
The Sigma 2 receptor promotes and the Sigma 1 receptor inhibits mu-opioid receptor-mediated antinociception

By Sánchez-Blázquez, P., Cortés-Montero, E., Rodríguez-Muñoz, M. et al.
Excerpt from the article published in Mol Brain 13, 150 (2020). https://doi.org/10.1186/s13041-020-00676-4
Editor’s Highlights
- σ2R is essential for the antinociceptive effects of exogenous and endogenous ligands of MOR but not for the antinociceptive effects of other families of G-receptors that also mediate analgesia, such as DOR, CB1R and α2AR.
- σ1R inhibits and σ2R facilitates MOR-mediated analgesia these receptors exchange their roles when regulating neuropathic pain perception
Abstract
The Sigma-1 receptor (σ1R) has emerged as an interesting pharmacological target because it inhibits analgesia mediated by mu-opioid receptors (MOR), and also facilitates the development of neuropathic pain. Based on these findings, the recent cloning of the Sigma-2 receptor (σ2R) led us to investigate its potential role as a regulator of opioid analgesia and of pain hypersensitivity in σ2R knockout mice. In contrast to σ1R deficient mice, σ2R knockout mice developed mechanical allodynia following establishment of chronic constriction injury-induced neuropathic pain, which was alleviated by the σ1R antagonist S1RA. The analgesic effects of morphine, [D-Ala, N-MePhe, Gly-ol]-encephalin (DAMGO) and β-endorphin increased in σ1R−/− mice and diminished in σ2R−/− mice. The analgesic effect of morphine was increased in σ2R−/− mice by treatment with S1RA. However, σ2R−/− mice and wild-type mice exhibited comparable antinociceptive responses to the delta receptor agonist [D-Pen2,5]-encephalin (DPDPE), the cannabinoid type 1 receptor agonist WIN55,212-2 and the α2-adrenergic receptor agonist clonidine. Therefore, while σR1 inhibits and σ2R facilitates MOR-mediated analgesia these receptors exchange their roles when regulating neuropathic pain perception. Our study may help identify new pharmacological targets for diminishing pain perception and improving opioid detoxification therapies.
Introduction
Sigma receptors (σRs) are unique transmembrane proteins expressed throughout the central nervous system and in certain peripheral tissues. Based on current classifications, there are two types of these receptors, namely, the sigma-1 receptor (σ1R) and the sigma-2 receptor (σ2R) [1,2,3,4]. The σ1R was initially identified in 1976 as a member of the plasma membrane opioid receptor family [5], while σ2R was not discovered until later. For many years, σRs were described to bind to radioligands in preparations of brain synaptosomes. [3H]( +)-pentazocine exhibits a high affinity for σ1R, whereas [3H]DTG binds with equal affinity to both σ1R and σ2R. Subsequent studies have revealed that these proteins are also involved in intracellular ion regulation and neuron survival [1, 4, 6,7,8].
The σ1R was purified, sequenced and cloned from guinea pig brain in 1996, and it bears little sequence homology to any known mammalian receptor [9]. On the other hand, it has been postulated that σ2R complexes with progesterone receptor membrane component 1 (PGRMC1). The recent molecular cloning of σ2R identified this protein as the TMEM97 protein [10,11,12]. Some evidence suggest that σ2R is also involved in cholesterol trafficking and homeostasis [13] and in the regulation of intracellular calcium levels [14]. Notably, σ2R is involved in several disease states, and the utility of its exogenous ligands as cancer therapeutics and diagnostic tools has been reported [15,16,17]. Additionally, σ2R has been implicated in multiple neurodegenerative and neurological disorders [18, 19]. Similar to the ligands of σ1R, certain molecules that bind to σ2R also reduce mechanical hypersensitivity in a spared nerve injury model [20].
The availability of σ2R−/− (σ2R knockout) mice, deficient in TMEM97/σ2R, have allowed us to investigate the potential role of this receptor in pain sensitivity. Because σ1R participate in a tonic anti-opioid system [21, 22], we also evaluated the capacity of σ2R to modulate opioid induced analgesia. We observed that σ2R-deficient mice do not exhibit overt physical or behavioral abnormalities. Most importantly, we found that σ2R contributes to the analgesic effects of MOR agonists but not those of delta opioid or cannabinoid type 1 receptor agonists.
…
Results
Characterization of σ2R −/− mice
We confirmed that σ2R−/− mice (KOMP Repository, MMRRC Stock #: 050148-UCD, Davis, CA, USA) did not express σ2R mRNA in brain tissue (Fig. 1a). Targeted deletion of the σ2Rgene was not accompanied by compensatory changes in the levels of mRNAs encoding critical proteins in our study, such as σ1R, MOR or MOR- and σ1R-regulated histidine triad nucleotide-binding protein 1 (HINT1) (Fig. 1b). σ2R-deficient mice bred normally and did not present evident physical or behavioral abnormalities at birth. At weaning (3 to 6 weeks old), σ2R−/− mice were smaller than WT mice (p < 0.05). However, by week 8, the differences in body weight were no longer significant. The locomotor performance of the mice was then evaluated by analyzing three basic parameters: horizontal activity, time spent in the center area, and rearing. While σ2R−/−mice exhibited a similar exploratory behavior and rearing activity as control, they exhibited increased activity and spent more time in the center area (Fig. 1c). The motor coordination of both groups of mice was comparable when evaluated with an accelerating rotarod.
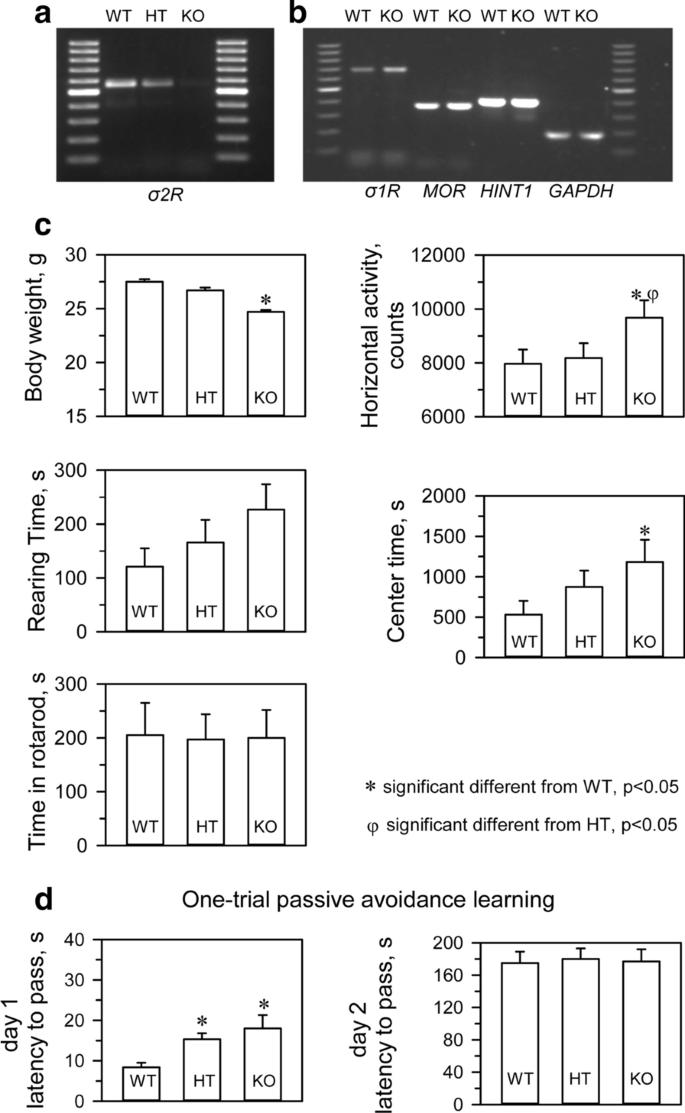
The WT and σ2R−/− mice were also subjected to an inhibitory avoidance paradigm that tests cognition/memory. A retention trial was conducted 24 h after the training trial. No significant differences were observed between WT and σ2R−/− mice in the retention trial. It should be noted that both groups did show an increase in latency in the retention trial compared to the training trial, which was interpreted as learning (Fig. 1d).
Chronic constriction injury in WT and σ2R −/− mice
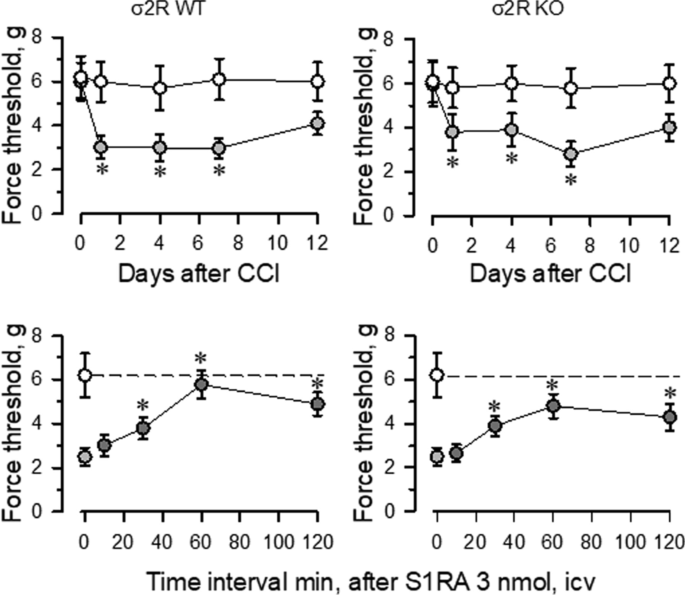
Mice with CCI-induced neuropathic pain display a series of behavioral and molecular changes that are diminished upon treatment with antiallodynic substances such as σ1R antagonists [29]. Thus, we assessed the possible relevance of σ2R in the development of neuropathic pain. Nerve-injured WT and σ2R−/− mice maintained a healthy appearance and were well groomed. The body weights of both groups of mice decreased after surgery but returned to preoperative values within 2–4 days. Before surgery (day 0), WT and σ2R−/− mice displayed similar responses to the mechanical nociceptive stimulus (Fig. 2). Seven days after surgery, sham-operated and CCI-exposed σ2R−/− mice displayed similar responses of the contralateral paw as WT animals. On the other hand, from 1 to 7 days after surgery both groups of CCI mice showed identical levels of allodynia in the ipsilateral nerve-injured legs, and on days 12 to 15, the nociceptive responses of both groups of CCI mice returned to presurgery levels. Icv administered S1RA (E-52862), a highly selective σ1R antagonist [30], reduced the allodynia induced by the CCI model in WT and σ2R−/− mice. The peak antiallodynic effect was observed 60 min after the administration of S1RA (Fig. 2).
Fig. 2
Influence of σ2R on the antinociceptive response to morphine
Icv administered morphine produces a dose-dependent antinociceptive effect when evaluated by the thermal tail-flick test (Fig. 3). In WT mice, the antinociceptive effect peaked approximately 30 min after injection and decreased after 120 min. The effect of morphine in σ2R−/− mice was significantly lower than in WT animals (Fig. 3a). The apparent ED50 of icv-administered morphine was 4.84 nmol (95% confidence interval: 3.63–6.43) for control mice and 22.10 nmol (19.34–24.72) for σ2R−/− mice (Fig. 3b). Basal latencies were not different between σ2R−/− mice and WT mice (1.61 ± 0.14 and 1.74 ± 0.13, respectively; n = 10).
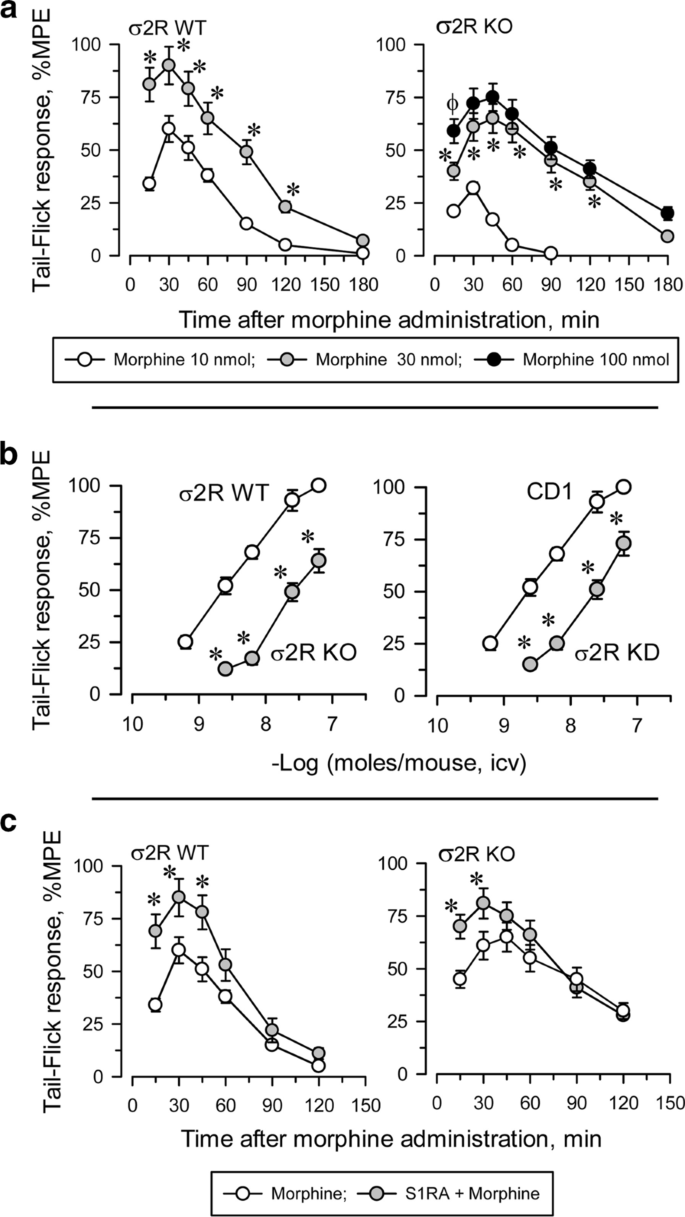
Antisense oligodeoxynucleotides are useful tools for reducing neural protein expression, and their selectivity in terms of related signaling proteins has been described elsewhere [26, 31]. We observed that the response of σ2R−/− mice and σ2R knockdown mice to morphine were identically decreased (Fig. 3b). It is known that in naïve mice, the administration of S1RA increases morphine antinociception [22, 30]. The ED70 of icv morphine in our analgesic paradigm was 10 nmol in WT mice and 30 nmol in σ2R−/− mice. Icv administered S1RA (3 nmol) increased the analgesic activity of morphine in both groups of mice (Fig. 3c).
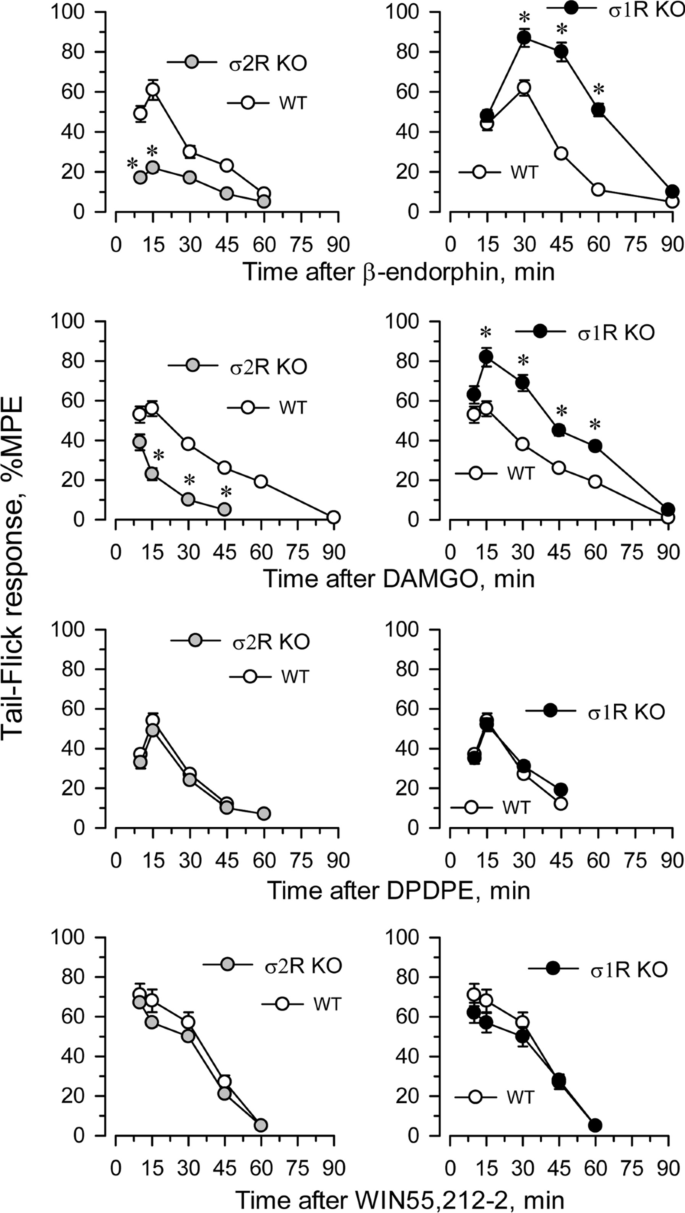
The influence of targeted deletion of σ1R gene on MOR-induced antinociception is a known issue [22]. While the antinociceptive effects of DAMGO and β-endorphin were diminished in σ2R−/− mice, they were increased in σ1R−/− mice (Fig. 4). The ability of these σ receptors to regulate analgesia promoted by activation of G receptors other than MOR was explored. The deletion of either form of σ receptor did not alter the analgesic activity of representative agonists of other G-receptors implicated in analgesia, such as the delta opioid receptor (DOR) agonist DPDPE, the cannabinoid receptor type 1 (CB1R) agonist WIN55,212-2 (Fig. 4) and the α2-adrenergic receptor (α2AR) agonist clonidine (not shown).
Fig. 4
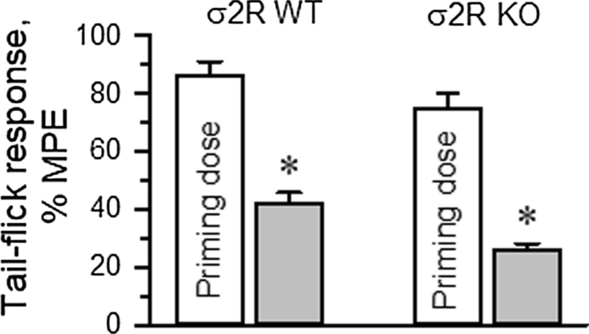
The influence of σ2R on the production of opioid-induced acute tolerance was also investigated. Mice received either saline (control) or morphine, and 24 h later, the analgesia evoked by a second injection of morphine was evaluated. Since mice showed a low analgesic response to morphine, to obtain comparable analgesic effects in both experimental groups, the dose of morphine administered to the σ2R−/− mice was increased to promote approximately 80% of the maximum possible effect (MPE) in our paradigm. A priming dose of morphine was icv injected into WT (10 nmol) and σ2R−/− mice (30 nmol), and the effect of their respective morphine ED80s was evaluated 24 h later. In WT mice, the analgesic effect of the ED80 decreased from 86 ± 5% MPE to 42 ± 4% MPE 24 h after the priming dose of 10 nmol morphine. Deletion of σ2R did not prevent the development of acute tolerance, and ED80 antinociception dropped from about 75 ± 5% to 26 ± 4% MPE (Fig. 5).
Fig. 5
Discussion
Because there are currently no reliable antibodies (with sensitivity and selectivity) testing the σ2R protein in neuronal tissue, PCR was used to confirm the absence of σ2R mRNA in the knockout animals provided by UC Davis KOMP Repository. σ2R−/− mice showed exploratory behavior, locomotor performance, motor coordination and cognitive abilities comparable to those of WT mice. Furthermore, like naïve WT mice, naïve σ2R−/− mice responded to a wide range of mechanical stimulus intensities (from innocuous to noxious). Consequently, targeted deletion of the σ2R gene did not affect normal mechanical stimulus perception or the motor response necessary to produce paw withdrawal. Nerve-injured WT and σ2R−/− mice subjected to CCI showed similar levels of allodynia on day 7 after surgery. Then, the absence of the σ2R receptor did not lead to significant alterations in the pathogenesis of neuropathic pain.
Several studies have demonstrated that σ1R−/− mice do not develop allodynia in different animal models of neuropathic pain such as CCI [33], paclitaxel [34], spinal cord contusion injury [35], or spare nerve injury [36]. Accordingly, σ1R antagonists reduce nerve injury-induced mechanical hypersensitivity in WT mice [30, 32]. Consistent with this idea, we observed that administration of the selective σ1R antagonist S1RA reduced allodynia in WT and σ2R−/− mice. On the other hand, molecules that bind to σ2R/Tmem97 as putative agonists reduce mechanical hypersensitivity in a spared nerve injury model with a duration of action and potency that is superior to that of gabapentin [20]. Thus, σ2R activation or σ1R antagonists may promote comparable antiallodynic effects, which suggests that both types of σRs are involved in regulating neuropathic pain but have opposing effects.
Interestingly, our study suggests that σ2R is involved in the analgesic effects of MOR agonists such as morphine, DAMGO and β-endorphin. In initial experiments, no differences in baseline latencies were observed among the WT (σ2R + / +), heterozygous (σ2R + / −), and homozygous (σ2R−/−) groups in the warm-water tail-flick test for analgesia. Therefore, the absence of a functional σ2R did not alter thermal nociception. However, the antinociceptive effects of morphine were impaired in σ2R−/− mice; the ED50 was 5 nmol in WT mice but more than 20 nmol in mice lacking σ2R. To explore the possibility that phenotypic modifications exhibited by σ2R−/− mice are a consequence of compensatory mechanisms assuming the physiological functions of σ2R, we analyzed the expression of proteins implicated in the processes evaluated in our study. The mRNA expression levels of σ1R, HINT1 and MOR were similar in WT and σ2R−/− mice. Most importantly, treatment with oligos to reduce the expression of σ2R mRNA diminished the responses of the mice to levels similar to those of σ2R−/− mice. Because oligo treatment promotes temporary reductions in target proteins, it is unlikely that compensatory changes resulting from the absence of this protein caused the diminished response of σ2R−/− mice to morphine.
Thus, our study suggest that σ2R is essential for the antinociceptive effects of exogenous and endogenous ligands of MOR but not for the antinociceptive effects of other families of G-receptors that also mediate analgesia, such as DOR, CB1R and α2AR. σ2R likely plays a relevant role in the regulation of MOR-mediated analgesia, sharing a physiological function with σ1R and glutamate N-methyl-D-aspartate receptor (NMDAR). The cytosolic C-terminus of MOR binds to the HINT1 protein, facilitating the interactions of σ1R and NMDAR with the MOR [22]. Notably, a lack of σ2R did not interfere with the beneficial effects of the selective σ1R antagonist S1RA on MOR-mediated analgesia. MOR agonists such as morphine increase the activity of NMDARs and then trigger a negative feedback on MOR signaling. S1RA promotes the inhibition of NMDARs by removing the σ1R from NMDAR NR1 subunits facilitating the binding of its inhibitor, calcium-activated calmodulin [22, 37]. As a result, morphine analgesia is increased and the perception of neuropathic pain is diminished [22]. As expected, this regulatory mechanism is absent in σ1R−/− mice [37], but our study showed that deletion of σ2R preserved the enhancement of morphine analgesia induced by S1RA. Thus, disruption of σ1R-mediated negative control of NMDARs on MOR activity seems to account for the enhancement of the antinociceptive effects of clinically relevant opioids such as morphine, fentanyl, oxycodone, codeine, buprenorphine, and tramadol [21, 38]. Accordingly, morphine shows an enhanced capacity to produce antinociception in σ1R−/− mice; 3 nmol morphine produces the same antinociceptive effect in σ1R−/− mice as 10 nmol morphine does in WT mice [22]. We report here that deletion of σ1R or σ2R mostly affects MOR function but does not alter antinociception promoted by either DOR or CB1R agonists. Therefore, while σ1R inhibits and σ2R facilitates MOR-mediated analgesia these receptors exchange their roles when regulating neuropathic pain perception. Our study may open new avenues for the identification of pharmacological targets for diminishing pain perception and improving handling of opioid detoxification therapies.