
An open-like conformation of the sigma-1 receptor reveals its ligand entry pathway
By Meng, F., Xiao, Y., Ji, Y. et al.
Excerpt from the article published in Nat Commun 13, 1267 (2022). https://doi.org/10.1038/s41467-022-28946-w
Editor’s Highlights
- Data suggest that access to the σ1R ligand binding site is likely achieved by protein conformational changes that involve the carboxy-terminal two-helix bundle, rather than structural changes in the cupin-fold domain.
- The PATH2 access hypothesis infers that the σ1R ligand enters from, and exits to, the membrane rather than an aqueous environment.
- Since σ1R is primarily localized within cells, it has been suggested that a synthetic ligand would need to partition into and pass through, at least the plasma membrane. Drug delivery studies also show that hydrophobic molecules do partition and enrich into the cellular membranes. Therefore, a membrane pathway would not be a prohibitory factor for ligand entry and exit in σ1R.
Abstract
The sigma-1 receptor (σ1R) is a non-opioid transmembrane receptor which has been implicated in many diseases, including neurodegenerative disorders and cancer. After more than forty years of research, substantial progress has been made in understanding this unique receptor, yet the molecular mechanism of its ligand entry pathway remains uncertain. Published structures of human σ1R reveal its homotrimeric organization of a cupin-fold β-barrel body that contains the ligand binding site, a carboxy-terminal V-shaped two-helix bundle, and a single amino-terminal transmembrane helix, while simulation studies have suggested a ligand entry pathway that is generated by conformational rearrangements of the cupin-fold domain. Here, we present multiple crystal structures, including an open-like conformation, of σ1R from Xenopus laevis. Together with functional binding analysis our data suggest that access to the σ1R ligand binding site is likely achieved by protein conformational changes that involve the carboxy-terminal two-helix bundle, rather than structural changes in the cupin-fold domain.
Introduction
The sigma-1 receptor (σ1R) is a small, unique integral membrane receptor that is localized primarily in the endoplasmic reticulum (ER)1,2,3. σ1R responds to a structurally diverse array of synthetic ligands such as (+)-pentazocine (agonist) and haloperidol (antagonist)2. It interacts with various effector proteins1, including ion channels4,5,6 and G-protein coupled receptors1,7,8,9, and it is implicated in many diseases, including neurodegenerative disorders10,11,12 and cancer13,14. Identified in 197615 and cloned in 199616, its first atomic structure was solved in 201617. The structural information, together with information gathered from functional studies, has provided insight into the key mechanistic elements of σ1R’s function2,3,18. However, critical mechanistic details of how ligands access their binding site in σ1R remain enigmatic.
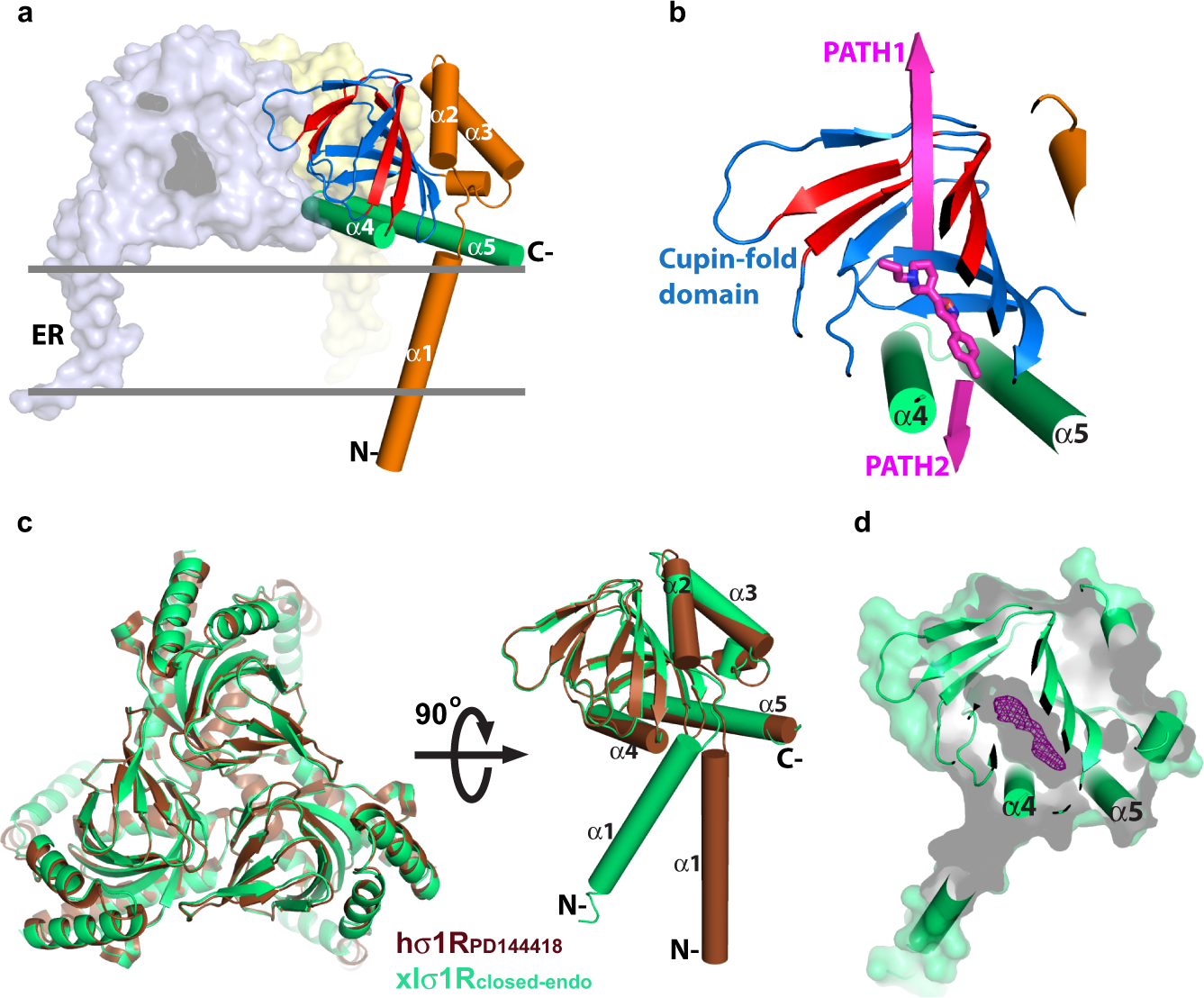
Recent crystal structures of human σ1R (hσ1R) show that hσ1R adopts a homotrimeric configuration, with each protomer comprising a single transmembrane helix (α1) at the amino-terminus, followed by a cupin-fold19 β-barrel body containing the ligand-binding site, and a membrane-adjacent V-shaped two-helix bundle (α4/α5) at its carboxy-terminus covering the cupin-fold domain like a lid17,20 (Fig. 1a). Currently, there are mainly two pathways proposed for ligand entry in σ1R17,20,21. The first proposed pathway (PATH1) would allow a ligand to enter σ1R through structural rearrangements of the cupin-fold domain, which involves unfolding and refolding of the β barrel; the second proposed pathway (PATH2) would allow a ligand to access its binding site in σ1R through the opening between α4 and α5 (Fig. 1b).
Recent molecular dynamics simulations support PATH1, as the simulations revealed two drastic conformational changes in hσ1R that break hydrogen bonds of multiple β sheets of the cupin-fold domain to expose the ligand-binding site20 (Fig. 1a). However, considering the energy expenditure required to disrupt the β barrel, and the entropic penalty incurred when exposing the largely hydrophobic interior of the cupin-fold domain to the aqueous environment, PATH1 seems rather energetically unfavorable. The results of another computational study involving steered molecular dynamics revealed that pulling a ligand (PD144418) out of the binding pocket of hσ1R required less initial force in PATH121. However, such a simulated conformational change of σ1R may not represent a physiological condition. Furthermore, the unfavorable energy expense would also accompany the pulling process in PATH1 when it breaks the β barrel.
To provide experimental evidence to assess ligand entry to σ1R we present multiple crystal structures of σ1R from Xenopus laevis (xlσ1R). Together with mutagenesis and binding analysis, the structural data support the notion that ligand entry to σ1R is achieved through PATH2.
Results
The closed conformation of xlσ1R
To better define the ligand entry pathway in σ1R, our initial strategy was to capture a σ1R structure in its open conformation. To achieve this goal, we screened expression for ten σ1R homologs from different species and crystallized well-behaved clones without adding any known ligand for structure determination. In the end, we solved a structure for the wild-type σ1R from Xenopus laevis (xlσ1R), which shares 67% sequence identity and 89% sequence homology with human σ1R (hσ1R) (Supplementary Fig. 1), to 3.20 Å, and termed it xlσ1Rclosed-endo.
The xlσ1Rclosed-endo structure was solved in the space group P21 with each asymmetric unit containing 12 protomers (Supplementary Fig. 2a), which deviate little from each other with all-atom RMSDs of only ~0.2 Å between the protomers. Like all reported structures for hσ1R, xlσ1R also crystallizes as a homotrimer in xlσ1Rclosed-endo, which resembles closely the hσ1R structures, especially the ones complexed with an antagonist such as PD144418 (hσ1RPD144418, PDB 5HK1), except the orientation of transmembrane helix α1 (Fig. 1c). Superposition of individual protomers of xlσ1Rclosed-endo and hσ1RPD144418 excluding α1 yielded an all-Cα RMSD of 0.35 Å, indicating a high degree of structural conservation among σ1Rs from different species (Fig. 1c). However, xlσ1Rclosed-endo failed to achieve our initial goal as it was captured in a closed conformation similarly to hσ1RPD144418 (Fig. 1d). Surprisingly, though no known ligand was added during purification or crystallization of xlσ1R, a clear electron density within the ligand-binding pocket indicates that xlσ1Rclosed-endo was bound by an unknown molecule (Fig. 1d and Supplementary Fig. 2b), which may have an endogenous origin. Unfortunately, the current resolution of xlσ1Rclosed-endo (3.20 Å) provides only limited details about the shape of the electron density (Fig. 1d and Supplementary Fig. 2b), making it impossible to reveal readily the molecular identity of this unknown molecule, and therefore the electron density was not modeled in xlσ1Rclosed-endo.
The crystal packing of xlσ1Rclosed-endo prohibits the PATH1 formation during ligand binding
In each asymmetric unit of xlσ1Rclosed-endo there are four homotrimers of xlσ1R, which are organized in a diamond shape with the cupin-fold domains packing against each other, whereas the transmembrane helices (α1s) mediate crystal contacts between adjacent asymmetric units (Supplementary Fig. 2a). In such a packing mode, each trimer has one protomer whose cupin-fold domain is tightly buried by two adjacent cupin-fold domains (Fig. 2a). For example, the tip region of the cupin-fold domain (Trp133–Tyr144) of Protomer C is in close contact with Protomer A (Thr106–Thr110) and Protomer D (Lys139–Tyr144) (Fig. 2b). As a result, the steric hindrance from Protomers A and D would prevent the tip region of Protomer C from moving away from the rest of its cupin-fold body, a conformational change that is required for the PATH1 formation in σ1R as suggested in previous simulations17,21. Therefore, one protomer in every trimer would not be able to open in this packing mode if PATH1 is the ligand entry pathway in σ1R. Then if a ligand was added to these packed crystals, it would not be able to access the ligand-binding site in one-third of the protomers.

o test this hypothesis experimentally, we soaked two known ligands of σ1R, PRE084 (agonist) or S1RA (antagonist), directly into the xlσ1Rclosed-endo crystals, and solved both structures, xlσ1Rclosed-PRE084 and xlσ1Rclosed-S1RA, respectively. Soaking of either ligand changed little on the xlσ1R crystal packing. Both xlσ1Rclosed-PRE084 and xlσ1Rclosed-S1RA are almost identical to xlσ1Rclosed-endo with all-atom RMSDs < 0.25 Å when aligning the three structures (Supplementary Fig. 2c). However, in both xlσ1Rclosed-PRE084 and xlσ1Rclosed-S1RA, each of the three protomers in the trimeric assembly features a ligand (PRE084 or S1RA) bound in the ligand-binding pocket (Fig. 2c and Supplementary Fig. 2d, e). This result demonstrates that soaking of the tightly packed crystals of xlσ1Rclosed-endo with the ligands led to the replacement of the unidentified molecule in each protomer, indicating that ligand access via proposed PATH1 is highly unlikely and that the conformational changes proposed for PATH2 seem more likely to facilitate ligand entry to each individual binding site.
An open-like conformation of xlσ1R that is consistent with PATH2
In our continued search for an open structure of xlσ1R, crystals with an appearance (cube-like) different from the xlσ1Rclosed-endo crystals (cuboid-like) were obtained and their structure was determined to 3.56 Å (xlσ1Ropen-endo). The xlσ1Ropen-endo structure was solved in the P212121 space group, with each asymmetric unit containing 12 protomers. These protomers are nearly identical to each other with all-atom RMSDs < 0.2 Å between the protomers. Similar to all σ1R structures, xlσ1Ropen-endo also assembles as homotrimers (Fig. 3a). Each asymmetric unit contains four trimers, which are packed in a tetrahedron shape through the α1–α1 contacts (Supplementary Fig. 3a).

Structural comparison between xlσ1Ropen-endo and xlσ1Rclosed-endo shows that the two structures are very similar, with the exception of two regions. One region consists of the transmembrane helix α1, which swings ~18° away from the trimer center in xlσ1Ropen-endocompared to xlσ1Rclosed-endo (Fig. 3a). Structural alignment between the two structures excluding the α1 region yielded an all-atom RMSD of 0.34 Å. This orientation difference of α1 may be due to crystal packing, as α1 in the hσ1R structures exhibits different orientations even between different protomers of the same structure17,20, e.g., hσ1RPD144418 (Fig. 3b). The other major difference lies in the carboxy-terminal two-helix bundle, α4/α5. In xlσ1Ropen-endo, the α4 helix rotates slightly away from α5, and the side chain of Tyr203 from α5 adopts a gauche+χ1 rotamer to point to the cupin-fold domain (Fig. 3c), thus creating an opening between α4 and α5 that generates a direct pathway for the ligand from the outside milieu to the ligand-binding site (Fig. 3d). Such an access pathway is reminiscent of that proposed in PATH2 (Fig. 1b). This opening spans an area of more than 10.3 Å × 5.5 Å (Fig. 3d), which is sufficiently large for the σ1R ligands such as PRE084 (~7.5 Å × 5.5 Å measured transversely) and S1RA (~7.6 Å × 5.2 Å transversely) to pass through. Therefore, the xlσ1Ropen-endo structure may represent an open-like conformation for σ1R and is consistent with PATH2 that predicts the ligand entry between α4 and α5.
Like xlσ1Rclosed-endo, the xlσ1Ropen-endo crystals were grown in the absence of any known ligand, but the xlσ1Ropen-endo structure also has an unidentifiable electron density within its ligand-binding pocket (Supplementary Fig. 3b). To investigate if the identified opening between α4 and α5 was induced randomly by the unknown molecule, or whether it is indeed relevant for ligand access to the xlσ1R binding site, we further determined two structures for the xlσ1R-PRE084 complex in the same crystallization condition as xlσ1Ropen-endo, by either co-crystallizing PRE084 with xlσ1R (xlσ1Ropen-PRE084-co, 3.10 Å) or soaking PRE084 directly into the xlσ1Ropen-endo crystals (xlσ1Ropen-PRE084-soak, 2.85 Å). As expected, both xlσ1Ropen-PRE084-co and xlσ1Ropen-PRE084-soak structures are nearly identical to xlσ1Ropen-endo(Supplementary Fig. 3c), and one PRE084 molecule binds in every protomer of the two structures (Supplementary Fig. 3d). Interestingly, we also observed some dynamics in the Tyr203 side chain in these two PRE084-containing structures, possibly due to their relatively high resolutions. In xlσ1Ropen-PRE084-co, some protomers have Tyr203 in a gauche+ χ1 rotamer as in xlσ1Ropen-endo, whereas others have Tyr203 in a trans χ1 rotamer as in xlσ1Rclosed-endo(Fig. 3e). In xlσ1Ropen-PRE084-soak, while most protomers have Tyr203 adopt a gauche+ χ1rotamer, a few protomers have Tyr203 in both gauche+ and trans χ1 rotamers with a partial occupancy for each (Fig. 3f). This result indicates that the σ1R ligand PRE084 can bind σ1R concurrently with an entrance formation between α4 and α5, consistent with the PATH2 hypothesis as a mechanism of ligand entry.
Blocking the entrance in PATH2 hinders ligand binding in σ1R
To functionally validate the PATH2 hypothesis of ligand entry to the σ1R binding site, we sought ways to either block the opening between α4 and α5 or prevent its formation, and assess whether either approach would impair ligand binding. The identified opening between α4 and α5 is surrounded by residues mainly from α4/α5, including Pro176, Leu179, Leu183 from α4 and Leu196, Val200, Tyr203, Leu207 from α5, as well as Leu97 from a loop region of the cupin-fold domain (Fig. 4a). These residues outline an ‘egg’ shape, with Leu183, Leu196, and Val200 at the narrower pointed end, Leu179 and Tyr203 in the middle, and Leu97, Pro176, and Leu207 at the wider rounded end (Fig. 4a). We reasoned that alterations of the middle residues (Leu179 and Tyr203) would probably have the most profound effect on the entrance formation. Therefore, we designed two experiments targeting Leu179 and Tyr 203 to obstruct PATH2-hypothesized ligand entry.
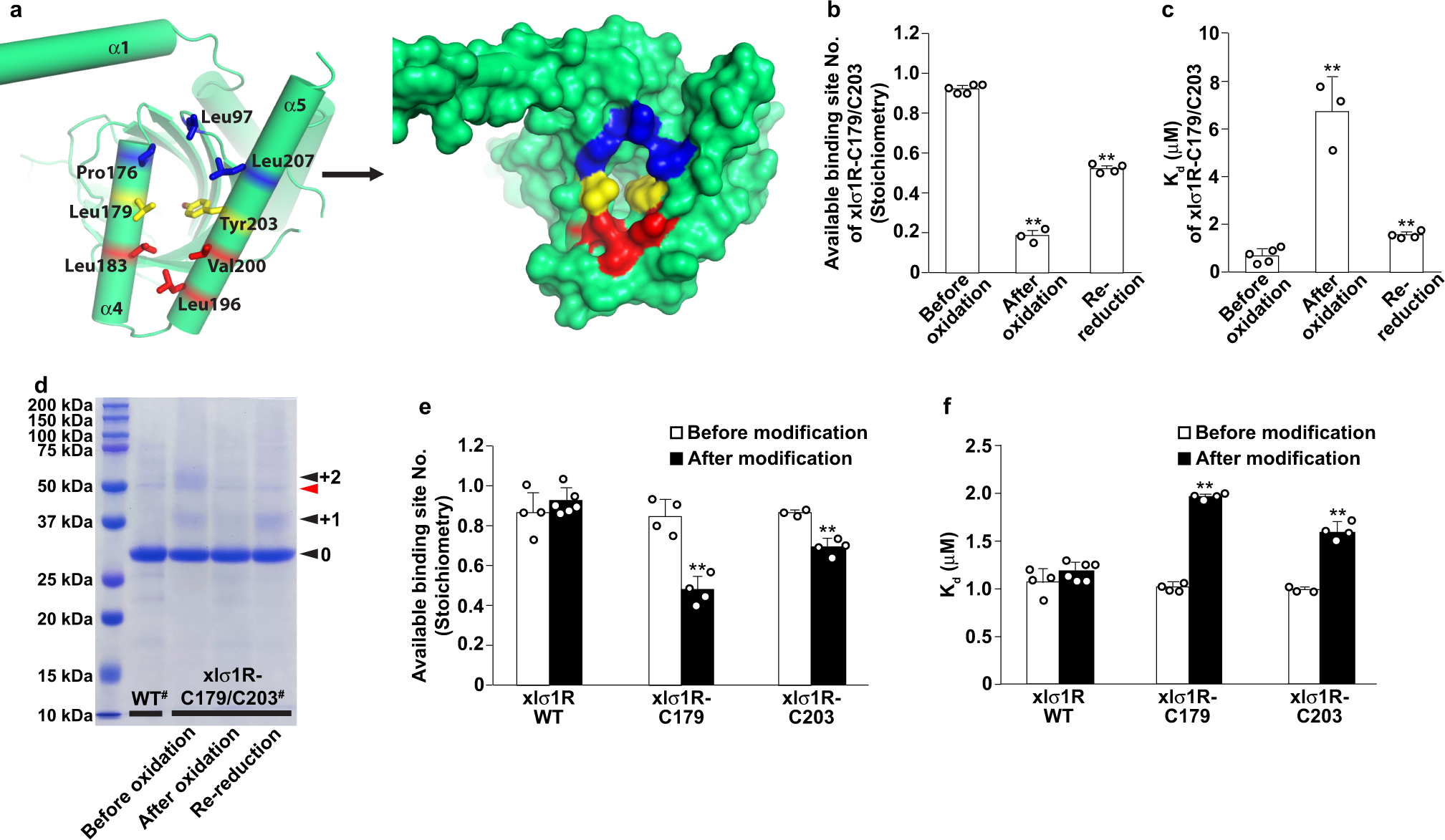
The first idea was to create a disulfide bridge between residues 179 and 203 to prevent the entrance from opening (Supplementary Fig. 4a), leading to a reduction in available binding sites. Therefore, only a fraction of the xlσ1R molecules would bind its ligand (e.g., PRE084), which can be evaluated by stoichiometry measurement using isothermal titration calorimetry (ITC). After introducing a pair of cysteine mutations at residue positions 179 and 203 (xlσ1R-C179/C203), the formation of the disulfide bond was catalyzed by oxidation. All ITC isotherms measured in this study were best fitted by a one-site model. As expected, the number of the available binding sites per protomer (stoichiometry) in xlσ1R-C179/C203 decreased to 0.18 ± 0.02 after oxidation compared to 0.92 ± 0.01 before oxidation (Fig. 4band Supplementary Fig. 4b, c), indicating an ~78% efficiency for the disulfide bond formation. Furthermore, the stoichiometry was partially reverted to 0.52 ± 0.02 by re-reducing the oxidized sample of xlσ1R-C179/C203 with β-mercaptoethanol to break the disulfide bond (Fig. 4b and Supplementary Fig. 4d). A partial recovery in the available binding site number after the re-reduction may result from the limited accessibility of β-mercaptoethanol to the disulfide bond between α4 and α5, which are likely covered by detergents in the protein sample due to the hydrophobic exterior face of α4/α517. This data supports that the opening between α4 and α5 mediates ligand entry and binding. Interestingly, the equilibrium dissociation constant (Kd) of xlσ1R-C179/C203 for PRE084 also increased by ~9-fold to 6.71 ± 1.44 μM after oxidation compared to 0.68 ± 0.24 μM before oxidation, and was partially reverted to 1.54 ± 0.16 μM after re-reduction (Fig. 4c and Supplementary Fig. 4b–d). These data indicate that the disulfide-bonded xlσ1R-C179/C203 protomers, in which the movement of α4 and α5 is constrained by the disulfide bridge, negatively affected ligand binding in the other, unbonded xlσ1R-C179/C203 protomers. This result is consistent with a previous report that suggests a cooperative ligand binding mechanism in σ1R20.
To assess the disulfide formation in xlσ1R-C179/C203 more directly, we utilized a bulky reagent, methoxypolyethylene glycol maleimide 5000 (mPEG-Mal-5K), to modify the free thiol groups of Cys179 and Cys203. The only native cysteine (Cys91) was mutated to serine to facilitate this analysis (see “Methods” for details). Modification of mPEG-Mal-5K adds 5 kDa per modification to the modified protein, which causes a shift of the relative mobility of the protein in a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel. Considering that the access of mPEG-Mal-5K to Cys179 or Cys203 would likely be impeded by the nearby detergents that cover the hydrophobic exterior face of α4/α5, we expected a relatively limited efficiency for modification, and therefore this result was only used for correlative analysis. As expected, the modification percentage of xlσ1R-C179/C203 before oxidation reached merely 21.9% (Fig. 4d), though the majority of the receptors are thought to be disulfide-free (Fig. 4b). However, consistent with the disulfide bond formation and breakage, the modification percentage of xlσ1R-C179/C203 decreased to 3.7% after oxidation and returned to 15.9% after re-reduction with β-mercaptoethanol (Fig. 4d). This result correlates well with the ITC data for xlσ1R-C179/C203.
The second idea was to modify the residue 179 or 203 with a bulky reagent to sterically block the opening between α4 and α5, which would also decrease the number of the available ligand binding sites. To do so, we generated a Leu179-to-cysteine mutant (xlσ1R-C179) and a Tyr203-to-cysteine mutant (xlσ1R-C203), and chose mPEG-Mal-5K as the blocking modifier. This reagent is bulky and has been shown to successfully modify the free thiol group of Cys179 or Cys203 in xlσ1R (Fig. 4d), and has the advantage to allow quick assessment of the relative modification levels for the ITC samples. Since xlσ1R contains a native cysteine (Cys91), we first tested the effect of mPEG-Mal-5K modification of Cys91 on PRE084 binding. The stoichiometry (number of available binding sites per protomer) and affinity (Kd) changed little for wild-type xlσ1R after mPEG-Mal-5K treatment (Fig. 4e, f), indicating that the mPEG-Mal-5K modification of Cys91 would not interfere with the following analysis for Cys179 or Cys203. Therefore, Cys91 was not mutated in this analysis. We then measured PRE084 binding by ITC for xlσ1R-C179 or xlσ1R-C203 after mPEG-Mal-5K modification. As expected, the number of the available binding sites per protomer (stoichiometry) decreased to 0.48 ± 0.07 from 0.85 ± 0.09 for xlσ1R-C179, and to 0.69 ± 0.04 from 0.87 ± 0.01 for xlσ1R-C203 (Fig. 4e). The blockade percentage was ~44% for xlσ1R-C179 and ~21% for xlσ1R-C203, which correlates with the SDS-PAGE analysis with more modification in xlσ1R-C179 (~23%) than in xlσ1R-C203 (~13%) (Supplementary Fig. 4e), possibly due to the location of Cys203 being in a more occluded region compared to Cys179 when the receptor adopts an open conformation (Fig. 4a). Similarly, the equilibrium dissociation constant (Kd) increased from 1.02 ± 0.05 μM to 1.95 ± 0.05 μM for the modified xlσ1R-C179, and from 0.99 ± 0.03 μM to 1.61 ± 0.09 μM for the modified xlσ1R-C203 (Fig. 4f), again indicating that the modified xlσ1R protomers impaired ligand binding in the other, unmodified protomers. Meanwhile, we are aware of the limitations associated with the modification experiment, such as the limited accessibility to Cys179 or Cys203, the relatively low efficiency of modification by mPEG-Mal-5K, and the extended shape of the mPEG-Mal-5K molecule that might have affected more xlσ1R protomers than the one being modified. Therefore, this result needs to be interpreted with caution. Nevertheless, the simplest explanation for the xlσ1R-C179 or xlσ1R-C203 data is that the mPEG-Mal-5K modification physically blocks the entrance between α4 and α5, providing supplementary support for the PATH2 hypothesis of ligand access.
A structure for the xlσ1R-C179/C203 double mutant
To further validate the structural integrity of the cysteine mutants of xlσ1R, we solved a structure for the xlσ1R-C179/C203 double mutant (xlσ1RC179/C203-S1RA, 3.80 Å) to ensure that the folding of xlσ1R was not disrupted by the cysteine mutation(s). The xlσ1RC179/C203-S1RAstructure was solved in the P21 space group, with 24 protomers in each asymmetric unit (Supplementary Fig. 5a). The same homotrimeric organization was observed in xlσ1RC179/C203-S1RA, which resembles closely other xlσ1R structures. Superposition of the xlσ1RC179/C203-S1RA homotrimer and other xlσ1R structures excluding α1 yielded all-atom RMSDs of 0.2–0.3 Å (Fig. 5a). The α4/α5 region of xlσ1RC179/C203-S1RA appeared also similar to the closed and the open-like conformations (Fig. 5a). The xlσ1RC179/C203-S1RA crystals were grown in the presence of PRE084, and S1RA was later soaked in after the crystals had formed. Each protomer of the xlσ1RC179/C203-S1RA structure contained one S1RA molecule in the ligand-binding pocket, which adopts a similar pose to the ligand in the xlσ1Rclosed-S1RA structure (Fig. 5b). As expected, no disulfide bond was formed between Cys179 and Cys203 in this structure (Fig. 5b), thus allowing S1RA access to and binding in the ligand-binding site. This result indicates that substitution of the residues 179 and 203 with cysteine did not perturb the structural integrity of xlσ1R, and the resulting xlσ1R mutants retained the ligand-binding activity.

Discussion
The combinatory approach of structural and functional studies has gained critical insight into the molecular underpinnings of σ1R function. However, how ligands access the ligand-binding site in σ1R remains elusive. In this study, we focused on this important question and interrogated two ligand entry pathways proposed for σ1R, PATH1 and PATH2, and our structural and functional data favor the PATH2 hypothesis for ligand access. Moreover, our data offered some new information for further discussion.
First, within the ligand-binding pocket of xlσ1Rclosed-PRE084 or xlσ1Rclosed-S1RA, the ligand is coordinated through a salt bridge between its cationic nitrogen and the carboxy group of Glu169 (the equivalent of Glu172 in hσ1R), and through hydrophobic interactions with Val81, Trp86, Met90, Tyr100, Leu102, Phe104, Tyr117, Ile121, Trp161, Ile175, Leu179, Phe181, Ala182, and Tyr203 (Supplementary Fig. 6), which is nearly identical to the ligand-binding pattern in hσ1R17. The highly conserved structures of σ1R and their ligand binding sites from two different species further support the single-transmembrane-helix model16 for σ1R as revealed by the crystal structures17,20.
Second, the structures of the xlσ1R-PRE084 complex in both closed and open-like conformations provide a unique opportunity to discuss how a ligand may enter and exit σ1R, which are likely reversed processes. Superposition of xlσ1Rclosed-PRE084 and xlσ1Ropen-PRE084-co indicates that PRE084 adopts slightly different poses in these two states (Supplementary Fig. 7), and it is feasible to propose the following exit process. From the closed state to the open-like state, the phenyl ring of PRE084 moves ~2 Å closer to α4 to avoid a steric clash with the side chain of Tyr203 (α5), which points toward the ligand-binding site. As a result, the PRE084 phenyl ring pushes against part of α4, causing it to rotate and move slightly away from α5. Interestingly, the steric pressure between PRE084 and α4 has also been observed in previous simulations20,22. Afterward, PRE084 may exit σ1R through the opening formed between α4 and α5. Similarly, PRE084 may enter σ1R in an inversed process. However, though we have observed both closed and an open-like conformation for σ1R, the mechanism that regulates the opening and closing of σ1R remains unknown. Meanwhile, the conformational change of α4 has also been suggested in σ1R agonism20, but its relationship with the conformational changes that lead to the opening of the receptor remains to be investigated.
Third, the PATH2 access hypothesis infers that the σ1R ligand enters from, and exits to, the membrane rather than an aqueous environment (Figs. 1a, 1b). Given the largely hydrophobic nature of most synthetic σ1R ligands2, it would be reasonable to assume that the ligand would partition into the membrane, or at the surface of the membrane (as the ligand becomes amphipathic when protonated). Since σ1R is primarily localized within cells, it has been suggested that a synthetic ligand would need to partition into and pass through, at least the plasma membrane22. Drug delivery studies also show that hydrophobic molecules do partition and enrich into the cellular membranes23. Therefore, a membrane pathway would not be a prohibitory factor for ligand entry and exit in σ1R.
Fourth, one major difference among the σ1R structures is the orientation of the transmembrane helix α1 (Fig. 3b). It is interesting to speculate whether the different orientation bears physiological relevance. Several residues from α1, including Trp26 and Leu27, form hydrophobic interactions with a loop region of the cupin-fold domain (Supplementary Fig. 8a), including Leu97, which is like a plug between α4 and α5 and is one of the residues surrounding the putative ligand entrance (Fig. 4a). Since α1 pulls away from Leu97 in the open-like conformation of xlσ1R (Fig. 3a), it is tempting to postulate that the Leu97-containing loop may have space to swing slightly away from the membrane (Supplementary Fig. 8b), so that a ligand dissolved at the membrane surface may slide in and out between α4 and α5 to access the ligand-binding site from within the membrane (Supplementary Fig. 8b). Unfortunately, this structural feature may be a very dynamic process and was not captured in the open-like xlσ1R structures in this study.
Finally, it is worth mentioning another structural effort that we attempted to capture an open conformation of σ1R by soaking a ligand of extended length into the xlσ1R crystals. We anticipated that the ligand would extend through the ligand entrance and cause the receptor to adopt an open state. Such an extended ligand, N1,N3-bis(1-((R)-3-([1,1′-biphenyl]-4-yl)butyl)-piperidin-4-yl)malonamide (DIM-3C)21, was kindly provided by Dr. Collina and Dr. Rossino for soaking the xlσ1R crystals. Unfortunately, the highly hydrophobic DIM-3C could only be dissolved with organic solvents such as methanol and dimethyl sulfoxide, which damaged the xlσ1R crystals, and no structure was obtained for the DIM-3C soaking experiment. However, interestingly, in one of the 24 protomers of the xlσ1RC179/C203-S1RAstructure, an extra electron density resembling the shape of S1RA was observed near the ligand entrance in addition to the S1RA molecule in the ligand-binding site (Supplementary Fig. 5b). The density was readily and best modeled with an S1RA molecule. Its morpholine ring sits between α4 and α5 and is surrounded by residues 179, 203, and Leu97, whereas the rest of the ligand remains outside of the receptor, possibly representing an entering (or exiting) pose for the ligand. Meanwhile, due to the relatively low resolution (3.80 Å) of the xlσ1RC179/C203-S1RA structure, this extra density needs to be interpreted with caution, and we used it only for supplementary discussion.