
Alzheimer’s disease: Ablating single master site abolishes tau hyperphosphorylation

By Kristie Stefanoska, Mehul Gajwani, Amanda R. P. Tan, Holly I. Ahel, Prita R. Asih, Alexander Volkerling, Anne Poljak, and Arne Ittner
Excerpt from the article published in SCIENCE ADVANCES, 6 Jul 2022, Vol 8, Issue 27
DOI: 10.1126/sciadv.abl8809
Editor’s Highlights
- Alzheimer’s disease (AD) is characterized by extracellular β-amyloid (Aβ) plaques and the intraneuronal accumulation of neurofibrillary tangles consisting of tau, a microtubule-associated neuronal protein that regulates the cytoskeleton of all neurons.
- Aβ-induced cognitive deficits are dependent on tau and reconstituting tau reinstates cognitive deficits.
- Tau has a large potential for combinatorial phosphorylation.
- Phosphorylation at serine (S) or threonine (T) residues followed by proline (P) is the most common form of phospho-epitope in tau, particularly within the proline-rich region (PRR) and C-terminal region (CTR) of tau, and includes 17 distinct SP or TP phosphorylation sites, many of which are prominently phosphorylated in aggregated tau in neurofibrillary tangles.
- Lack of master site T181 in human tau was clearly associated with reduced Aβ-induced impact on learning and memory and correlates with lower multi-site phosphorylation of tau at SP/TP and non–SP/TP sites.
- Master site T181 phosphorylation bears strong potential as diagnostic biomarker.
Abstract
Hyperphosphorylation of the neuronal tau protein is a hallmark of neurodegenerative tauopathies such as Alzheimer’s disease. A central unanswered question is why tau becomes progressively hyperphosphorylated. Here, we show that tau phosphorylation is governed by interdependence— a mechanistic link between initial site-specific and subsequent multi-site phosphorylation. Systematic assessment of site interdependence identified distinct residues (threonine-50, threonine-69, and threonine-181) as master sites that determine propagation of phosphorylation at multiple epitopes. CRISPR point mutation and expression of human tau in Alzheimer’s mice showed that site interdependence governs physiologic and amyloid-associated multi-site phosphorylation and cognitive deficits, respectively. Combined targeting of master sites and p38α, the most central tau kinase linked to interdependence, synergistically ablated hyperphosphorylation. In summary, our work delineates how complex tau phosphorylation arises to inform therapeutic and biomarker design for tauopathies.
INTRODUCTION
Alzheimer’s disease (AD) is a fatal neurodegenerative disease, for which no cure exists (1, 2). AD is characterized by extracellular β-amyloid (Aβ) plaques and the intraneuronal accumulation of neurofibrillary tangles consisting of tau, a microtubule-associated neuronal protein that regulates the cytoskeleton of all neurons (3–5). Tau pathology, the progressive deposition of tau, correlates with cognitive decline in AD (3, 5). Thus, tau pathology is predictive of disease progression, supporting a central role of tau (6), which makes tau a prime target of AD diagnostic and therapeutic developments (4). Pathologic tau occurs in a hyperphosphorylated state, i.e., excessive modification with phospho-groups at multiple amino acid side chains simultaneously (7, 8).
Tau has a large potential for combinatorial phosphorylation (7, 9). Phosphorylation at serine (S) or threonine (T) residues followed by proline (P) is the most common form of phospho-epitope in tau, particularly within the proline-rich region (PRR) and C-terminal region (CTR) of tau (8), and includes 17 distinct SP or TP phosphorylation sites, many of which are prominently phosphorylated in aggregated tau in neurofibrillary tangles (5, 7). These sites are targeted by proline-directed SP/TP kinases, the main kinases involved in hyperphosphorylation and therefore pathologic tau (7, 10, 11). Tau phosphorylation is linked to aggregation into multimers and fibrils (12) and modulates cognitive deficits induced by pathogenic tau or by Aβ (13, 14). Despite these clear associations of phosphorylated tau with disease, phosphorylated tau also occurs in physiologic states (15), and site-specific tau phosphorylation mediates functions other than disease progression, including modulation of signaling complexes or lowering of microtubule binding affinity of tau (13, 16). It remains an unresolved question how site-specific and hyperphosphorylation of tau are mechanistically linked.
To fill this gap in knowledge, we screened tau phosphorylation with a focus on SP/TP sites, the most common type of tau phospho-epitope, for mechanistic relations between site-specific and multi-site phosphorylation. We identify central sites, which we term “master sites,” whose phosphorylation governs overall multi-site tau phosphorylation, and we validate these master sites in experiments in vitro and in mouse brain. Thus, tau phosphorylation is regulated by an intrinsic mechanism that modulates relations between phospho-sites and determines how tau arrives at a multi-site phosphorylated state. Overall, this study builds a hitherto unknown basis for the mechanistic connection between site-specific and hyperphosphorylation of tau with broad implications for tauopathies and tau-mediated neurologic disorders.
RESULTS
We propose that tau phosphorylation is mechanistically interdependent, i.e., initial phosphorylation at one site modulates the phosphorylation state at another site (Fig. 1A). We propose and define the term “interdependence” as the mechanistic relationship that modulates tau phosphorylation at a given site by prior phosphorylation at another site.
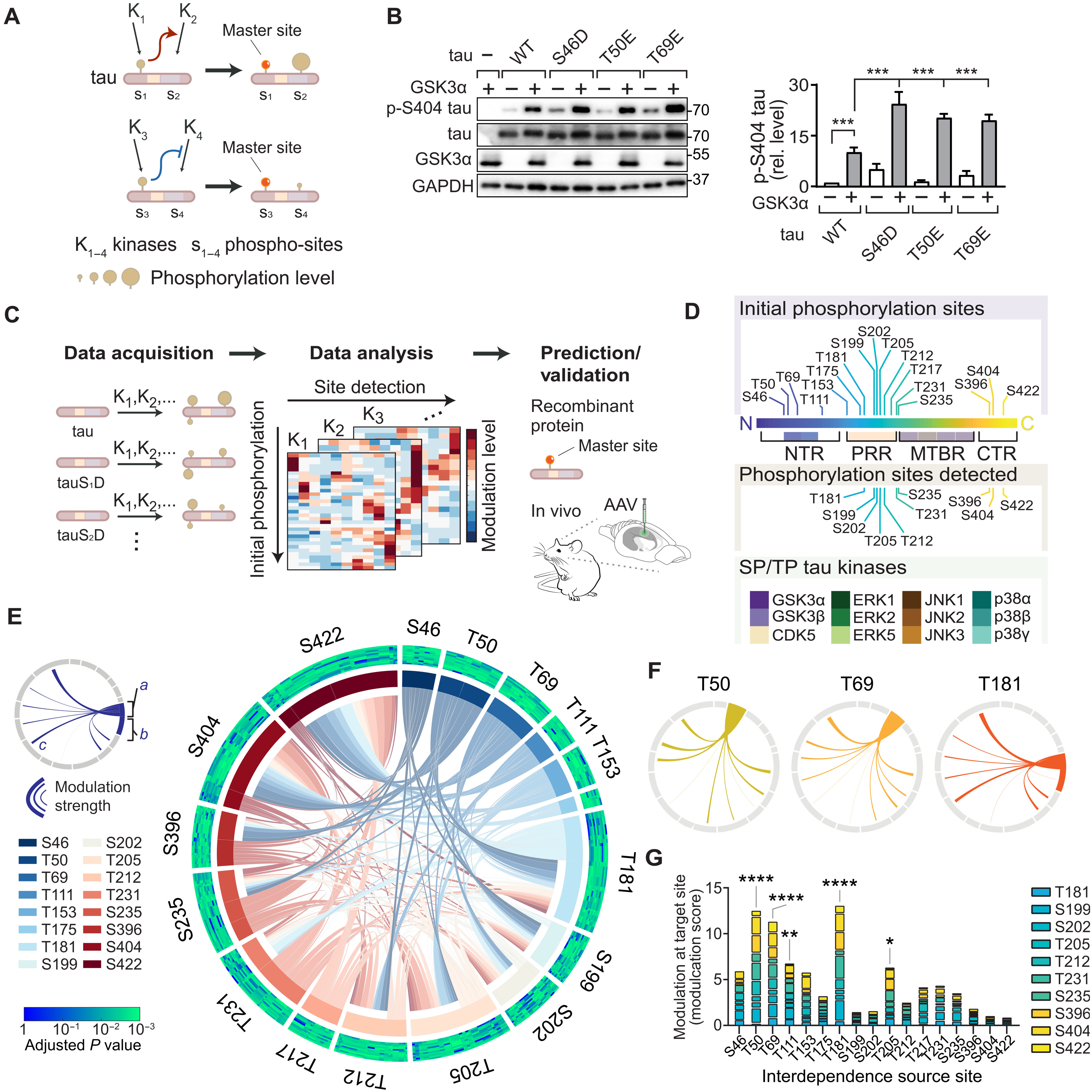
Interdependence defines master sites integral to tau multi-site phosphorylation.
(A) Schematic of site interdependence. Phosphorylation at one site (e.g., S1) by kinase K1 disproportionately affects phospho-state at another site (e.g., S2). (B) Site interdependence enhances distal tau phosphorylation. Immunoblots from 293T cells expressing human wild-type tau (WT) or indicated variants ± glycogen synthase kinase 3α (GSK3α). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), loading control. n = 3. Note that positive interdependence promotes higher pS404 levels in tau variants. (C) Mapping tau SP/TP phosphorylation interdependence. Expression of tauWT or variants with phospho-mimetic conversions, representing initial phosphorylation state, in cells ± kinases (K1, K2,…). Phospho-status is quantified with epitope-specific antibodies and mapped onto multidimensional dataset. Data analysis identifies interdependence patterns and master sites, followed by validation. (D) Seventeen initial phosphorylation sites, 10 sites detected, and 12 SP/TP kinases in screen. (E) Integrated plot of tau SP/TP phosphorylation interdependence for 10 target and 17 source sites. Modulation a, at source site; b, at source site; c, toward target site. Adjusted P values for modulation are plotted for each site (Spearman’s correlation). Cumulative modulation is represented by sector width. (F) Interdependence for master sites. (G) Cumulative modulation scores (factor of correlation coefficient and relative phosphorylation level). Means ± SEM. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05 [analysis of variance (ANOVA)].
To systematically explore interdependence for SP/TP phosphorylation of tau, we set up a site interdependence screen based on a tractable experimental paradigm in cultured cells that combined initial phosphorylation, increased kinase activity, and assessment of resulting tau phosphorylation at specific epitopes. We used 293T cells expressing tau variants defining a site-specific initial phosphorylation state (based on phospho-mimicking side chains, i.e., serine → aspartate; threonine → glutamate). We raised activity of one tau kinase by concomitant expression of a SP/TP kinase (Fig. 1B and fig. S1). Controls without expression of kinase (i.e., tau alone) served to determine the effect that fixed phosphomimetic residue has on modifications at other SP/TP sites (Fig. 1B and fig. S1). Mimicking initial phosphorylation at distal SP/TP sites resulted in instances of significant modulation of site-specific tau phosphorylation, i.e., significantly different levels of detectable phospho-tau epitopes, which were quantified through densitometric analysis of immunoblots and normalized to total tau and loading control [glyceraldehyde-3-phosphate dehydrogenase (GAPDH)] (Fig. 1B and fig. S1). Integrating these experimental conditions facilitated data acquisition across three parameters: (i) pre-SP/TP site phosphorylation, (ii) increased SP/TP kinase activity, and (iii) SP/TP site phosphorylation status (Fig. 1C). Resulting multidimensional data built the framework for definition of tau phosphorylation interdependence, to be further validated in vivo and explored mechanistically (Fig. 1C and fig. S2). Our experimental setup based on SDS–polyacrylamide gel electrophoresis (PAGE) and immunoblot for detection was superior to dot blot detection of phosphorylated tau (fig. S3). Validated phospho-epitope–specific tau antibodies were used for detection (fig. S4 and table S1). Phospho-mimicking tau variants did not induce aggregation or unfolded stress response (see Materials and Methods and fig. S5). Analogous tau expression in cultured cells served previously to identify tau kinase and phosphorylation-dependent mechanisms relevant to neurons in vitro and in vivo (16, 17).
Expanding this approach across 17 SP/TP initial phosphorylation sites and 12 kinases produced a multidimensional dataset of steady-state levels of tau phosphorylation at 10 different phospho-sites in relation to different kinase activities and initial phosphorylation (Fig. 1, C and D, fig. S2, and table S1). Notably, multi-site phosphorylation exhibits distinct steady-state forms for tau (18) and other proteins (19). Global data analysis across all 12 SP/TP kinases demonstrated interdependence across individual sites and different SP/TP kinases (fig. S6), exhibiting predominantly positive modulation, likely due to enhanced kinase activities in our experimental approach or limited effects of the kinases studied on tau phosphatase activity (20–22). Clustering the data along initial phosphorylation and kinase activity showed that sites T181, T205, T212, S404, and S422 were hotspots within the PRR and CTR for modulation by tau phosphorylation interdependence (fig. S6). Thus, SP/TP initial phosphorylation differentially and distally modulates phosphorylation across tau. These results identify site interdependence as a key feature of tau phosphorylation in living cells and show that interdependence effects are quantifiable in direction, strength, and extent.
Comparing data from different SP/TP kinases revealed similarities, as well as unique patterns in site interdependence (fig. S7). Mitogen-activated protein (MAP) kinase p38α showed most extensive interdependence effects (i.e., numerous SP/TP site modulation) (fig. S7), whereas few interdependence effects were apparent in extracellular signal–regulated kinase 1 (ERK1) and p38γ activities toward tau (fig. S7). Thus, initial phosphorylation can alter the site-specific activity of kinases toward tau and site interdependence is notably kinase specific.
We aimed to determine positional characteristics and magnitude of interdependence in tau. To visualize phosphorylation interdependence, we plotted modulation effects along the primary sequence of tau, based on the parameters of initial phosphorylation and target site modulation, including directionality and strength of effects (fig. S8). This showed proximal and distal interdependence effects for each tau kinase between phosphorylation events in the N-terminal region (NTR), PRR, and CTR (fig. S8).
To identify significant interdependence links between tau phosphorylation sites, we analyzed levels of epitope phosphorylation by cross-correlation depending on (i) the phosphorylation site targeted, (ii) initial phosphorylation, or (iii) increased kinase activity in our experimental system (fig. S9, A and B). This permitted effect comparisons of individual phosphorylation events for all and for individual SP/TP kinases to identify significantly similar or dissimilar behavior (fig. S9, A and B). Resulting Spearman’s correlation coefficients enabled calculation and ranking of modulation scores to determine most impactful initial phosphorylation sites and performing hierarchical clustering of sites or kinases to visualize dis/similarities. Integrated analysis for strength, target, and significance of modulation produced an overview of the complexity of tau phosphorylation within paradigm parameters (i.e., 17 initial phosphorylation, 12 SP/TP kinases, and 10 phosphorylation detection sites) and revealed relations of broadest impact across SP/TP phosphorylation events and kinases (Fig. 1E). NTR sites T50 and T69 presented with numerous significant interdependence connections onto other tau phosphorylation sites (Fig. 1, F and G). T181 and T205 within tau’s PRR had multiple high-significance relations with other sites across most kinases tested (Fig. 1, F and G). Notably, CTR sites (i.e., S396, S404, and S422) contributed significantly less to tau phosphorylation interdependence across all kinases tested when compared to sites within the NTR and the PRR (Fig. 1G and fig. S10). Sites markedly governing a broad range of tau SP/TP epitopes were termed master sites. Initial modification at master sites was a strong predictor of subsequent tau phosphorylation (Fig. 1, E and G, and fig. S10). Across all SP/TP kinases, master sites in the NTR (T50 and T69) as well as PRR’s T181 and T205 formed clusters of similar modulation relations (fig. S9D). S396 and S404 clustered separately with PRR sites (e.g., T231 and S235) and with CTR site S422, respectively, suggesting that proximity within tau’s primary sequence is not a dominant driver of site interdependence in tau phosphorylation (fig. S9D). Ranking all phospho-sites by impact and significance of modulation revealed cumulative modulation by prior phosphorylation status at T181, S199, and S422 (fig. S9C). These sites are prominent epitopes of pathological hyperphosphorylated tau detected in human brain samples and mouse models of AD and other tauopathies (15, 23, 24). Modulation of S422, a hallmark of advanced tau pathology (25), was prominently associated with prior phosphorylation and kinase activities of p38α, glycogen synthase kinase 3β (GSK3β), and c-Jun N-terminal kinase 3 (JNK3) (fig. S9C). Identified master sites T50, T69, T181, and, to a lesser extent, T111 and T205 made significant contributions to initial phosphorylation-dependent modulation of tau phosphorylation across tau kinases tested (Fig. 1G and fig. S10). These results suggest that a critical contribution to sequential tau phosphorylation arises from initial phosphorylation at specific master sites in the NTR and PRR.
We next confirmed predictions of master sites by proof-of-concept experiments in systems of progressive complexity. In cultured cells, tau phosphorylation by kinases GSK3β and p38α was modulated by T50, T69, T181, and T205 (fig. S11). We wondered whether tau phospho-site interdependence is a physiologic mechanism that occurs during healthy brain function. To test this idea, we focused on master site T205. T205 is of interest because (i) it is a critical residue within the AT8 epitope of pathological tau, a hallmark histological marker of Alzheimer’s tauopathy (26–28) and (ii) T205 phosphorylation of endogenous tau conveys site-specific functions (13, 16). To address the physiologic relation of T205 phosphorylation and multi-site tau phosphorylation in vivo, we used mice, in which the endogenous Mapt gene was CRISPR-Cas9–edited at the T205 codon to a phospho-deficient [threonine → alanine (A)] or phospho-mimetic side chain [→ glutamate (E)] at this position (Fig. 2A) (13). CRISPR tauT205A and tauT205E mice allow for testing for requirement and sufficiency of this master site in physiologic phosphorylation in murine tau, respectively. Brains of tauT205E/E, tauT205A/A and tauT205T/T control mice showed markedly different levels of tau phosphorylation at multiple epitopes including T181, S199, T212, T231, S396, and S422 in tauT205E/E and tauT205A/A as compared with tauT205T/T controls, despite overall similar total tau levels (Fig. 2, B and C, and fig. S12). This was confirmed in immunoprecipitated murine tau followed by phospho-peptide detection (fig. S12). Differential tau phosphorylation in tauT205E/E and tauT205A/A brains significantly correlated with predicted modulation by T205 phosphorylation in vitro, particularly for T181 and S422 (Fig. 2, D and E). These results show that modification of endogenous tau at T205 modulates physiologic tau phosphorylation at multiple epitopes and that predicted site interdependence in cultured cells applies to endogenous tau in brain tissue.
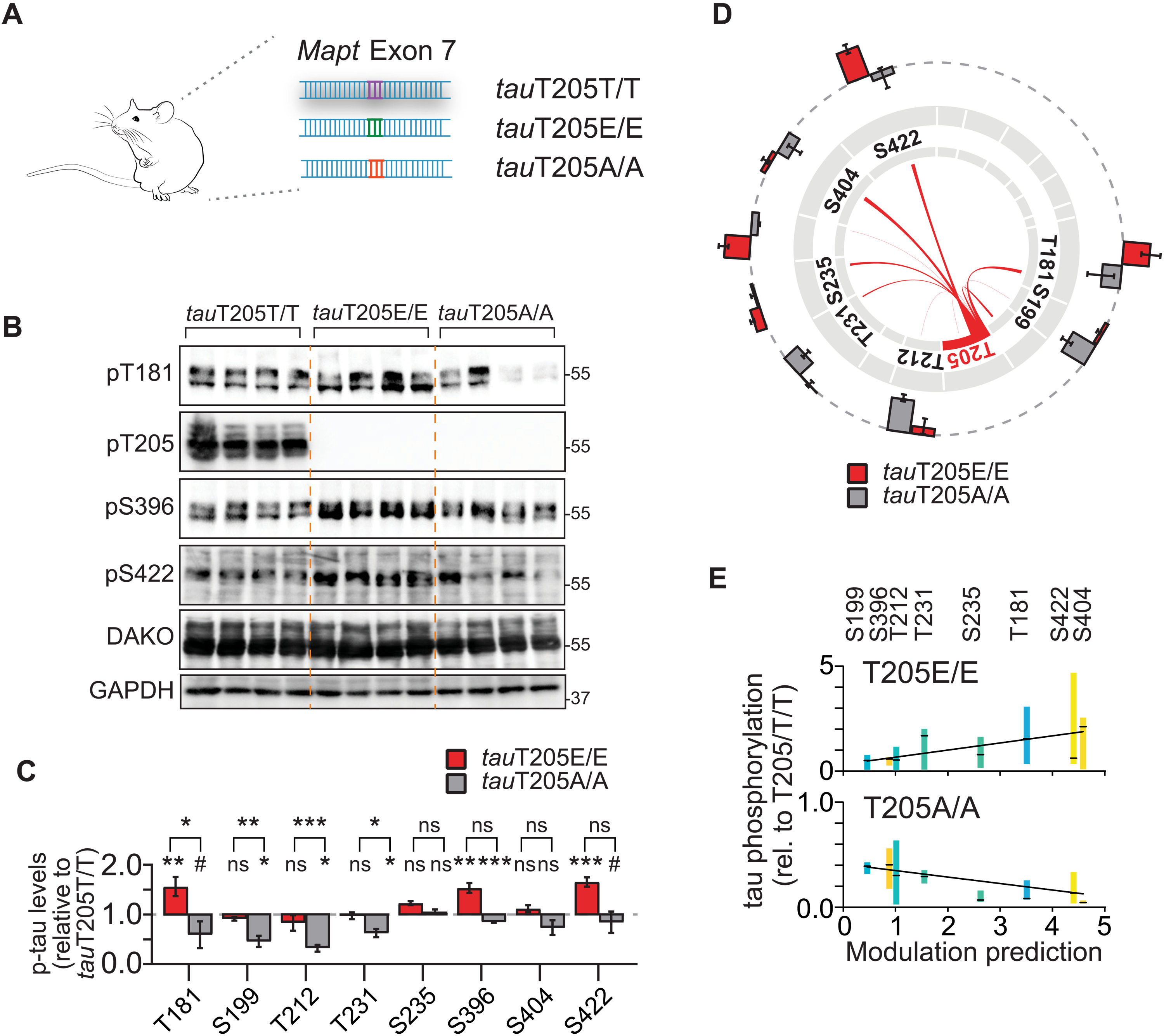
Master site interdependence controls endogenous tau phosphorylation in vivo.
(A) CRISPR-Cas9 genome-edited mice with conversion of codon in Mapt gene encoding for master site T205 to phospho-deficient alanine (tauT205A) or phospho-mimicking (tauT205E) codon. (B and C) Gene editing of master site T205 in mice modulates levels of site-specific tau phosphorylation of endogenous murine tau predicted by interdependence analysis. Levels of indicated phospho-tau levels in cortex of tauT205A/A, tauT205E/E relative to control tauT205T/T mice, and total tau levels. n = 5 to 6. (D) Visual correlation of T205 interdependence determined in cultured cells with tau phosphorylation levels at different epitopes in CRISPR T205 mice. Differences in tau phosphorylation in tauT205A/A and tauT205E/E as compared with tauT205T/T cortex mapped onto T205-dependent modulation at SP/TP sites. n = 5 to 6. (E) Regression analysis of T205-dependent modulation strength predicted at indicated sites in tau with differences in cortical phosphorylation levels of tauT205A/A and tauT205E/E mice. Values are means ± 10 to 90 percentile relative to tauT205T/T. tauT205A/A (n = 12), P < 0.0001; tauT205E/E (n = 11), P = 0.0002 (linear regression with F test). Data are presented as means ± SEM unless indicated otherwise. ***P < 0.001; **P < 0.01; *P < 0.05; ns, not significant (ANOVA).
Murine and human tau share a high degree of phospho-epitope identity (15, 29). This suggests that master site interdependence is transferable to human tau, including in the context of AD. Notably, tau phosphorylation—at multiple and individual sites—is involved in tau-dependent AD mouse models, including downstream of Aβ in Aβ precursor protein (APP) transgenic models (13, 16, 30, 31). We investigated master sites T50, T69, and T181 in an AD mouse model by expressing adeno-associated virus (AAV)–delivered wild-type human tau (human tauWT) or phospho-deficient tau variants in the brains of APP23.tau−/− mice, in which the influence of endogenous tau is precluded (Fig. 3A) (13, 32). We addressed master site–dependent tau phosphorylation in this APP transgenic mouse model because tau is critical for phenotypes and signaling downstream of Aβ (33, 34). The reconstitution approach by AAV-mediated neuronal expression of tau variants permitted assessment of site interdependence solely in human tau in living brain tissue, including in the context of a disease model. We focused in these experiments on assessing requirement of these sites for multi-site phosphorylation of human tau as an approach complementary to the phospho-mimicking tauT50E, tauT69E, and tauT181E variants used in cultured cell experiments (fig. S11). AAV-mediated expression of phospho-deficient tau variants showed that T181 and, to a lesser extent, T50 and T69 control multi-site phosphorylation of human tau in the context of high Aβ levels in AD mice (Fig. 3, B and C). Notably, levels of phosphorylation of T205, S396, S404, S422 and others were markedly lower when tau with ablated T50, T69, or T181 site was expressed in APP mice, confirming predictions of phosphorylation modulation by these master sites (Fig. 3, B and C, and fig. S13). These results were supported by mass spectrometry and phospho-peptide enrichment of tau immunoprecipitated from APP23.tau−/− and tau−/− mice expressing tau variants, which also indicated that occurrence of non-SP/TP phosphorylation was lower in tauT50A, tauT69A, or tauT181A as compared with tauWT (fig. S13). To assess the consequence of master site ablation on cognitive phenotypes of AD mice, APP23.tau−/− mice expressing tauWT or tauT181A variant after intracranial AAV delivery performed spatiotemporal learning paradigm (Morris water maze) (Fig. 3, D to G, and fig. S14). TauWT-expressing APP23.tau−/− mice showed strong impairment of learning and cognitive flexibility in the acquisition and reversal phase, respectively, as well as of memory in the probe trails, while tauT181A-expressing APP23.tau−/− mice showed significantly better learning and memory. All experimental groups had comparable swim speeds and visual acuity (fig. S14). To control for effects on locomotor activity, mice performed open field tests, which did not show differences between tauWT and tauT181A-expressing APP23.tau−/− nor tau−/− mice (fig. S14). These results show that Aβ-associated phosphorylation of human tau at multiple pathologically relevant epitopes is regulated by master sites in line with our interdependence predictions. Furthermore, ablation of master site T181 results in modulation of Aβ-associated cognitive deficits.
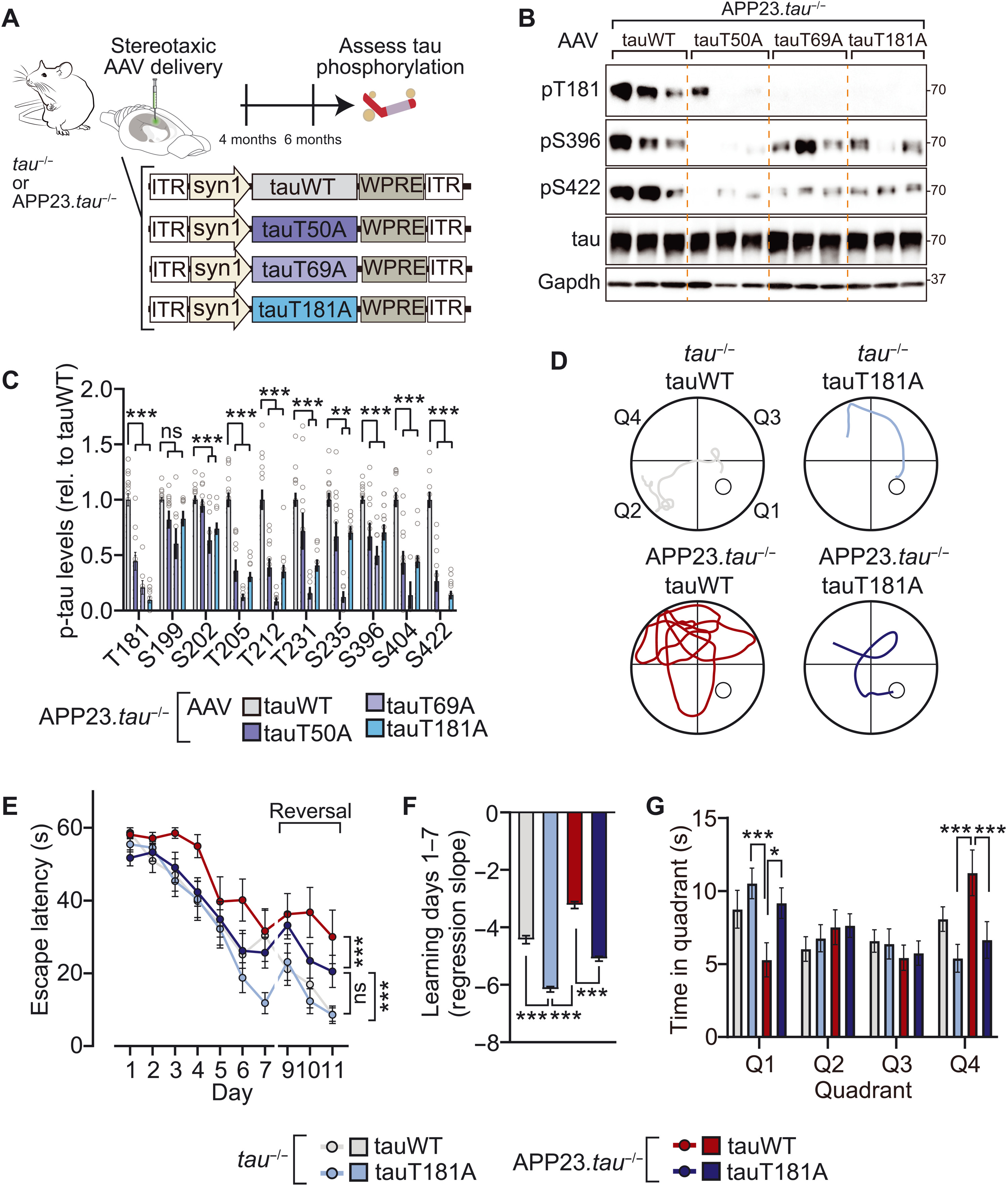
Master site interdependence governs human tau phosphorylation and Aβ-tau–induced cognitive impairment in vivo.
(A) Schematic of AAV-mediated expression of tauWT or indicated phospho-deficient tau variants in tau−/−or APP23.tau−/− (B and C) Master site–governed SP/TP phosphorylation of human tau in an AD mouse model. Tau-deficient human APPswe transgenic mice (APP23.tau−/−) were injected intracerebrally with AAV-driving neuronal expression of tauWT or indicated tau variants (tauT50A, tauT69A, and tauT181A). Phosphorylation levels of tau variant or tauWT-expressing mice relative to total tau levels were assessed by immunoblot. n = 8 to 16. (D to G) Morris water maze spatiotemporal learning paradigm with APP23.tau−/− and tau−/− mice injected intracerebrally with AAV-driving neuronal expression of tauWT or tauT181A. (D) Morris water maze escape paths on day 7. Quadrants as indicated with hidden platform in Q1. (E) Escape latency during acquisition phase (days 1 to 7) and reversal phase (days 9 to 11). (F) Acquisition phase learning curve slopes. (G) Probe trials on day 8. Q1, target quadrant. Data are presented as means ± SEM unless indicated otherwise. ***P < 0.001; **P < 0.01; *P < 0.05 (ANOVA).
We next asked which tau SP/TP kinases are governed most extensively by interdependence. Plotting modulation by initial tau phosphorylation for kinases individually corroborated kinase-specific interdependence in cultured cells (Fig. 4A and fig. S15). Cross-correlation of all phosphorylation events depending on initial phosphorylation returned varying degrees of similarity and dissimilarity of tau kinases in terms of interdependence of tau phospho-sites (Fig. 4B). Hierarchical clustering of tau kinases placed p38α in proximity with ERK2 and ERK5 and removed from p38 family members p38β and p38γ (Fig. 4B), despite a high degree of sequence identity between p38 kinases (35). We next addressed whether p38α made critical contributions to multi-site tau phosphorylation in vivo. We probed for murine tau phosphorylation at a range of epitopes in neuron-specific p38α knockout (p38αΔneu) and control p38αloxP/loxP mouse hippocampus (Fig. 4C), a brain region with high neuronal p38α expression (36). We found a marked reduction in tau phosphorylation at multiple sites in p38αΔneu as compared with p38αloxP/loxP hippocampus (Fig. 4, D and E, and fig. S16). Tau phosphorylation in p38β−/− and p38γ−/− mice was comparable to control mice for almost all sites tested (fig. S16), suggesting that within the p38 family, p38α rather than p38β or p38γ is a high-impact, multi-epitope tau kinase in vivo. These results show that p38α is a hippocampal tau master kinase in neurons in vivo, likely due to interdependence mechanisms.
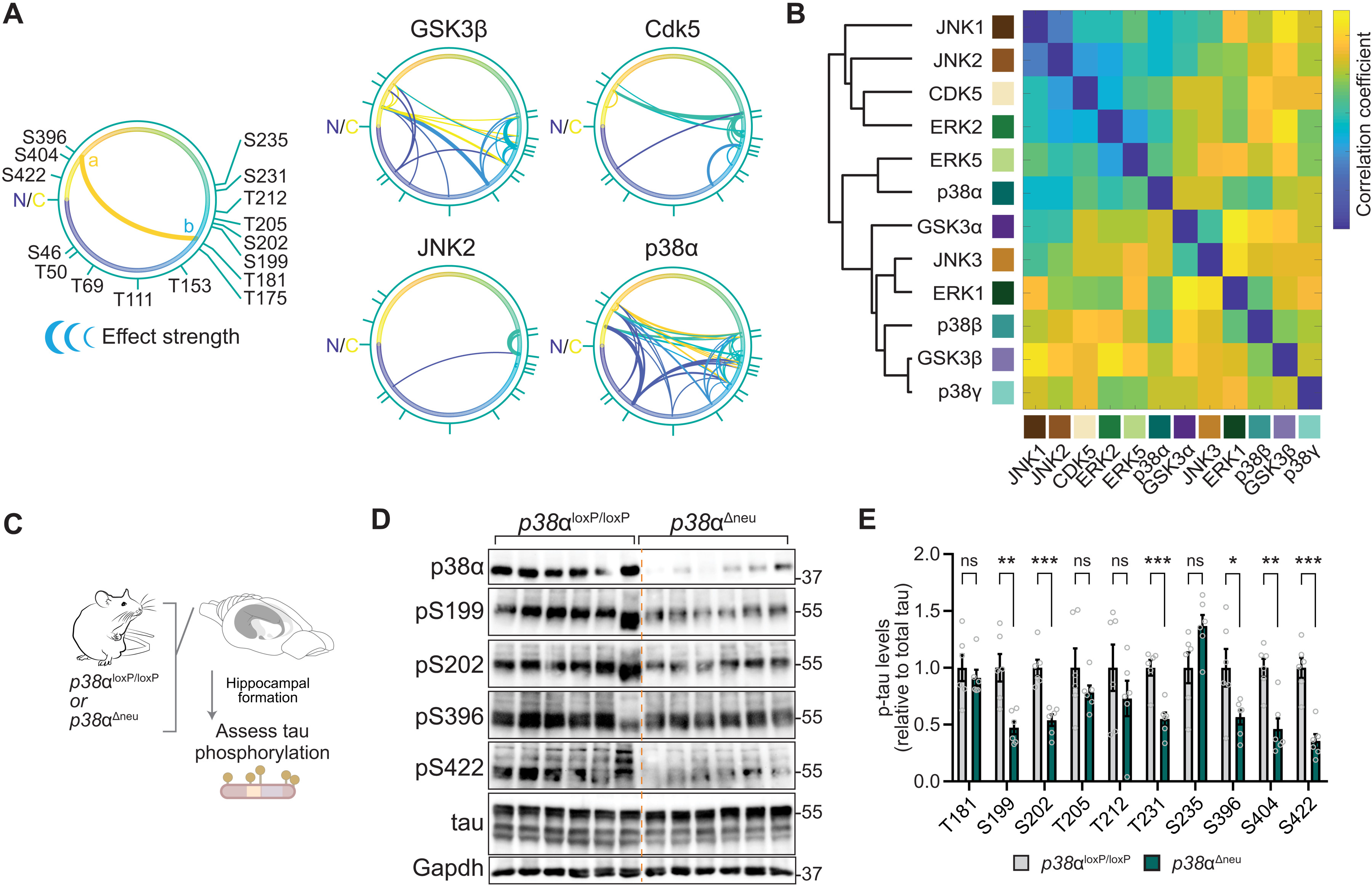
MAP kinase p38α is a multi-site tau kinase in vivo.
(A) Tau SP/TP phosphorylation interdependence is kinase specific. Directionality of interdependence relations between initial phosphorylation (a) and modulated phosphorylation (b). Connecting line thickness indicates significance and modulation strength. Kinases with extensive high-significance interdependence (GSK3β and p38α) and kinases with sparse relations (Cdk5 and JNK2) are shown. n = 4. N, N terminus; C, C terminus of tau (441 amino acids) primary sequence. (B) Correlation matrix of tau phosphorylation interdependence relations associated with 12 SP/TP kinases (n = 4), including hierarchical clustering by pair-wise Pearson’s correlations. Note that tau kinase similarity differs from evolutionary relation of kinase families for GSK3 and p38 kinases. (C to E) Phosphorylation of endogenous tau is reduced at multiple epitopes in hippocampus of mice with neuron-restricted p38α deficiency (p38αΔneu) as compared with control p38αloxP/loxP mice. (D) Representative immunoblots probed with antibodies for pS199 tau, pS202 tau, pS396 tau, pS404 tau, pS422 tau, tau, and p38α are shown. n = 6. Residual p38α in p38αΔneu is due to expression in glial cells (36). (E) Immunoblots in (D) were quantified relative to Gapdh and tau. Data are expressed as means ± SEM. ***P < 0.001; **P < 0.01; *P < 0.05. Note that most phosphorylation sites tested show markedly lower levels in p38αΔneu as compared with control p38αloxP/loxP mice.
Interdependence in tau phosphorylation can arise from intrinsic (i.e., solely between substrate and kinase) or extrinsic mechanisms (i.e., requirement for additional factors). To explore intrinsic mechanisms of interdependence, we performed in vitro kinase reactions (16). We focused on comparisons of p38α with the prominent tau kinases GSK3β (37) and Cdk5 (38, 39). p38α showed extensive phosphorylation of recombinant tau in vitro at multiple sites (fig. S17A and table S5). Phosphorylation of recombinant tau when incubated with CDK5 was limited to specific sites, which correlated with interdependence relations predicted for this kinase (fig. S17B and table S5). Direct tau phosphorylation by GSK3β did not correlate with interdependence identified for this kinase (fig. S17B), suggesting that GSK3β requires extrinsic factors in the cellular context such as engagement of priming kinases for further tau phosphorylation (40). Levels and significance of recombinant tau phosphorylation and differential phosphorylation in p38α-deficient mice correlated with interdependence predicted for p38α (Fig. 5A and fig. S17B).
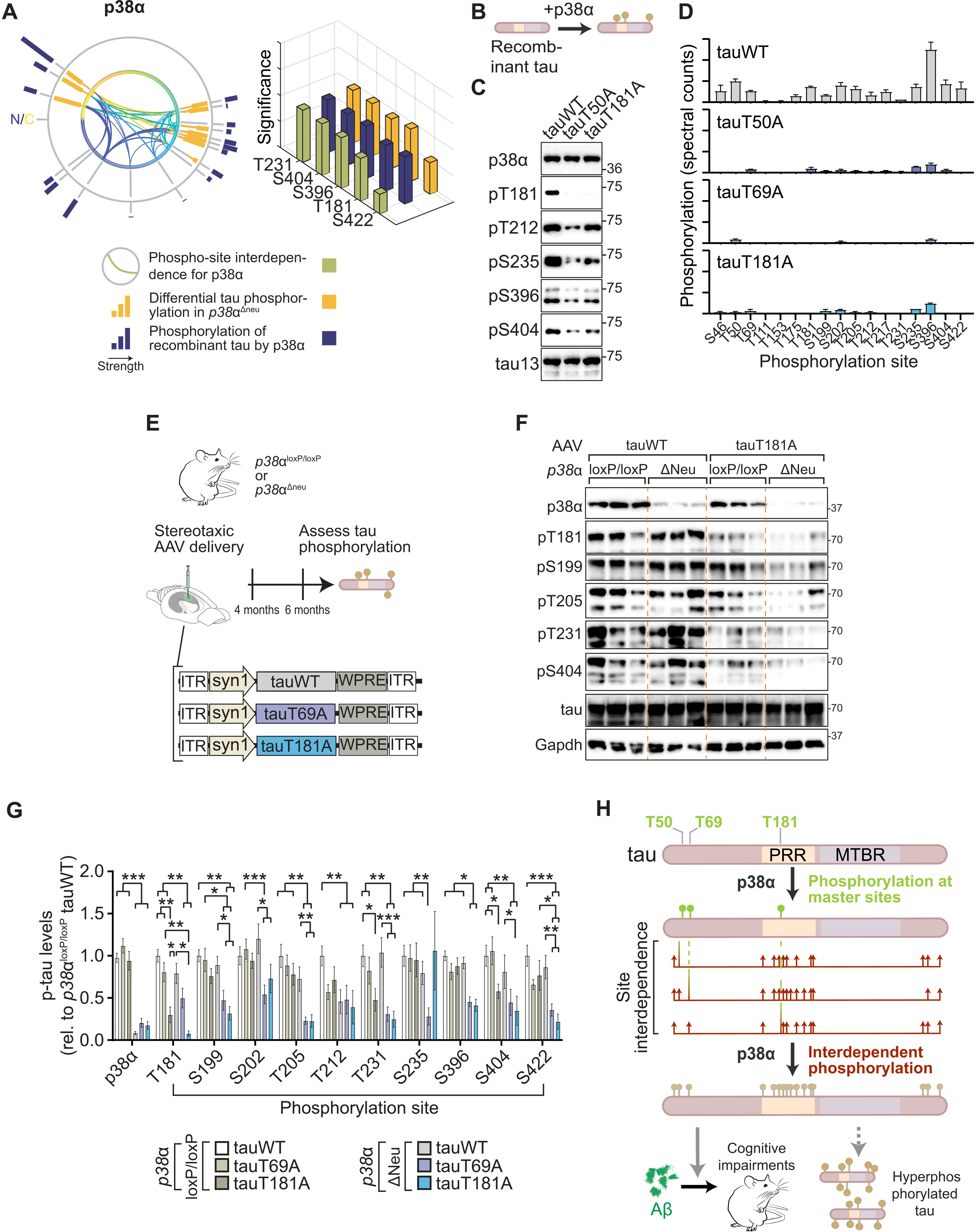
Removal of single master site and kinase p38α abolishes tau multi-site hyperphosphorylation.
(A) Mapping of tau site interdependence and differential levels of p38α-mediated tau phosphorylation. Levels (circular graph) and significance (bar chart) of differential tau phosphorylation in brains of p38α-deficient mice (p38αΔneu) and of recombinant tau by p38α correlates with predicted interdependence for p38α. (B) Schematic of in vitro kinase reactions of recombinant tauWT, tauT50A, tauT69A, or tauT181A with p38α to delineate intrinsic basis of tau phosphorylation interdependence. (C) Immunoblots (n = 6) and (D) phospho-proteomic quantification (n = 3) of kinase reactions of p38α with indicated tau variants showing markedly lower phosphorylation of recombinant tau variants at multiple epitopes. (E to G) Combination of master sites T50 and T181 and master kinase p38α governs phosphorylation of human tau. (E) Schematic of stereotaxic delivery of AAVs expressing human tau (tauWT) or indicated tau variants in hippocampus of p38αΔneu and control p38αloxP/loxP mice. (F) Representative immunoblots for tauWT- and tauT181A-expressing mice probed with phospho-tau epitope, tau, and p38α antibodies. n = 10 to 12. (G) Immunoblots were quantified relative to total tau and tauWT. Note that the absence of master sites T69 and T181 in human tau ablates the majority of tau phosphorylation in p38αΔneu compared with p38αloxP/loxP controls. (H) Hypothetical model of tau multi-site hyperphosphorylation governed by site interdependence and mediated by p38α. Initial phosphorylation by p38α at master sites, T50, T69, or T181, promotes subsequent multi-site phosphorylation by p38α. Thus, interdependence between master sites and other phospho-sites may promote tau hyperphosphorylation and contribute to Aβ-induced cognitive deficits. MTBR, microtubule-binding region. Data are presented as means ± SEM unless indicated otherwise. ***P < 0.001; **P < 0.01; *P < 0.05 (ANOVA).
To explore master sites in intrinsic p38α-tau mechanisms, we phosphorylated master site–deficient tau (i.e., tauT50A, tauT69A, and tauT181A) or WT tau with p38α (Fig. 5Band fig. S18). Ablation of T50, T69, or T181 rendered tau less amenable to phosphorylation by p38α at multiple sites (Fig. 5, C and D, fig. S18, and table S6), showing that modulation depending on these sites contributes to direct tau phosphorylation by p38α due to an intrinsic kinase-substrate mechanism. T181 phosphorylation by p38α preceded subsequent multi-site phosphorylation at PRR, and CTR epitopes in kinase reactions stopped at different time points (fig. S19). We next addressed whether these sites were decisive for multi-site phosphorylation by p38α specifically in neurons in vivo. While AAV-mediated delivery of human tauWT to p38αΔNeu brains did not result in lowered tau phosphorylation, likely due to presence of murine tau (13, 16), expression of human tauT69A or tauT181A in p38αΔNeu resulted in markedly lower phosphorylation at multiple sites, with pronounced synergism of kinase and master site ablation (Fig. 5, E to G, and fig. S20). In summary, phosphorylation of master sites acting in synergy with p38α is required for extensive and interdependent tau phosphorylation at multiple epitopes, accelerates aggregation of human tau in vitro, and contributes to Aβ-induced cognitive deficits in vivo (Fig. 5H and fig. S21).
DISCUSSION
In summary, our work details how interdependence mechanistically links site-specific tau phosphorylation to hyperphosphorylation and defines master sites that govern phosphorylation across the entire tau molecule. Site interdependence is a physiologic mechanism and is, notably, intrinsic to the tau protein. This may not only govern physiologic behavior of tau but may also promote hyperphosphorylation and gain of pathologic function. Moreover, it provides a concept for synergism in posttranslational modifications with specific master sites critically controlling global changes in multi-site substrates.
Our screen in 293T cells based on expression of human tau phospho-site variants and SP/TP kinases revealed a complex network of site interdependence. This proposes a hierarchy of tau phosphorylation events that includes long-range effects between sites in distal regions of tau. It surpasses simple directional priming of neighboring sites (41, 42). Long-range site interdependence between phospho-sites in the NTR, PRR, and CTR of tau differs from priming phosphorylation in tau and other multi-site kinase substrates (43, 44), as priming phosphorylation is limited by both proximity and directionality (45). Our results implicate T50, T69, T181, and T205 as key sites that drive multi-site phosphorylation of human tau. Site interdependence is deterministic of tau multi-site phosphorylation and, therefore, of critical interest for diagnostic and therapeutic approaches. Our study purposefully focusses on SP/TP phosphorylation of tau—the most abundant form of tau modification—to delineate the concept of site interdependence. However, detection limitations of phospho-tau immunoblots due to unavailability of antibodies for all SP/TP sites and of mass spectrometry due to low peptide abundance will entail future studies about interdependence of N-terminal SP/TP and non–SP/TP sites. Moreover, our work does not extend directly to tau kinases other than the main proline-directed kinases or to tau phosphatase, which both likely contribute to multi-site phosphorylated tau states (5, 7).
The principal concept of site interdependence applies to physiologic tau phosphorylation. Results from tauT205A and tauT205E mice present T205 as a physiologic master site of tau phosphorylation. They also show that site interdependence is a driving mechanism in multi-site phosphorylation of murine tau. Notably, T205 phosphorylation dampens neurotoxic Aβ signaling and maintains cognitive function (13, 16). However, engaging T205 phosphorylation may eventually promote physiologic multi-site and potentially hyperphosphorylation through site interdependence. It remains unclear, however, why interdependence mechanisms exist that promote tau multi-site modification in the healthy brain.
Aβ-induced cognitive deficits are dependent on tau (33, 34) and reconstituting tau reinstates cognitive deficits (13). APP23.tau−/− mice expressing tau lacking master site T181 did not show significant cognitive deficits and resulted in learning performance comparable to nontransgenic mice. A site-specific function of T181 cannot be excluded at this point. However, lack of master site T181 in human tau was clearly associated with reduced Aβ-induced impact on learning and memory and correlates with lower multi-site phosphorylation of tau at SP/TP and non–SP/TP sites. These results suggest that multi-site phosphorylation, as well as concomitant negative impact on cognitive function, can be alleviated by “breaking” individual master sites within tau.
Synergism of master sites and kinase activity converts p38α into a “master” tau kinase in vitro with isolated tau and in vivo in hippocampal neurons. Normally, p38α targets substrates at stringent consensus epitopes (46) in conjunction with docking motifs (47), neither of which are present in tau. Notably, removal of a single master site was sufficient to ablate p38α-mediated multi-site tau phosphorylation in isolated protein and brain. Thus, site interdependence can direct protein kinase activities toward multiple epitopes in multi-site substrates, such as tau. p38 kinases are highly similar, sharing ~60 to 70% sequence identity. However, they act differently toward tau in vivo (this study) and in short-term kinase reactions [(16) and this study]. This is unexpected because p38 kinases can phosphorylate conventional substrates with equal efficiency (48, 49). We compared tau phosphorylation patterns in p38αΔneu mice with those from p38β and p38γ knockout mice. Genetic deletion of p38β and p38γ does not markedly reduce multi-site tau phosphorylation, but rather at individual sites. This and our data on master site ablation in p38α kinase assays and in the living brain support that interdependence is a driving mechanism for p38α. Other prominent tau SP/TP kinases GSK3β and Cdk5 appear to differ in their mechanism of tau phosphorylation, potentially due to priming site effects by non–SP/TP kinases (50). Future studies will be needed to resolve the relation of p38α, GSK3β, and Cdk5 in tau phosphorylation.
Notably, expression of tau master site variants in p38αloxP/loxP control brains did not results in pronounced reduction of tau phosphorylation at multiple epitopes, unlike expression in p38αΔNeu or in APP23.tau−/− brains. This is likely due to presence of murine tau in the tau-competent genetic backgrounds and its impact on transgenic human tau (51). Mouse tau follows interdependence principles comparably to human tau as our results from tauT205 CRISPR mice indicate. Influence of murine tau on tau phosphorylation of human tau in tau-competent mice also raises the possibility that both intra- and intermolecular mechanisms determine interdependence and govern tau phosphorylation of a pool of tau proteins, at least in living cells.
Site interdependence can create stable species of multi-phosphorylation substrates, similar to other proteins with multi-site modification (19). Notably, multi-site phosphorylation of isolated tau by p38α strictly required master sites T50, T69, or T181 individually. However, these sites may not act independently in vivo as ablation of T205 in tauT205 CRISPR mice and expression of tau variants result in reciprocal changes in their phosphorylation abundance (determined by phospho-peptide mass spectrometry). T50 is not present in murine tau and may therefore promote multi-site phosphorylation specifically in human tau. Our results in cells and with isolated tau indicate that tau phosphorylation at master sites is mutually subject to interdependence effects by prior phosphorylation. This is also the case for T181 in human tau in vivo based on immunoblot and mass spectrometry data. In isolated tau, T181 phosphorylation preceded multi-site phosphorylation, indicating that master site phosphorylation may be an early event under experimental conditions with reduced parameters. Detection of phospho-T50 and phospho-T69 epitopes is limited by low levels in mass spectrometry data to conclude on interdependence between these two sites that specifically could not be determined. In recombinant phosphorylation of tau by p38α, T50 and T69 phosphorylation is reduced in tauT181A, indicating that under isolated reaction conditions, these sites are mutually interdependent.
Site interdependence may explain the abundance of specific tau phospho-epitopes in pathological tau within and outside the central nervous system (24), providing a novel rationale for phospho-tau–based biomarker design in tauopathies. Master site T181 phosphorylation bears strong potential as diagnostic biomarker (24). Ablating T181 resulted in marked reduction of tau multi-site phosphorylation. Thus, T181 in tau may serve as both a diagnostic and therapeutic target. Targeting tau master sites is likely applicable to neurological disorders beyond AD, such as frontotemporal dementia, Parkinson’s disease, stroke, and chronic traumatic encephalitis, in which pathologically phosphorylated tau is abundant (4, 52).
Overall, site interdependence may answer a central question in Alzheimer’s research: Why and how does the neuronal tau protein get progressively hyperphosphorylated? This raises the exciting possibility that therapeutic inhibition of master sites will block intrinsic augmenting mechanisms of tau phosphorylation, decrease levels of pathologic tau, and, thus, maintain healthy cellular function in AD and other tauopathies.