
Aβ mediates Sigma receptor degradation via CaN/NFAT pathway

By Min Fang, Pei Zhang, Yanxin Zhao, Aiping Jin, and Xueyuan Liu
Excerpt from the article published in American Journal of Translational Research 2016; 8(8): 3471–3481. Published online 2016 Aug 15. PMCID: PMC5009399
Editor’s Highlights
- Extracellular Aβ accumulation has been found as a major molecular mechanism underlying the pathogenesis of Alzheimer disease (AD).
- The abnormal Aβ accumulation may disrupt the intracellular Calcium homeostasis of neurons, resulting in neurotoxicity. The calcium influx may increase the calcineurin(CaN) activity.
- Then, CaN may increase the intracellular calcium concentration via homocysteine dependent way.
- Sigma-1R (σ-1 receptor) binds to BiP, forming the sigma-1R/BIP complex.
- BIP is a member of HSP70 family in the endoplasmic reticulum and may maintain the normal protein conformation, regulate protein spatial localization and recruit E3 ubiquitin ligase for ubiquitination.
- Aβ accumulation may induce calcium influx in neurons and activate CaN/NFAT signaling pathway to initiate BIP expression, which recruits ubiquitin ligase E3, leading to the ubiquitination and degradation of sigma σ-1 receptor.
- Thus, the neuroprotection of SigmaR is compromised, resulting in neuronal apoptosis.
Abstract
Sigma receptor is an endoplasmic reticulum protein and belongs to non-opioid receptor. Increasing evidence shows that Sigma receptor activation can significantly attenuate AD induced neurological dysfunction and the functional deficiency of Sigma receptor plays an important role in the Aβ induced neuronal loss. This study aimed to investigate the influence of extracellular accumulation of Aβ on the Sigma receptor expression. Our results showed the increase in extracellular Aβ had little influence on the mRNA expression of Sigma receptor, but gradually reduced its protein expression. Co-immunoprecipitation was employed to evaluate the interaction of Sigma receptor with other proteins. Results showed BIP could bind to Sigma receptor to affect the ubiquitination of Sigma receptor. Further investigation showed there was a NFAT binding site at the promoter of BIP. Then, Western blot assay was performed to detect NFAT expression. Results showed extracellular Aβ affected the nuclear translocation of NFAT and the CaN activity of NFAT also increased with the accumulation of extracellular Aβ. In this study, NFAT-BIP luciferase reporter gene system was constructed. Results showed NFAT was able to regulate the transcription of BIP. Thus, we speculate that extracellular Aβ accumulation may activate CaN/NFAT signaling pathway to induce chaperone BIP expression, which results in Sigma receptor ubiquitination and its degradation.
Introduction
Alzheimer disease (AD) is a common neurodegenerative disease in old people [1–3]. Progressive cognition impairment may cause the loss of social function and life skills in AD patients [4,5]. Extracellular Aβ accumulation has been found as a major molecular mechanism underlying the pathogenesis of AD [6–8]. The abnormal Aβ accumulation may disrupt the intracellular Calcium homeostasis of neurons, resulting in neurotoxicity. The calcium influx may increase the calcineurin (CaN) activity [9–12]. Then, CaN may increase the intracellular calcium concentration via homocysteine dependent way. Our results showed CaN activity increased significantly, and inhibition of CaN activity could significantly reduce the homocysteine induced cell injury [13]. In clinical trials, studies have shown that CaN expression increases dramatically in the brain of AD patients. Our in vitro experiment also reveals that Aβ is able to significantly increase CaN activity of hippocampal neurons.
Sigma receptor is an endoplasmic reticulum protein and belongs to non-opioid receptor family. Neurons, astrocytes and microglia have sigma receptor expression. In recent years, increasing evidence shows that sigma receptor activation is able to markedly attenuate the neurological dysfunction in AD patients, and patients with early AD have reduced expression of sigma receptor [14,15]. Under normal condition, sigma receptor may bind to BiP, which is essential for its location on the endoplasmic reticulum. Sigma receptor can regulate multiple cellular processes and the target proteins of sigma protein include ion channels (such as potassium, sodium and calcium ion channels on cell membrane), neurotransmitter receptors and protein kinases [16]. Available studies have confirmed that one of important functions of BiP is to recruit E3 ubiquitin ligase, leading to the ubiquitination and subsequent degradation of BiP, which is crucial for the maintenance of normal cell function. Whether Aβ induced degradation of sigma receptor is also dependent on this pathway has never been reported.
Further investigations of our group indicated the nuclear factor of activated T cells (NFAT) binding site at the transcription factor binding site of promoter of BIP gene. NFAT is a transcription factor with multiple regulatory activities. Some studies have shown that the pathogenesis of acute and chronic diseases such as AD, bronchial asthma, inflammatory bowel disease, diabetes, osteoporosis, arthritis and myocarditis is closely related to the abnormal activation of NFAT. The NFAT activity is regulated by the calcium/calmodulin-dependent protein phosphatase C. The abnormal Aβ accumulation induced calcium influx may cause abno.
Taken together, we hypothesize that Aβ accumulation may activate CaN/NFAT signaling pathway to induce chaperone BIP expression and then E3 ubiquitin ligase is recruited to induce the ubiquitination of sigma receptor, leading to its degradation, which may compromise the neuroprotection of sigma receptor. Our findings may provide new evidence for the degradation of sigma receptor in AD.
…
Results
Aβ25-35 activates CaN/NFAT pathway and reduces sigma-1R/BIP expression in N2A cells
Western blot assay of whole, nuclear and cytoplasmic proteins was used to investigate whether Aβ25-35treatment resulted in the activation of CaN/NFAT signaling and inhibition of Sigma-1R expression in N2A cells. Exposure of N2A cells to 5 μmol/L or 10 μmol/L Aβ25-35 for 48 h increased CaN activity in a concentration-dependent manner, as compared to untreated N2A cells (Figure 1A). Aβ25-35 (5 μmol/L or 10 μmol/L) also caused a concentration-dependent reduction in the cytoplasmic NFAT protein expression and a concentration-dependent increase in the nuclear NFAT protein expression (Figure 1B). This suggests that Aβ25-35 increases CaN activity, leading to the translocation of NFAT from the cytoplasm to the nucleus. Interestingly, the sigma-1R expression in N2A cells was reduced by Aβ25-35 in a concentration-dependent manner (Figure 1C) and FK506 (a CaN inhibitor) was able to alleviate the inhibitory effect of Aβ25-35 on sigma-1R expression (Figure 1D). These indicate that the Aβ25-35 induced inhibition of sigma-1R expression in N2A cells is mediated, at least in part, by the CaN/NFAT pathway. Target prediction showed the BiP promoter sequence was a potential transcription regulation target of NFAT. Consistent with this prediction, exposure of N2A cells to 5 μmol/L or 10 μmol/L Aβ25-35 for 48 h resulted in a concentration-dependent increase in both mRNA (Figure 3A) and protein expression (Figure 3B) of BiP. This implies that Aβ25-35 may induce the translocation of NFAT from the cytoplasm to the nucleus (Figure 1B) and then it binds to BiP promoter to induce BIP expression.
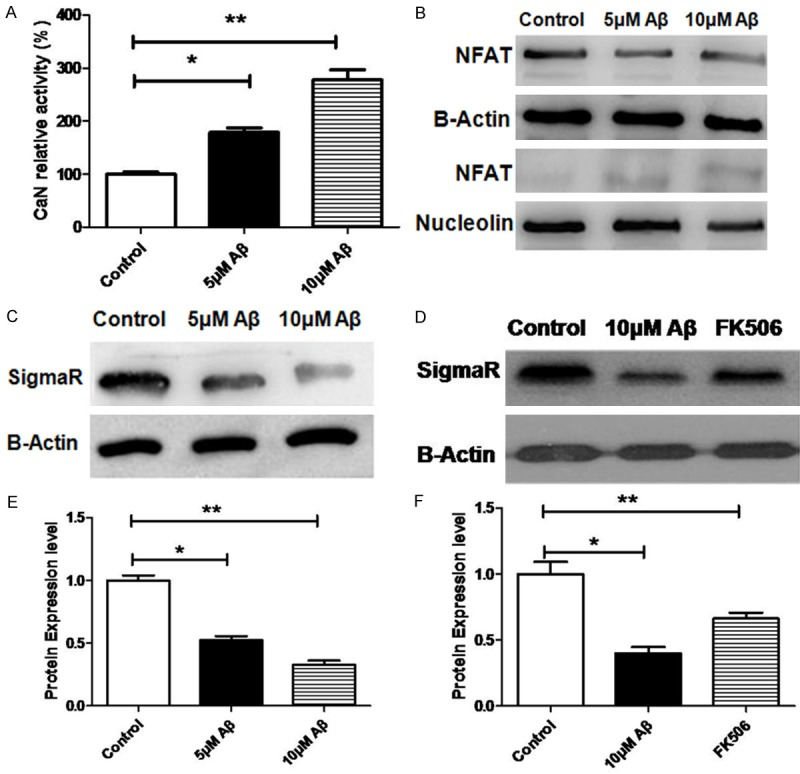
Aβ25-35 activates CaN/NFAT pathway and reduces Sigma-1R/BIP expression in N2A cells. A: CaN activity after treatment with Aβ at different concentrations. B: NFAT protein expression in the cytoplasm and nucleus after treatment with Aβ at different concentrations. C: SigmaR protein expression after treatment with Aβ at different concentrations. D: Influence of CaN inhibitor FK506 on SigmaR expression in N2A cells after treatment with 10 uM Aβ. C, E: Optical density in Western blot assay; D, F: Optical density in Western blot assay. All data are expressed as means ± SD (n=5/each group). *P<0.05, **P<0.01.
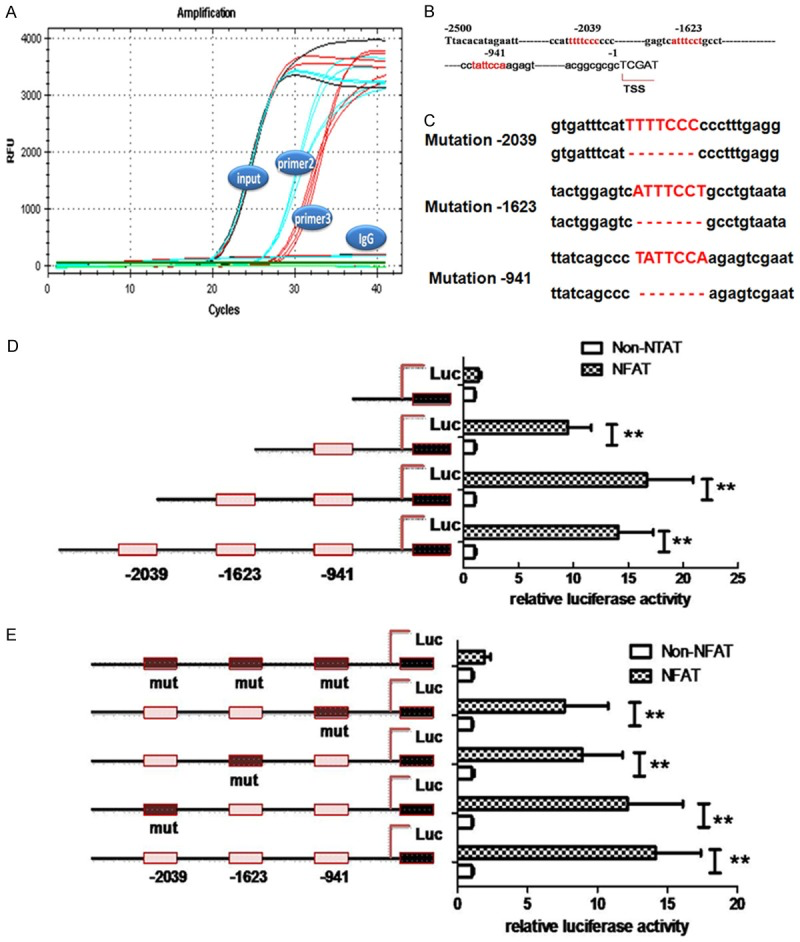
Transcriptional regulation of BIP by NFAT. (A) ChIP-PCR was used to detect the binding site of NFAT in BIP promoter. NFAT1 specific antibody was used to precipitate the corresponding genomic DNA segments and then real-time PCR was done to amplify the binding segments of NFAT1. input: DNA segments that were broken by ultrasound and not immunoprecipitated, but amplified with 3 pairs of primers. IgG: IgG was used to immunoprecipitate DNA segments that were broken by ultrasound, followed by amplification with 3 pairs of primers. primer2 and primer3: after immunoprecipitation with anti-NFAT1 antibody, the binding segments were amplified with 3 pairs of primers. Primer 2 and primer 3 were designed for the amplification of binding site at -1623 bp and -941 bp in (B), respectively. (B) Prediction of NFAT binding site in the BIP promoter. (C) Mutation binding sites of luciferase expression vector. In the synthesis of BIP promoter, deletion mutation was designed at 3 binding sites. (D) Effect of NFAT on the transcriptional activity of BIP promoters. BIP promoters with different length were cloned and contained different binding sites of NFAT. In 293T cells, the BIP promoter was cotransfected with NFAT1 gene, and then luciferase activity was detected. (E) Effect of NFAT on the transcriptional activity of BIP promoters with mutation at different sites. In the synthesis of BIP promoter, the deletion mutation was designed at the NFAT binding site. In 293T cells, the BIP promoter was cotransfected with NFAT1 gene, and then luciferase activity was detected. All data are expressed as means ± SD (n=5/each group). *P<0.05, **P<0.01.
BiP/sigma-1R complex influences sigma-1R ubiquitination and degradation in N2A cells
Co-IP was employed to investigate the interaction between BiP and Sigma-1R, and its possible effect on the Sigma-1R ubiquitination. In N2A cells treated with Aβ25-35 (5 or 10 μmol/L) for 48 h, sigma-1R and ubiquitin were both found to exist in the form of a complex that contained BiP (Figure 2A). To explore whether sigma-1R ubiquitination influenced sigma-1R expression, degradation of ubiquitin-conjugated proteins was inhibited using MG132 (a ubiquitin-proteasome inhibitor) or HRD-1 siRNA (silencing of E3 ubiquitin-protein ligase). Both MG132 and HRD-1 siRNA partially decreased the inhibitory effect of Aβ25-35 on sigma-1R protein expression in N2A cells (Figure 2B). This suggests that the Aβ25-35 induced inhibition of sigma-1R protein expression involves, at least in part, the ubiquitination and degradation of sigma-1R, and that this may be mediated by BiP.
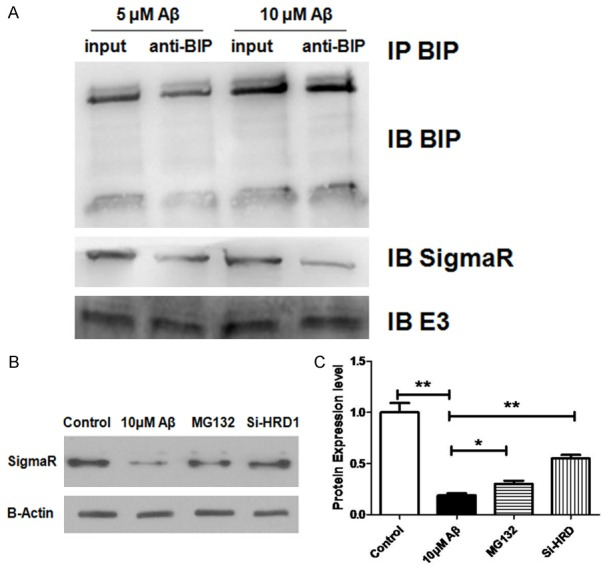
BiP/Sigma-1R complex influences Sigma-1R ubiquitination and degradation in N2A cells. A: BiP/Sigma-1R complex co-immunoprecipitation after treatment with Aβ at different concentrations. input: no beads and antibody after Aβ treatment; anti-BIP: binding protein in the presence of BIP antibody. Immunoprecipitation was performed with anti-BIP antibody and then Western blotting to detect the expression of BIP, SigmaR and E3 ligase in the complex. B: Effect of E3 ligase on SigmaR protein expression: E3 ligase inhibitor MG132 and E3 ligase specific siRNA were used to treat N2A cells after treatment with 10 uM Aβ, and then Western blotting was performed to investigate the influence of E3 ligase on SigmaR expression. B, C: Optical density in Western blotting. All data atre expressed as means ± SD (n=5/each group). *P<0.05, **P<0.01.
NFAT regulate BIP expression at transcription level in N2A cells
After Aβ treatment, genomic DNA was extracted from N2A cells and sonicated. Then, precipitation was done with NFAT specific antibody. According to the NFAT binding site determined by bioinformatics analysis, 3 pairs of primers were designed with input as a positive control and IgG as a negative control, followed by fluorescence PCR. As shown in Figure 3A, the target segments could be expanded in input group with 3 pairs of primers, but no signal was observed in IgG group. The second pair and third pair of primers could be used to expand target segment in input group, suggesting that there is a transcriptional binding site in the promoter of BIP (Hspa5) and its binding may initiate the BIP (Hspa5) transcription. We further constructed fluorescent reporter system of NFAT induced BIP transcription. As shown in Figure 3B, in the presence of complete BIP promoter, 1500 bp BIP promoter and 1000 bp BIP promoter, NFAT was able to induce the transcription of luciferase, but luciferase transcription was not induced in the presence of 500 bp BIP promoter, suggesting that there is no NFAT binding site in the 500 bp BIP promoter.
CaN/NFAT pathway is essential for the effects of Aβ25-35 on sigmaR/BIP expression
In Aβ25-35 treated N2A cells siRNA was used to silence CaN expression and then the CaN/NFAT pathway, sigma receptor and BIP expression were detected. Results showed CaN silencing significantly reduced the CaN expression and activity (Figure 4Aand and4B)4B) and also decreased the nuclear translocation of NAFT (Figure 4A). This indicates that siRNA mediated silencing of CaN is effective to block CaN/NFAT pathway. We further detected the mRNA and protein expression of BIP and sigma receptor. Results indicated that siRNA mediated silencing of CaN dramatically reduced the mRNA and protein expression of BIP, but sigma receptor expression increased markedly. This confirms that CaN/NFAT pathway is essential for the Aβ25-35 induced degradation of sigma receptor.
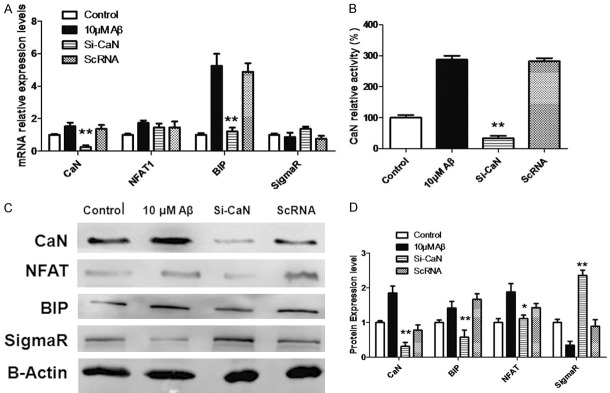
Effects of CaN/NFAT pathway on Sigma and BIP expression in N2A cells after Aβ25-35 treatment. A: Detection of mRNA expression. N2A cells were treated with both 10 uM Aβ and CaN specific siRNA. Normal cells served as a control and scramble ScRNA as a negative control. B: Detection of CaN activity. Specific siRNA was used to down-regulate CaN mRNA expression and then CaN activity was detected with corresponding kit. C: Western blotting was done to detect protein expression. After silencing of CaN expression, Western blotting was done to detect the protein expression of CaN, NFAT, BIP and SigmaR. C, D: Optical density was detected after Western blotting. All data are expressed as means ± SD (n=5/each group). *P<0.05, **P<0.01.
Blocking of CaN/NFAT pathway affects sigmaR expression in N2A expression
Immunofluorescence staining was done to detect the sigmaR expression in Aβ25-35 treated N2A in which the nucleus was blue (DAPI) and sigmaR was green. As shown in Figure 5, more blue fluorescence was observed in normal cells. After treatment with 10 μmol/L Aβ25-35, SigmaR expression reduced significantly, suggesting extracellular Aβ accumulation may affect sigmaR expression. After blocking CaN in CaN/NFAT pathway, sigmaR expression increased dramatically as compared to Aβ treated cells, but the sigmaR expression remained unchanged in random scRNA group as compared to Aβ treated cells. This indicates that extracellular Aβ accumulation may affect the sigmaR protein expression via CaN/NFAT pathway.
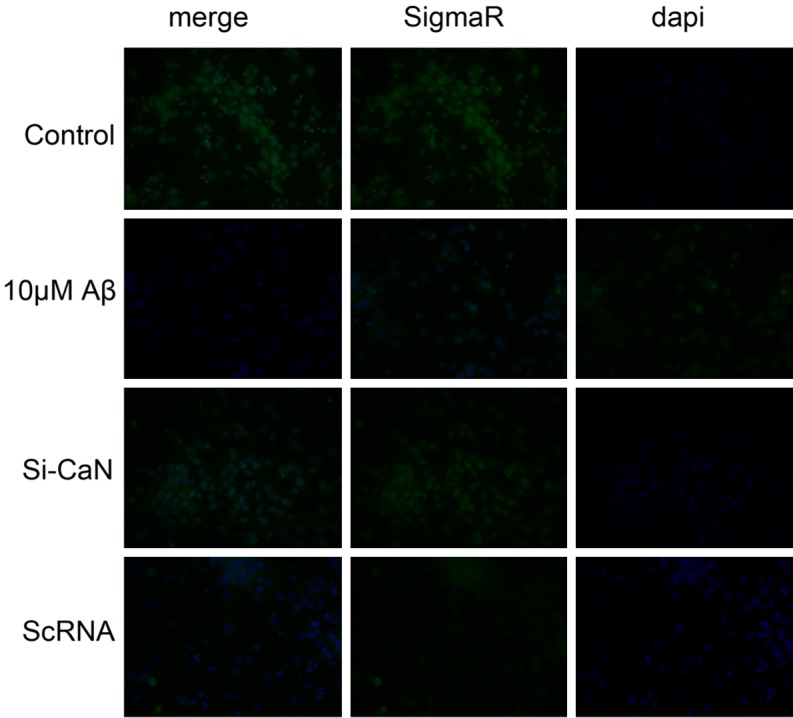
Effects of CaN/NFAT pathway interfering on SigmaR expression in N2A cells. N2A cells were treated with both 10 uM Aβ and specific CaN siRNA. Normal cells served as a control and scramble ScRNA as a negative control. After 48-h treatment, fluorescence intensity was measured. Blue: Dapi positive nucleus; Green: SigmaR.
Discussion
AD is a neurodegenerative disease characterized by progressive cognition impairment. Currently, there are no measures for the early prevention, diagnosis and effective treatment of AD, and it is estimated that there are more than 90 million people affected by AD worldwide in 2050 [17]. AD is pathologically characterized by neurofibrillary tangles, senile plaque formation and neuron loss. Aβ is a major component of senile plaque. It may selectively increase N-methy-D-aspartate (NMDA) receptor activity and induce the calcium influx via the NMDA receptor channel, resulting in calcium overload and subsequent neuronal death [18].
Sigma-1 (σ1) receptor is a G protein-coupled receptor containing 223 amino acid residues and has a high expression in the limbic system including the hippocampus. Recent studies indicate that Sigma receptor play important roles in the neuroprotection, improvement of learning disability and psychiatry treatment [19]. Some investigators employ PET for the detection of Sigma-1 (σ1) receptor, and results showed the Sigma-1 (σ1) receptor density and activity in the hippocampus and cortex of AD patients are significantly lower than in subjects
without cognition impairment. Under normal condition, Sigma-1 (σ1) receptor localizes the membrane of mitochrondion related endoplasmic reticulum and forms calcium sensitive chaperone structure with BIP. In the presence of specific activator (such as cocaine and pentazocine), Sigma-1 (σ1) receptor separates from BiP, translocates to other compartments in cells and then regulates IP3 receptor, NMDA receptor or dopamine receptor on endoplasmic reticulum or sodium, potassium and calcium ion channels [20–22]. In addition, it may also affect intracellular TCA cycle and ATP production, regulate oxidative stress and mitochondrial function, and modify the secretion of some neurotransmitters (such as glutamate, dopamine, norepinephrine, acetylcholine, γ-aminobutyric acid). Thus, it is generally accepted that Sigma-1 (σ1) receptor is crucial for the pathogenesis and development of AD. A variety of studies confirm that Sigma-1 (σ1) receptor is able to regulate PI3K-Akt-mTOR signaling pathway to attenuate Aβ toxicity, promote neuronal growth in the hippocampus and regulate the expression of apoptosis related genes (such as Bax, Bcl-2 and caspase-3) to inhibit neuronal apoptosis [23,24].
BIP is a member of HSP70 family in the endoplasmic reticulum and may maintain the normal protein conformation, regulate protein spatial localization and recruit E3 ubiquitin ligase for ubiquitination. BIP on the endoplasmic reticulum may bind to sigma σ-1 receptor, which is important for the maintenance of normal function of sigma σ-1 receptor [25,26]. In our study, real time PCR and Western blot assay were employed to detect BIP expression. Results showed the mRNA and protein expression of BIP reduced significantly. In addition, Western blot assay was employed to detect the protein expression of BIP and its binding protein. Results confirmed the binding between BIP and sigma σ-1 receptor, and ubiquitin group was also detectable in the complex. This phenomenon was reversed after treatment with ubiquitin ligase inhibitor and siRNA. Thus, we speculate that, after Aβ treatment, BIP recruits ubiquitin ligase E3, leading to the ubiquitination and degradation of sigma σ-1 receptor.
To further explore the mechanism underlying the Aβ induced BIP expression, bioinformatics was employed to analyze the promoter sequence of BIP gene. Results showed there were NFAT binding sites in the promoter of BIP gene. NFAT is a transcription factor family and plays important role in the gene transcription during immune response. The NFAT activation is regulated by calcium/calmodulin-dependent protein phosphatase C. NFAT is a factor that may regulate multiple cell processes such as T cell activation, differentiation and auto-tolerance. Several studies have confirmed that the pathogenesis of some acute and chronic diseases (such as bronchial asthma, Alzheimer’s disease and diabetes) is closely related to the NFAT activation. In our study, results showed Aβ induced CaN activation was able to induce the nuclear translocation of NFAT,
leading to the increase in transcriptional activity of NFAT. Bioinformatics analysis was employed to predict BIP (HSPA5) as a potential target of NFAT. Detection of BIP expression indicated that BIP expression was related to the nuclear translocation of NFAT. To further confirm whether NFAT is able to initiate BIP expression, NFAT-BIP, luciferase reporter gene expression system was constructed. In the presence of full length BIP promoter, the NFAT1 induced transcription of luciferase reduced. When the promoter was only 500 bp in length, NFAT failed to induce the transcription of luciferase. Experiment on the mutation of NFAT binding site in BIP promoter further confirmed the deficiency of NFAT binding site in the BIP promoter, which displayed a reduced transcription of BIP. When the three binding sites became mutant, NFAT failed to induce the transcription of luciferase. These findings indicate that NFAT is able to bind to the promoter of BIP gene to regulate BIP expression.
To investigate the role of CaN/NFAT pathway in the Aβ induced degradation of SigmaR, CaN was inhibited in vitro. In the presence of Aβ at a high concentration, siRNA target CaN was able to inhibit CaN and block CaN/NFAT signaling pathway. After inhibition of CaN mRNA expression, the CaN activity reduced in N2A cells, accompanied by reductions in NFAT expression and BIP expression and increase in sigmaR expression. This was further confirmed by Western blot assay and immunofluorescence. This suggests that CaN/NFAT is important for the Aβ induced SigmaR degradation. These findings provide evidence for the therapy of AD targeting SigmaR.
Taken together, our findings indicate extracellular Aβ accumulation may induce calcium influx in neurons and activate CaN/NFAT signaling pathway to initiate BIP expression, which recruits ubiquitin ligase E3, leading to the ubiquitination and degradation of sigma σ-1 receptor. Thus, the neuroprotection of SigmaR is compromised, resulting in neuronal apoptosis.