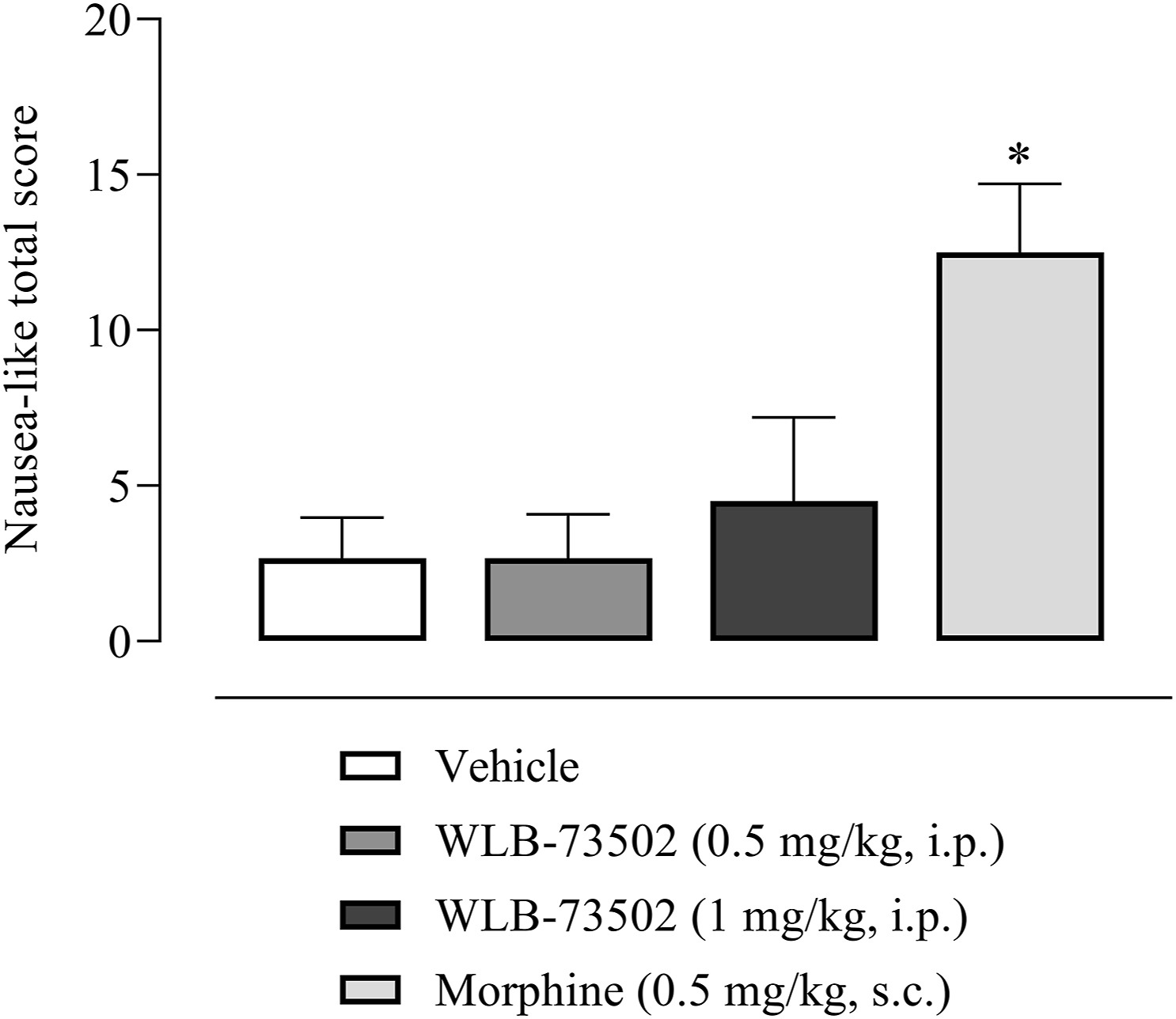
Effect of single administration of WLB-73502 or morphine on emetic response in male ferrets. Values in bars are mean ± SEM. nausea-like total scores (sum of retching, vomiting, and other nausea-related behaviours including licking, gagging, chewing, backward walking, head burying in cage shavings, wet dog shake, mouth clawing and prolonged typical ventral recumbency) recorded for 4 h following compound administration. n = 6 per treatment group. ∗P < 0.05 vs. vehicle group (one-way ANOVA followed by Bonferroni’s post hoc test).
3.7.2. Pharmacokinetics
We estimated the PK profile of WLB-73502 in plasma samples from male ferrets to verify if tested doses (0.5 and 1 mg/kg, i.p) provided the plasma exposure needed to elicit analgesia in rat pain models. Plasma exposure after single i.p. administration of WLB-73502 was higher in ferrets (Table S4) than in rats (Table 3). Both Cmax(620 ± 98 ng/mL) and AUC0–∞ (2463 ± 625 ng·h/mL) in ferrets at 1 mg/kg exceeded by far values obtained in rats administered with the active 3 mg/kg dose (Cmax = 441 ± 73 ng/mL, AUC0–∞ = 136 ± 15 ng·h/mL), suggesting that analgesia by WLB-73502 would be achievable in ferrets without accompanying emesis.
4. Discussion
The current study investigated the intrinsic activity on MOR and S1R, brain exposure, target engagement and ultimately the benefit-risk (efficacy-safety) balance of WLB-73502 in comparison with morphine and oxycodone.
WLB-73502 binds to human MOR (Ki = 64 nmol/L) and S1R (Ki = 118 nmol/L) but it is selective against a panel of more than 180 targets including receptors, transporters, ion channels and enzymes28. The activation of MOR induces distinct intracellular signalling pathways that can be differentially stimulated, including i) activation of the Gαi subunit, which inhibits adenylyl cyclase and reduces cAMP intracellular levels; and ii) recruitment of β-arrestins, scaffolding proteins that hinder the G-protein coupling of agonist-activated GPCRs, ultimately resulting in receptor desensitization43. Biased opioid agonists showing a preference for activating the G-protein versus the β-arrestin pathway (i.e., G-protein biased MOR agonists) were proposed to be safer opioid analgesics11,44. WLB-73502 behaved as a partial agonist at the MOR G-protein pathway and was inactive in the β-arrestin-2 signalling pathway. This is in contrast with conventional opioids such as morphine, oxycodone or fentanyl, that behaved as full agonists at the MOR G-protein pathway and recruited β-arrestin signalling to some extent. Responses and operational τvalues were lower for all MOR agonists in β-arrestin than in cAMP determinations. Indeed, reduced intrinsic efficacy, ceiling effects and distortions by analysis of amplified (G-protein) versus linear (arrestin) signalling mechanisms may lead to bias overestimation, the weaker the G-protein partial agonism is the greater the apparent bias6,45,46. Despite these constrains, biased agonism favouring G protein signalling was supported for some MOR partial agonists47. Is WLB-73502 a G-protein-biased MOR agonist? Bias is suggested, but it can neither be established nor ruled out based on the inactivity of WLB-73502 in the β-arrestin recruitment assay. Independent on bias, WLB-73502 had lower intrinsic efficacy than oxycodone, morphine and fentanyl at both G-protein and β-arrestin pathways. This confirms its partial MOR agonist nature, which together with its bispecific ability to block S1R may explain its safety superiority.
The antagonistic activity on S1R is a key differential feature of WLB-73502. The S1R, a non-opioid receptor, was first identified as a tonically active antiopioid system by Chien and Pasternak48, who reported that S1R agonists lower and S1R antagonists enhance opioid analgesia22,48. They also first noticed that S1R functionally regulate opioid analgesia without affecting other opioid-related effects, particularly morphine’s effects on gastrointestinal transit or lethality22. Now, S1R antagonism is recognized as a well-grounded opioid adjuvant strategy based on numerous studies reporting antinociceptive enhancement when combining multiple MOR and selective S1R ligands25,49, and when using opioids in S1R knockout mice50,51. Clinical data also support S1R-mediated modulation of opioid analgesia. Postoperative pain intensity was reported to be reduced when combining a selective S1R antagonist with morphine respect to morphine alone52. Patients receiving add-on administration of haloperidol (a potent, albeit non-selective S1R antagonist) also required less opioid administration and rescue therapy to treat abdominal pain than patients treated only with opioids53. Supraspinal and peripheral, but not spinal S1R blockade enhanced opioid antinociception25,51,54. At the molecular level, S1R physically associates with MOR and this interaction allows S1R ligands to modulate opioid transduction without influencing MOR binding55,56. Indeed, S1R is an integral membrane chaperone protein that normally reside at the mitochondrion-associated endoplasmic reticulum membrane, but when cells are stimulated or undergo stress it translocates to the endoplasmic reticulum reticular network and plasma membrane to regulate a variety of receptors and ion channels57,58. S1R activation increased the interaction of S1R with GPCRs at the cell surface and inhibit GPCR signalling59. Accordingly, S1R ligands work as allosteric modulators of MOR function, S1R antagonists blocking tonic inhibition exerted by S1R in the heteromeric complex56.
The antinociceptive activity of WLB-73502 in comparison with morphine and oxycodone was explored in several rat pain models. The increase of mechanical (paw pressure) and thermal (tail-flick) nociceptive thresholds is characteristic of opioids. Dose–response curves demonstrated that WLB-73502 had similar potency to oxycodone and near 4-fold more potency than morphine in reducing both thermal and mechanical nociception. This is not surprising, despite its partial MOR agonist activity, taking into consideration its bivalent nature. S1R antagonists lack activity in acute nociception when administered alone but enhanced opioid-induced thermal23 and mechanical50 antinociception. The antinociceptive effect of WLB-73502 in the paw pressure test in mice was partially reverted by the co-administration of the S1R agonist PRE-084, and blocked when co-administered with the MOR antagonist naloxone28, thus supporting the S1R-mediated enhancement of MOR analgesia.
Maximal efficacy after single administration was also achieved by WLB-73502 in reducing mechanical hypersensitivity in carrageenan-induced inflammatory, MIA-induced OA and SNI-induced neuropathic pain models. Potency was similar to oxycodone and 2-fold superior to morphine in inflammatory and OA pain, but superior to both oxycodone (3-fold) and morphine (8-fold) in the neuropathic pain model. Here, besides potentiation of its opioid activity, the contribution per se of the non-opioid S1R component of WLB-73502 needs to be considered. Unlike opioids, S1R antagonists do not modify the normal sensory mechanical and thermal perception thresholds but they exert antiallodynic and antihyperalgesic effects in sensitizing conditions (i.e., nerve injury), enabling the reversal of diminished nociceptive thresholds back to normal values25,60. Studies in S1R knockout mice61,62and using S1R antagonists62, 63, 64, 65, 66 have consistently demonstrated antiallodynic and antihyperalgesic effects. Not only behavioural but also electrophysiological (inhibition of spinal wind-up and nerve injury-induced enhanced axonal activity in peripheral recordings)66,67, neurochemical (increased noradrenaline but reduced glutamate release in the spinal dorsal horn)68 and molecular (downregulation of spinal NMDAR function)69 studies support a role for S1R antagonists in inhibiting augmented excitability secondary to sustained afferent nociceptive input. Thus, especially in partially opioid-refractory pain such as neuropathic pain70, where stand-alone opioids including morphine and oxycodone required higher doses to exert antinociception, the activity of WLB-73502 likely benefits more from its bivalent inhibitory activity on S1R.
Repeated administration of opioid analgesics results in pharmacodynamic tolerance. Tolerance to the antiallodynic effect of WLB-73502 did not develop in the OA pain model following repeated b.i.d. administration for 28 days at active doses (0.5–3 mg/kg). Time-dependent accumulation of WLB-73502 during repeated administration was discarded as plasma concentration–response curves revealed no differences in EC50 values between Day 1 and 28. In contrast to WLB-73502, tolerance developed with opioid comparators (morphine and oxycodone), their antiallodynic effect almost disappearing after dosing for 21 days. Tolerance has been reported both for full and partial (and biased) agonists such as buprenorphine71; and it was described to develop, or not, or depending on MOR numbers in the case of partial, biased MOR agonist such as SR-17018, PZM21 or TRV-13072, 73, 74, 75. Knockout mice lacking β-arrestin-2 failed to develop antinociceptive tolerance after chronic morphine treatment in one first study76, but morphine tolerance was reported to develop, with a delayed onset and to a lesser degree than in wild-type mice in a second study77. The low intrinsic activity at MOR including undetected β-arrestin recruitment by WLB-73502 could thus explain why tolerance did not develop. However, its antagonistic activity at S1R probably offers a more plausible explanation. Repeated administration of S1R antagonists in combination with opioids disrupt or delay opioid tolerance in numerous pain models, including neuropathic78, inflammatory27 and OA79 pain. Moreover, when administered to morphine-tolerant mice, S1R antagonists rescued opioid analgesia23,27,80. Among mechanisms underlying opioid tolerance, the involvement of N-methyl-ᴅ-aspartate receptors (NMDAR) and modulation of the crosstalk between MOR and NMDAR (i.e., anti-opioid glutamate/NMDAR system) seem the most plausible mechanisms for a S1R antagonist to inhibit opioid tolerance81, 82, 83. Repeated opioid treatment activates pain facilitatory pathways through specific phosphorylation of NMDAR that limit opioid analgesia83, 84, 85. In turn, activated NMDAR stimulates kinase cascades that phosphorylate MOR and disrupt its coupling to G-proteins, which also limits opioid analgesia during tolerance development83,85. Indeed, tolerance to opioid-analgesia is prevented by inhibiting NMDAR82,86, and that is what S1R antagonists indirectly do. S1R binds to the C-terminal part of NR1 subunit of NMDAR at a site precluding the binding of calcium–calmodulin, the negative regulator of NMDAR function. When a S1R antagonist binds to S1R, the affinity of S1R for the NR1 subunit of NMDAR diminishes, S1R detaches and allows the entrance of calcium–calmodulin, that inhibit NMDAR, this reducing excitatory nociceptive transmission and the detrimental signalling from NMDAR to MOR80.
Opioid-induced side effects may be dose-limiting, thus affecting pain control, and reduce patients’ quality of life. Constipation is one of the most common (approximately 40% of opioid users) debilitating side effects of opioids87,88. Nausea and vomiting are also common opioid-induced gastrointestinal adverse effects (40% of patients may experience nausea and 15%–25% vomiting), highly distressing and often leading to poor adherence to opioid therapy89. Respiratory depression is also common but often under-diagnosed in patients receiving opioid analgesia and it is regarded as the main hazard of opioid use by clinicians90. It results in excessive sedation and respiratory impairment only in 0.5% or less of the cases, but fatalities are regularly reported91. WLB-73502 did not induce gastrointestinal transit inhibition or respiratory depression at doses exerting full antinociceptive effect in rats (5 mg/kg). At higher doses it induced partial intestinal transit inhibition and its effect on respiratory depression reached a plateau of ∼30% reduction, with no differences between the 10 and 20 mg/kg doses. In contrast, oxycodone induced remarkable/maximal inhibitory effects on intestinal transit and lung ventilation at doses overlapping those required for analgesia. Similarly, and contrary to morphine, WLB-73502 did not induce emetic response (retching or vomiting) in ferrets at doses that largely exceed the exposure needed to achieve analgesia in rats (i.e., AUC was 18-fold higher in ferrets dosed at 1 mg/kg for the emesis study than in rats receiving 3 mg/kg, a dose active in antinociceptive studies), suggesting that analgesia by WLB-73502 would be achievable also in ferrets without accompanying emesis. In the case of morphine, plasma exposure known to be associated with antinociception in rats92 exceeds the exposure attained in ferrets93 dosed similarly to this study, supporting overlapping of the proemetic and analgesic effects of morphine. Reduced respiratory depression, constipation, vomiting and need for rescue antiemetics, and reinforcing activity were reported for biased MOR agonists44,94,95. Favourable oliceridine safety profile over morphine when considering emesis96 and respiratory depression97 was also reported in human. But other studies applying genetic98,99, pharmacological72 or mixed100,101 approaches reported severe adverse effects associated with G-protein biased MOR agonists. Thus, it is unclear if no/reduced efficacy for β-arrestin-2 recruitment by WLB-73502 accounts for its improved side effect profile. Its low intrinsic activity at MOR together with its antagonistic activity at S1R may also explain its improved safety. S1R blocking is known to inhibit opioid-induced intestinal transit inhibition, sedation, mydriasis (mice) or lethality22,23,51,78, and reduced respiratory depression compared to fentanyl at equianalgesic dose was reported with a bifunctional S1R/MOR piperidinamide derivative102. S1R has also been involved in the control of emesis: agonists enhance whereas S1R antagonists inhibit emetic response103. Interestingly, a reduction of pain and opioid-related side effects, including nausea, vomiting and need for concomitant antiemetic medication was found in the clinic, when comparing the combination of a selective S1R antagonist S1RA (E-52862; MR309) plus morphine with morphine alone in the acute postoperative setting52.
Addiction is a major drawback of opioids. Apparently, analgesia, rewarding and dependence come together as they all depend upon actions at MOR mainly through the G-protein/cAMP second messenger pathway104. The role played by the β-arrestin pathway here, if any, is unclear13,72,73,105,106. We did not evaluate rewarding, tolerance to reward or dependence in this study. WLB-73502 has low intrinsic efficacy at MOR, which would be potentially associated with reduced abuse potential respect to full MOR agonists. Its bivalent S1R antagonism could also potentially contribute due to the opioid-sparing effect (i.e., reduction of opioid load while maintaining efficacy). The reduced effect on respiratory depression would potentially reduce the risk for overdose. As it regards to dependence, we previously published that, contrary to oxycodone, naloxone-precipitated withdrawal signs were not detected following repeated b.i.d. administration of WLB-73502 for 10 days in mice28. Finally, S1R antagonists have been proposed as medications for drug abuse as they inhibit rewarding by opioids23 and other abused substances including cocaine, methamphetamine, ethanol, and nicotine107, 108, 109. Opioid addiction is a truly sensitive and delicate matter. Further studies are warranted.
Target engagement supports activity findings. When administered systemically (i.p.), WLB-73502 crossed the BBB (higher exposure in brain than in plasma) and, according to its bivalent nature, bound to both brain MOR and S1R. Preliminary PK/PD analysis revealed a consistency between brain exposure, MOR and S1R affinities, brain MOR and S1R occupancy, and antinociceptive efficacy. Efficacy in vivo at 3 mg/kg (average 75%) was a bit higher than expected based on a direct translation of receptor occupancies obtained by ex vivo autoradiography (average 50 and 60% for MOR and S1R, respectively). Non-exact linear correspondence (i.e., slope ≠ 1) between the cause (binding to receptors) and the effect (antinociceptive response) is not surprising, particularly if a superior effect is expected when both S1R and MOR are recruited.
5. Conclusions
WLB-73502 is a bispecific S1R antagonist and MOR partial agonist with analgesia comparable (nociceptive and mixed inflammatory and OA pain) or superior (neuropathic pain) to full MOR agonists, but it does not induce tolerance and causes no/less constipation, respiratory depression, and nausea/vomiting than strong opioids. WLB-73502 benefits from its partial MOR agonist nature (low intrinsic efficacy at Gi/o-protein activation and undetectable β-arrestin-2 recruitment) and its bivalent S1R antagonist activity, responsible for opioid-dependent and -independent effects, to increase its therapeutic index. This makes WLB-73502 a promising alternative for treating chronic refractory pain, potentially neuropathic cancer pain, where regular stand-alone opioids do not achieve satisfactory outcomes and are limited by drug tolerance and adverse effects110. In agreement with findings with WLB-73502, other bifunctional S1R antagonist/MOR agonist derivatives including piperidinamides102, benzylpiperazines111 and 4-aryl-1-oxa-4,9-diazaspiro[5.5]undecanes112 also produced fewer opioid-like side effects, thus highlighting dual S1R antagonism/MOR agonism as a hopeful avenue for the development of potent and safer analgesics113.