
Activation of Sigma-1 Receptor Alleviates ER-Associated Cell Death and Microglia Activation in Traumatically Injured Mice
By Mingming Shi, Liang Liu, Xiaobin Min, Liang Mi, Yan Chai, Fanglian Chen, Jianhao Wang, Shuyuan Yue, Jianning Zhang, Quanjun Deng, and Xin Chen
Excerpt from the article published in Journal of Clinical Medicine 2022, 11, 2348 Doi: https://doi.org/10.3390/jcm11092348
Editor’s Highlights
- The sigma-1 receptor (Sig-1R) is a ubiquitous ER-resident chaperone localized acting as a dynamic pluripotent modulator of fundamental cellular processes at the mitochondria-associated membranes (MAMs).
- It is involved in the pathogenesis of CNS diseases, especially in neurodegenerative diseases, such as Alzheimer’s Disease (AD), Huntington’s Disease (HD), Parkinson’s Disease (PD), and amyotrophic lateral sclerosis (ALS).
- Sig-1R is constitutively engaged in the modulation of UPR, in which Sig-1R directly or indirectly regulates three ER sensors and their downstream signalling pathways.
- Activation of Sig-1R attenuated ER stress-associated apoptosis, pyroptosis, and neuroinflammation, as well as restored cerebrovascular function and neurological function in mice subjected to traumatic brain injury (TBI).
- The inflammasome-associated protein expression of the NLR family, pyrin domain-containing 1 (NLRP1) and pyrin domain-containing 3 (NLRP3) were significantly increased post-TBI.
- Thus, Sig-1R activation may provide a promising strategy for translating into clinical therapeutic approaches targeting pharmacological treatment in patients with TBI.
Abstract
Background: Endoplasmic reticulum (ER) stress and unfolded protein response (UPR) are associated with neuroinflammation and subsequent cell death following traumatic brain injury (TBI). The sigma-1 receptor (Sig-1R) acts as a dynamic pluripotent modulator of fundamental cellular processes at the mitochondria-associated membranes (MAMs). The activation of Sig-1R is neuroprotective in a variety of central nervous system diseases, but its impact on ER stress-induced by traumatic brain injury is not known. This study investigated the role of Sig-1R in regulating the ER stress-mediated microglial activation and programmed cell death (apoptosis and pyroptosis) induced by TBI. Methods: Ten human brain tissues were obtained from The Tianjin Medical University General Hospital. Four normal brain tissues were obtained from patients who underwent surgery for cerebral vascular malformation, through which peripheral brain tissues were isolated. Six severe TBI tissues were from patients with brain injury caused by accidents. None of the patients had any other known neurological disorders. Mice with Sig-1R deletion using CRISPR technology were subjected to controlled cortical impact-induced injury. In parallel, wild-type C57BL/6J mice were analyzed for outcomes after they were exposed to TBI and received the Sig-1R agonist PRE-084 (10 mg/kg daily for three days) either alone or in combination with the Sig-1R antagonist BD-1047 (10 mg/kg). Results: The expression of Sig-1R and the 78 kDa glucose-regulated protein, a known UPR marker, were significantly elevated in the injured cerebral tissues from TBI patients and mice subjected to TBI. PRE-084 improved neurological function restored the cerebral cortical perfusion, and ameliorated and brain edema in C57BL/6J mice subjected to TBI by reducing endoplasmic reticulum stress-mediated apoptosis, pyroptosis, and microglia activation. The effect of PRE-084 was abolished in mice receiving Sig-1R antagonist BD-1047. Conclusions: ER stress and UPR were upregulated in TBI patients and mice subjected to TBI. Sig-1R activation by the exogenous activator PRE-084 attenuated microglial cells activation, reduced ER stress associated programmed cell death, and restored cerebrovascular and neurological function in TBI mice.
1. Introduction
Traumatic brain injury (TBI) is a major cause of mortality and long-term disability worldwide and a heavy burden to the economy [1]. Secondary brain injury occurs in hours and days following TBI, involving complex and interrelated pathologies that include cellular apoptosis [2], glutamate excitotoxicity [3], ferroptosis [4], pyroptosis [5], and neuroinflammation [6]. Extensive basic and clinical research on TBI in the past has not been translated into successful pharmacological interventions [7].
Secondary cerebral injuries resulting in persistent ER stress have been increasingly recognized for contributing to neuroinflammation and uncontrolled cell death [8,9]. Three signaling pathways of unfolded protein response (UPR) are extensively reported to regulate inflammatory and cell death signaling [10–12].
Mitochondria-associated membranes (MAMs) serve as a scaffold for the crosstalk between endoplasmic reticulum (ER) and mitochondria, thus playing a pivotal role in the signaling pathways that maintain cellular health. The sigma-1 receptor (Sig-1R) is a ubiquitous ER-resident chaperone localized at MAMs [13] of the central nervous system (CNS). It is involved in the pathogenesis of CNS diseases, especially in neurodegenerative diseases, such as Alzheimer’s Disease (AD) [14], Huntington’s Disease (HD) [15], Parkinson’s Disease (PD) [16], and amyotrophic lateral sclerosis (ALS) [17]. Sig-1R modulates the rate of cell apoptosis. For example, activating Sig-1R reduced ER stress following cerebral ischemic injury by preventing the protein kinase RNA-like ER kinase (PERK) and Inositol-requiring enzyme 1α (IRE1α)-mediated neural apoptosis [18]. Sig-1R also inhibited the production of inflammatory cytokines and reduced mortality by blocking the endonuclease activity of IRE1α in a preclinical rodent model of sepsis [19]. It has been reported previously that a Sig-1R agonist effectively inhibited microglia activation after neural injury [20], but the molecular mechanism of Sig-1R in the context of TBI remains unclear.
Here, we reported results from a study designed to investigate the role of Sig-1R in regulating TBI-induced ER stress and its biological and neurological consequences by analyzing cerebral tissue collected from TBI patients during decompressive craniotomy and by studying mouse models of TBI, including Sig-1R null mice. This study tested the hypothesis that activation of Sig-1R by an exogenous agonist inhibited ER stress-associated microglial activation, apoptosis, and pyroptosis to reduce neuronal cell death and improve cerebrovascular and neurological functions in the mouse model.
…
3. Results
3.1. TBI Upregulated the Expression of Sig-1R and GRP78 in TBI Patients and Mice Subjected to TBI
The clinical study comprised of six TBI patients and four cerebrovascular malformation patients. We detected significantly more Sig-1R and the UPR marker (GRP78) in the brain homogenates from TBI patients than those from AVM (0.401 ± 0.1177) (Figure 2A). Consistent with the patient study, the expression of Sig-1R in the ipsilateral cerebral hemispheres was increased in a time-dependent manner when compared with sham mice, reaching the peak level at 3d after TBI followed by decreasing to the level comparable to that of sham mice at day 7 post-surgery (Figure 2B). Sig-1R expression was significantly reduced in TBI mice infected with AAV9-Sig-1R (−0.293 ± 0.0751), but not changed in the TBI mice infected with the control vector (Figure 2C), suggesting that the Sig-1R gene was suppressed. The expression of GRP78 was significantly increased in the cerebral tissue from Sig-1R knockout mice 3 days after TBI as compared with control mice (AAV-9-NC) (0.425 ± 0.0515) (Figure 2C). Immunofluorescence staining showed that Sig 1R was primarily localized in astrocytes (GFAP) of the sham mice. In contrast, Sig-1R was not only abundantly expressed in astrocytes, but also in neurons (NeuN) and microglia (Iba-1) of TBI mice (Figure 2D).
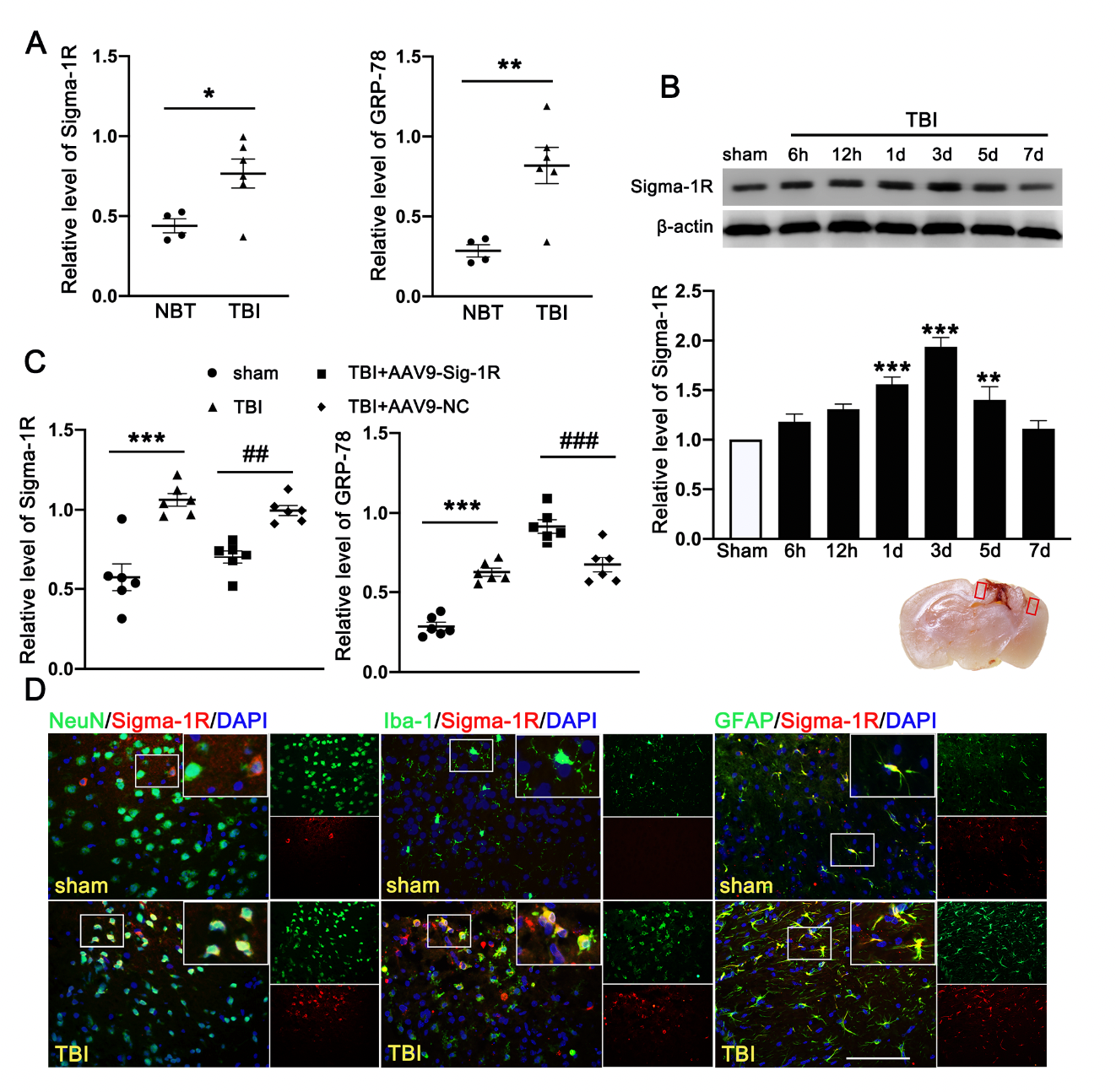
Time course expression and cellular localization of endogenous Sig-1R in mice subjected to TBI. (A) Quantitative analysis of the relative protein expression of Sig-1R and GRP78 in cerebral tissues from TBI (n = 6) and AVM patients (NBTs) (n = 4). (B) The time-dependent expression of Sig-1R in the ipsilateral cerebral hemispheres after TBI. n = 6 per group. (C) Quantitative analysis of the expression of Sig-1R and GRP78 in the ipsilateral cerebral hemispheres from TBI mice indicated that knocking out the expression of Sig-1R promoted increase of GRP78 expression after TBI. n = 6 per group. (D) Double immunofluorescence staining of Sig-1R (red) with neurons (NeuN, green), microglia (Iba-1, green), astrocytes (GFAP, green) showed that Sig-1R was primarily localized in astrocytes in the sham groups, whereas Sig-1R was abundantly expressed in neurons, microglia, and astrocytes in the ipsilateral peri-lesion cortex after TBI. Nuclei were stained with DAPI (blue). Two small red squares within the coronal section of the brain indicated the areas where the microphotographs were taken. n = 6 per group Scale bar = 100 μm. Data are represented as mean ± SEM. * p < 0.05, ** p < 0.01 and *** p < 0.001 vs. sham group; ## p < 0.01 and ### p < 0.001 vs. TBI +AAV9-NC group; one-way ANOVA, Tukey’s post hoc test.
3.2. Activation of Sig-1R Improved Neurological Outcomes and Cerebrovascular Function after TBI
Neurological function defined by mNSS was progressively and significantly impaired in TBI mice, being the worst on day 1 post-TBI and then gradually improving (Figure 3A). In contrast, mNSS was significantly improved in TBI mice receiving PRE-084, especially 5 days after TBI, as compared with those receiving the vehicle buffer (Figure 3A). The neuroprotective effects of PRE-084 were again reversed by BD-1047 from day 7 after TBI (Figure 3A). Next, we evaluated the performance improvement in PRE-084 treated mice. PRE-084 treated mice moved to the platform with a shorter latency when compared to vehicle or BD-1047 treated mice (Supplementary Figure S1A). Similarly, PRE-084 treated mice exhibited a significant performance for the correct quadrant and spent more time in correct quadrant (Supplementary Figure S1B). The effects of PRE-084 were blocked by the Sig-1R antagonist BD-1047 (Supplementary Figure S1B). PRE-084-treated TBI mice significantly reduced the volume of cerebral lesion at 14 d after TBI (−4.975 ± 1.334), as compared with TBI mice receiving the vehicle buffer, whereas BD-1047 reversed the protective effect of PRE-084 (4.475 ± 1.334) (Figure 3B).
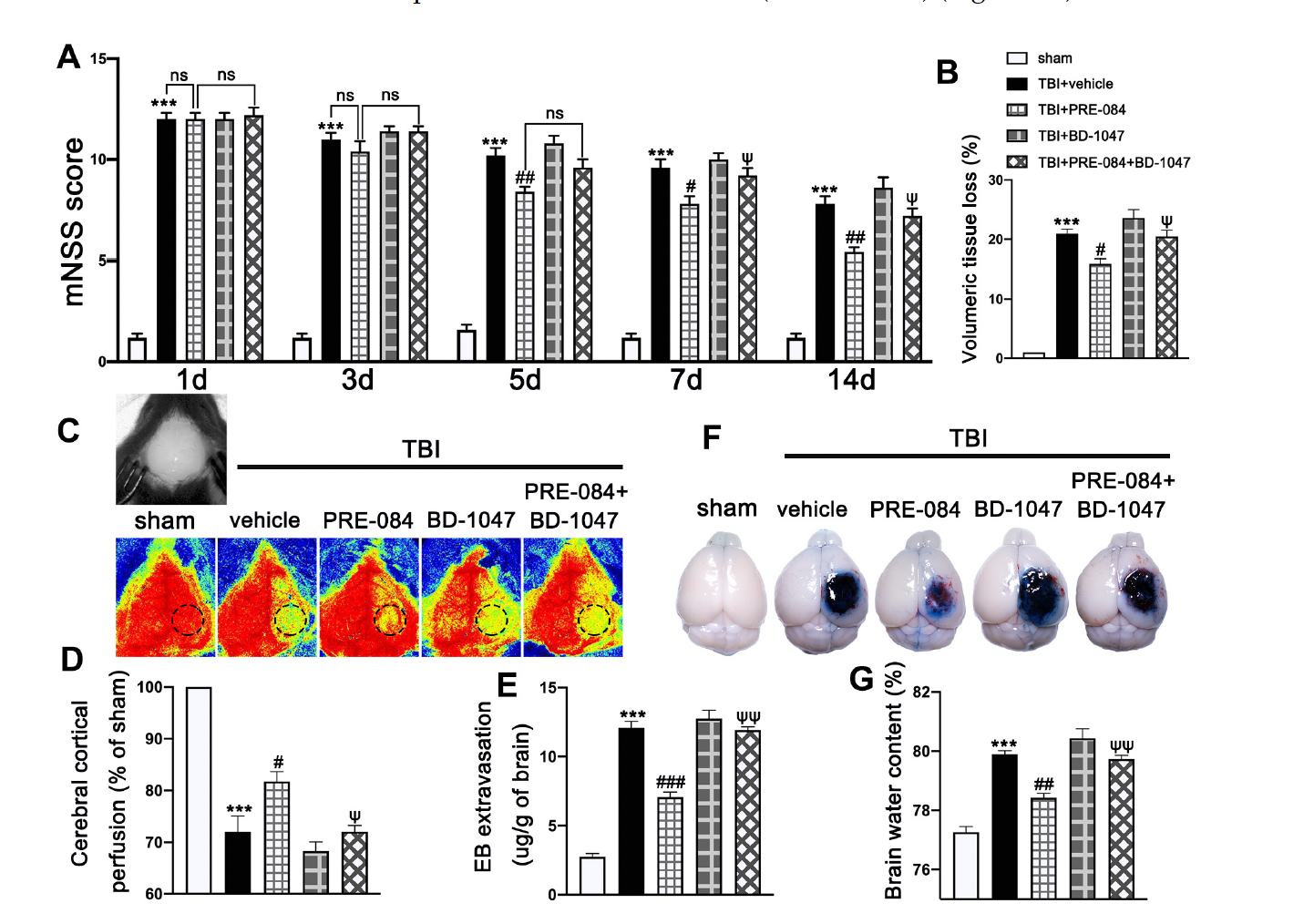
The effects of Sigma-1 receptor (Sig-1R) agonist PRE-084 and antagonist BD-1047 on neurological outcomes and cerebrovascular function post-injury. (A) Neurological function score of mice were assessed using modified neurological severity score (mNSS) at 1 d, 3 d, 5 d, 7 d, and 14 d after TBI. n = 12 per group. (B) Quantitative analysis of lesion volume at 14 d post-injury. n = 6 per group. (C) Representative cerebral cortical perfusion images and (D) quantitative analysis of cerebral cortical perfusion in cortical injury area (small black and dotted circle) after TBI. n = 6 per group. (E) Quantitative analysis of brain water content of mice in different groups at 3 d after TBI. n = 6 per group. (F) Representative images of Evans blue (EB) leakage and (G) quantitative analysis of EB extravasation at 3 d after TBI. n = 6 per group. Data are represented as mean ± SEM. *** p < 0.001 vs. sham group; # p < 0.05, ## p < 0.01 and ### p < 0.001 vs. TBI + Vehicle group; ψ p < 0.05, ψψ p < 0.01 vs. TBI + PRE-084 group; ns, no significance; one-way ANOVA, Tukey’s post hoc test.
The cerebral cortical perfusion surrounding the injury area was significantly reduced at 3 d after TBI compared with that of sham mice (−29.131 ± 2.699) (Figure 3C,D). Treatment with PRE-084 significantly increased the cerebral cortical perfusion when compared with the vehicle treatment. (11.600 ± 2.699). The Sig-1R antagonist BD-1047 given alone further reduced cerebral perfusion of TBI mice and also reversed the protective effect of PRE-084 (−10.370 ± 2.699) (Figure 3C,D). Similarly, cerebral water content in vehicle-treated mice was significantly higher than those treated with PRE-084 (1.467 ± 0.2176) (Figure 3E). Finally, BBB permeability qualified by Evans blue extravasation at 3 d after TBI was significantly reduced in PRE-84-treated mice (−5.025 ± 0.5812), but not in those receiving PRE-084 together with BD-1047 (Figure 3F,G).
Double immunofluorescence staining showed that TBI mice receiving the vehicle had less Claudin-5-positive vessels (labeled by the endothelial marker CD31) in lesioned boundary than sham mice at 3 d post-injury (−50.650 ± 5.762) (Figure 4A,B). PRE-084 restored the Claudin 5 expression (24.940 ± 5.762) and its effect was blocked by BD-1047 (−26.920 ± 5.762) (Figure 4A,B). This observation was further supported by the Western blots, showing that expression of Claudin-5 after TBI was significantly reduced (−0.334 ± 0.0647) (Figure 4C), which was rescued in mice receiving PRE-084 (0.210 ± 0.0647) and suppressed again in mice treated with PRE-084 and BD-1047(−0.319 ± 0.0647).
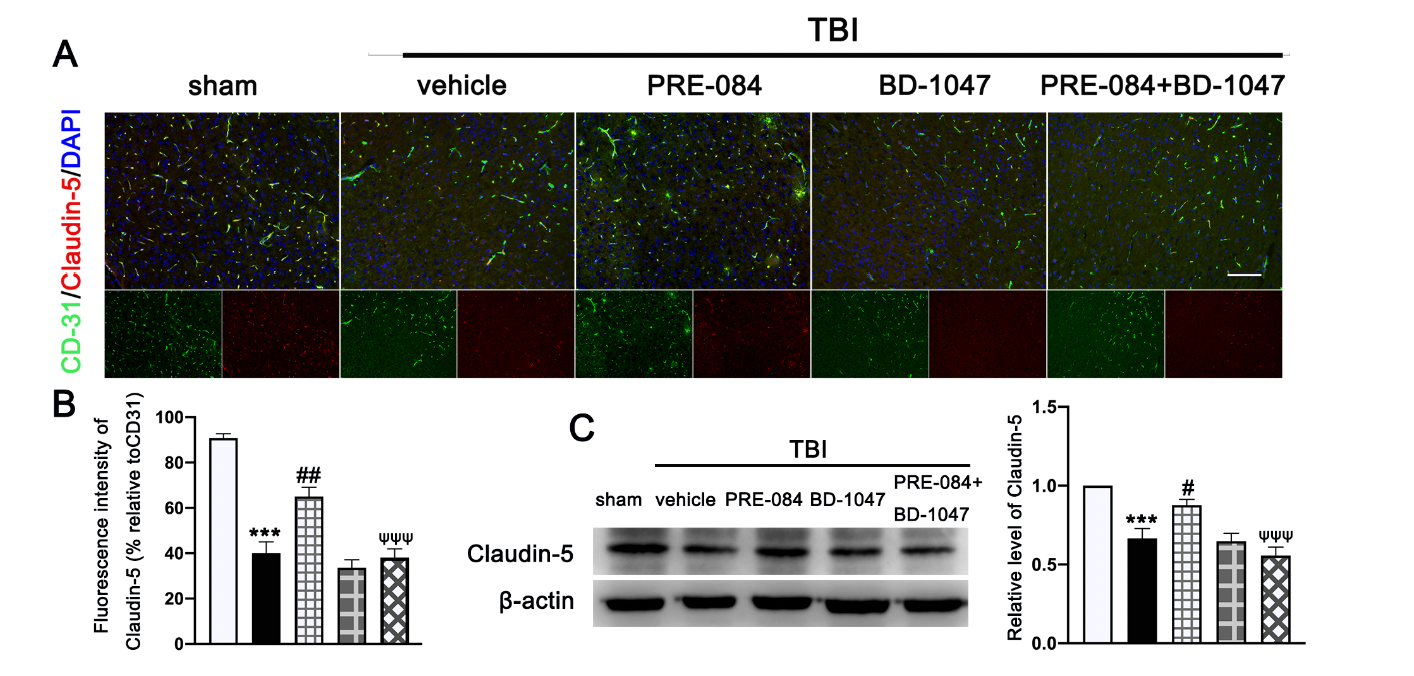
The effects of Sigma-1 receptor (Sig-1R) agonist PRE-084 and antagonist BD-1047 on blood
brain barrier (BBB) permeability after traumatic brain injury (TBI). (A) Representative double immunofluorescence staining of tight junction protein Caludin-5 (red) with platelet endothelial cell adhesion molecule 1 (CD-31) (green) and (B) quantitative fluorescence intensity analysis of Caludin 5 (relative to CD31) in the ipsilateral peri-lesion cortex after TBI. Nuclei were stained with DAPI (blue). n = 6 per group. Scale bar = 100 μm. (C) Representative Western blot band and quantitative analysis of the relative expression of Claudin-5 at 3 d after TBI. n = 6 per group. Scale bar = 100 μm. Data are represented as mean ± SEM. *** p < 0.001 vs. sham group; # p < 0.05, ## p < 0.01 vs. TBI + vehicle group; ψψψ p < 0.001 vs. TBI + PRE-084 group; one-way ANOVA, Tukey’s post hoc test.
3.3. PRE-084 Suppressed Mitochondrial Dysfunction and ER Stress-Mediated Neuronal Apoptosis after TBI
The protein expression of GRP-78 (0.693 ± 0.0431), p-PERK/PERK (1.387 ± 0.1366), p IRE1α/IRE1α (3.900 ± 0.591), and C/EBP-homologous protein (CHOP) (19.370 ± 1.335) were significantly increased at 3 d post-TBI (Figure 5A). PRE-084 prevented the increased expression of these factors and the inhibitory effects of PRE-084 were abolished by BD 1047 (Figure 5A). We also detected significant a decrease in the expression of B-cell lymphoma-2 (Bcl-2)/Bcl-2-associated X (Bax) (−0.760 ± 0.058) and an increase in cleaved Caspase-3 (0.7133 ± 0.0506) in ipsilateral cerebral hemispheres of TBI mice at 3 d post-injury (Figure 5B), indicating severe apoptosis and the production of mitochondrial ROS. Both processes were reduced in mice receiving PRE-084 after TBI when compared with TBI mice receiving the vehicle buffer (Figure 5B). The effects of PRE-084 were blocked by the Sig-1R antagonist BD-1047 (Figure 5B). Consistent with findings from immunoblots, flow cytometry analysis revealed that the mitochondrial ROS was markedly reduced in the ipsilateral cerebral hemispheres of TBI mice treated with PRE-084 as compared with those with the vehicle buffer (−0.215 ± 0.060), whereas BD-1047 treatment reversed the effect of PRE-084 (0.175 ± 0.060) (Figure 5C). Finally, TUNEL staining detected less TUNEL-positive neurons in the lesioned boundary of TBI mice receiving PRE-084 at 3 d post-TBI (−16.133 ± 3.500) (Figure 5D). This anti-apoptosis effect of PRE-084 was abolished in mice receiving PRE-84 and BD-1047 together (−22.075 ± 3.500).
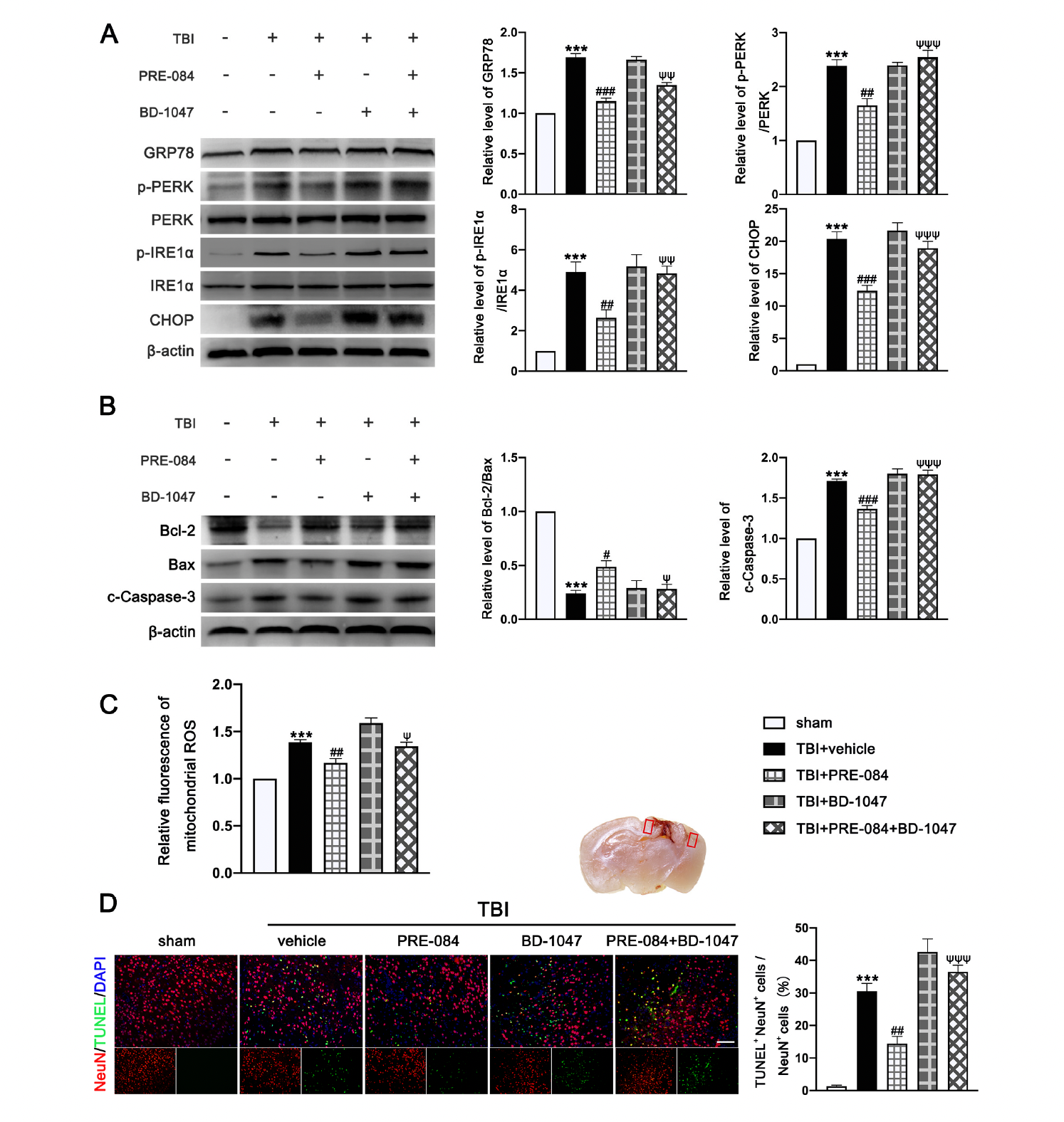
3.4. PRE-084 Attenuated Inflammasome-Mediated Pyroptosis
We first evaluated the expression of cleaved-caspase-1 p20 in the ipsilateral cerebral hemispheres by Western blot at 0 h (sham), 6 h, 12 h, 1 d, 3 d, 5 d, and 7 d after TBI. We found that c-caspase-1 p20 increased its expression in a time-dependent manner and peaked at 3 d after TBI mice (Figure 6A,B), followed by a gradual decrease, but it remained higher than that in sham mice at 7 d after TBI. Double immunofluorescence staining detected caspase-1-mediated pyroptosis in neurons (NeuN+), microglia (Iba-1+), and astrocytes (GFAP+) in the lesioned boundary at 3 d after TBI (Figure 6C). The necrotic and dead cells were visualized by PI staining at 3 d after TBI. PI-positive cell death was detected in neurons (NeuN), microglia (Iba-1), and astrocytes (GFAP) (Figure 6C), consistent with the result of cellular distribution of c-caspase-1 p20.
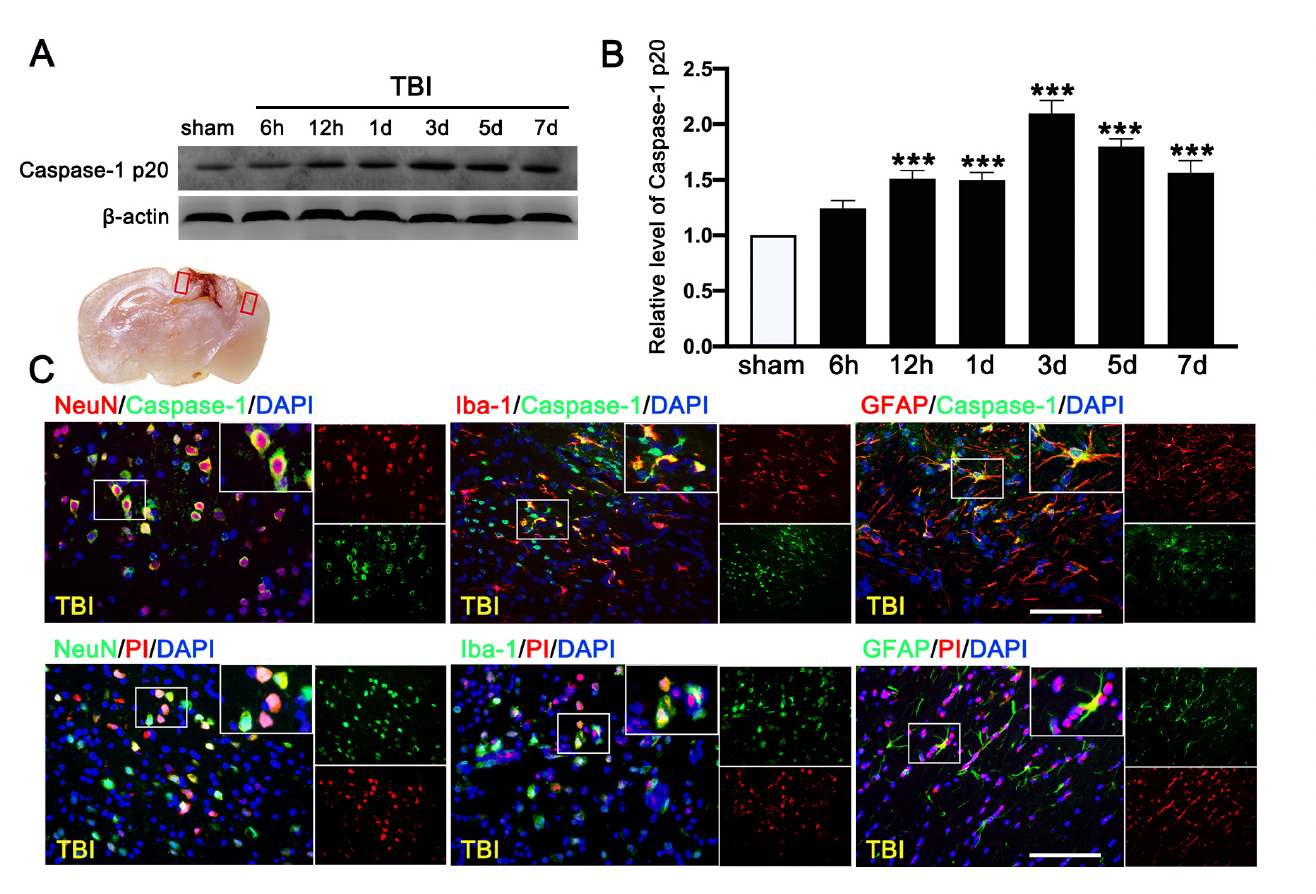
Time course and cellular location of Caspase-1-mediated pyroptosis after traumatic brain injury (TBI). (A) Representative Western blot and (B) quantitative analysis of the time-dependent expression of Caspase-1 p20 in the ipsilateral cerebral hemispheres after TBI. n = 6 per group. (C) Double immunofluorescence staining of Caspase-1 (green) with neurons (NeuN, red), microglia (Iba-1, red), astrocytes (GFAP, red) in the lesioned boundary of cortex after TBI and staining of propidium iodide (PI) (red) with neurons (NeuN, green), microglia (Iba-1, green), and astrocytes (GFAP, green) in the lesioned boundary of cortex after TBI. Nuclei were stained with DAPI (blue). n = 6 per group. Scale bar = 100 μm. Data are represented as mean ± SEM. *** p < 0.001 vs. sham group; one-way ANOVA, Tukey’s post hoc test.
The PRE-084 treatment significantly reduced the percent of Caspase-1+/PI+ cells and this anti-pyroptosis effect of PRE-084 was abolished in mice also receiving BD-1047 (Figure 7A). Moreover, the inflammasome-associated protein expression of the NLR family, pyrin domain-containing 1 (NLRP1) (2.533 ± 0.2357), NLR family, pyrin domain-containing 3 (NLRP3) (3.733 ± 0.3018), absent in melanoma-2 (AIM2) (3.633 ± 0.2757), adaptor protein apoptosis-associated speck-like protein-containing a caspase recruitment domain (ASC) (2.812 ± 0.2268), Caspase-1 p20 (1.050 ± 0.1651), Caspase-11 p20 (1.127 ± 0.1136), amino terminal-domain gasdermin D (GSDMD-N) (0.7997 ± 0.08744), interleukin 1β (IL-1β) (1.533 ± 0.2323), and interleukin 18 (IL-18) (1.057 ± 0.0726) were significantly increased post-TBI (Figure 7B). The upregulation of these proinflammatory markers was blocked by PRE-084 and enhanced by BD-1047 (Figure 7B). BD-1048 also reversed the PRE-084 effect.
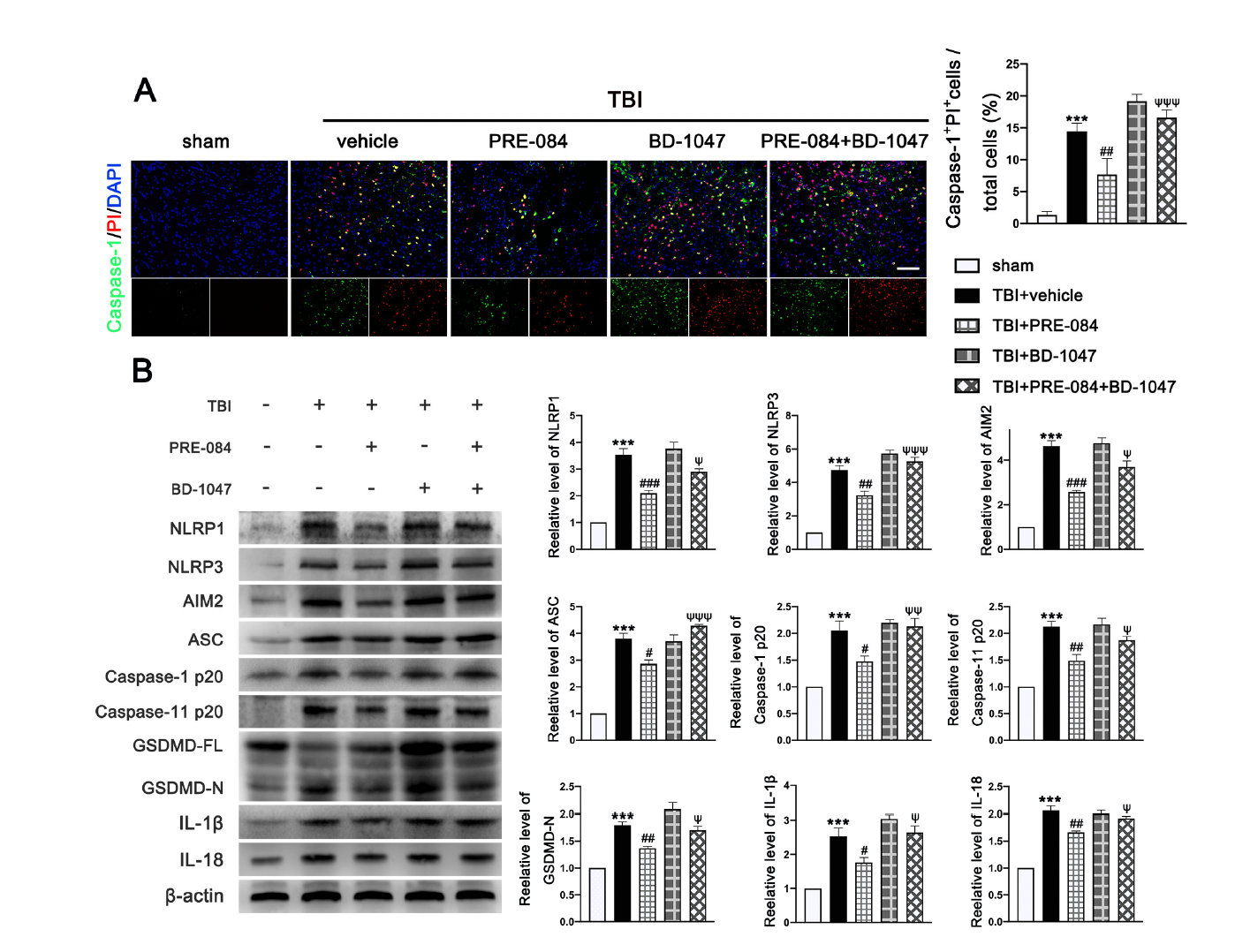
The effects of Sigma-1 receptor (Sig-1R) agonist PRE-084 and antagonist BD-1047 on inflammasome-mediated pyroptosis after traumatic brain injury (TBI). (A) Representative double immunofluorescence staining of Caspase-1 (green) with propidium lodide (PI) (red) in the lesioned boundary of cortex after TBI. Nuclei were stained with DAPI (blue) and quantitative analysis of Caspase-1+ PI+ cells/total cells in the lesioned boundary of cortex after TBI. n = 6 per group. Scale bar = 100 μm. (B) Representative western blot bands of NLR family, pyrin domain-containing 1 (NLRP1), NLR family, pyrin domain-containing 3 (NLRP3), absent in melanoma-2 (AIM2), adaptor protein apoptosis-associated speck-like protein-containing a caspase recruitment domain (ASC), Pro-Caspase-1, Caspase-1 p20, full-length gasdermin D (GSDMD-FL), amino terminal-domain gasdermin D (GSDMD-N), interleukin 1β (IL-1β), and interleukin 18 (IL-18) as well as quantitative analyses of the relative expression of proteins NLRP1, NLRP3, AIM2, ASC, Caspase-1 p20, GSDMD N, IL-1β, and IL-18 in the ipsilateral cerebral hemispheres after TBI. n = 6 per group. Data are represented as mean ± SEM. *** p < 0.001 vs. sham group; # p < 0.05, ## p < 0.01 and ### p < 0.001 vs. TBI + vehicle group; ψ p < 0.05, ψψ p < 0.01, ψψψ p < 0.001 vs. TBI + PRE-084 group; one-way ANOVA, Tukey’s post hoc test.
3.5. PRE-084 Promoted Microglia/Macrophages Activation and Inhibited Release of Inflammatory Cytokines following TBI
It has been well established that Sig-1R expressed at MAMs is involved in the activation of microglia in stroke [26], Parkinson’s disease [16], and Amyotrophic Lateral Sclerosis [27]. We found that the activated microglia/macrophages labelled with inducible nitric oxide synthase (iNOs) (a potential pro-inflammatory mediator) (40.750 ± 1.945) were significantly increased after TBI (Figure 8A). The microglial/macrophages activation was not significantly reduced in TBI mice receiving PRE-084 (Figure 8A). In contrast, the activated microglia stained with Arginase-1 (a potential anti-inflammatory mediator) were increased after TBI (17.450 ± 1.640) and remarkably accelerated by PRE-084 treatment (25.41 ± 1.640) (Figure 8A). This observation was further validated by immunoblotting of cerebral tissue homogenates (Figure 8B). More importantly, the Sig-1R antagonist BD 1047 also reversed this effect of PRE-084 (Figure 8B). Consistent with the microglial/macrophage cells undergoing activation, ipsilateral cerebral cells from TBI mice secreted significantly more pro-inflammatory mediators, tumor necrosis factor α (TNF-α) (3.357 ± 0.3739), and interleukin 6 (IL-6) (3.120 ± 1.962) than sham mice measured at 3 d post-TBI (Figure 8B). The development of this pro-inflammatory phenotype was prevented in TBI mice receiving PRE-084, the effect of which was blocked in mice that also received BD-1047 (Figure 8B).
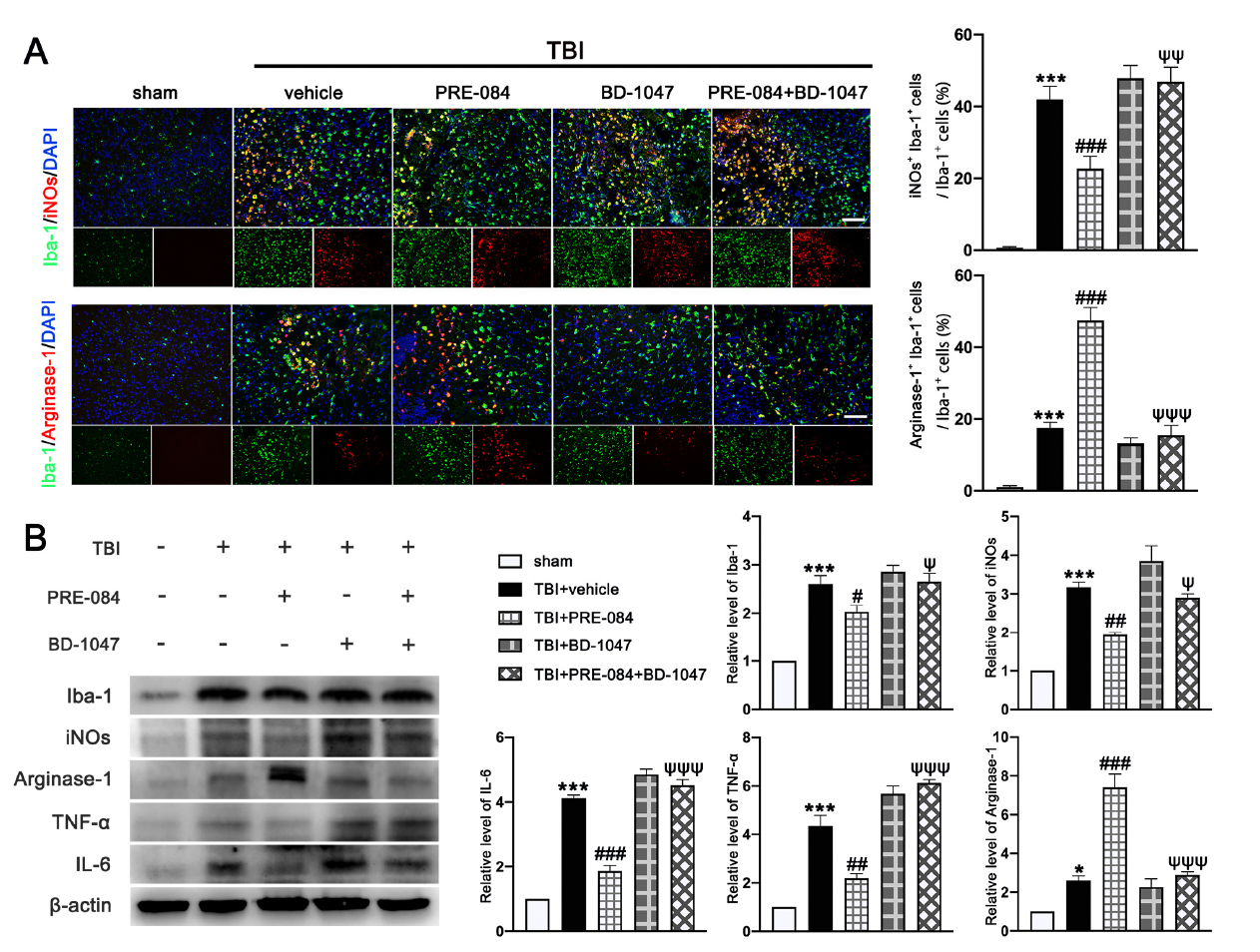
The effects of Sigma-1 receptor (Sig-1R) agonist PRE-084 and antagonist BD-1047 on regulating microglia/macrophages activation after traumatic brain injury (TBI). (A) Representative double immunofluorescence staining of microglia (Iba-1, green) with a potential pro-inflammatory phenotype marker inducible nitric oxide synthase (iNOs) (red) and a potential anti-inflammatory phenotype (M2) marker Arginase-1 (red) in the lesioned boundary of cortex after TBI and quantitative analysis of iNOs+ Iba-1+ cells/Iba-1+ cells and Arginase-1+ Iba-1+ cells/Iba-1+ cells in the lesioned boundary of cortex after TBI. (B) Representative Western blot bands and quantitative analyses of Iba-1, iNOs, Arginase-1, tumor necrosis factor α (TNF-α), and interleukin 6 (IL-6) in the ipsilateral hemispheres at 3 d after TBI. Data are represented as mean ± SEM. * p < 0.05 and *** p < 0.001 vs. sham group; # p < 0.05, ## p < 0.01 and ### p < 0.001 vs. TBI+ vehicle group; ψ p < 0.05, ψψ p < 0.01, ψψψ p < 0.001 vs. TBI + PRE-084 group; one-way ANOVA, Tukey’s post hoc test.
4. Discussion
In the current study, we first demonstrated that Sig-R and GRP-78 expression were significantly increased in TBI patients’ brain tissues, indicating that ER stress in the brain after TBI was evoked and meanwhile neural expression of Sig-1R as a part of the home defense mechanism was up-regulated. Consistently, we detected the same results in the brain of mice subjected to TBI. As previous studies described that Sig-R plays an important role in neuroprotection [20] but the molecular mechanism of Sig-1R in the context of TBI remains unclear. We then investigated the effects of Sig-1R on secondary brain injury following TBI and whether the mechanism by which Sig-1R exerts such strong neuroprotective was through inhibiting ER stress. First, we found that knockout of Sig-R by CRISPR/CAS9 in mice remarkably aggravated ER-stress after TBI. Then, we also detected that activation of Sig-1R with PRE-084 significantly alleviated the ER stress-induced cell death and microglia activation, and rescued the lesion volume, neurological deficits, and cerebrovascular dysfunction after TBI (Figure 9A,B). Conversely, these neuroprotective effects of PRE-084 were mostly blocked in the presence of Sig-1R antagonist BD-1047. All these results together indicate that activation of Sig-1R may provide strong neuroprotective and anti-inflammatory effects by inhibiting ER stress following TBI.
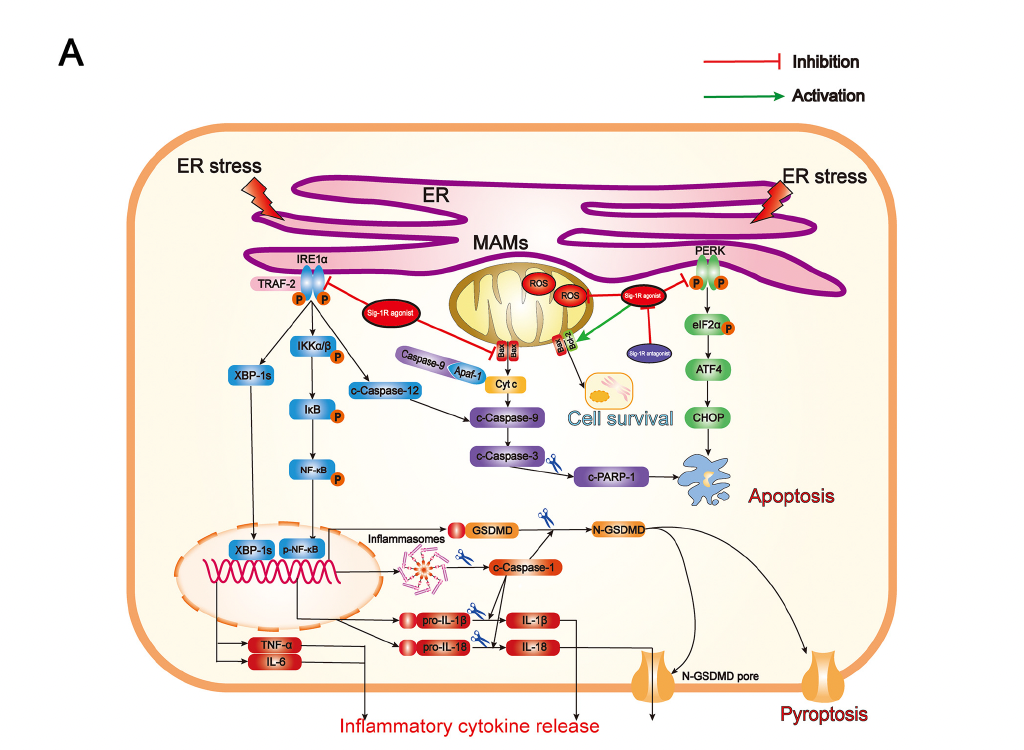
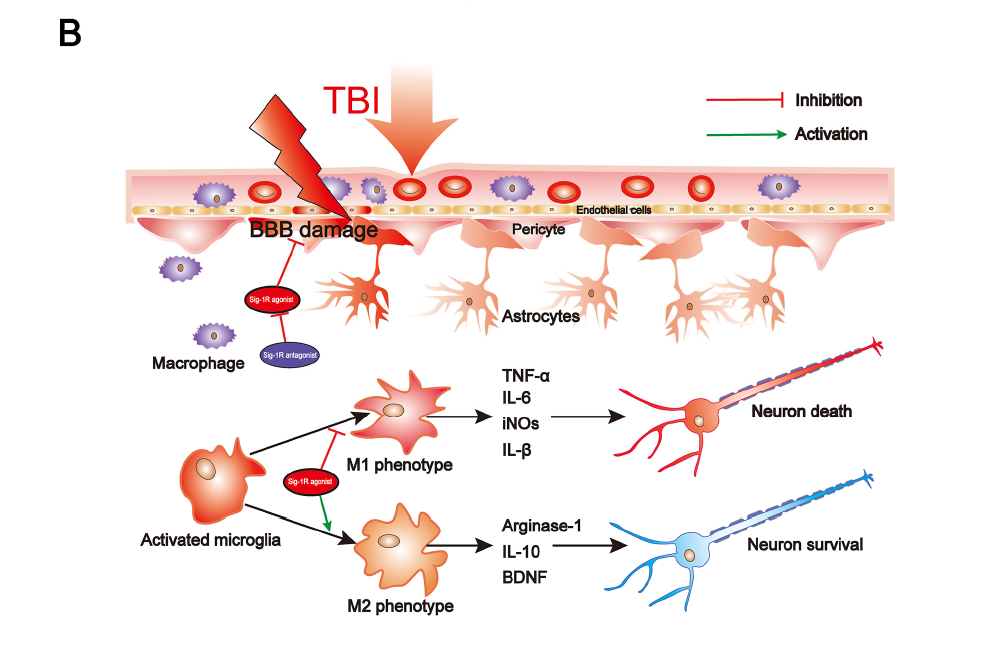
Summary of the underlying mechanisms of the protective effect of Sigma-1 receptor against endoplasmic reticulum stress-mediated apoptosis, pyroptosis, and inflammatory response in mice subjected to traumatic brain injury. (A) Sigma-1 receptor agonist inhibits endoplasmic reticulum stress-mediated and mitochondria-mediated apoptotic, pyroptotic and inflammatory pathways activation. (B) Sigma-1 receptor agonist confer neuroprotection via amelioration of blood-brain barrier damage, microglial activation and polarization, and neuronal death after TBI.
Secondary cell death and neuroinflammation are two major pathological hallmarks of TBI and are associated with cerebrovascular dysfunction and neurological deficits post-injury. Extensive findings have elucidated that persistent and devastating ER stress is involved in neuronal apoptosis after TBI and inhibition of ER stress significantly reduced neuronal apoptosis and improved neurological function post-injury [11]. Growing evidence has elucidated that, after TBI, inflammasome-mediated pyroptosis plays a critical role in determining cell fate and regulating immune response [5,28] and ER stress plays important roles in regulating inflammasome-mediated pyroptosis [29–31]. Neuroinflammation orchestrated by activated microglia plays an extremely critical role in aggravating secondary brain injury and deterring brain repair after TBI. Recently, it has been broadly demonstrated that ER stress plays a critical role in modulating microglia polarization and neuroinflammation, in which inhibition of ER stress, especially IRE1α and PERK-associated downstream signaling pathways, efficiently attenuates microglia-mediated neuroinflammation. As previous studies have identified that microglia/macrophages not only divide to M1 and M2 phenotypes, but also include Mhem, MHb, Mox, and M4 phenotypes [32–34], it is complicated to explore microglial activation phenotypes transformation during TBI in vivo. In the present study, we only detected that Sig-1R effectively inhibited microglia-mediated potential pro-inflammatory mediators release (iNOs, TNF-α, IL-6), but promoted microglia-mediated potential anti-inflammatory mediator expression (Arginase-1). Thus, the approach targeting the excessive ER stress-mediated cell death (apoptosis and pyroptosis) and microglia-mediated neuroinflammation could provide a promising therapeutic strategy for TBI.
Sig-1R is constitutively engaged in the modulation of UPR, in which Sig-1R directly or indirectly regulates three ER sensors and their downstream signaling pathways (Figure 9A). Previous studies have confirmed that, in response to ER stress, Sig-1R is mainly involved in regulating both UPR branches (PERK-mediated and IRE1α-mediated branches). Although numerous studies have revealed that activating Sig-1R efficiently suppressed apoptosis via the PERK/CHOP pathway [13,18,35–37], our present study is the first to document that activation of Sig-1R with the selective agonist, PRE-084, in mice subjected to TBI significantly decreases PERK/CHOP pathway-related neuronal apoptosis. In addition, in a previous in vitro study using immunoprecipitation assay, it was found that, upon ER stress, Sig-1R directly interacted with IRE1α and regulated its dimerization and phosphorylation [38]. It has been confirmed that Sig-1R activation efficiently reduced the expression of p-IRE1α in response to severe ER stress following cerebral ischemia injury [18,36]. Collectively, these observations first unraveled the underlying mechanism by which Sig-1R activation may modulate ER stress-associated apoptosis, pyroptosis, and microglia-mediated neuroinflammation in a mouse model of TBI.
Several limitations of the present study need to be acknowledged. First, since the clinical data of TBI patients were mostly patients with polytrauma, even if we obtained the insult brain for research, it is not enough to prove that the up-regulation of ER stress responses is simply due to brain trauma, which may be due to other system/organ injuries. Second, since the double immunofluorescence staining showed that Sig-1R was extensively expressed in neurons, microglia, and astrocytes, further investigations are necessary to reveal the roles of Sig-1R in astrocytes after TBI. In addition, we did not specifically demonstrate the mechanisms by which Sig-1R modulates IRE1α and PERK. Although the transient interaction between Sig-1R and IRE1α has been elucidated in a previous study, the dynamical interaction was not unraveled with the administration of diverse Sig-1R ligands. Similarly, the interaction between Sig-1R and PERK remains poorly understood. Thus, further studies using co-immunoprecipitation (Co-IP) and/or other assays are needed to determine the detailed interactions between Sig-1R, IRE1α, and PERK. Last but not least, in this study, the mechanism by which Sig-1R exerts such profound anti-inflammatory and neuroprotective activities was not demonstrated. Further studies are needed to explore other Sig-1R-mediated signaling pathways that modulate cell death and immune responses in the context of brain insult.
5. Conclusions
In conclusion, the present study first demonstrated that GRP-78 expression was significantly increased in both TBI patients brain samples and TBI mice brain samples, indicating that ER stress in brain after TBI was evoked. Then, we detected that the neural expression of Sig-1R as a part of the home-defense mechanism was up-regulated in brain tissues from both TBI patients and TBI mice. Then, we found that activation of Sig-1R with PRE-084 attenuated ER stress-associated apoptosis, pyroptosis, and neuroinflammation, as well as restored cerebrovascular function and neurological function in mice subjected to TBI. Thus, Sig-1R activation may provide a promising strategy for translating into clinical therapeutic approaches targeting pharmacological treatment in patients with TBI.