
Amyloid β, Tau, and α-Synuclein aggregates in the pathogenesis, prognosis, and therapeutics for neurodegenerative diseases
By Urmi Senguptaab, and Rakez Kayedab
Excerpt from the article published in Progress in Neurobiology, Volume 214, July 2022, 102270, ISSN 0301-0082, DOI: https://doi.org/10.1016/j.pneurobio.2022.102270
Autors’ Highlights
- Co-occurring proteinopathies confound disease diagnosis and treatment development.
- Overlapping Aβ, tau, and α-Syn pathologies are observed in AD and related diseases.
- Aβ, tau, and α-Syn converge directly or indirectly in vitro and in vivo.
- We review recent advancements in biomarkers and neuroimaging for Aβ, tau, and α-Syn.
- A combined therapeutic approach might be more successful for disease intervention.
Abstract
Aggregation of specific proteins are histopathological hallmarks of several neurodegenerative diseases, such as, Amyloid β (Aβ) plaques and tau neurofibrillary tangles in Alzheimer’s disease (AD); morphologically different inclusions of ratiometric 3 repeat (3 R) and 4 repeat (4 R) tau isoforms in progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and Pick’s disease (PiD); α-Synuclein (α-Syn) containing Lewy bodies (LBs) and dystrophic Lewy neurites (LNs) in Parkinson’s disease (PD) and dementia with Lewy bodies (DLB). However, mixed brain protein pathologies have been frequently observed in many of these diseases and in normal aging brains, among which Aβ/tau and tau/α-Syn crosstalks have received increased attention. Interestingly, studies have also shown synergistic interplay among Aβ, tau, and α-Syn in several neurodegenerative diseases, suggesting a protein triumvirate. In this review, we summarize the emerging evidence of Aβ, tau, and α-Syn aggregation in pathophysiology, and their overlap in a spectrum of neurodegenerative diseases including AD, PSP, PiD, CBD, PD and DLB. We discuss the prognostic advancements made in biomarker and imaging techniques in the triumvirate proteinopathies. Finally, we discuss the combined therapeutic modality involving biomarkers and imaging techniques for future combinatorial immunotherapeutic targeting more than one protein aggregates. We hope that such a multitarget therapeutic approach will have synergistic or additive effects to manage neurodegenerative diseases with two or more protein pathologies that might uncover a promising strategy for personalized combination therapies. Managing neurodegenerative diseases by optimizing the diagnostic criteria and the correct combination of immunotherapies will be a key factor in the success of future treatment.
1. Introduction
Neurodegenerative diseases encompass a wide range of clinically and pathologically diverse diseases that are exponentially growing with extended lifespan (Hou et al., 2019, Wyss-Coray, 2016). In the past decades, our knowledge about protein aggregation and its role in these diseases have significantly increased. Although histopathological examination of diseased brain tissues show large insoluble protein inclusions, numerous studies reported the importance of smaller aggregating species of amyloid β (Aβ), tau, α-Syn and several other amyloid proteinsin disease initiation/progression (Gerson et al., 2014a, Ingelsson, 2016, Sengupta et al., 2016). Tauopathies, a group of neurodegenerative diseases including Alzheimer’s disease (AD), progressive supranuclear palsy (PSP), Pick’s disease (PiD) and corticobasal degeneration (CBD) are characterized by the ratiometric presence of aggregated 3 repeat (3 R) and 4 repeat (4 R) tau isoforms (Gotz et al., 2019). PSP, PiD and CBD associate with clinical features of frontotemporal dementia (FTD), and the pathological diagnosis of tauopathies relies on specific tau inclusions in cell/brain region-specific distribution (Kovacs, 2015). Both PSP and CBD show 4 R tau-containing inclusions: tufted astrocytes and oligodendroglial coiled bodies in PSP, and astrocytic plaques in CBD (Kovacs, 2019b). PiD is characterized by filamentous 3 R tau, known as Pick’s bodies in the neurons with occasional tau-positive ramified astrocytes and rare oligodendroglial globular inclusions (Gotz et al., 2019, Kovacs, 2019b). While pure tauopathies embody a wide-ranging heterogeneity in clinical symptoms and morphology of tau aggregates, AD, a ‘secondary tauopathy’ is characterized by Aβ senile plaques and tau neurofibrillary tangles (NFTs) (Braak and Braak, 1991, DeTure and Dickson, 2019, Silva and Haggarty, 2020). Such a heterogenous tau pathology can be resulted from the variability in tau posttranslational modifications (PTMs) and protein-protein interactions (Colin et al., 2020, Silva and Haggarty, 2020). Abnormal deposition of α-Syn into fibrillar Lewy bodies (LBs) and dystrophic Lewy neurites (LNs) characterize another group of diseases, known as synucleinopathies that include Parkinson’s disease (PD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA) (Goedert et al., 2017). Despite presenting α-Syn aggregates as the hallmark feature, the pattern of α-Syn aggregates and their cellular location are different in these clinically distinct synucleinopathies (Coon and Singer, 2020, Goedert et al., 2017). Several physiological cellular processes such as proteostasis, senescence, mitochondrial function, and genomic stability are often impaired in protein aggregation related neurodegenerative diseases (Ferrucci et al., 2020, Hohn et al., 2020, Kwon et al., 2020). Although, these diseases exhibit specific protein aggregates as hallmarks, it is becoming increasingly clear that multiple protein crosstalks may underlie the essential information in these diseases. Mixed brain protein pathologies (co-pathologies) have been frequently observed not only in many of the neurodegenerative diseases, but also in normal aging brains so that so the presence of even one additional low-grade protein pathology can significantly increase the risk of developing dementia (McAleese et al., 2021). Besides their disease-specific pathologies, Aβ, tau and α-Syn aggregates often coexist in many diseases (Irwin and Hurtig, 2018, Jellinger, 2011) (Fig. 1A). Nonetheless, overlapping late-stage clinical symptoms and postmortem protein pathologies stance a huge challenge to their accurate diagnosis (Das et al., 2020, DeTure and Dickson, 2019, Dugger and Dickson, 2017, Kovacs, 2019a). These challenges underscore the necessity of developing biomarkers, neuroimaging studies, and aggregated protein-targeted immunotherapies, along with their successful clinical trials (Kovacs, 2019b). In this lieu, better understanding of complex molecular cross-talks in multiple protein pathologies as observed in histopathological studies of human postmortem brain tissues and animal models is the crux of successful therapeutics (Bittar et al., 2018, Kwon et al., 2020).
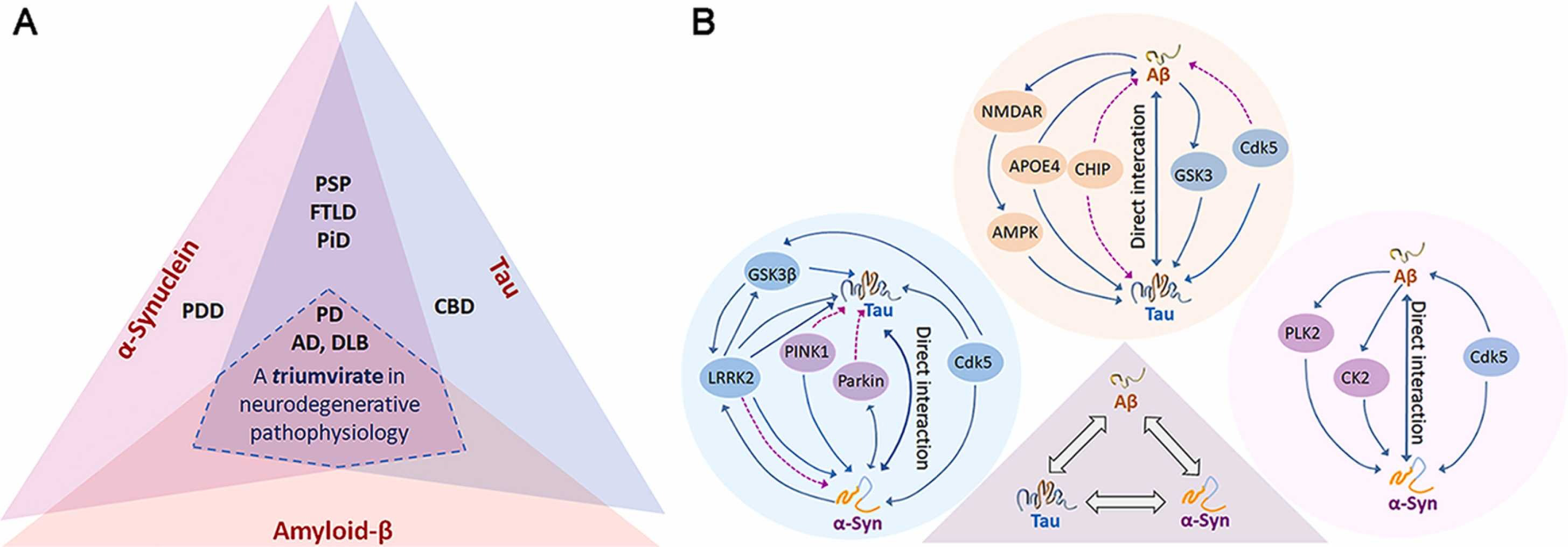
Aβ, tau and α-Syn proteinopathies.
(A) Overlapping protein pathologies of Aβ, tau and α-Syn in several neurodegenerative diseases. (B) Molecular cross-talks among the trio depicting Aβ/tau, α-Syn/tau and Aβ/α-Syn interactions and their plausible convergence into a combined effect. Each interacting protein pair has direct and indirect effects culminating into impaired cellular processes. Blue continuous line represents direct interaction and purple dotted line represents indirect interaction.
With compelling preclinical evidence suggesting the trio Aβ/tau/α-Syn crosstalks via multiple molecular mechanisms, several questions remain unanswered. For instance, it is still unknown to what extent these protein cross-talks exacerbate deleterious effects in humans. This gap of knowledge illustrates the need to explore the protein triumvirate with a focus on their combined toxic effects. Utilizing available literatures, here we aim to blend the current molecular understandings of the three proteins cross-talks in neurodegenerative diseases to inform the design of a combined therapeutic modality. We briefly discuss their aggregation, neurotoxicity, their overlapping pathology, and the current advances on their fluid biomarkers and PET-imaging. We then propose an innovative combined therapeutic approach where imaging techniques and biomarkers will be crucial for early diagnosis and prognosis, eventually leading to an effective combinatorial immunotherapy with a multi-targeted passive immunotherapeutic strategy. This approach would lead to personalized combination therapies, a promising future treatment modality.
2. Aβ, tau and α-Syn in neuronal pathogenesis
The modified amyloid cascade hypothesis highlights the dynamic and heterogeneous soluble Aβ oligomers (AβO) as the primary species causing neurotoxicity and altering non-neuronal cell functions (Cline et al., 2018). Amyloid oligomers adopt a generic sequence-independent conformation, indicative of a similar mechanism of toxicity existing in all amyloid diseases (Campioni et al., 2010, Chiti and Dobson, 2006, Kayed and Glabe, 2006). Low-molecular weight assemblies, such as oligomers are more heterogeneous and dynamic in nature (Giuffrida et al., 2010). Several lines of evidence argued whether and how neurotoxicity is affected by the size of Aβ assemblies. Soluble AβO of different molecular weights were detected in human AD and transgenic mouse brain tissues, suggesting their heterogeneity (Sengupta et al., 2016). Synthetic AβO and proto-fibrillar Aβ caused endoplasmic reticulum (ER) and oxidative stress, neuroinflammation, synaptic, and neuronal hyperactivity often leading to neuronal death (Grochowska et al., 2017, Hector and Brouillette, 2020, Ono and Tsuji, 2020, Penke et al., 2020). Such proto-fibrillar Aβ was also detected in human AD and APP transgenic mouse brains, while AβO caused memory deficits in APP transgenic mice (Lasagna-Reeves et al., 2011b, Ono and Tsuji, 2020). AβO-induced neurotoxicity is mediated by different mechanisms including direct interaction with membranes or membrane-bound vesicles and receptors (Beretta et al., 2020, Kayed and Lasagna-Reeves, 2013, Kotler et al., 2014, Sengupta et al., 2016). Although the heterogeneity of aggregated Aβ and its patho-mechanism related to AD have been extensively studied, the exact nature of the neurotoxic Aβ species in other neurodegenerative diseases remains largely unexplored.
Accumulating evidence suggest that in vitro prepared and brain tissue-derived tau oligomers (TauO) as the potent neurotoxic species causing synaptic dysfunction, impaired memory, and seed for intracellular tau aggregation in cellular models and in vivo (Castillo-Carranza et al., 2014, Fa et al., 2016, Lasagna-Reeves et al., 2011a, Lasagna-Reeves et al., 2012b, Puzzo et al., 2017, Wang et al., 2017). We showed that TauO isolated from AD, PSP, DLB and TBI brain tissues seeded tau monomers in vitro (Bittar et al., 2019, Gerson et al., 2014b, Lasagna-Reeves et al., 2012, Puangmalai et al., 2020). The seeding potency of AD brain-derived high-molecular wight TauO correlated with the clinical severity in AD patients (Dujardin et al., 2020). Different oligomerization states and PTMs indicate the extent of intra-disease variability in tau aggregation (Dujardin et al., 2020). Brain-derived TauO when injected into wild-type (WT) mice, spread from the site of injection to distant brain regions, eliciting behavioral deficits (Clavaguera et al., 2009, Lasagna-Reeves et al., 2012a, Lasagna-Reeves et al., 2012b, Wu et al., 2013). Studies from human AD brains and animal models corroborate a transcellular tau pathology propagation via anatomically connected brain regions that is reminiscent of prion protein pathology (Mudher et al., 2017, Vasili et al., 2019, Vogel et al., 2020, Wu et al., 2016). Though it is hard to pinpoint the exact mechanism(s) for in-and-out transfer of tau aggregates among neighboring cells, several routes are implicated including membrane-bound receptors, exosomes, ectosomes and tunneling nanotubes (TNTs) (Abounit et al., 2016, Dujardin et al., 2014, Puangmalai et al., 2020, Tardivel et al., 2016).
According to the α-Syn cascade hypothesis, aggregates of this protein are formed long before the clinical symptoms are manifested (Ingelsson, 2016). Although, the initiating factors of in vivo α-Syn aggregation are still unknown, in vitro α-Syn aggregation is influenced by chemical variabilities such as pH and salt concentrations (Buell et al., 2014). Endogenous α-Syn aggregation specifically the formation of variable-sized oligomers (α-SynO), their neurotoxicity, and propagation in multiple neurodegenerative diseases have been recently reviewed (Kayed et al., 2020). α-SynO can cause nuclear and mitochondrial dysfunction, synaptic impairment, proteostasis imbalance, ER stress, and apoptosis (Du et al., 2020, Ingelsson, 2016). MSA brain-derived or recombinant α-SynO strains induced brain-region/cell-type-specific α-Syn pathology in TgM83 mice (Lau et al., 2020). Apart from neuronal α-Syn pathology (Kayed et al., 2020), astrocytic α-Syn aggregates are also observed in PD, DLB, and less frequently in MSA (Sorrentino et al., 2019). Internalized α-Syn aggregates impair lysomal degradation and autophagy , and cause mitochondrial abnormalities in astrocytes (Lindstrom et al., 2017, Sorrentino et al., 2019). Although how α-Syn pathology spreads and triggers neurodegeneration are still unclear, several mechanisms have been put forward including the role of membrane-associated receptors, TNTs and exosomes (Abounit et al., 2016, Guo et al., 2020, Hijaz and Volpicelli-Daley, 2020, Masaracchia et al., 2018). Altogether, structural, and functional studies highlight the significance of the patho-mechanism of cytotoxic α-Syn aggregates for developing therapeutics.
3. Aβ, Tau and α-Syn in inflammation
Neuroinflammation is commonly observed in several neurodegenerative diseases including AD, PSP, PD, DLB and ALS, where microglia and astrocytes play important roles in neuroinflammation (Alster et al., 2020, Amin et al., 2020, Bachiller et al., 2018, Guzman-Martinez et al., 2019, Lindstrom et al., 2017). Several studies have highlighted the role of oligomeric aggregates of Aβ, tau and α-Syn in initiating both glial and neuronal inflammation via the activation of several inflammation-associated components. NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3), a component of NLRP3 inflammasome co-localizes with neuritic plaques and is elevated in AD (Heneka et al., 2013). Fibrillar, proto-fibrillar and oligomeric Aβ activated NLRP3 inflammasome, indicating an innate immune response in the beginning of pathogenesis (Luciunaite et al., 2020). Also, both tau monomers and oligomers, but not fibrils directly affected microglia via activation of NLRP3 (Ising et al., 2019). Previously, we reported TauO-associated neuroinflammation in tauopathy mouse model and FTD and AD postmortem brain tissues, suggesting a toxic feedback cycle between TauO and inflammation (Nilson et al., 2017). In our recent study, we reported TauO-associated astrocyte senescence and inflammation in a tauopathy mouse model through the release of high mobility group box 1 (HMGB1) protein, a nuclear protein that is released by glia and neurons upon inflammasome activation (Gaikwad et al., 2021, Paudel et al., 2018). A recent study showed that induced peripheral inflammation potentiated the adverse effects of α-Syn oligomers mediating central neuroinflammation and cognitive deficits in TgG2.3 synucleinopathy mice (La Vitola et al., 2021). Neurons, astrocytes, and microglia express a group of pattern recognition receptors, the cell membrane-bound Toll-like receptors (TLRs) that are pathologically involved in AD, PD and ALS (Fiebich et al., 2018). Oligomeric Aβ and α-Syn caused increased production of pro-inflammatory cytokine by binding to TLR4 mediated by MyD88 (a TLR adapter protein) in murine microglia cell line or rat primary astrocytes (Hughes et al., 2020, Hughes et al., 2019). Although, the direct involvement of TLR4/MyD88 signaling pathway in TauO-induced inflammation is not reported yet, but this signaling pathway has been implicated in AD (Fiebich et al., 2018). An activating immune cell receptor TREM2 (triggering receptor expressed on myeloid cells 2) is dominantly expressed in brain microglial cells (Ulland and Colonna, 2018). The involvement of TREM2 and its variant TREM2R47H in Aβ, tau, and α-Syn pathologies has been documented in several neurodegenerative diseases including AD, PD, and FTD (Finelli et al., 2015, Guerreiro et al., 2013, Rayaprolu et al., 2013, Ulland and Colonna, 2018). TREM2 deletion led to intensified neuritic dystrophies, diffused Aβ plaques, compromised microglial clustering and even elevated soluble AβO resulting from the diffused plaques in PS2APP mice of amyloid plaques (Meilandt et al., 2020). On the other hand, chronic activation of TREM2 caused decreased amyloid deposition, recruitment of microglia to Aβ plaques, reversed amyloid-associated gene expression, and improved spatial learning and novel object recognition memory in 5XFAD mice developing Aβ plaques and gliosis (Oakley et al., 2006, Price et al., 2020). Studies on TREM2 deficiency yielded conflicting results in the setting of tau pathology such as increased tau phosphorylation and aggregation in young hTau mice, and brain atrophy or neurodegeneration upon aging (Bemiller et al., 2017), whereas enhanced tau pathology in THY-Tau22 tauopathy mice only at late stage (Vautheny et al., 2021). Additionally, TREM2 deficiency or TREM2R47H expression provided protection from tau-driven neurodegeneration with attenuated neuroinflammation in PS19 tauopathy mice expressing human mutant tau P301S (Gratuze et al., 2020, Leyns et al., 2017). The functional role of TREM2 contributing to disease pathogenesis is well studied in Aβ and tau pathology together; TREM2 deletion, expression of TREM2R47H variant or even microglia depletion led to reduced Aβ plaque-associated microgliosis with enhanced neuritic plaques and associated pathological tau spreading in APPPS1–21 mice developing Aβ plaques, 5XFAD mice, and in human AD (Gratuze et al., 2021, Jonsson et al., 2013, Leyns et al., 2019, Oakley et al., 2006, Prokop et al., 2019). Further, TREM2 depletion also caused brain atrophy in the late stages in TauPS2APP mice, a triple transgenic mouse model depositing Aβ plaques and NFT pathology (Grueninger et al., 2010, Lee et al., 2021). Nonetheless, a comparatively less studied area is the role of TREM2 in α-Syn pathology where TREM2 deficiency exacerbated α-Syn pathology and associated neuroinflammation and neurodegeneration in PD cell models and transgenic animals (Guo et al., 2019). However, with all the confounding results of TREM2 in Aβ, tau, and α-Syn pathologies, it is still debatable whether targeting TREM2 would be beneficial for disease management, and if yes, then at which stage it can be targeted. Interestingly, TREM2 acts with apolipoprotein E (APOE), another strong genetic risk factor for late-onset AD causing inflammation (Kim et al., 2009, Qin et al., 2021, Strittmatter et al., 1993). AD patients carrying APOE ε4 allele showed enhanced accumulation of Aβ and tau in a longitudinal study compared to patients without the allele (Baek et al., 2020). APOE ε4-driven tau-mediated neurodegeneration independent of Aβ pathology was noted in both tauopathy animal models and human AD patients (Shi et al., 2017, Therriault et al., 2020, Wang et al., 2018), and APOE ε2-associated tau accumulation was reported in PSP patients (Zhao et al., 2018). Remarkably, APOE ε4 genotype was also associated with exacerbated α-Syn pathology, independent of its effects on Aβ and tau in transgenic mouse model and PD patients (Davis et al., 2020). The role of TREM2 and APOE on Aβ, tau, and α-Syn pathologies in neurodegenerative diseases has been substantially reviewed (Adalbert et al., 2007, Gratuze et al., 2021, Li and Zhang, 2021, Rodriguez-Vieitez and Nielsen, 2019, Singh et al., 2019a, Udeochu et al., 2018, Ulland and Colonna, 2018). Nevertheless, an in-depth understanding of how the relationship between TREM2/APOE and these protein pathologies can be utilized for therapeutic intervention, is still unclear.
4. Aβ, tau and α-Syn: converged pathology and molecular cross-talks
The co-occurrence of multiple protein aggregates even as hybrid aggregates are frequently observed in a wide range of diseases, corroborating their molecular cross-talks (Moussaud et al., 2014, Nguyen et al., 2021) (Fig. 1B). Despite the emerging studies indicating crosstalks among Aβ, tau, and α-Syn, much of the in-depth understanding of their mechanistic coupling and its subsequent pathogenicity are still lacking. When considering functional cross-talks of the trio, it is important to acknowledge the cross-seeding mechanism (Ren et al., 2019). Here, we limit our discussion to Aβ, tau and α-Syn interactions both in vitro and in vivo with an emphasis on their in vitro cross-seeding.
4.1. Aβ/Tau interaction
The mechanistic linking of Aβ and tau in AD pathophysiology is well documented (Eckert et al., 2011, Nisbet et al., 2015). Previously, we showed that TauO-targeted passive immunotherapy improved cognitive deficits in an AD mouse model, suggesting a toxic combination of Aβ and tau (Castillo-Carranza et al., 2015). Simulation modeling in AD patients showed accelerated tau propagation in the brain regions with higher Aβ burden across the spectrum of Braak stages (Vogel et al., 2020). At synaptic level, AβO can activate extra-synaptic glutamate receptors, N-methyl-D-aspartate receptors (NMDARs), eventually activating AMP-activated protein kinase (AMPK), which in turn, phosphorylates dendritic tau causing its dissociation from microtubule and aggregation (Nisbet et al., 2015). At proteomelevel, AβO exerted widespread effects on tau, TAR DNA-binding protein 43 (TDP-43) and heterogeneous nuclear ribonucleoproteins in human induced pluripotent stem cells (Sackmann and Hallbeck, 2020). Indirect Aβ/tau interaction can also be mediated by APOE4/Aβ direct interaction and activating kinases (Fig. 1B) (Busche and Hyman, 2020, Vasilevskaya et al., 2020). Aβ/tau toxic synergy caused mitochondrial dysfunction, neurodegeneration in AD patients and impaired neuronal circuits in AD mouse model (Busche and Hyman, 2020, Eckert et al., 2011, Iaccarino et al., 2018). Their synergistic cooperation also caused hyperactivity phenotype and suppression of synaptic gene expression in TgAPP/PS1 mouse model conditionally expressing human tau in a mouse tau-deficient background (Pickett et al., 2019). In vitro, a bidirectional cross-seeding mechanism between Aβ and tau and their synergistic effects in promoting each other’s aggregation are proposed (Ren et al., 2019). In line with this, inhibitors designed for Aβ core inhibited Aβ-mediated tau aggregation and reduced seeding of Aβ and tau. These observations indicate a direct interaction facilitating cross-seeding of tau by Aβ via a shared epitope (Tripathi and Khan, 2020), a concept offering great potential for designing inhibitors against aggregation of these two interacting proteins.
4.2. α-Syn/Tau interaction
The pathological association of α-Syn and tau was supported by their genome-wide association study and the co-occurrence of their fibrillar aggregates in different diseases (Abounit et al., 2016, Moussaud et al., 2014). Our group provided the first line of evidence showing in vivo co-occurrence of α-SynO and TauO in PD and DLB postmortem brain tissues (Sengupta et al., 2015). Additionally, PD brain-derived α-SynO and TauO complexes accelerated tau pathology and memory deficits compared to PSP brain-derived pure TauO in a tauopathy mouse model (Castillo-Carranza et al., 2018). We observed that TauO-targeted passive immunotherapy improved cognitive and motor deficits in TgM83 synucleinopathy mice (Gerson et al., 2018). Concordantly, another group showed tau-mediated hippocampal neurotransmission and memory deficits in TgG2.3 synucleinopathy mice where tau knocking out improved α-Syn A53T-associated cognitive dysfunction and synaptic deficits (Peters et al., 2020, Singh et al., 2019b). In past years, direct and indirect cross-talks between α-Syn and tau were extensively studied. Several groups including ours demonstrated α-Syn/tau direct interaction forming aggregates reflecting cross-seeding mechanism in vitro (Gerson et al., 2018, Giasson et al., 2003, Hojjatian et al., 2021, Lasagna-Reeves et al., 2010, Sengupta et al., 2020). We demonstrated that compared to self-aggregated TauO, α-SynO-induced TauO has a more deleterious effect on different cell lines and primary cortical neurons (Castillo-Carranza et al., 2018). Polymorphic α-SynO-dependent TauO heterogeneity is associated with distinct biological properties (Sengupta et al., 2020). Interestingly, α-Syn/tau fibrillar copolymers triggered robust pathology transmission in rodent model of tauopathy (Williams et al., 2020). Indirectly, tau and α-Syn were functionally linked through mitochondrial dysfunction in PD via several mitochondria-associated kinases such as glycogen synthase kinase-3β (GSK-3β), leucine-rich repeat kinase 2 (LRRK2) and cyclin-dependent kinase 5 (Cdk5) (Fig. 1B) (Feng et al., 2020, Outeiro et al., 2019, Takaichi et al., 2020, Wilkaniec et al., 2016). In Tg4510 mice overexpressing human mutant P301L tau, phosphorylated (p) GSK-3β co-localized with p-α-Syn and p-tau (Takaichi et al., 2020). Although in vitro LRRK2/α-Syn interaction is yet to be established (Outeiro et al., 2019), LRRK2/tau interaction is well documented. On the other hand, Cdk5 directly interacts with LRRK2, α-Syn, and tau, and plays a role in α-Syn/tau interaction (Wilkaniec et al., 2016), suggesting α-Syn/tau indirect interaction via Cdk5 and LRRK2. Collectively, these comprehensive studies suggest i) a direct effect of α-SynO in tau aggregation, and ii) α-Syn/tau combined deleterious effect in pathogenesis.
4.3. Aβ/α-Syn interaction
A significant amount of LBs and LNs are reported in AD patients (Koehler et al., 2013, Mattsson et al., 2013), thus recognizing AD also as a synucleinopathy (Uchikado et al., 2006). More than half of AD patients usually exhibit concurrent pathologies among which, vascular dementia and DLB are the most common (DeTure and Dickson, 2019). Hybrid oligomers of Aβ and α-Syn were detected in brain tissues of AD/PD patients and APP/ α-Syn transgenic mice, suggesting a direct interaction between them (Tsigelny et al., 2008). A potential cross-talk between fibrillar Aβ and α-Syn was shown by preformed α-Syn fibrils induced accelerated α-Syn pathology in 5xFAD mice, a transgenic mouse model with abundant Aβ plaques, suggesting the effetcs of Aβ deposits on α-Syn pathology in a ‘feed-forward’ mechanism (Bassil et al., 2020, Wilkaniec et al., 2016). A bidirectional effect between α-SynWT or mutant α-SynA30P expression and Aβ plaque burden was observed in two different TgAPP mice (Bachhuber et al., 2015, Khan et al., 2018). These in vivo observations are supported by recent in vitro studies showing the co-aggregation of Aβ and α-Syn and their direct interaction through the C-terminal region of Aβ (Candreva et al., 2020, Koppen et al., 2020). Both monomeric and oligomeric α-Syn cross-seeded Aβ oligomerization and inhibited its fibrillation (Candreva et al., 2020). Co-aggregation of the two proteins as hybrid oligomers was also predicted by molecular dynamics simulation (Iljina et al., 2018) and exogenously added Aβ42 at sub-lethal concentration induced increased α-Syn aggregation in neuronal cells (Wilkaniec et al., 2016). An indirect Aβ/α-Syn interaction is mediated by Aβ-induced phosphorylation of α-Syn by several kinases such as Cdk5, casein kinase 2 and polo-like kinase 2 (PLK2) (Marsh and Blurton-Jones, 2012, Wilkaniec et al., 2016) (Fig. 2).
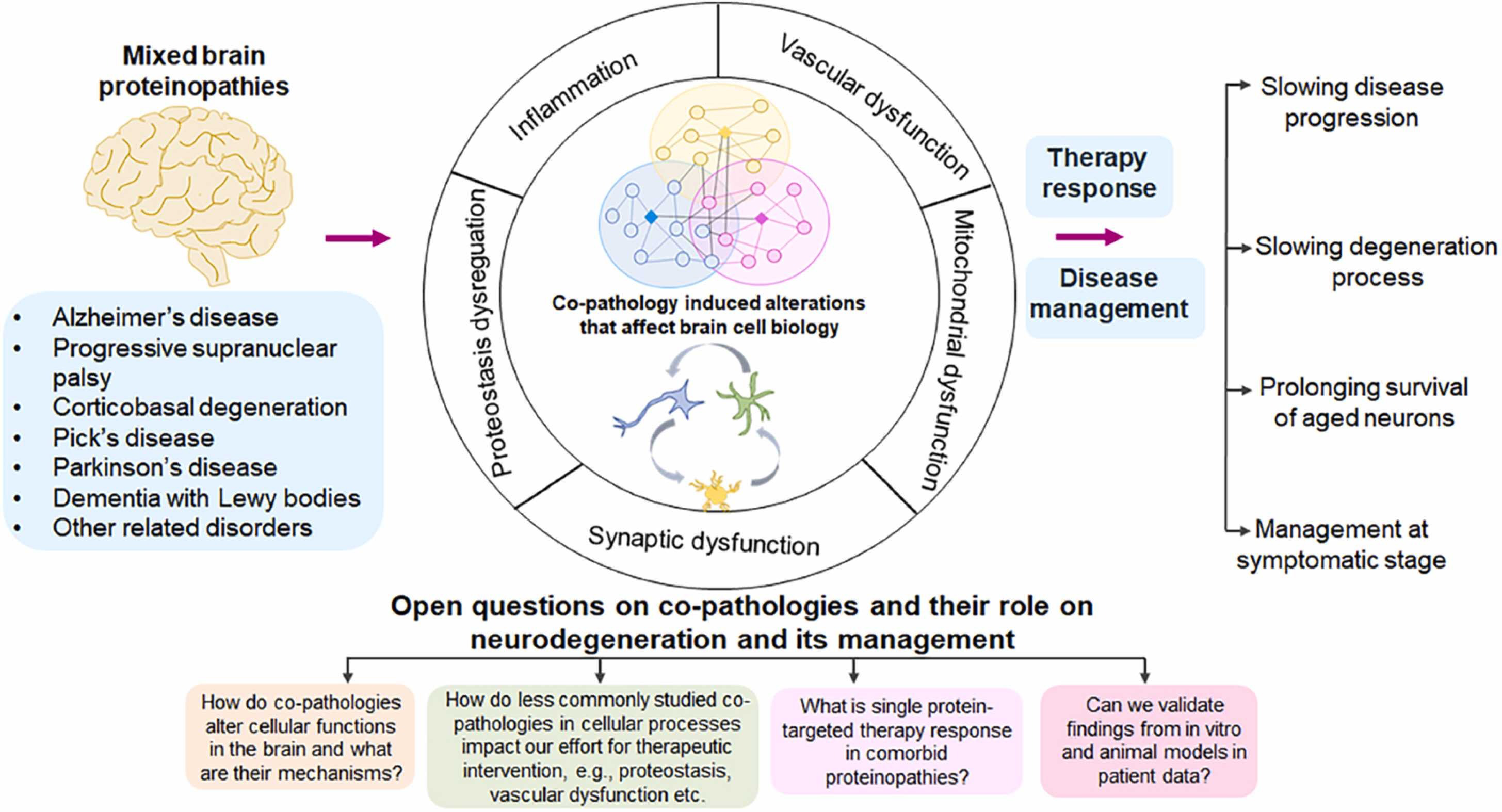
Mixed brain protein pathologies in several neurodegenerative diseases.
The three most widely studied protein pathologies of Aβ, tau and α-Syn interact mechanistically in AD and related disorders where they often co-exist. Studies show that interacting networks of co-pathologies affect neuron/glia brain cell biology leading to several cellular dysfunctions such as proteostasis, synaptic, mitochondrial, and vascular dysfunctions and inflammation. While the observation of co-pathologies in these diseases opens up various questions that remain to be resolved, more tailored approach might lead to better therapy responses, thus allowing us to manage these diseases at various stages.
4.4. Aβ/tau/α-Syn interaction
Earlier studies showed that Aβ and tau pathology strongly co-existed in PD, PDD, and DLB brains and AD CSF (Irwin and Hurtig, 2018). Co-oligomer formation of α-Syn, Aβ, and tauK18 was more favorable than that of α-Syn self-oligomers as shown by single-molecule two-color coincidence detection (sm-TCCD) technique and statistical modeling (Iljina et al., 2018). Co-occurrence of multiple protein aggregates in a single disease or across several diseases was suggested to be due to the cross-seeding among these proteins (Robinson et al., 2018). In addition to tau pathology, Aβ, α-Syn and TDP-43 pathologies were observed in PiD, CBD and PSP, while majority of AD cases showed prevalent α-Syn or TDP-43 co-pathologies. Mostly Aβ, and to some extent Aβ/TDP-43 co-pathologies were noticed in DLB and MSA cases. In ALS and FTD with TDP-43 inclusions, co-pathologies of Aβ and α-Syn were noticed at almost similar rates as of TDP-43 pathology (Robinson et al., 2018). These studies highlight the extent of co-pathologies in neurodegenerative diseases justifying the requirement to encompass more than one protein pathology while considering them as therapeutic targets. A careful analysis of the intricate relationship among the three proteins (Fig. 1B) and its role in cellular processes will be informative. So, the main insight to arise from these studies is, if targeting one protein is not sufficient to combat the disease progression, then why not aiming at multiple proteins for designing an effective immunotherapeutic intervention? This section highlights the cumulative impact of aggregated Aβ, tau and α-Syn altering cellular processes, which is essential to the designing of successful interventions intended to target these proteins. In the following sections, we discuss the advancements in fluid biomarker and imaging for prognostic purposes and how these developments aid in better diagnosis.
5. Disease prognosis
5.1. Fluid biomarkers in neurodegenerative diseases
Although disease-specific protein aggregates are observed at later stages of the diseases, the real risk lies in the quantity of active species capable of seeding at the initial stages, thus emphasizing the necessity of early intervention. Hence, sensitive, and reliable biomarkers are of paramount importance for early diagnostic/prognostic purposes for developing efficient immunotherapeutic. Latest technological advancements in preclinical research have enabled the development of fluid biomarkers. In AD and CBD, CSF Aβ42 amount is inversely related to that of total-tau (t-tau) and phosphorylated-tau (p-tau, Thr231) (Constantinides et al., 2017, Nguyen et al., 2020). Recent data suggest CSF p-tau Thr217 as a better candidate marker distinguishing AD from non-AD dementias and controls (Barthelemy et al., 2020, Janelidze et al., 2020). These studies independently showed that CSF p-tau Thr217 strongly correlated with tau positron emission tomography (PET) tracer [18F]flortaucipir (discussed later), neocortical amyloid β PET measures, and more accurately distinguished AD dementia from other non-AD neurodegenerative disorders compared to p-tau Thr181 (Barthelemy et al., 2020, Janelidze et al., 2020). Several O-glycosylated Aβ species were detected from AD CSF among which Aβ1–15 and Aβ1–17 peptides were most abundant (selectively glycosylated at tyrosine 10 residue on Aβ sequence). However, glycosylated Aβ40 or Aβ42 were not identified in AD CSF, implying for further investigations on the role of tyrosine 10 glycosylationon glycopeptide production and the effects of these glycopeptides on amyloid deposition (Halim et al., 2011, Kummer and Heneka, 2014). Aggregated Aβ and tau including TauO were also detected in the CSF and blood plasma of patients with AD and mild cognitive impairment (MCI) as well as serum, oral fluids, ocular fluids, and nasal fluids of AD patients (Kolarova et al., 2017, Lee et al., 2019, Sengupta et al., 2017).
Decreased CSF total α-Syn (t-α-Syn) and increased α-SynO levels were found in DLB and PD compared to AD and other neurodegenerative diseases (Liguori et al., 2019, van Steenoven et al., 2018). The ratio of α-SynO to t-α-Syn could distinguish DLB and PDD patients from AD patients (Hansson et al., 2014). Increased level of α-Syn in plasma and plasma exosome correlated with the severity of motor symptoms in PD patients (Chang et al., 2019, Ding et al., 2017, Niu et al., 2020). Nonetheless, future longitudinal studies will be essential to establish exosome-derived α-Syn as a potential biomarker for early diagnosis of PD. Another study suggested that the ratio of CSF p-tau/α-Syn alone or in combination with CSF tumor necrosis factor α(TNF-α) or plasma interleukin-6 (IL-6) can distinguish PD from controls (Delgado-Alvarado et al., 2017). Increased phosphorylated-α-Syn (p-α-Syn, Ser129) in CSF and peripheral erythrocytes could distinguish PD patients from controls (Fayyad et al., 2019, Tian et al., 2019). Furthermore, p-Tyr125-, nitrated-Tyr39-, and ubiquitinated-α-Syn were also detected in the plasma of PD patients (Foulds et al., 2011, Vicente Miranda et al., 2017). However, further characterization is needed to corroborate the implication of these α-Syn PTMs in the body fluids. Salivary α-SynO also showed a promise in differentiating PD patients from healthy controls (Bougea et al., 2019). Together, these studies suggest that like in AD, biomarkers development in synucleinopathies has taken a leap in the past few decades, while several other neurodegenerative diseases including FTD, CBD, PSP and PiD require further investigation. Moreover, CSF α-Syn was also reported in AD and MCI (Korff et al., 2013), strengthening the CSF α-Syn biomarker role in multiple diseases. Regardless of all the advances made in the fluid biomarker field, the prognostic values of these biomarkers have yet to be achieved. Contemplating the prevalence of overlapping neurodegenerative diseases, relying not on only one, but on more than one protein biomarker, may have better impact on disease prognosis.
5.2. Neuroimaging
Magnetic resonance imaging (MRI) and positron emission tomography (PET) are the widely used imaging techniques in neurodegenerative diseases. The advances in structural and functional MRI in AD, DLB, PD, and FTLD have been reviewed elsewhere (Agosta et al., 2017). Aβ aggregation has been shown by PET imaging in distant but functionally connected brain regions in cognitively normal and MCI individuals (Pascoal et al., 2019). Until now, US Food and Drug Administration (FDA) has approved three Aβ-imaging tracers to be used in human brain tissue: [18F]-Florbetapir (Amyvid™), [18F]-Flutemetamol (Vizamyl™), and [18F]-Florbetapen (Neuraceq™) (Anand and Sabbagh, 2017). Also, the advent of tau-PET imaging tracers has brought new hopes for early disease diagnosis, which is a rapidly growing field. In a recent study, 18F-Flortaucipir PET (AV-1451) imaging showed a correlation between the tau-burdened brain regions and their postmortem tau pathology, corresponding to Braak stages V and VI in individuals with or without dementia (Fleisher et al., 2020). Two Flortaucipir clinical trials (NCT03809351 and NCT02605785) in UAB Alzheimer’s Disease Center Cohort and PSP patients are underway. Another promising tau-PET tracer for AD and other tauopathies is 18F-PI-2620, a second generation tracer (Kroth et al., 2019, Mormino et al., 2020). This tracer binds to 3 R/4 R tau fibrils and can distinguish AD patients from healthy controls (Kroth et al., 2019, Mueller et al., 2020). In a recent multicenter study, this PET tracer differentiated PSP patients from controls (Beyer et al., 2020). Interstingly, it also acted as a surrogate biomarker for neuronal injury (Beyer et al., 2020). Another recent collaborative longitudinal study on 1343 participants from cognitively unimpaired, MCI, FTD, probable DLB, and Alzheimer clinical syndrome, determined that both Aβ and tau aggregates co-occurred in majority of the cases by using Pittsburgh Compound B (PIB) and Flortaucipir for Aβ and tau PET imaging (Gordon and Tijms, 2019). This study supported a bivariate biomarker model, where amyloidosis preceded tau pathology. Early in 2020, FDA approved tau-PET tracer Flortaucipir and α-Syn PET tracer Fluorodopa-F18 for tauopathies and PD. In a longitudinal PET study on MCI patients for fibrillar Aβ and tau, a biphasic course of inflammation was observed, suggesting that a rational approach should be considered to either stimulate or suppress the inflammation depending on the disease stage (Ismail et al., 2020). Several PET imaging tracers for neuroinflammation are also under investigation (Kreisl et al., 2020). Evaluation of several Aβ and tau imaging traces for AD, tracers for dopaminergic neurons in PD and DLB, and neuroinflammation in AD and PD are in different phases of trials (II-IV) (ClinicalTrials.gov) (Table 1). These studies together highlight the impact this protein triumvirate has on the disease diagnosis.
| Target protein/process | Related Disease type | Antibody name | Developer/Sponsor/Responsible party | Clinical trial identifier | Clinical trial | Status |
|---|---|---|---|---|---|---|
| Aβ PET | AD | [11 C] Pittsburgh Compound-B (PIB) | Tammie L. S. Benzinger, MD, PhD | NCT04134923 | Phase II | Active, not recruiting |
| AD | [C-11]PiB-PET/MRI | Jonathan E McConathy, University of Alabama at Birmingham | NCT03503331 | Phase II | Recruiting | |
| AD | amyloid PET 11 C-PiB | National Taiwan University Hospital | NCT04604600 | Phase III | Recruiting | |
| AD | F-18-AV45 | Chang Gung Memorial Hospital | NCT04174287 | Phase II | Recruiting | |
| AD | Amyloid PET 11 C-PiB | National Taiwan University Hospital | NCT03969732 | Phase III | Recruiting | |
| AD | [18 F]AV-1451 | Neil M Rofsky, MD, MHA, University of Texas Southwestern Medical Center | NCT04080544 | Phase II | Recruiting | |
| Tau PET | AD | 18 F-PM-PBB3 (APN-1607 or MNI-958) | Chang Gung Memorial Hospital | NCT04169126 | Phase II | Recruiting |
| AD | 18-F-Flortaucipir | Mayo Clinic | NCT03821857 | Phase IV | Recruiting | |
| AD | F 18 T807 | Tammie L. S. Benzinger, MD, PhD, Washington University School of Medicine | NCT02414178 | Phase II | Active, not recruiting | |
| AD | MK 6240 | Tammie L. S. Benzinger, MD, PhD, Washington University School of Medicine | NCT04104659 | Phase II | Active, not recruiting | |
| AD | [18 F]PI-2620 | Jae Seung Kim, Asan Medical Center | NCT03903211 | Phase II | Recruiting | |
| AD | [18 F]APN-1607 | Aprinoia Therapeutics Inc. | NCT04141150 | Phase II | Recruiting | |
| Aβ and tau PET | AD, FTD | AV45 (amyloid) 18 F-PM-PBB3 (APN-1607 or MNI-958) (tau) | Chang Gung Memorial Hospital | NCT04309253 | Phase II | Recruiting |
| Aβ and tau PET | AD | Amyloid PET 11 C-PiB MK-6240 (tau | Michael Yassa, University of California, Irvine | NCT03860857 | Phase III | Recruiting |
| Dopaminergic nerve terminals | PD | [F-18] Fluorodopa (FDOPA) | Thomas Jefferson University | NCT04459052 | Phase II | Active, not recruiting |
| PD | 18 F-DOPA | National Taiwan University Hospital | NCT03973502 | NA | Recruiting | |
| PD | 18 F-DOPA | University of Alberta | NCT03042416 | Phase III | Active, not recruiting | |
| PD | [18 F]DPA-714 | Nantes University Hospital | NCT03230526 | Phase II | Recruiting | |
| PD | DaTSCAN™ Ioflupane (123I) | GE Healthcare | NCT04193527 | Phase III | Recruiting | |
| PD | (11 C)PE2I | Weill Medical College of Cornell University | NCT03675282 | Phase II | Recruiting | |
| PD | Ioflupane | Ken Marek, MD, Michael J. Fox Foundation for Parkinson’s Research | NCT01141023 | Phase II | Active, not recruiting | |
| PD, DLB | 18 F-DOPA | University of Alberta | NCT04706910 | Phase III | Recruiting | |
| DLB | 18 F-fluoroethoxybenzovesamicol (18 F-FEOBV) | University of Aarhus | NCT04291144 | NA | Recruiting | |
| AD, PD, LBD, FTD, CBD, PSP and others | 18 F-Flutemetamol | Oskar Hansson, Skane University Hospital | NCT03174938 | NA | Recruiting | |
| Aβ related inflammation | AD | HB-adMSCs | Hope Biosciences | NCT04228666 | Phase II | Active, not recruiting |
| Brain inflammation | AD | Neflamapimod (VX-745) | University Hospital, Toulouse | NCT03435861 | Phase II | Recruiting |
| Neuroinflammation | AD | Montelukast | Ihab Hajjar, Emory University | NCT03991988 | Phase II | Recruiting |
| Brain amyloid and vascular effects | AD | icosapent ethyl (IPE) | VA Office of Research and Development | NCT02719327 | Phase II, Phase III | Recruiting |
| Neuroinflammation | PD | [18 F]DPA-714 | Jonathan E McConathy, University of Alabama at Birmingham | NCT03457493 | Phase I, Phase II | Recruiting |
Aβ-, tau-PET tracers in phase II-IV of clinical trials at the time of this review (clinical trials.gov). Phase I trials have not been included here. PET tracers for neuroinflammation in AD and PD, and dopaminergic neurons in PD and DLB have also been documented here. Interventional studies in these phases that are either recruiting or active but not recruting have been listed.
6. Disease-modifying therapy
6.1. Therapeutic premise and recent advances in monotherapies
Development of novel therapies to reduce the severity of disease progression, if not entirely halting the neurodegeneration process, is warranted. Despite all the preclinical studies and clinical attempts, the identification of effective therapeutics has not been possible, which might have resulted from an inadequate understanding of complex multiple proteinopathies (Weninger et al., 2020). Several strategies, including small molecule-, antisense oligomer-, gene therapy/editing-, and cell-based therapies are being used in AD (Weninger et al., 2020). A stable isotope labeling kinetics (SILK)-based clinical trial (NCT03545126) has begun in FTD, CDB, and PSP patients to evaluate the kinetics of tau aggregation and clearance (Paterson et al., 2019). The clinical trials of pharmacologic agents in AD has been extensively reviewed (Cummings et al., 2020). The detection of aggregated proteins in the biological fluids has provided the rationale for developing Aβ-, tau- and α-Syn-targeted passive immunotherapy in AD, and PD (Valera et al., 2016) (Table 2), where targeting extracellular protein aggregates will reduce their internalizationin the neighboring cells and spreading. Antibody BAN2401 (Lecenemab) targeting Aβ protofibrils is in phase III trial (NCT03887455). In the mid-2021, FDA approved Aβ-targeting Aducanumab (Aduhelm, Biogen Inc) as the first drug to treat AD patients. In a recently published investigation, Aducanumab showed adverse side effects in two of its phase III clinical trials, which included amyloid-related imaging abnormalities (ARIA) in 40% of the participants and almost one fourth of them experienced drug-associated symptoms such as headache, confusion, dizziness, and nausea (Salloway et al., 2021). Gantenerumab (AβO and plaques) and solanezumab(Aβ monomer) are also in different phases of clinical trials for AD (Cummings et al., 2020). Failure in multiple Aβ phase III clinical trials has spurred the interest in tau-directed immunotherapy (Ng et al., 2020). The synergistic effects of Aβ and tau in AD might explain the failure of Aβ monotherapy (Busche and Hyman, 2020). Recent studies indicate that tau is an enticing target for immunotherapy not only in primary tauopathies such as PSP and CBD, but also in secondary tauopathies such as AD and DLB (VandeVrede et al., 2020). Tau immunotherapy in AD shows promise of biomarkers and combination therapy (Bittar et al., 2020, Sandusky-Beltran and Sigurdsson, 2020). Antibody 8E12 targeting N-terminal tau has been tested in PSP patients (Shin et al., 2020) and is being evaluated in an early AD clinical trial (NCT03712787). Antibody BIIB092 (Gosuranemab) against an N-terminal fragment of tau was expected to target extracellular toxic tau fragments. Despite its success in phase I trial (NCT02460094), phase II trial (NCT03068468) was terminated due to unmet primary endpoint (PASSPORT study). In a phase I randomized trial (NCT04185415), humanized antibody UCB0107 (Bepranemab) targeting central region of tau was well-tolerated among PSP patients with successful safety profile and phase II study is under consideration (Albert et al., 2019). In the middle of 2020, UCB and Roche have started clinical testing of this antibody in AD (https://www.alzforum.org). Antibodies PRX002 and BIIB054 targeting oligomeric and N-terminal α-Syn, respectively are being evaluated in phase II trial (Shin et al., 2020). The limited success in monotherapies indicate a more complex scenario than that was initially anticipated, suggesting revisiting the monotherapy efforts.
| Target protein/process | Related Disease type | Antibody name | Developer/Sponsor/Responsible party | Clinical trial identifier | Clinical trial | Status |
|---|---|---|---|---|---|---|
| Tau-targeted passive immunotherapy | AD | ABBV-8E12 (Tilavonemab) | AbbVie | NCT02880956 | Phase II | Active, not recruiting |
| AD | ABBV-8E12 (Tilavonemab) | AbbVie | NCT03712787 | Phase II | Enrolling by invitation | |
| PSP | BIIB092 (Gosuranemab) | Biogen | NCT03352557 | Phase II | Active, not recruiting | |
| PSP | UCB0107 (Bepranemab) | UCB Pharma ( UCB Biopharma SRL) | NCT04867616 | Phase II | Recruiting | |
| AD, tauopathies | ACI-35.030 | AC Immune SA | NCT04445831 | Phase I/II | Recruiting | |
| AD | JNJ-63733657 | Janssen Research & Development, LLC | NCT04619420 | Phase II | Recruiting | |
| Mild AD | IONIS-MAPTRx | Ionis Pharmaceuticals, Inc. | NCT03186989 | Phase I/II | Active, not recruiting | |
| Early symptomatic AD | LY3303560 | Eli Lilly and Company | NCT03518073 | Phase II | Recruiting | |
| Aβ-targeted passive immunotherapy | AD | BIIB037 (Aducanumab) | Biogen | NCT04241068 | Phase III | Enrolling by invitation |
| Early AD | Lecanemab (BAN2401) | Eisai Inc. | NCT03887455 | Phase III | Recruiting | |
| AD | LY3002813 (Donanemab) | Eli Lilly and Company | NCT03367403 | Phase II | Active, not recruiting | |
| Aβ-targeted passive immunotherapy on tau burden | AD | Crenezumab | Hoffmann-La Roche | NCT03977584 | Phase II | Recruiting |
| α-Syn targeted passive immunotherapy | PD | RO7046015 (PRX002) | Hoffmann-La Roche | NCT03100149 | Phase II | Active, not recruiting |
| PD | BIIB054 (Cinpanemab) | Biogen | NCT03318523 | Phase II | Active, not recruiting |
Aβ-, tau- and α-Syn-targeted passive immunotherapies in phase II-IV of clinical trials at the time of this review (clinical trials.gov). Phase I trials have not been included here. Interventional studies in these phases that are either recruiting or active but not recruting have been listed.
Although new exciting data are incessantly revealing the identity of aggregated protein crosstalks and their effects in different model systems, questions still remain (Fig. 2): a) how mixed protein pathologies can alter the synergy in brain cell communications (neuron/glia)? b) how cellular functions like proteostasis, mitochondrial, synaptic, and vascular processes are affected by the mixed protein pathologies? c) what is the success of single protein-targeted therapeutic approach in mixed protein pathologies? d) can we validate the findings from in vitro, cellular, and animal models in patient data? The multifaceted nature of neurodegenerative diseases and complex effects of multiple protein aggregates on cellular functions might justify undertaking a more tailored approach to receive therapy response for disease management while using our contemporary knowledge on protein crosstalks.
6.2. Combinatorial therapeutics
Given the complex multifactorial nature and overlapping proteinopathies in neurodegenerative diseases, combination therapy is believed to be the next logical step for their treatment (Kwon et al., 2020). Immunotherapy targeting α-Syn along with tau, Aβ, prion, or TDP-43 has been proposed on the basis of α-Syn’s interaction with these proteins (Visanji et al., 2019). Aβ/tau-targeted phase II trial of Crenezumab is ongoing in individuals carrying an autosomal-dominant mutation E280A in Presenilin1 (NCT01998841). Another phase II trial (NCT03977584) has been initiated to evaluate the effect of Crenezumab on longitudinal tau burden in the same individuals enrolled in the previous study. The consolidation of all the interactions associated with these three proteins in multiple cellular dysfunctions may allow us to broaden the clinical perception of these overlapping neurodegenerative diseases. Therefore, in line with this vision, we propose a combinatorial model that integrates prognosis by biomarkers/imaging and immunotherapies directed toward more than one of the three key proteins for an effective disease-management intervention regimen (Fig. 3).
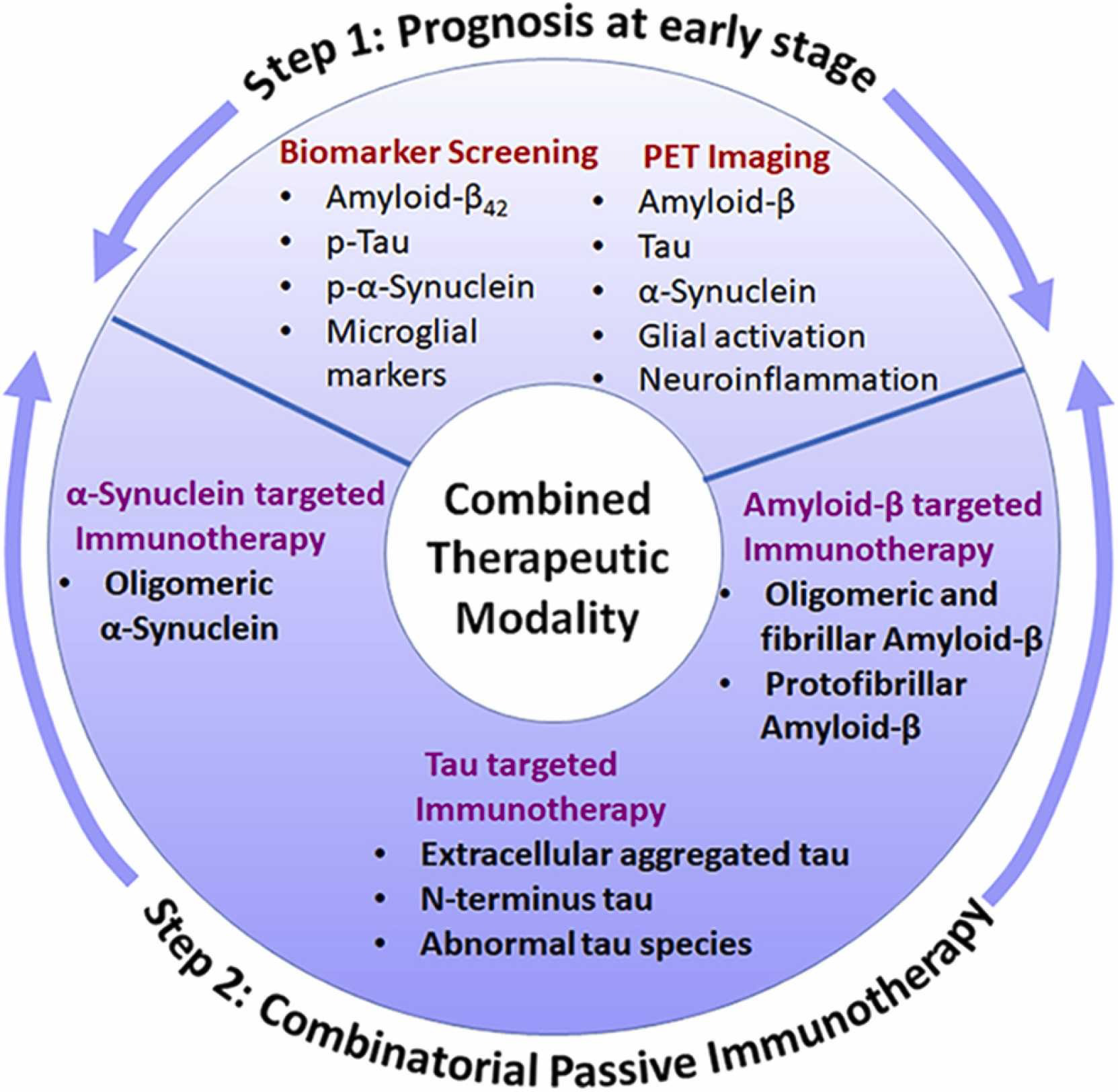
Opportunities for combined therapeutic modality enhancing disease intervention.
Step 1: Prognosis, where any of the successful measures from biomarker screening and/or PET imaging can lead to step 2. Step 2: A combinatorial passive immunotherapy targeting Aβ, tau and α-Syn can be adopted based on the outcomes from step 1.
7. Conclusion and future projection
There is an increasing effort to understand how different amyloids and neurodegenerative processes interact in vivo, as co-occurring pathologies are frequently observed, which have complicated both pathophysiological investigations, as well as treatment development. Understanding the mechanisms of the roles of Aβ, tau, and α-Syn in the neurodegenerative disease landscape is critical for developing therapeutics by influencing the dynamics among these three proteins. Therapeutics targeting other impaired cellular processes, such as proteostasis, inflammation, and cell senescence might be essential as well for the management of these diseases. In this review, we have integrated our knowledge of overlapping Aβ, tau, and α-Syn proteinopathies as connected steps to highlight the spectrum of these aggregate-driven interactions leading to the validation for multiprotein Aβ/tau/α-Syn-targeted immunotherapy with examples of AD and other related disorders. Such multitarget therapeutics might have synergistic or additive effects, which can be adapted to any disease with two or more protein pathologies. This possibility would open avenues for future studies on developing antibodies to specifically target multi-protein aggregates in a multi-immunotherapeutic modality. The low success rate of the ongoing clinical trials indicates a wide variability among patients, implying that a generalized approach for treating patients of the same neurodegenerative disease might not be logical. Thus, personalized combination therapy would be the next promising approach, in which disease management by identifying the correct combination of passive immunotherapies and applying them in patient-specific optimized regimens will be the goal for treatment success.