
Sigma-1 receptor and seizures

By Edijs Vavers, Liga Zvejniece, and Maija Dambrova
Excerpt from the article published in Pharmacological Research, Volume 191, May 2023, 106771, ISSN 1043-6618, DOI: https://doi.org/10.1016/j.phrs.2023.106771.
Editor’s Highlights
- As a chaperone protein, sigma-1 receptor (Sig1R) has already demonstrated its potential as a multidimensional drug target and has been recognized as a valid target for the management of seizures.
- Sig1R antagonists seem to be useful for the acute treatment of intoxication-induced seizures due to overdoses of various drugs, especially psychostimulants and drugs of abuse.
- Agonists and positive allosteric modulators seem to act as a promising tool for the management of seizures and seizure-related comorbidities by reducing undesired side effects such as significant CNS depressio.
Abstract
Over the last decade, sigma-1 receptor (Sig1R) has been recognized as a valid target for the treatment of seizure disorders and seizure-related comorbidities. Clinical trials with Sig1R ligands are underway testing therapies for the treatment of drug-resistant seizures, developmental and epileptic encephalopathies, and photosensitive epilepsy. However, the direct molecular mechanism by which Sig1R modulates seizures and the balance between excitatory and inhibitory pathways has not been fully elucidated. This review article aims to summarize existing knowledge of Sig1R and its involvement in seizures by focusing on the evidence obtained from Sig1R knockout animals and the anti-seizure effects of Sig1R ligands. In addition, this review article includes a discussion of the advantages and disadvantages of the use of existing compounds and describes the challenges and future perspectives on the use of Sig1R as a target for the treatment of seizure disorders.
1. Introduction
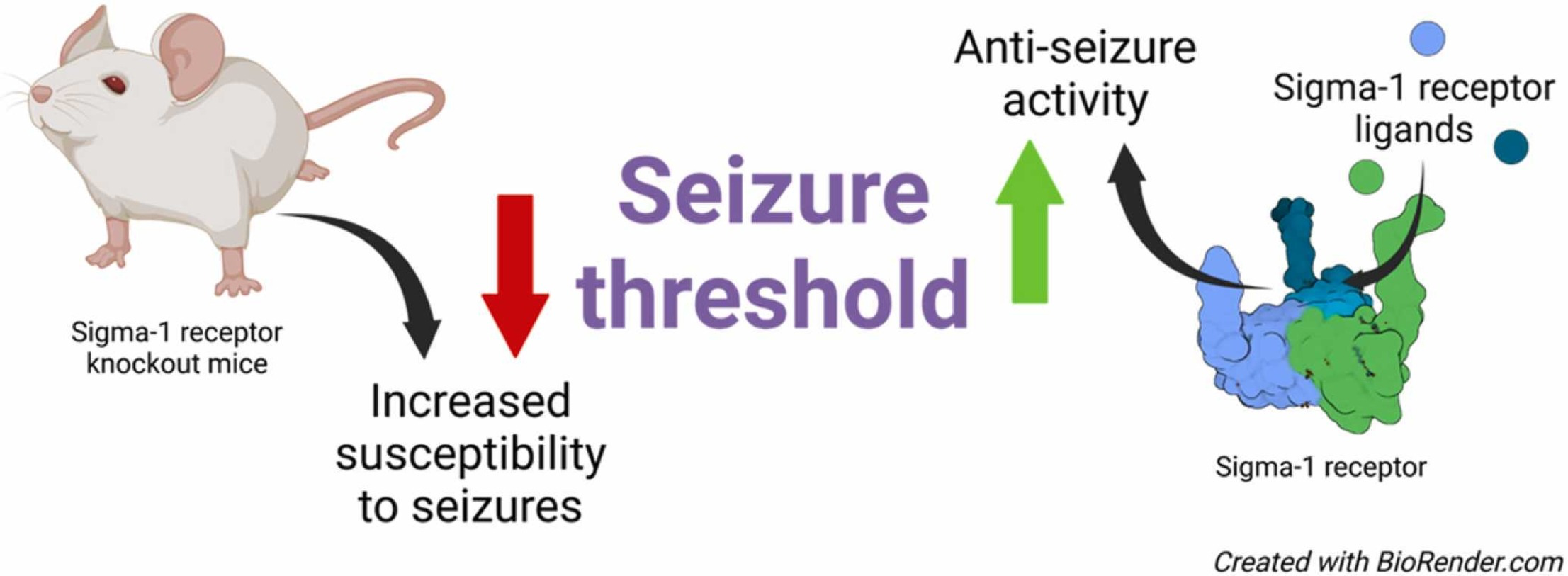
Currently, almost one-third of patients with seizures cannot be cured due to drug resistance to available anti-seizure medications [1]. Due to the heterogeneity of seizures and existence of several seizure-related comorbidities, seizure management is challenging and sometimes even impossible [2]. This critical situation and the high number of patients who are refractory to treatment have necessitated the search for new drug targets and the development of effective anti-seizure drugs. Among several novel targets proposed and investigated to date, sigma-1 receptor (Sig1R) has recently been recognized as a novel and promising drug target for the management of seizures and seizure-related comorbidities [1], [3], [4].
The first recorded study on the involvement of Sig1R in modulating seizure threshold was published in 1979 [5]. This particular study, called “Classification of Opioids on the Basis of Change in Seizure Threshold in Rats”, by Cowan et al. demonstrated that the anticonvulsant effects could be associated with so-called sigma receptor agonists [5]. At that time, Sig1R was thought to be a sigma opioid receptor, which had been proposed by Martin et al. in 1976 [6]. However, it was not until several decades later that Sig1R was recognized as a valid and independent entity [7], and only in 2013 was Sig1R included in a list of receptors as “sigma non–opioid intracellular receptor 1” [8]. Sig1R is demonstrated to be a Type II endoplasmic reticulum-resident single transmembrane domain protein [9], [10] localized at high densities in the interface between the endoplasmic reticulum and mitochondria [11]. Sig1R is known to form direct interactions between several cellular proteins and thus is designated an intracellular chaperone protein [12]. A number of ligands can bind to Sig1R with high affinity and demonstrate significant anti-seizure effects not only in animal models but also in clinical trials [1], [13]. Therefore, the aim of this review is to provide an overview of the existing knowledge on Sig1R and seizures by focusing on the possible mechanisms involved based on studies of Sig1R knockout (KO) animals and Sig1R ligand effects in vivo.
2. Evidence from Sig1R knockout animals
Sig1R KO mice were developed in the early 2000’s [14]. Both inbred C57BL/6J and outbred CD-1 background KO mice are available for studying the physiological and pathological role of Sig1R. Overall, Sig1R KO animals demonstrate no overt phenotype and are viable and fertile [14]. However, detailed phenotyping of these animals over time has revealed a link between Sig1R malfunction and diverse health problems, including cognitive, psychiatric, and specific motor and cardiac dysfunctions [15], which are age- and gender-dependent [15], [16], [17]. Notably, no spontaneous seizures have been reported in Sig1R KO animals [14], [15]. However, by using pentylenetetrazol (PTZ) and (+)bicuculline (BIC) as chemoconvulsants, it was revealed that Sig1R KO mice are more susceptible to tonic seizures than wild-type (WT) animals [18]. Therefore, this study provided significant in vivo evidence confirming the direct involvement of Sig1R in the control of the seizure threshold and thus the biological necessity of Sig1R for maintaining resistance to seizures. Abnormal excessive synchronous discharge, i.e., the imbalance between excitation and inhibition of neurons, plays an essential role in the generation of seizures [19]. Both ionotropic and metabotropic glutamate and gamma-aminobutyric acid (GABA) receptors, as well as voltage-gated ion channels are known to be key players in maintaining the balance between excitation and inhibition of neurons [20], [21], [22]. Therefore, in this review, we focus on studies investigating glutamate and GABA receptor and voltage-gated ion channel expression and function in Sig1R KO animals.
2.1. Ionotropic and metabotropic glutamate receptors
N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) and kainate receptors are ionotropic glutamate receptors, and increased activation of these receptors can elicit seizures [23], [24] . Sig1R has been shown to form direct protein-protein interactions between the C-terminus of the GluN1 (NR1) subunit of NMDA receptors in transfected cells [25], [26] and in samples from CD-1 mouse midbrains [27]. By increasing phosphorylation of NMDA receptors, intracellular influx of Ca2+ into the neuron and NMDA receptor currents, activation of Sig1R can facilitate NMDA receptor signaling [22], [28], [29], [30]. Although direct interaction between Sig1R and NMDA receptors is observed, mRNA and total protein levels of different subunits of NMDA receptors in the brain in both C57BL/6J and CD-1 background Sig1R KO mice are unchanged when compared to WT animals [18], [26], [31], [32], [33]. However, in Sig1R KO mice, NMDA receptor function is downregulated due to decreased phosphorylation of NMDA receptors. Decreased phosphorylation of the GluN2B (NR2B) subunit of NMDA receptors has been observed in the dentate gyrus, basolateral amygdala and midbrain of C57BL/6J background Sig1R KO mice [31], [33], [34] and the spinal cord of CD-1 background Sig1R KO mice [35].
. Sig1R has been shown to form direct protein-protein interactions between the C-terminus of the GluN1 (NR1) subunit of NMDA receptors in transfected cells [25], [26] and in samples from CD-1 mouse midbrains [27]. By increasing phosphorylation of NMDA receptors, intracellular influx of Ca2+ into the neuron and NMDA receptor currents, activation of Sig1R can facilitate NMDA receptor signaling [22], [28], [29], [30]. Although direct interaction between Sig1R and NMDA receptors is observed, mRNA and total protein levels of different subunits of NMDA receptors in the brain in both C57BL/6J and CD-1 background Sig1R KO mice are unchanged when compared to WT animals [18], [26], [31], [32], [33]. However, in Sig1R KO mice, NMDA receptor function is downregulated due to decreased phosphorylation of NMDA receptors. Decreased phosphorylation of the GluN2B (NR2B) subunit of NMDA receptors has been observed in the dentate gyrus, basolateral amygdala and midbrain of C57BL/6J background Sig1R KO mice [31], [33], [34] and the spinal cord of CD-1 background Sig1R KO mice [35].
The phosphorylation status of the NR2B subunit of NMDA receptors regulates its interaction with Ca2+/calmodulin-dependent protein kinase II [36], which has an important role in mediating learning and memory [37]. Indeed, long-term potentiation, a cellular correlate of learning and memory, is reduced in the hippocampus [38] and basolateral amygdala [33] of C57BL/6J background Sig1R KO male mice. By using patch-clamp recordings, reduced NMDA receptor currents were found in the dentate gyrus [31]and basolateral amygdala [33] of C57BL/6J background Sig1R KO male mice, while no significant differences were found in the action potential firing rate, paired-pulse ratio, miniature excitatory postsynaptic current and number of glutamatergic ion channels in hippocampal CA1 pyramidal neurons when compared to WT mice [38]. Seizures were induced in CD-1 background Sig1R KO male mice after an in vivo intracerebroventricular (i.c.v.) administration of NMDA (1 nmol) [39], [40]. No significant differences were observed between WT and Sig1R KO animals, including in comparisons of the latency of the first seizure, duration of seizures [39] and number of animals with clonic and tonic seizures after administration of NMDA [40]. Therefore, the increased susceptibility to seizures of Sig1R KO animals cannot be explained by the direct involvement of NMDA receptors.
Of all glutamate receptors, the molecular interaction between Sig1R and NMDA receptors has been the most studied to date. The expression level and function of kainate receptors as well as that of metabotropic G protein-coupled glutamate receptors have not been demonstrated in Sig1R KO animals. Patch-clamp measurements in brain slices of C57BL/6J background Sig1R KO mice showed that the function of postsynaptic AMPA receptors as well as the AMPA/NMDA receptor ratio in the hippocampus is not changed relative to WT mice [38]. Since the expression and function of glutamate receptors is brain region specific, additional studies are necessary to fully elucidate the involvement of glutamate receptors in the modulation of seizure thresholds in Sig1R KO animals.
2.2. Ionotropic GABA receptors
Synaptic plasticity involves both long-term potentiation and long-term depression [41]. Consistent with findings of decreased long-term potentiation [33], [38], patch-clamp recordings in mouse brain slices have shown that long-term depression in the basolateral amygdala is impaired in C57BL/6J background Sig1R KO male mice due to a possible decline in GABA-A receptor-mediated inhibition [33]. Impaired long-term depression was observed in the nucleus accumbens of male ICR mice after repeated inhibition of Sig1R with the Sig1R antagonist NE-100 [42]. NE-100 treatment was shown to decrease the expression levels of the α1, α2, β2 and β3 subunits of the GABA-A receptor in the nucleus accumbens [42]. However, these observations should be interpreted with caution, since NE-100 also caused a decrease in the levels of mRNA encoding the γ2 subunit of GABA-A receptor in Sig1R KO animals [18]. Although changes in the expression levels of subunits of GABA-A receptors can be directly associated with seizures [43], [44], in Sig1R KO animals, the mRNA levels of α4, α5, β3, γ2 and δ subunits of GABA-A receptors have been shown to be similar to the levels observed in WT mice [18], [33]. In addition, direct patch-clamp measurements of GABA-activated currents in granular cells in the dentate gyrus showed no differences between C57BL/6J male WT and Sig1R KO animals [31]. Therefore, GABA-A receptors do not seem to directly account for the differences in seizure thresholds observed between WT and Sig1R KO animals.
2.3. Metabotropic GABA receptors
Detailed molecular and immunohistochemical analyses of Sig1R KO mouse brain samples have resulted in the identification of in vivo interactions between Sig1R and metabotropic GABA-B receptors [18]. Functional GABA-B receptors are heterodimerswhose function depends on the dimerization of the GABA-B R1 and GABA-B R2 subunits [45], [46], [47]. While increased mRNA levels of the R2 subunit of GABA-B receptors (Gabbr2) were found in the cortex of CD-1 background Sig1R KO mice, no difference in Gabbr2 levels was observed in the prefrontal cortex, striatum, hippocampus, hypothalamus, or midbrain [17]. However, it was found that genetic inactivation of Sig1R significantly reduced the protein expression of GABA-B R2 in the hippocampus and habenula [18]. The most significant decrease in the expression of GABA-B R2 in Sig1R KO mice was observed in the lateral habenula and ventral part of the medial habenula [18]. The habenula is considered a key brain region for motivation and decision-making, and it is also a critical functional hub regulating spatial memory, mood and fear, circadian rhythms, pain and addiction [48]. Therefore, GABA-B receptors might be responsible for several specific habenula-dependent phenotypic characteristics of Sig1R KO mice due to decreased GABA-B R2 expression. Although Sig1R KO mice show decreased GABA-B R2 expression, increased immunostaining of R1 subunit of GABA-B receptors was observed in the hippocampus in these animals [18]. In addition, GABA-B R2 KO mice also exhibit very prominent GABA-B R1 immunostaining in some scattered hippocampal interneurons, suggesting that these neurons express high levels of GABA-B R1 even in the absence of GABA-B R2 [49], which could indicate that some compensatory mechanism is also activated in Sig1R KO animals.
There are several similarities between the phenotypes of CD-1 background Sig1R KO and Balb/c background GABA-B R2 KO animals that can be observed. GABA-B R2 KO mice demonstrate elevated anxiety-related behavior in the light-dark box paradigm and antidepressant-like behavior in the forced swim test [50]. Recently, it was found that CD-1 background Sig1R KO male mice demonstrate antidepressant-like behavior in the tail-suspension test [17], [51] and have increased serotonin levels in the frontal cortex [17]. The habenula sends direct glutamatergic and cholinergic projections not only to the dopaminergic ventral tegmental area and substantia nigra pars compacta, GABAergic interpeduncular nucleus, cholinergic laterodorsal tegmentum and noradrenergic locus coeruleus but also to the serotonergic dorsal and median raphe [48], [52]. Therefore, it is highly possible that a decrease in habenular control of basally active serotoninergic neurons in the raphe nucleus might result in the increased serotonin levels observed in Sig1R KO mice.
It should be noted that the expression level of GABA-B R2 determines the severity of the phenotype observed in Sig1R KO and GABA-B R2 KO animals. For example, full GABA-B R2 KO mice exhibit spontaneous epileptiform activity [53], while for Sig1R KO animals, no spontaneous seizures have been reported. However, decreased expression of GABA-B R2 seems to be responsible for the increased susceptibility to seizures found in Sig1R KO mice [18]. GABA-B receptors are known to play an important role in seizure modulation and have been studied as a possible target for treating absence seizures [21]. Metabotropic GABA-B receptor stimulation results in a prolonged decrease in neuronal excitability via the inhibition of adenylyl cyclase and voltage-gated Ca2+ channels and the activation of G protein-coupled inward rectifying potassium channels [54]. GABA-B receptors can modulate excitability by engaging pre- and postsynaptic GABA-B heteroreceptors as well as presynaptic GABA-B autoreceptors [55]. The activation of presynaptic GABA-B receptors is known to suppress the release of several neurotransmitters, and depending on whether this action is exerted in GABAergic or glutamatergic neurons, anticonvulsant or proconvulsant effects may emerge [21]. Therefore, GABA-B receptors can have variable functions that modulate circuit excitability in the brain [55] and seem to be responsible for the increased susceptibility to tonic seizures in Sig1R KO mice.
2.4. Voltage-gated ion channels
Sig1R has been shown to modulate the activity of several ion channels, including voltage-gated Na+, K+, and Ca2+ channels, which play a critical role in controlling neuronal excitability [22]. Disfunction of these ion channels, either due to acquired or genetic channelopathies, is a common cause of seizures and developmental epileptic encephalopathies [56], [57]. Sig1R has been shown to directly interact with Nav1.5 voltage-gated Na+ channels in transfected tsA201 cells [58]. The association between Sig1R and several voltage-gated K+ channels has been demonstrated by coimmunoprecipitation using transfected cells, Xenopus laevis oocytes, and mice brain samples [59], [60], [61]. Coimmunoprecipitation studies have also demonstrated an association between Sig1R and voltage-gated Ca2+ channels in rat primary retinal ganglion cells [62]. However, using brain synaptosomes isolated from C57BL/6J background Sig1R KO mice, it was concluded that Sig1R does not regulate voltage-gated Ca2+ channels [63], and both Sig1R agonists and antagonists have been shown to inhibit potassium chloride-induced depolarization in Sig1R KO mice tissue samples [18], [63]. In neonatal cardiac myocytes isolated from Sig1R KO mice, Sig1R ligands have been shown to inhibit Na+ channel function [64]. Additionally, Sig1R-independent effects of Sig1R ligands on voltage-gated K+ channels have been demonstrated [59], and should be kept in mind.
Despite observed Sig1R-independent activities of Sig1R ligands, Sig1R can alter ion channel function in the absence of ligands [59], [64], and thus it has been suggested to function as an atypical auxiliary subunit of voltage-gated ion channels [59], [65]. Sig1R has been demonstrated to bind to alpha-2-delta subunit of voltage-gated Ca2+ channels, thus facilitating the interaction of alpha-2-delta with NMDA receptors [66]. However, it should be noted that the expression levels of alpha-2-delta subunits of voltage-gated Ca2+ channels are not changed in brain and spinal cord samples isolated from CD-1 background Sig1R KO mice [66]. Although Sig1R is shown to be colocalized with the Kv2.1 voltage-gated K+ channels in postsynaptic sites of cholinergic synapses in C57BL/6J mice ventral horn motoneurons, the expression levels and distribution of Kv2.1 voltage-gated K+ channels are not changed in Sig1R KO animals [67]. Nevertheless, by using whole-cell patch clamp measurements, an increased excitability of motoneurons has been observed in amyotrophic lateral sclerosis model mice SOD1-G93A lacking Sig1R [68]. Therefore, the loss of Sig1R might indicate for some increased neuronal excitability due to disturbed voltage-gated channel function, which has been already discussed previously [69]. Given that the susceptibility to seizures is influenced by various factors, including cell type and brain region specificity, basal activity of the neuronal system, and age [22], it is necessary to conduct a detailed analysis of the expression and function of voltage-gated ion channels in Sig1R KO animals to better understand the possible involvement of these channels in modulating seizure threshold.
2.5. Possible molecular cornerstones and role of astrocytes
Cell surface expression of GABA-B receptors is a dynamic process that is regulated through several mechanisms [70]. The balance of recycling and degradation of GABA-B receptors is controlled by phosphorylation/dephosphorylation events [70]. It has been shown that activation of protein phosphatase-2A (PP2A) dephosphorylates Ser783 of the R2 subunit of GABA-B receptors and shifts the recycling/degradation equilibrium toward degradation so that the majority of GABA-B receptors are no longer recycled but instead degraded in lysosomes [70], [71], [72]. Indeed, increased protein levels of PP2A were found in Sig1R KO mice [73]. In addition, increased activity of PP2A is also known to induce dephosphorylation of the Ser1303 of the NR2B subunit of NMDA receptors [36]. Both decreased expression of the R2 subunit of GABA-B receptors and decreased phosphorylation of NR2B of NMDA receptors have been observed in Sig1R KO animals [18], [31], [33]. Interestingly, the regulation of PP2A activity and signaling can be mediated through direct interaction between ceramides and inhibitor-2 of PP2A, also known as SET protein [74]. A recent metabolomics study of plasma and brain cortex samples from adult and elderly CD-1 background Sig1R KO mice demonstrated increased ceramide, especially C18-ceramide, concentrations in the cortex [17]. In addition, it has been shown that SET preferentially binds C18-ceramide [74], [75]. High ceramide levels in the cortex of Sig1R KO mice may indicate increased PP2A activity due to the binding of ceramide to the SET protein with the subsequent release of active PP2A. In contrast to KO conditions, it has been shown that inhibition of PP2A activity can upregulate Sig1R in the endoplasmic reticulum [76]. Therefore, dephosphorylation of the NR2B subunit of NMDA receptors and decreased expression levels of the R2 subunit of GABA-B receptors, likely due to increased dephosphorylation of GABA-B R2 at Ser783 and its lysosomal degradation, might be ultimately attributed to increased PP2A activity in Sig1R KO animals (Fig. 1).
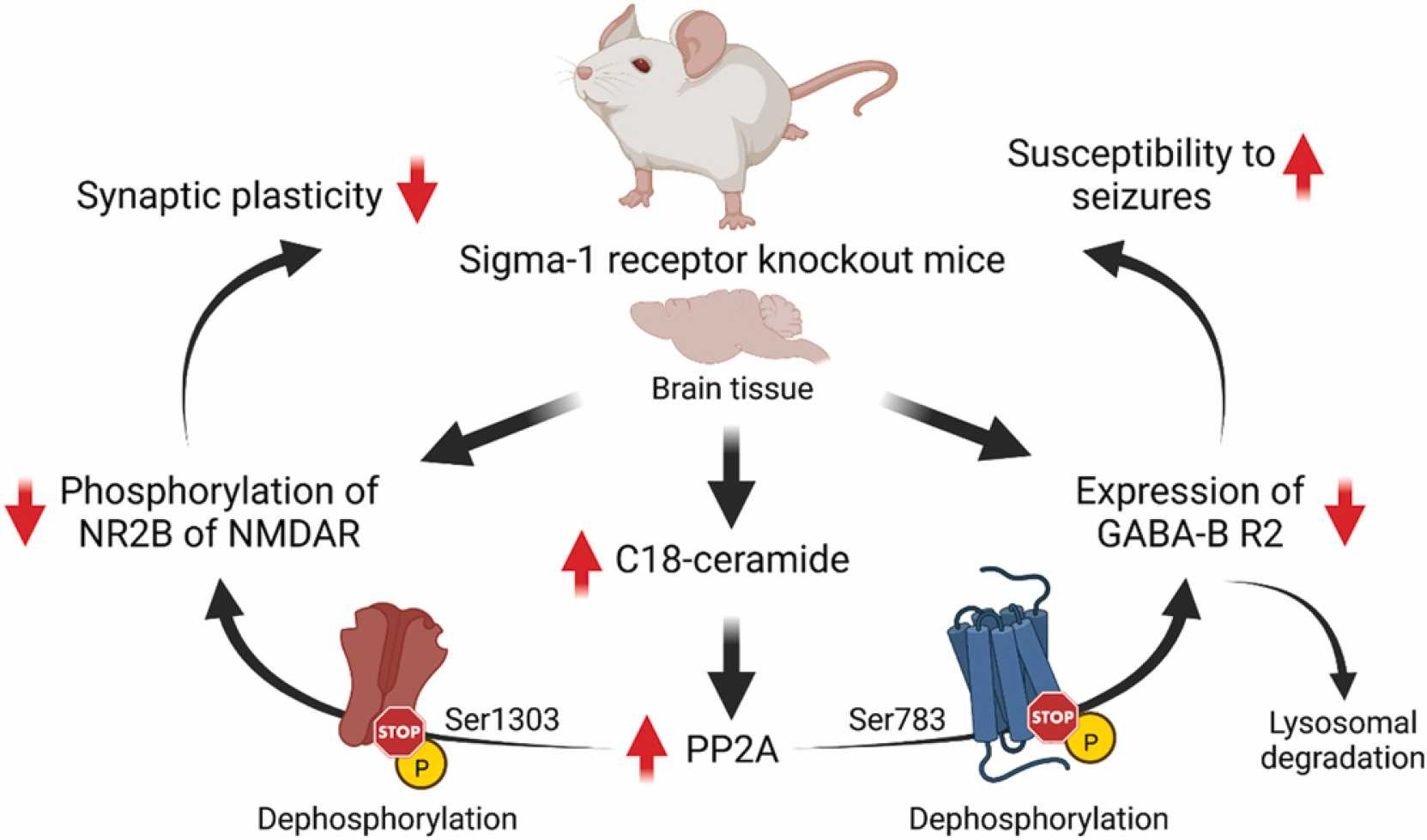
Possible molecular mechanisms underpinning some phenotypic characteristics of Sig1R KO mice. GABA-B – metabotropic gamma-aminobutyric acid receptors; NMDAR – N-methyl-D-aspartate receptors; NR2B – GluN2B subunit of NMDAR; R2 – subunit of GABA-B receptors; Ser – serine; PP2A – protein phosphatase-2A. Created with BioRender.com.
With regards to the modulation of seizure threshold the function of astrocytes in Sig1R KO animals currently is underestimated. Since astrocytes regulate the extracellular levels of neurotransmitters, such as glutamate and GABA, and ions, especially K+ by potassium spatial buffering [77], [78], astrocytic dysfunction contributes to pathological changes in synaptic transmission and can lead to hyperexcitability and seizures [77], [79]. Astrocytes isolated from C57BL/6J background Sig1R KO mice were not able to fully promote ganglion cell growth and survival in vitro than astrocytes which were isolated from WT animals [80]. In addition, astrogliosis has been observed in primary neuron-glia culture isolated from C57BL/6J background Sig1R KO mice [81]. In contrary, decreased expression of glial fibrillary acidic protein, an astrocytic cytoskeletal protein and marker of astrogliosis, has been found in the cerebellum of CD-1 background Sig1R KO mice [51]. These observations might indicate to different astrocyte subtype-specific functions of Sig1R. Disfunction of blood-brain barrier is one of the hallmarks of seizures [82], and it has been shown that astrocytes contribute to blood-brain barrier stability through direct contact of perivascular endfeet with endothelial cells and pericytes [83]. Knockdown of Sig1R with siRNA in human umbilical vein endothelial cells has been shown to reduce the endothelial barrier function in vitro [84]. It has been demonstrated that after brain ischemia-reperfusion C57BL/6J background Sig1R KO male mice are more prone to degradation of structural proteins of blood-brain barrier than WT animals [85]. In addition to all the above mentioned, the astrocyte function in Sig1R KO animals should be considered in line with age-dependent metabolic background [17]. For example, lipid accumulation in astrocytes leads to the formation of lipid-accumulated reactive astrocytes that can promote neuronal hyperactivity [86]. Therefore, more focused studies of the function of astrocytes in Sig1R KO animals with regards to seizures could provide important novel clues for the mechanisms of Sig1R in the control of seizure threshold.
In summary, studies with Sig1R KO animals have provided insights specifically on the involvement of Sig1R-dependent processes in the modulation of seizure thresholds. Decreased expression of GABA-B R2, especially in the cholinergic ventral medial habenula, together with disturbed function of ion channels, some specific metabolic changes and elevated levels of serotonin have been observed in Sig1R KO mice, thus increasing the understanding of the direct role of Sig1R in the modulation of several molecular pathways (both directly and indirectly) and brain regions that might be involved in the regulation of seizure thresholds and the development of several seizure-related comorbidities.
3. Modulation of seizures by Sig1R ligands
3.1. Anti-seizure effects
There are several Sig1R ligands that have demonstrated significant anti-seizure effects not only in preclinical animal models but also in clinical trials. Although several of these compounds have a high affinity for binding to Sig1R, their anti-seizure effects are not always directly attributed to Sig1R due to their high affinity for binding to other targets (Table 1). The binding affinity to a specific target does not always dictate the pharmacodynamics of that compound due to particular pharmacokinetic properties, especially biodistribution and metabolism in vivo. However, evidence from Sig1R KO animals indicates that seizure thresholds and susceptibility to seizures can be mediated through Sig1R, thus confirming that Sig1R is a valid molecular target for the development of novel anti-seizure drugs and might explain previously observed anti-seizure activities of Sig1R ligands. Anti-seizure effects have been observed for Sig1R ligands classified as Sig1R agonists, antagonists and positive allosteric modulators (3.1.1 Agonists, 3.1.2 Antagonists, 3.1.3 Allosteric modulators). Table 1 provides a comprehensive overview of preclinical anti-seizure effects that have been demonstrated for Sig1R ligands.
| Compound and affinity to Sig1R (nM) | Additional targets < 10 µM (Ki, nM) | Effective dose | Administration route, time before the seizures | Animals | Seizure model | References |
| Sig1R agonists | ||||||
| ( ± )Alazocine (( ± )SKF-10,047) Ki = 1800–4700 | Kappa opioid R(0.4) Mu opioid R (1.2) Delta opioid R(IC50 = 184) NMDA(2554) | |||||
| ED50 = 2.4–6.1 µM | in vitro | Wistar rats | NMDA (10–80 µM) in neocortical slices | [87] | ||
| 10–40 mg/kg | s.c., 30 min | Male Sprague-Dawley rats | Flurothyl (10%) | [5] | ||
| 10–40 mg/kg | i.p., 20 min | Male Sprague-Dawley rats | Amygdala-kindling | [88] | ||
| 0.1–10 mg/kg | s.c., 20 min | Male and female genetically sensitive Mongolian gerbils | Sensory-evoked epileptic seizures | [89] | ||
| 5 and 20 mg/kg | i.p., 15 min | Female Sprague-Dawley rats | MES (corneal) | [90] | ||
| 50 mg/kg | i.p., 10 min | Male Sprague-Dawley rats | Hyperbaric pressure-induced neurological syndrome | [91] | ||
| ED50 = 50 nmol (i.c.v.); ED50 = 24–69 µmol/kg (i.p.) | i.c.v., 15 min; i.p., 15–45 min | Male and female DBA/2 mice | Sound (109 dB) | [92] | ||
| 20 mg/kg | i.p., 40 min | Male C57BL/6J mice | MES-T (auricular) | [93] | ||
| 20 mg/kg | i.p., 40 min | Male C57BL/6J mice | PTZ (80 mg/kg, s.c.) | [93] | ||
| 20 mg/kg | i.p., 40 min | Male C57BL/6J mice | Kainic acid (30 mg/kg, i.p.) | [93] | ||
| (+)Alazocine ((+)SKF-10,047) Ki = 45–153 | NMDA (587) Kappa opioid R (1600) Mu opioid R (1900) TMEM97 (IC50 = 4543) | |||||
| 1–30 mg/kg | i.p., 10 min | Male Swiss-Webster mice | PTZ (125 mg/kg/ s.c.) | [94] | ||
| ED50 = 7.1 mg/kg | s.c., 30 min | Male CF-1 mice | MES (corneal) | [95] | ||
| ED50 = 17.4 mg/kg | i.v., 15 min | Male Swiss-Webster mice | NMDLA (500 mg/kg, s.c.) | [96], [97] | ||
| ED50 = 1.5 mg/kg | s.c. | Male Sprague-Dawley rats | MES (auricular) | [98] | ||
| ED50 = 28.2 mg/kg | s.c., 30 min | Male Swiss-Webster mice | NMDLA (500 mg/kg, s.c.) | [99] | ||
| 0.3–5.6 mg/kg | i.p., 15 min | Male C57BL/6J mice | (-)Cocaine (100 mg/kg, i.p.) | [100] | ||
| ANAVEX1–41 (AE14) Ki = 44 | M1 (19) M3 (50) M4 (77) M2 (114) TMEM97 (3924) | |||||
| 19 mg/kg | p.o. | Mice | PTZ | [101] | ||
| 45 mg/kg | p.o. | Mice | MES | [101] | ||
| Blarcamesine(ANAVEX®2–73, AE37) IC50 = 860 | M1-M4 (IC50 = 3300–5200) Na+channel (5100) NMDA (8000) | |||||
| 30 and 100 mg/kg | i.p., 1 h | Swiss male mice | MES | [102], [103] | ||
| 30 and 100 mg/kg | i.p., 1 h | Swiss male mice | PTZ (130 mg/kg, i.p.) | [102], [103] | ||
| 15, 30 and 60 mg/kg | i.p., 1 h | Swiss male mice | Semicarbazide | [102] | ||
| Caramiphen Ki = 16–26 | M1 (1.2) M2 (32) nAchR (IC50 = 300) DAT (5180–5250) | |||||
| ED50 = 5.7–14.0 µM | in vitro | Wistar rats | NMDA (10–80 µM) in neocortical slices | [87] | ||
| IC50 = 46 µM | in vitro | Male Wistar rats | Mg2+-free model in hippocampal slices | [104] | ||
| ED50 = 14 mg/kg | s.c., 30 min | Male Sprague-Dawley rats | MES (auricular) | [105] | ||
| 100 mg/kg | i.m. | Guinea pigs | Soman (2 x LD50) | [106] | ||
| ED50 = 52 mg/kg | i.p., 30 min | Male NSA/CF1 mice | MES (auricular) | [107] | ||
| ED50 = 1.5 mg/kg | i.m., 30 min | Male Crl CDBR Vaf/Plus Sprague-Dawley rats | Soman (180 µg/kg = 1.6 × LD50, s.c.) | [108] | ||
| ED50 = 3.1 mg/kg | s.c., 30 min | Male Sprague-Dawley rats | MES (auricular) | [109] | ||
| ED50 = 7.8 mg/kg | i.v., 10 min | Shanghai mice | Nicotine (1 mg/kg, i.v.) | [110] | ||
| 20 mg/kg | i.m., 5, 10 and 20 min (after) | Male Sprague-Dawley rats | Sarin (108 mg/kg = 1.2 × LD50, i.m.) | [111] | ||
| 100 mg/kg | i.m., 30 and 60 min (after) | Male Sprague-Dawley rats | Soman (154 µg/kg = 1.4 × LD50, s.c.) | [112] | ||
| 20 mg/kg | i.m., 10 min | Male Sprague-Dawley rats | Soman (132 μg/kg, s.c.) | [113] | ||
| *Carbetapentane(Pentoxyverine) Ki = 10–129 | 5HT3A (8–165) M1 (76) M2 (167) Alpha1 (IC50 = 1550) TMEM97 (56–1953) DAT (3090) | |||||
| ED50 = 10.2–38.2 µM | in vitro | Wistar rats | NMDA (10–80 µM) in neocortical slices | [87] | ||
| IC50 = 38 µM | in vitro | Wistar rats | Mg2+-free model in hippocampal slices | [104] | ||
| ED50 = 4.8 mg/kg | s.c. | Male Sprague-Dawley rats | MES (auricular) | [114] | ||
| ED50 = 48 µmol/kg | s.c., 30 min | Male Sprague-Dawley rats | MES (auricular) | [105] | ||
| ED50 = 215.3 mg/kg (p.o.); ED50 = 45 mg/kg (i.p.) | p.o. and i.p., 30 min – 4 h | Male CF-1 mice | MES (corneal) | [115] | ||
| – | – | Guinea pigs | Soman (2 x LD50) | [106] | ||
| ED50 = 10 mg/kg | i.m., 30 min | Male Crl CDBR Vaf/Plus Sprague-Dawley rats | Soman (180 µg/kg = 1.6 × LD50, s.c.) | [108] | ||
| * 12.5 and 25 mg/kg | i.p., 30 min | Male Sprague-Dawley rats | Kainic acid (10 mg/kg, i.p.) | [116] | ||
| ( ± )Cyclazocine Ki = 36–47 | Kappa opioid R(0.1–0.2) Mu opioid R (0.2–0.3) Delta opioid R(2.0) NOP R (157–7500) | |||||
| ED50 = 0.4–0.7 µM | in vitro | Wistar rats | NMDA (10–80 µM) in neocortical slices | [87] | ||
| 1–10 mg/kg | i.p., 20 min | Male Sprague-Dawley rats | Amygdala-kindling | [88] | ||
| (+)Cyclazocine Ki = 17 | Kappa opioid R(0.1–0.2) Mu opioid R (0.1–0.3) Delta opioid R(1.1) | |||||
| 5 mg/kg | s.c., 30 min | Male Sprague-Dawley rats | Flurothyl (10%) | [5] | ||
| Citalopram Ki = 167–404 | SERT (0.5–33.0) 5-HT2C(156–617) H1 (283–371) Alpha1 (711–1820) Ca2+channels (996–1588) 5-HT2B (1171) M1 (1430) Na+channels (2409) NET (2178–6190) TMEM97 (5410) K+channels (IC50 = 2600–3981) DAT (9270) 5-HT2A (9500) | |||||
| 30, 100 and 300 µM | in vitro | Zebrafish (Tg(fli1a:EGFP)y1) | PTZ (40 mM) | [117] | ||
| 30, 100 and 300 µM | in vitro | Zebrafish (Tg(fli1a:EGFP)y1) | Pilocarpine (20 mM) | [117] | ||
| 1 and 10 µM | i.c. | Male Wistar rats | Pilocarpine (intra-hippocampal perfusion with 10 mM pilocarpine for 40 min) | [118] | ||
| 1 mg/kg | i.p., 30 min | Male NMRI mice | PTZ (0.5%, i.v. infusion) | [119], [120] | ||
| 15 mg/kg | i.p. for 4 days (after) | Male Sprague-Dawley rats | Kainic acid (5 mg/kg, i.p. repeatedly) | [121] | ||
| 20 mg/kg | i.p., 30–60 min | Male and female C57BL/6J mice | Amygdala-kindling | [122] | ||
| *Dextrorphan Ki = 118–481 | SERT(401–484) NET (340) Mu oioid R (420) NMDAR (486–906) Kappa opioid R (5950) | |||||
| 1–250 µM | in vitro | Guinea pigs | Mg2+-free model in neocortical brain slices | [123] | ||
| ED50 = 3.5–5.6 µM | in vitro | Wistar rats | NMDA (10–80 µM) in neocortical slices | [87] | ||
| IC50 = 1.5 µM | in vitro | Male Wistar rats | Mg2+-free model in hippocampal slices | [104] | ||
| ED50 = 12 µmol/kg | s.c., 30 min | Male Sprague-Dawley rats | MES (auricular) | [124] | ||
| ED50 = 102 µmol/kg | i.p., approx. 30 min | Male CF-1 mice | NMDA (250 mg/kg, i.p.) | [125] | ||
| ED50 = 35 nmol (i.c.v.); ED50 = 19–37 µmol/kg (i.p.) | i.c.v., 15 min; i.p., 15–45 min | Male and female DBA/2 mice | Sound (109 dB) | [92] | ||
| ED50 = 9.6 mg/kg | s.c., 30 min | Male CF-1 mice | MES (corneal) | [95] | ||
| 30 mg/kg | i.p., 30 min | Female SPF-Fu mice | NMDA (1 nmol/µl, i.c.v.) | [126] | ||
| ED50 = 1.9 mg/kg | i.v., 15 min | Male ddY mice | NMDA (2 nmol, i.c.v.) | [127] | ||
| ED50 = 5.5 mg/kg | i.v., 15 min | Male ddY mice | AMPA (16 nmol, i.c.v.) | [127] | ||
| ED50 = 2.2 mg/kg | i.v., 15 min | Male ddY mice | Kainic acid (4 nmol, i.c.v.) | [127] | ||
| ED50 = 2.7 mg/kg | i.v., 15 min | Male ddY mice | BIC (2 nmol, i.c.v.) | [127] | ||
| ED50 = 6.9 mg/kg | i.v., 15 min | Male ddY mice | PTZ (20 µmol, i.c.v.) | [127] | ||
| 30 mg/kg | i.p., 15 min | Female Wistar rats | Amygdala-kindling | [128] | ||
| 25 and 50 mg/kg | – | Rats | NMDA | [129] | ||
| ED50 = 31 mg/kg | 30 min | Female NMR mice | MES (corneal) | [130] | ||
| 12.5 and 25 mg/kg | s.c., 30 min | Male Sprague-Dawley rats | Kainic acid (10 mg/kg, i.p.) | [131] | ||
| * 10 µg/0.5 µl | i.c.v. | Male DBA/2 mice | Kainic acid (0.07 µg/0.07 µl, i.c.v.) | [132] | ||
| ED50 = 20 mg/kg | i.p., 20 min | Male Sprague-Dawley rats | MES | [133] | ||
| ED50 = 5 µg | i.c.v., 15 min | Male Sprague-Dawley rats | NMDA (12.5 nM, i.c.v.) | [134] | ||
| *Dextromethorphan Ki = 10–652 | SERT (1.4–40.0) NET (240–6000) nAChR (IC50 = 700–8900) Alpha1 (830–3000) Mu opioid R (1280) Na+channels (4800) NMDAR (780–8945) M1 (5070) Kappa opioid R (7000) | |||||
| 100 µM | in vitro | Guinea pigs | Mg2+-free model in neocortical brain slices | [123] | ||
| ED50 = 11–15 µM | in vitro | Wistar rats | NMDA (10–80 µM) in neocortical slices | [87] | ||
| ED50 = 6.2 mg/kg | s.c. | Male Sprague-Dawley rats | MES (auricular) | [114] | ||
| 35 mg/kg | i.p., 30–45 min | Male Sprague-Dawley rats | Amygdala-kindling | [135] | ||
| ED50 = 106 µmol/kg | i.p., approx. 30 min | Male CF-1 mice | NMDA (250 mg/kg, i.p.) | [125] | ||
| ED50 = 11.8 mg/kg | s.c., 30 min | Male CF-1 mice | MES (corneal) | [95] | ||
| ED50 = 93.9 mg/kg (p.o.); ED50 = 45.7 mg/kg (i.p.) | p.o. and i.p., 30 min – 4 h | Male CF-1 mice | MES (corneal) | [115] | ||
| ED50 = 70 nmol (i.c.v.); ED50 = 28–36 µmol/kg | i.c.v., 15 min; i.p., 15–45 min | Male and female DBA/2 mice | Sound (109 dB) | [92] | ||
| 10 mg/kg | i.p., 30 min | Female SPF-Fu mice | NMDA (1 nmol/µl, i.c.v.) | [126] | ||
| – | – | Guinea pigs | Soman (2 x LD50) | [106] | ||
| ED50 = 59 mg/kg | i.p., 30 min | Male Sprague-Dawley rats | Amygdala-kindling | [136] | ||
| – | – | Mice | NMDLA | [137] | ||
| – | – | Mice | PTZ | [137] | ||
| – | – | Mice | MES | [137] | ||
| ED50(clonus) = 161 µmol/kg | i.p., 60 min | Genetically epilepsy-prone rats (GEPR-9 s) | Sound (109 dB) | [138] | ||
| 7.5–15 mg/kg | i.p., 30 min | Female Wistar rats | Amygdala-kindling | [128] | ||
| ED50 = 20 mg/kg | i.p., 30 min | Male CD-1 mice | NMDA (0.8 µg/mouse, i.c.v.) | [139] | ||
| ED50 = 38.6 mg/kg | 30 min | Female NMR mice | MES (corneal) | [130] | ||
| 12.5–75 mg/kg | p.o., 15 min | Rats | Kainic acid | [140] | ||
| 50 and 75 mg/kg | p.o., 15 min | Male Fischer rats | Kainic acid (8 mg/kg, i.p.) | [141] | ||
| 10, 20 and 40 mg/kg | i.p., 3 h | Male Wistar rats | Sound (100 dB) | [142] | ||
| 0.2 mg/kg | i.p., 25 min | Female mice | Theophylline (300 mg/kg, i.p.) | [143] | ||
| ED50 = 7.94 mg/kg | i.p., 30 min | Wistar rat pups | Sound (122–125 dB) | [144] | ||
| 12.5 and 25 mg/kg | s.c., 30 min | Male Sprague-Dawley rats | Kainic acid (10 mg/kg, i.p.) | [131] | ||
| 12.5 and 25 mg/kg | s.c., 30 min | Male Sprague-Dawley rats | BAY k-8644 (37.5 µg, i.c.v.) | [131] | ||
| * 5 and 10 µg/0.5 µl | i.c.v. | Male DBA/2 mice | Kainic acid (0.07 µg/0.07 µl, i.c.v.) | [132] | ||
| ED50 = 25 mg/kg | i.p., 20 min | Male Sprague-Dawley rats | MES | [133] | ||
| 12.5 mg/kg | i.p., 30 min | Mice | BAY k-8644 (37.5 µg, i.c.v.) | [145] | ||
| * 12 and 24 mg/kg | s.c., 30 min | Male Sprague-Dawley rats | Kainic acid (10 mg/kg, i.p.) | [146] | ||
| 25 mg/kg | s.c., 30 min | Male Fischer 344 rats | Trimethyltin (8 mg/kg, i.p.) | [147] | ||
| 10, 30 and 100 mg/kg | i.p., 30 min | Male NMRI mice | PTZ (i.v. infusion) | [148] | ||
| 25 and 50 mg/kg | i.p., 30 min | Male NMRI mice | PTZ (i.v., 5 mg/ml at a rate of 0.5 ml/min) | [149] | ||
| 25 mg/kg | s.c., from postnatal day 8–21 | C57BL/6NJ background Grin2a knock-in mice | Spontaneous seizures | [150] | ||
| ED50 = 168 µg | i.c.v., 15 min | Male Sprague-Dawley rats | NMDA (12.5 nM, i.c.v.) | [134] | ||
| *Dimemorfan Ki = 151 | TMEM97 (4421) | |||||
| ED50 = 70 µmol/kg | i.p., 30 min | Male ICR mice | MES (corneal) | [151] | ||
| 6.25 and 12.5 mg/kg | i.p., 30 min | Mice | BAY k-8644 (37.5 µg, i.c.v.) | [145] | ||
| * 12 and 24 mg/kg | s.c., 30 min | Male Sprague-Dawley rats | Kainic acid (10 mg/kg, i.p.) | [146] | ||
| Ditolylguanidine (DTG) Ki = 15–208 | TMEM97(12–107) M1 (744) VAChT (1134) ERG2 (2000) M2 (2960) Opioid R (3950) NMDAR (6690) EBP (6700) | |||||
| ED50 = 4.7–18.4 µM | in vitro | Wistar rats | NMDA (10–80 µM) in neocortical slices | [87] | ||
| IC50 = 15 µM | in vitro | Male Wistar rats | Mg2+-free model in hippocampal slices | [104] | ||
| ED50 = 5.25 nM | i.c. | Rats | BIC (i.c.) | [152] | ||
| Donepezil Ki = 15 | AChE (2–38) BchE (IC50= 20–15240) BACE1 (IC50 = 169–3200) H3 (IC50 = 350) K+channels (IC50 = 1995) BuChE (2140) | |||||
| 2.5 mg/kg | p.o. for 3 weeks (after) | Male Sprague-Dawley rats | Pilocarpine (25 mg/kg, i.p.) | [153] | ||
| 5.6 mg/kg | i.p., 1 h | Male CF1 mice, C56Bl/6 J background male and female heterozygous Scn1a knockout (Scn1a+/-) mice | MES (corneal) | [154] | ||
| 5.6 mg/kg | i.p., 1 h | Male and female C56Bl/6 J background heterozygous Scn1a knockout (Scn1a+/-) mice | PTZ (100 mg/kg, i.p.) | [154] | ||
| ( ± )Fluoxetine Ki = 191–240 | SERT (0.3–270.0) Alpha2(6–5012) DAT (11–6670) 5-HT2C(50–398) 5-HT2A(55–1820) Na+channel(82–90) NET (85–6670) AchE (130) M2 (512–2700) Alpha1 (605–5260) M1 (702–1030) Ca2+channels (736–1095) M3 (762–3100) 5-HT6 (771–1770) H1 (933–5400) M5 (976–2070) 5-HT7 (1000) Sig2R (1610) 5-HT1A (1820–8310) M4 (2090) K+channels (5012) 5-HT2B (5030) 5-HT1D (4270) 5-HT1B (6170) H3 (7300) | |||||
| 40 µM | in vitro | Male Sprague-Dawley rats | Picrotoxin (i.c. in hippocampal slices) | [155] | ||
| 1 µM | in vitro | Wistar rat embryos | High K+ (7 mM) in hippocampal and prefrontal cells | [156] | ||
| 100 µM | in vitro | Male Swiss mice | Mg2+-free model in olfactory bulb slices | [157] | ||
| 100 µM | in vitro | Male Wistar rat pups | Low Mg2+/high K+ in hippocampal slices | [158] | ||
| 100 µM | in vitro | Male Wistar rat pups | 4-AP in hippocampal slices | [158] | ||
| ED50 = 8.2–15.9 mg/kg | i.p., 4 h; i.p., 28-day treatment, 4 h before the sound | Genetically epilepsy-prone rats (GEPR-9 s) | Sound | [159] | ||
| 3.5 and 7.0 nmol | i.c., 15 min | Male Sprague-Dawley rats | BIC (118 pmol, i.c.) | [160] | ||
| 20 mg/kg | i.p., 1 h | Male Sprague-Dawley rats | BIC (118 pmol, i.c.) | [161] | ||
| 15 mg/kg | i.p., 1 h | Genetically epilepsy-prone rats (GEPR-9 s) | Sound (115 dB) | [162], [163] | ||
| 10 mg/kg | i.p., 1 week after 21-day pretreatment | Male Wistar rats | Electrical hippocampal stimulation | [164] | ||
| 20 mg/kg | i.p. for five days (after) | Male Sprague-Dawley rats | Pilocarpine (320 mg/kg, i.p.) | [165] | ||
| 20 mg/kg | i.p., 65 min | Male CBA mice | Picrotoxin (0.75 mg/ml at a rate of 1.1 ml/min, i.v.) | [166] | ||
| 20 mg/kg | s.c., 60 min | Male CFLP mice | PTZ (130 mg/kg, s.c.) | [167] | ||
| 10 mg/kg/day | 7 days | El mice | Handling-induced | [168] | ||
| 25 mg/kg | i.p., 2 h | Genetically epilepsy-prone rats (GEPR-9 s) | Sound (100 dB) | [169] | ||
| 20 mg/kg | i.p. | Groggy (GRY/Idr) rats | Absence-like seizures | [170] | ||
| 30 mg/kg/day | i.p. for 17 weeks | WAG/Rij rats | Absence-like seizures | [171] | ||
| 10 mg/kg | i.p. for 7 days (after) | Male rats | 4-AP (3 mg/kg, i.p.) | [172] | ||
| 5 and 10 mg/kg | i.p., 30 min | Male Wistar rats | Penicillin (500 IU/2.5 µl, i.c.) | [173] | ||
| 10 mg/kg | i.p., 30–60 min | Male and female C57BL/6J mice | Amygdala-kindling | [122] | ||
| 20 mg/kg | p.o. | Seizure-susceptible Wistar rats | Sound (90–110 dB) | [174] | ||
| 10 mg/kg | i.p., 30 min | Male Wistar rats | PTZ (45 mg/kg, i.p.) | [175] | ||
| Fluvoxamine Ki = 36 | SERT (2–540) DAT (2–9200) NET (299–5000) Alpha1 (1290–4800) Alpha2 (1900) K+channels (IC50 = 3100) Ca2+channels (IC50 = 4900) 5-HT2C (5790–6700) TMEM97 (8439) | |||||
| 20 mg/kg | i.p., 30 min | Male mice | PTZ-induced kindling (35 mg/kg, i.p.) | [176] | ||
| Ifenprodil (erythro diastereomer) Ki = 2–125 | TMEM97(5–98) EBP (1.0) NMDAR(8–94) Alpha1(12–140) 5-HT1A(46) 5-HT2B(66–95) DAT (363) 5-HT2A (421–510) 5-HT7 (513) 5-HT2C (748) Alpha2 (1600–2300) | |||||
| IC50 = 6.3 µM | in vitro | Male Wistar rats | Mg2+-free model in hippocampal slices | [104] | ||
| 3 µM | in vitro | Sprague-Dawley rats | BIC (10 µM) in hippocampal slice cultures | [177] | ||
| 10 µM | in vitro | Human | Mg2+-free model in slices from dysplastic human neocortex | [178] | ||
| 10 mg/kg | i.v., 30 min | Male To mice | NMDLA (85 mg/ml at a rate of 0.14 ml/min, i.v.) | [179] | ||
| ED50 = 61 mg/kg | i.p., 30 min | Male NSA/CF1 mice | MES (auricular) | [107] | ||
| 60 mg/kg | i.p., 30 min | Male NSA/CF1 mice | NMDA (250 mg/kg, i.p.) | [107] | ||
| ED50 = 40 mg/kg | i.p. | Male OF-1 mice | Handling-induced and strychnine- potentiated convulsions | [180] | ||
| ED50(clonus) = 1.1 µmol/kg | i.p., 60 min | Genetically epilepsy-prone rats (GEPR-9 s) | Sound (109 dB) | [138] | ||
| 50 pmol | i.c. | Male Wistar rats | Putrescine (10–20 nmol, i.c.) | [181] | ||
| 20 mg/kg | i.p., 30 min | ddY mice | PTZ (100 mg/kg, s.c.) | [182] | ||
| 100 µg | i.c.v. | Male Sprague-Dawley rats | Amygdala-kindling | [183] | ||
| 30 mg/kg | i.p. | Male Sprague-Dawley rats | Status epilepticus | [184] | ||
| 20, 40 and 60 mg/kg | i.p., 60 min | Male Wistar rats | PTZ (100 mg/kg, s.c.) | [185] | ||
| ED50 = 15.5 mg/kg | i.p., 30 min | Male Wistar rats | Lindane (8 mg/kg, i.p.) | [186] | ||
| 20 mg/kg | i.p. | Male Wistar rats | Electrical stimulation-induced cortical epileptic afterdischarges | [187] | ||
| 20 mg/kg | i.p. for one-two weeks every 8 h during epileptogenesis | C57BL/6J mice | Kainic acid (20 mM, i.c.) | [188] | ||
| Opipramol Ki = 0.2–50.0 | H1 (6) EBP (13) ERG2 (17) TMEM97 (110) 5-HT2A (120) D2 (120–300) Alpha1 (200) D1 (900) H2 (4470) Alpha2 (6100) | |||||
| IC50 = 52 µM | in vitro | Male Wistar rats | Mg2+-free model in hippocampal slices | [104] | ||
| 20 mg/kg | i.p., 30 min | Male Swiss mice | PTZ (100 mg/kg, i.p.) | [189] | ||
| ( ± )Pentazocine Ki = 15–135 | Kappa opioid R(2–75) Mu opioid R (3–7) NMDAR (25–2820) Delta opioid R (49) TMEM97 (1500–1900) D2 (3500) | |||||
| ED50 = 7.9–11.6 µM | in vitro | Wistar rats | NMDA (10–80 µM) in neocortical slices | [87] | ||
| 4, 8 and 16 mg/kg | s.c., 30 min | Male NMRI mice | MES (corneal) | [190] | ||
| ED50 = 6 mg/kg | s.c. | Male NMRI mice | PTZ (1% at a rate of 0.3 ml/min, i.v.) | [190] | ||
| 10–160 µg | i.c.v. | Male Sprague-Dawley rats | Flurothyl | [191] | ||
| ED50 = 63.3 mg/kg | s.c., 30 min | Male Swiss-Webster mice | NMDLA (500 mg/kg, s.c.) | [192] | ||
| 10–50 mg/kg | i.p. | Mice | MES | [193] | ||
| Sig1R antagonists* * | ||||||
| *BD-1063 Ki = 6–9 nM | TMEM97 (210–318) | |||||
| * 3 nmol | i.c.v. | Male CD-1 mice | NMDA (1 nmol, i.c.v.) | [39] | ||
| * ( ± )Fenfluramine (ZX008) Ki = 266 | SERT (108) 5-HT1A (327) 5-HT2B (400–5710) NET (740–1990) 5-HT2C (1410–3183) 5-HT2A (3800–5460) Na+channels (4840) | |||||
| 500 µM | in vitro | Wistar rats | Low Mg2+model in brain slices | [194] | ||
| 50 μM | in vitro, 24 h before recordings | Mutant Scn1Lab-/-zebrafish larvae | Spontaneous seizures | [195] | ||
| * 25 µM | in vitro | Mutant Scn1Lab-/-zebrafish larvae | Spontaneous seizures | [196], [197] | ||
| 20 mg/kg | i.p., 45 min | Male CD-1 mice | MES-T | [198] | ||
| * (+)Fenfluramine IC50 = 70 | SERT (150–855) NET (1286) 5-HT2C (362–6245) 5-HT2B (379–5099) 5-HT2A (2470–11107) | |||||
| * 3 nmol | i.c.v. | Male CD-1 mice | NMDA (300 pmol, i.c.v.) | [199] | ||
| 100 µM | in vitro | Mutant Scn1Lab-/-zebrafish larvae | Spontaneous seizures | [200] | ||
| *S1RA (E-52862, MR-309) Ki = 3–24 nM | 5-HT2B (328) TMEM97 (9300) | |||||
| * 3 nmol | i.c.v. | Male CD-1 mice | NMDA (300 pmol, i.c.v.) | [199] | ||
| (S)Safinamide (FCE 26743, NW 1015, PNU 151774, PNU 151774E) IC50 = 19 | MAO-B(2–450) TMEM97 (1590) | |||||
| ED50 = 4.1 mg/kg (i.p.); ED50 = 6.9 mg/kg (p.o.) | i.p.; p.o. | Mice | MES | [201], [202] | ||
| ED50 = 8.0 mg/kg (i.p.); ED50 = 11.8 mg/kg (p.o.) | i.p.; p.o. | Rats | MES | [201], [202] | ||
| ED50 = 26.9 mg/kg | p.o. | Mice | BIC | [201] | ||
| ED50 = 60.6 mg/kg | p.o. | Mice | Picrotoxin | [201] | ||
| ED50 = 21.5 mg/kg | p.o. | Mice | 3-MPA | [201] | ||
| ED50 = 26.8 mg/kg | p.o. | Mice | PTZ | [201] | ||
| ED50 = 104.1 mg/kg | p.o. | Mice | Strychnine | [201] | ||
| 10 and 30 mg/kg | i.p., 15 min | Male Wistar rats | Kainic acid (10 mg/kg, i.p.) | [203] | ||
| 10 and 30 mg/kg | i.p., 60 min | Male Wistar rats | Amygdala-kindling | [204] | ||
| 50 mg/kg (p.o.) | p.o. | Male cynomologus monkeys | Electrically induced afterdischarges | [205] | ||
| ED50 = 4.1 mg/kg | i.p. | Male Carworth Farms No. 1 mice | MES | [206] | ||
| ED50 = 12 mg/kg | p.o. | Sprague-Dawley rats | MES | [206] | ||
| Sertraline (1S-cis isomer) Ki = 29–57 | SERT (0.1–3.4) DAT (25–260) Alpha1 (36–335) Alpha2 (83–651) NET (159–978) M1 (310–430) M5 (365) M4 (367) 5-HT2C (567–2300) 5-HT2A (783–2200) M2 (984) Ca2+channels (1305–2084) 5-HT2B (2160) Na+channels (2334) MC5 (4313) 5-HT1A (3700) H1 (5000–6600) TMEM97 (5297) K+channels (6814) | |||||
| 15 and 30 mg/kg | i.p., 8 h | Genetically epilepsy-prone rats (GEPR-9 s) | Sound (115 dB) | [207] | ||
| 25 mg/kg | i.p., 4 h | Male Wistar rats | 4-AP (2.5 mg/kg, i.p.) | [208], [209] | ||
| 2.5 and 25 mg/kg | i.p., 4 h | Male Wistar rats | PTZ (50 mg/kg, i.p.) | [208] | ||
| Positive allosteric modulators of Sig1R | ||||||
| *E1R (4 R,5S-methylphenylpiracetam) | N/A | |||||
| * 10 and 50 mg/kg | i.p., 60 min | Male Swiss-Webster mice | PTZ (1% at a rate of 0.6 ml/min, i.v.) | [210] | ||
| 50 mg/kg | i.p., 60 min | Male Swiss-Webster mice | BIC (0.01% at a rate of 0.6 ml/min, i.v.) | [210] | ||
| 75 mg/kg | i.p., 30 min | Male Swiss-Webster mice | NE-100 (75 mg/kg, i.p.) | [210] | ||
| *SKF-83959 | D1 (1–8) 5-HT2A (266) Alpha2 (295) D3 (399) D2 (920) | |||||
| * 10, 20 and 40 mg/kg | i.p., 40 min | Male C57BL/6J mice | MES-T | [93] | ||
| * 20 and 40 mg/kg | i.p., 40 min | Male C57BL/6J mice | PTZ (80 mg/kg, s.c.) | [93] | ||
| * 40 mg/kg | i.p., 40 min | Male C57BL/6J mice | Kainic acid (30 mg/kg, i.p.) | [93] | ||
| *SOMCL-668 | N/A | |||||
| * 40 mg/kg | i.p., 40 min | Male C57BL/6J mice | MES-T | [93] | ||
| * 40 mg/kg | i.p., 40 min | Male C57BL/6J mice | PTZ (80 mg/kg, s.c.) | [93] | ||
| * 40 mg/kg | i.p., 40 min | Male C57BL/6J mice | Kainic acid (30 mg/kg, i.p.) | [93] | ||
| Ropizine (SC-13504) | N/A | |||||
| 4–8 mg/kg | i.v. | Papio papiobaboons | Photosensitive epilepsy | [211] | ||
| 3–6 mg/kg | – | Beagle dogs | MES | [212] | ||
| 3, 30 and 300 mg/kg | p.o., 30 min | Male Sprague-Dawley rats | Pre-kindled and partially kindled hippocampal seizures |
Preclinical anti-seizure effects of Sig1R ligands.
Affinities of compounds were obtained from databases PubChem and Binding Database (accessed from May 25, 2022 till June 28, 2022). Table demonstrates the binding affinities (Ki) to targets of compounds at concentrations below 10 µM (< 1 ×10-5). Bolded targets – equal or higher affinity than for Sig1R. N/A – not available. 3-MPA – 3-mercaptopropionic acid; 4-AP – 4-aminopyridine; AMPA – α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; BIC – bicuculline; MES – maximal electroshock-induced seizures; MES-T – maximal electroshock seizure threshold; NMDA – N-methyl-D-aspartic acid; NMDLA – N-methyl-DL-aspartic acid; PIC – picrotoxin; PTZ – pentylenetetrazole; STRYC – strychnine. Targets: 5-HT1-7 – serotonin receptor subtypes; alpha-1 and alpha-2: adrenergic receptors; AchE – acethylcholine esterase; DAT – dopamine transporter; EBP – emopamil binding protein; ERG2 – C8 sterol isomerase; MAO-B – monoamine oxidase B; Mu, Kappa, Delta – opioid receptor subtypes; nAChRs – nicotinic actehylcholine receptors; Na+, K+ and Ca2+ – corresponding ion channels; NET – norepinephrine transporter; NMDAR – N-methyl-D-aspartate receptors; NOP R – nociceptin opioid peptide receptor; SERT – serotonin transporter; TMEM97 – transmembrane protein 97; VAChT – vesicular acethylcholine transporter. *Sig1R-dependent activity confirmed. * *Sig1R antagonists demonstrate anti-seizure activity against seizures induced by psychostimulants and drugs of abuse (for more details see Section 3.1.2.).
The anti-seizure effects of Sig1R ligands have been shown in a wide range of preclinical in vivo animal models. These study paradigms included the administration of chemoconvulsants (Fig. 2) with the aim of inducing seizures due to the inhibition of the GABAergic system (PTZ, BIC, flurothyl, lindane, picrotoxin, penicillin), overactivation of the glutamatergic system (NMDA, AMPA, kainic acid) or cholinergic system (pilocarpine, nicotine, soman, sarin), as well as use of electrical stimulation-induced seizure models (6 Hz test, maximal electroshock seizures (MES, both auricular and corneal) and amygdala kindling), and use of specific animal models, studying sound-induced seizures in DBA/2 mice and genetically epilepsy-prone rats, absence-like seizures in Groggy (GRY/Idr and WAG/Rij) rats, and seizures in zebrafish (Scn1Lab+/-) and mice (Scn1a+/-) with Dravet syndrome (Table 1).
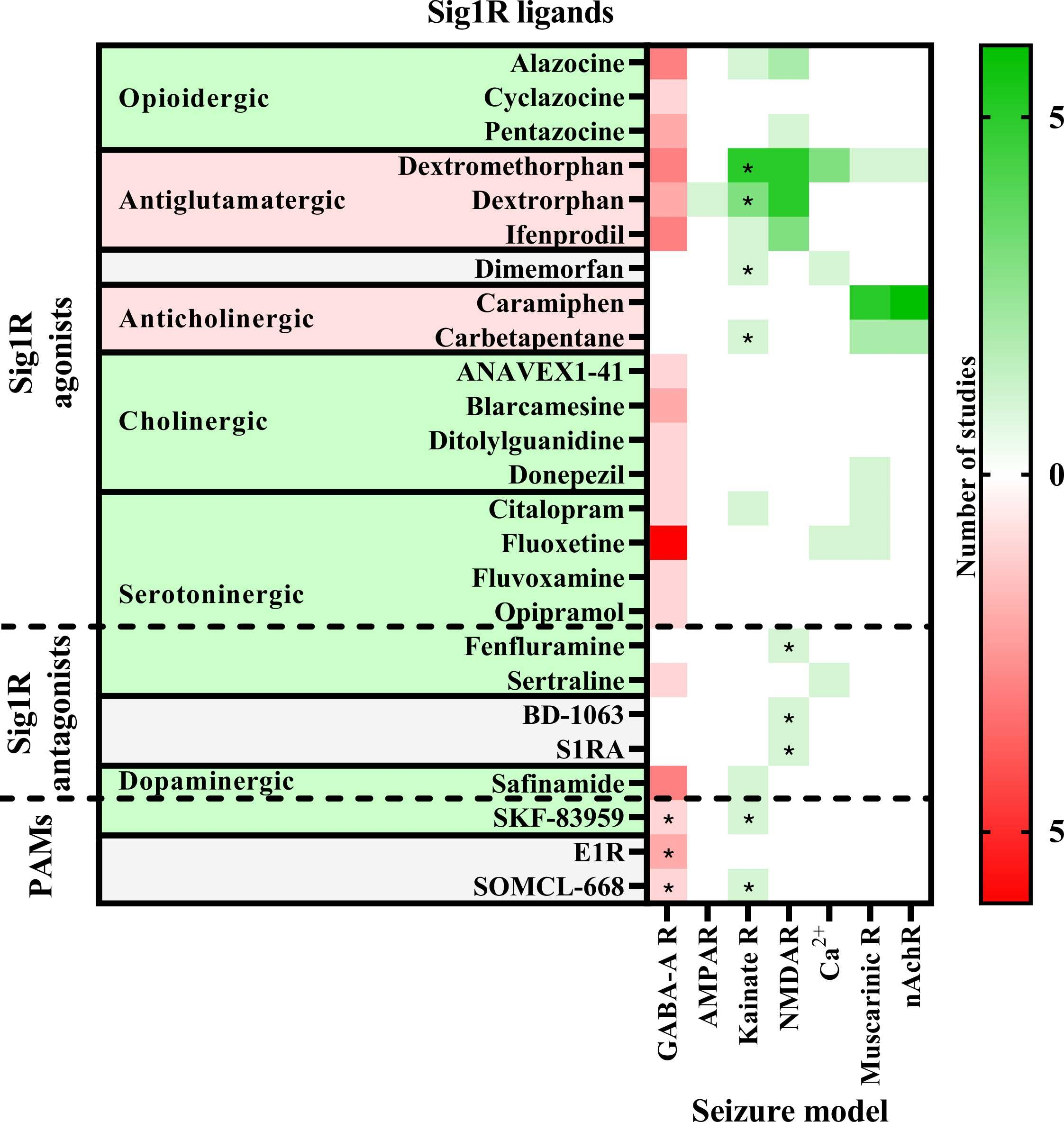
Anti-seizure activity profile of Sig1R ligands working against chemoconvulsant-induced seizures in vivo.
Chemoconvulsants have been used to induce seizures either by inhibition (red) of the gamma-aminobutyric acid (GABA)-A receptors or overactivation (green) of ionotropic glutamate receptors (AMPAR – α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors; Kainate R – kainate receptors; NMDAR – N-methyl-D-aspartate receptors), intracellular Ca2+ increase or acetylcholine receptors (Muscarinic R – muscarinic receptors; nAchR – nicotinic acetylcholine receptors). Asterisks indicate seizure models in which the anti-seizure effects of the Sig1R ligand has been confirmed to be Sig1R-dependent. PAMs – positive allosteric modulators of Sig1R. For more details see Table 1.
3.1.1. Agonists
Benzomorphans, prototypical Sig1R agonists, were the first Sig1R ligands for which anti-seizure effects were attributed to activity at Sig1R [5], [88]. Since racemic benzomorphans demonstrate high affinity to opioid receptors, the anti-seizure effects of the (+)isomer of alazocine (SKF-10,047), which shows higher affinity for Sig1R than for opioid receptors, was evaluated in 1985 by using the PTZ-induced seizure test in male Swiss-Webster mice (Table 1) [94]. (+)Alazocine, administered 10 min before PTZ, induced a significant dose-dependent (1–30 mg/kg, i.p.) decrease in the percent of animals with tonic seizures. At a dose of 30 mg/kg, (+)alazocine completely prevented the occurrence of tonic seizures (Table 1) [94]. Therefore, the potential of Sig1R as a target for the treatment of seizures was recognized and later demonstrated for several Sig1R agonists (Table 1). To date, Sig1R could be considered the unifying molecular entity for benzomorphans (opioids), cough suppressants, antidepressants and anxiolytics (e.g., selective serotonin reuptake inhibitors (SSRIs)) and some anti-dementia drugs (Fig. 3).
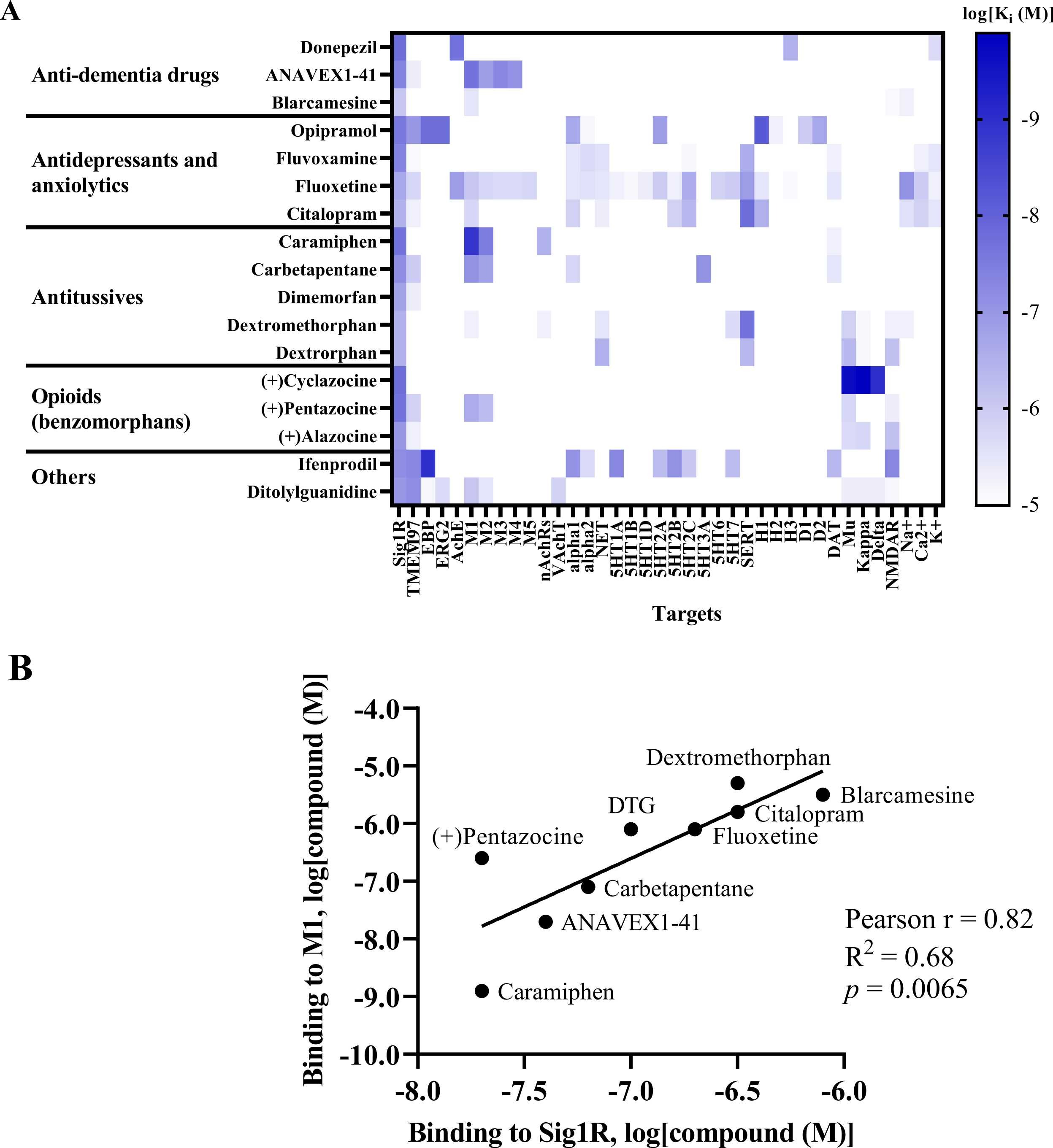
Sig1R agonists demonstrating anti-seizure effects and their corresponding high-affinity targets.
(A) Heatmap demonstrates the binding affinities (Ki) to targets of compounds at concentrations below 10 µM (< 1 ×10-5). Affinities of compounds were obtained from the PubChem and Binding Databases (accessed from May 25, 2022, to June 28, 2022). TMEM97 – transmembrane protein 97; EBP – emopamil binding protein; ERG2 – C-8 sterol isomerase; AchE – acetylcholine esterase; M1-M5 – muscarinic receptors; nAChRs – nicotinic acetylcholine receptors; VachT – vesicular acetylcholine transporter; NET – norepinephrine transporter; alpha-1 and alpha-2: adrenergic receptors; SERT – serotonin transporter; 5-HT1–7 – serotonin receptor subtypes; DAT – dopamine transporter; D1 and D2 – dopamine receptors; H1-H3 – histamine receptors; Mu, Kappa, Delta – opioid receptor subtypes; NMDAR – N-methyl-D-aspartate receptors; Na+, K+ and Ca2+ – corresponding ion channels. (B) Correlation of the binding of Sig1R agonists to Sig1R and to muscarinic M1 receptors. DTG – ditolylguanidine. Graphs were generated using GraphPad Prism software. For more details, see Table 1.
High-affinity binding to muscarinic, serotonin, opioid and NMDA receptors outlines the pharmacodynamic profile of Sig1R agonists (Fig. 3A). Interestingly, a strong positive correlation was observed between ligand affinities to Sig1R and muscarinic M1 receptors(Fig. 3B), which was previously described for several M1 receptor antagonists [214]. Overstimulation of muscarinic receptors, especially M1 receptors, is known to result in seizures in vivo [215], [216]. Therefore, the anticholinergic Sig1R agonists caramiphenand carbetapentane are effective against pilocarpine- and soman-induced seizures (Fig. 2). However, this finding prompts questions about the direct involvement of Sig1R in the anti-seizure effects of caramiphen and carbetapentane, since the anti-seizure effect of these ligands is probably achieved due to direct inhibition of muscarinic receptors. On the other hand, in vivo studies have shown anti-seizure effects induced by the dual Sig1R and muscarinic receptor agonists ANAVEX1–41 and blarcamesine (Table 1, Fig. 2), which also demonstrate anti-amnesic and neuroprotective effects [217], [218]. Currently, blarcamesine is in clinical trials for the treatment of Alzheimer’s disease (NCT04314934), and another trial has begun to test the efficacy of blarcamesine in patients with Rett syndrome (NCT04304482). While additional data are needed to fully understand the possible interaction between Sig1R and muscarinic receptors that leads to the anti-seizure effects of Sig1R agonists, the synergistic activity between both targets seems to indicate that they are a promising strategy for the treatment of seizures and disease-related comorbidities, e.g., decreased cognitive ability and memory problems.
Sig1R agonists and NMDA receptor antagonists, e.g., (+)alazocine, ifenprodil, dextromethorphan and its metabolite dextrorphan, are effective in reducing seizures induced by the overstimulation of glutamate receptors (Fig. 2). It has been hypothesized previously that the anti-seizure effects of antiglutamatergic Sig1R agonists is primarily due to the inhibition of NMDA receptors [134], [219], [220]. It has been shown that the anti-seizure effects of Sig1R-NMDA receptor complex ligands against NMDA-induced seizures both in vitro and in vivo correlates with the binding affinity of these ligands to NMDA [87], [134], [220]. However, it should be noted that in some of these studies, the binding affinity to Sig1R was determined using [3H](+)SKF-10,047 ([3H](+)alazocine), which is not a Sig1R selective agonist per se and binds to NMDA receptors with nanomolar affinity (Table 1). Nevertheless, PPCC, a highly potent Sig1R agonist with no binding to NMDA receptors [221], was not able to prevent or modulate NMDA-induced seizures in mice [39], [40], [199]. Currently, the Sig1R-dependent anti-seizure effects of some Sig1R agonists and positive allosteric modulators has been confirmed against kainic acid- (Fig. 2, Fig. 4A) but not NMDA-induced seizures (Table 1, Fig. 2) and might suggest the involvement of other glutamate receptors. However, whole-cell voltage-clamp measurements in mouse hippocampal neurons have shown that antiglutamatergic Sig1R agonists are not able to attenuate AMPA- or kainate-induced currents [219]. While direct inhibition of NMDA receptors seems to account for the anti-seizure effects of antiglutamatergic Sig1R agonists, simultaneous activation of Sig1R, by facilitating NMDA receptor signaling, might be considered a valuable strategy to maintain synaptic plasticity, learning and memory.
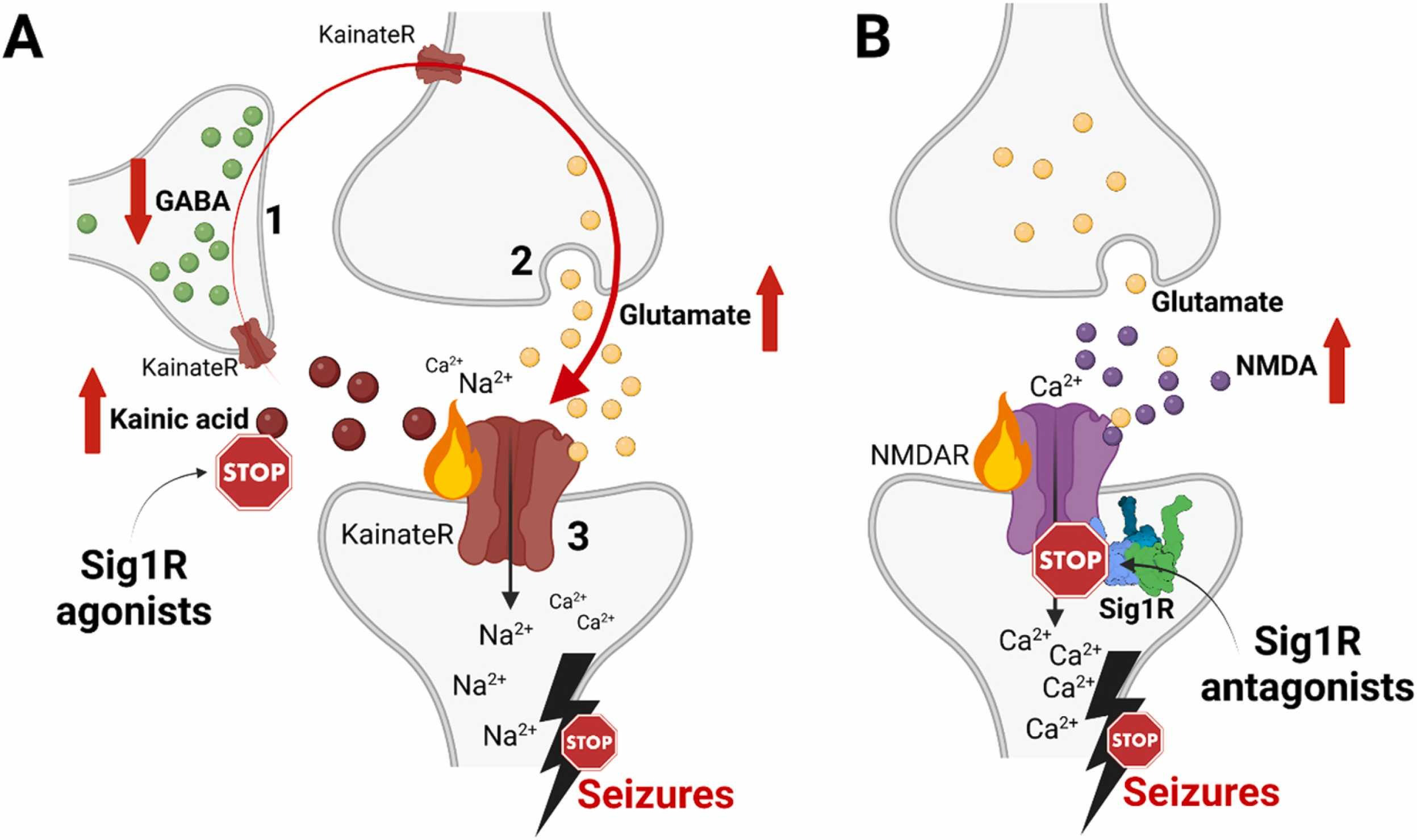
Confirmed Sig1R-dependent mechanisms of anti-seizure activity of Sig1R agonists and antagonists.
(A) High doses of kainic acid suppress the release of GABA from GABAergic interneurons (1) and enhance presynaptic release of glutamate from glutamatergic neurons (2), which, together with kainic acid, activate postsynaptic KainateR (3) and leads to seizures [265]. Sig1R-dependent anti-seizure activity of Sig1R agonists (dextromethorphan, dextrorphan, dimemorfan and carbetapentane) has been pharmacologically confirmed against kainic acid-induced seizures by using Sig1R antagonists (Table 1). Therefore, based on the activity of kainic acid, Sig1R agonists either increase GABA release from GABAergic interneurons (1), block presynaptic glutamate release (2) and/or attenuate KainateR activity (3). (B) High doses of NMDA activate NMDA receptors and induce seizures. Sig1R antagonists (BD-1063, S1RA) can alleviate NMDA-induced seizures. However, they are ineffective against NMDA-induced seizures in Sig1R KO animals thus confirming Sig1R-dependent activity (Table 1). KainateR – kainate receptors; NMDAR – N-methyl-D-aspartate receptors. Created with BioRender.com.
While the exact serotonin-related mechanisms in the modulation of seizures remain elusive, serotoninergic drugs are promising candidates for the treatment of seizures, especially those caused by rare and severe epilepsy syndromes [222]. Although a direct molecular interaction between Sig1R and serotonin receptors has not been identified, recent findings in Sig1R KO mice indicate that Sig1R might be involved in the regulation of both plasma and brain serotonin levels [17]. Several serotoninergic Sig1R agonists have demonstrated anti-seizure effects not only in preclinical seizure models (Table 1) but also in patients. Therefore, Sig1R has been suggested as a target that might mediate the anti-seizure effects of serotoninergic drugs.
One of the most selective Sig1R agonists [223] tested in preclinical seizure models is PRE-084 [210]. The anti-seizure effects of PRE-084 was evaluated in chemoconvulsant-induced seizure models [210]. PRE-084 administered at doses of 3, 10 and 50 mg/kg (i.p.) was shown to be ineffective against PTZ- and BIC-induced seizures in ICR male mice [210]. However, PRE-084 was able to alleviate high-dose Sig1R antagonist NE-100-induced seizures [210]. To elucidate the full potential of Sig1R agonists as anti-seizure drugs, more studies with selective Sig1R agonists are needed. Direct interaction of a drug with Sig1R is considered an advantage in treating central nervous system (CNS) diseases through a multitarget-directed ligand approach [224]. Therefore, based on the pharmacodynamic profile of Sig1R agonists demonstrating anti-seizure effects, a multitargeted drug-based approach should be considered in the development of novel anti-seizure medications for the treatment of seizures and disease-related comorbidities.
3.1.2. Antagonists
Sig1R antagonists have been suggested as promising drug candidates for treating seizures that are caused by an overdose of psychostimulants or drugs of abuse. Several laboratories have demonstrated that Sig1R ligands can attenuate cocaine-induced seizures and lethality of these seizures in animals [225], [226], [227]. It was revealed that activation of Sig1R can facilitate the toxic effects of cocaine, while inhibition of Sig1R can alleviate and even block them [227], [228], [229], [230]. In addition, Sig1R antagonists were found to attenuate methamphetamine-induced seizures [231], [232]. The effect of pharmacological Sig1R antagonist protection against cocaine-induced seizures was confirmed by an in vivo study using antisense oligodeoxynucleotides against Sig1R [229]. Based on this, a number of Sig1R antagonists with high affinity and selectivity have been synthesized and evaluated against cocaine-induced seizures and include several derivatives of rimcazole, ethylenediamine analogs of BD-1008, a derivative of piperidineAC-927, derivatives of piperazine (e.g., AC-928, YZ-069, SN79, CM-156), panamesine (EMD-57445) and others [233], [234], [235]. Preclinical activity testing of Sig1R antagonists has confirmed that inhibition of Sig1R is a powerful strategy for blocking the seizures and lethality induced by psychostimulants and drugs of abuse.
Cocaine-induced seizures, which are usually generalized, mainly occur after long-term abuse of cocaine or cocaine overdose [236]. In addition, cocaine-induced seizures are very often difficult to manage with existing antiepileptic drugs [237]. Cocaine is known to block the transporters that are responsible for the uptake of dopamine (DAT), serotonin (SERT), and norepinephrine (NET) by the presynaptic nerve terminals [238]. It was thought that the direct binding of cocaine to serotonin, dopamine, muscarinic or sigma receptors might also have an important role in cocaine-induced toxic effects [228], [239], [240]. The interaction of cocaine between serotoninergic, cholinergic and sigma neuronal systems was concluded to be critical for the occurrence of cocaine-induced seizures [239]. The direct interaction between cocaine and Sig1R was found in 1988 when it was demonstrated that cocaine binds to Sig1R with micromolar affinity (2–7 µM) [241], [242]. It should be noted that administration of cocaine at high doses can easily result in micromolar concentrations in vivo [243]. Therefore, the interaction of cocaine with some low affinity targets, including Sig1R, can become physiologically relevant and thus directly linked to mechanisms of cocaine-induced toxicity.
In transfected cells, Sig1R has been shown to form direct protein-protein interactions with DAT [244]. In addition, it was demonstrated that activation of Sig1R can promote an outward-facing conformation of DAT, thus potentiating the response of cocaine (1–10 µM) by enhancing cocaine binding to DAT [244]. In addition, Sig1R has been shown to form heteromers with dopamine D1 [245] and D2 receptors in transfected cells [246]. At high cocaine concentrations (30–150 µM), consistent with a severe cocaine overdose in vivo, cocaine can increase the heterodimerization between the Sig1R and dopamine D1 receptors [246]. In contrast, this has not been observed in Sig1R KO animals [246]. Several lines of evidence indicate that increased dopamine D1 and decreased D2 receptor activation can result in seizures [247]. Activation of dopamine D1 receptors has been shown to potentiate glutamate responses, particularly those mediated by NMDA receptors [248]. Therefore, it seems that Sig1R could serve as an amplifier of dopamine D1 receptor-mediated signaling [249], which, in the case of an overdose of cocaine, can result in severe seizures.
Interestingly, by using whole-cell voltage-clamp recordings in mouse brain slices, it was shown that cocaine and methamphetamine can inhibit presynaptic GABA release in the ventral tegmental area, the origin of the dopaminergic neurons of the mesocorticolimbic dopamine system [250], [251], [252]. The inhibitory effect of cocaine and methamphetamine on GABA release was demonstrated as a decrease in slow inhibitory postsynaptic currents (sIPSCs) resembling the activity of GABA-B receptors [250], [252], which are one of the key systems for controlling the excitability of dopaminergic neurons [251]. It was also demonstrated that the inhibitory effect of cocaine (10 µM) on sIPSCs was significantly reduced when performing recordings in brain slices isolated from male C57BL/6J background Sig1R KO mice [252]. In the same study, the Sig1R antagonists NE-100 (2 µM) and BD-1063 (2 µM) blocked the cocaine-induced inhibition of sIPSCs [252], thus confirming the direct involvement of Sig1R.
Inhibition of NMDA receptors has been suggested as a possible strategy to block cocaine-induced seizures [243], [253], [254], [255], [256], [257]. Direct molecular interactions between Sig1R and NMDA receptors have been demonstrated [22], [29], and inhibition of the NMDA receptor complex by Sig1R antagonists has already been proven to be a promising strategy to treat chronic neuropathic pain conditions [258], [259], [260]. Recently, the significant interplay between Sig1R, histidine triad nucleotide-binding protein 1, alpha-2-delta subunit of voltage-gated Ca2+ channels and the subunit of NR1 of NMDA receptors was shown to be an important molecular complex that participates in the regulation of NMDA receptor-mediated responses, especially in neuropathic pain [66]. The involvement of Sig1R in the modulation of nociceptive responses was revealed in Sig1R KO mice [261], [262]. The pain-attenuated phenotype of Sig1R KO mice opened a new era for the search for novel compounds with inhibitory activity at Sig1R that could be used to treat several pain conditions [258]. An excessive influx of Ca2+ via activated NMDA receptors can cause abnormal electrical activity, which is also one of the main reasons for the development of seizures and excitotoxicity [20]. It should be noted that two of the most selective Sig1R antagonists known to date, BD-1063 and S1RA [263], [264], have demonstrated anti-seizure effects against seizures induced by acute and direct i.c.v. administration of NMDA in mice [39], [199]. In addition, the anti-seizure effects of BD-1063 were confirmed to be Sig1R-dependent (Fig. 2; Fig. 4B) since the effect was abolished in CD-1 background Sig1R KO mice [39]. Therefore, the Sig1R-NMDA receptor complex could be viewed as a possible target for anti-seizure drugs when signal transduction through NMDA receptors is significantly increased. Downregulation of NMDA receptor-mediated excitatory activity either by direct inhibition of Sig1R (Fig. 4B) or NMDA receptors seems to be beneficial in acute situations.
3.1.3. Allosteric modulators
The allosteric modulators of Sig1R form a special group of Sig1R ligands. Despite the lack of specific screening assays, several positive allosteric modulators [266], as well as negative allosteric modulators of Sig1R [267], have been identified and described. Why have positive allosteric Sig1R modulators attracted so much interest in the field? Almost all known positive allosteric modulators of Sig1R demonstrate significant anti-seizure effects in preclinical studies (Table 1) [266]. The neuroprotective [268], [269], [270], [271], antidepressant [269], [270], [272], anxiolytic [272], [273], anti-inflammatory [270], [274] and even antipsychotic-like [271] effects of positive allosteric modulators have also been demonstrated in animal models. With several advantageous properties, this group of compounds is viewed as a source of promising novel drug candidates for the treatment of seizures and disease-associated comorbidities (Fig. 5).
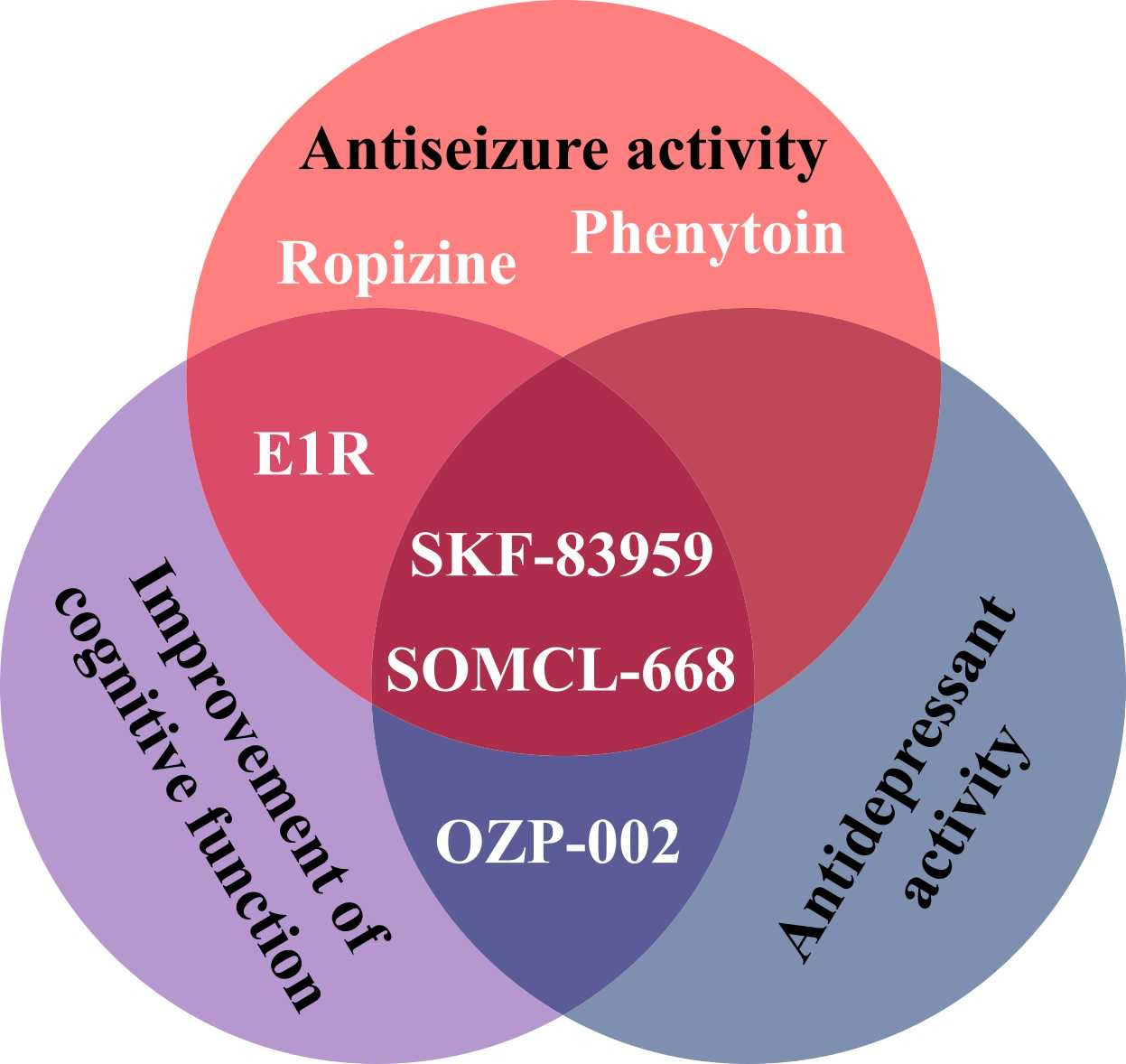
Effects of positive allosteric modulators of Sig1R demonstrated by preclinical in vivo studies.
The discovery of positive allosteric Sig1R modulators is associated with specific binding properties of radiolabeled Sig1R agonist dextromethorphan observed in the beginning of the 1980’s [275]. The binding of antitussive dextromethorphan was found to be enhanced by the antiepileptic drug phenytoin [275], which became known as the first positive allosteric modulator of Sig1R. It should be noted that at that time, the ability of dextromethorphan to bind with Sig1R was not discovered until several years later [276], [277], [278]. It was shown that ropizine, a compound with similar anti-seizure effects to that of phenytoin, can also enhance the binding of dextromethorphan to Sig1R [278], [279], [280]. This observation allowed to propose the hypothesis that dextromethorphan might possess some inherent anti-seizure property, which was confirmed in later studies (Table 1). The discovery of several new (second generation) positive allosteric modulators and demonstration of their anti-seizure effects [210], [281] confirmed that modulation of the activity of Sig1R is a valid strategy to reduce the number of seizure episodes and alleviate seizure severity.
Although pharmacological effects of allosteric modulators have been demonstrated preclinically in vivo, the underlying mechanisms are not fully elucidated. Several possible mechanisms have previously been discussed [266]. One of the main characteristics of allosteric modulators of Sig1R is that they do not compete with prototypic Sig1R ligands to bind to the Sig1R orthosteric site and can enhance or decrease the activity of Sig1R ligands in various in vitro assays and animal models. The direct allosteric Sig1R binding site is not yet known. However, the findings of an article published by Rossino et al. include novel speculations about the possible binding site, at least for some of the positive allosteric Sig1R modulators [282]. A transient binding site, also known as a metastable binding site, was identified at the entrance of the occluded orthosteric Sig1R ligand binding site [282]. Binding to metastable sites induces conformational changes in the receptor structure, exposing the occluded orthosteric binding site [282]. This metastable binding site, which was demonstrated by using specifically designed bitopic Sig1R ligands, was suggested as an allosteric binding site of benzazepine derivatives, e.g., SOMCL-668 [282]. However, this publication did not discuss how the binding of allosteric modulators to this putative allosteric site would impact the binding of Sig1R agonists or antagonists. Therefore, experimental confirmation of this hypothesis is needed and may provide important insights into the mechanisms of allosteric Sig1R modulators.
Negative allosteric Sig1R modulators include some benzamide derivatives, such as venetoclax (brand name: Venclexta®) and navitoclax, which have been studied in clinical trials for the treatment of acute myeloid leukemia, chronic lymphocytic leukemia, small lymphocytic lymphoma and myelofibrosis [267]. Venclexta®, a prescription orphan drug, was approved by the U.S. Food and Drug Administration (FDA) in 2016. As negative Sig1R modulators, these compounds were able to reduce the specific binding of [3H](+)pentazocine, decrease the radioligand affinity and shift right the competitive binding curves of several Sig1R-related ligands [267]. Although Sig1R antagonists are known for their anti-seizure properties, negative allosteric modulation of Sig1R has not yet been linked with seizure-modulating properties.
Selective allosteric modulators, such as E1R and SOMCL-668, have been proposed [266]. However, for both positive and negative modulators, other primary mechanisms and high affinity targets exist. For example, SKF-83959 demonstrates high affinity for dopamine D1 receptors [283] and alpha-2 adrenoreceptors [284], while phenytoin mainly acts on voltage-gated Na+ channels [285]. Fenfluramine (brand name: Fintepla®), an FDA-approved orphan drug for the treatment of seizures associated with Dravet syndrome (2020) and recently also Lennox-Gastaut syndrome (2022), was found to act as a positive allosteric modulator of Sig1R in preclinical animal models of disturbed neurocognitive function [13], [286]. In addition to fenfluramine’s serotonergic profile (Table 1) [222], [286], fenfluramine demonstrates high affinity (Ki = 0.27 µM) binding to the orthosteric Sig1R binding site (Table 1) [286]. Therefore, by directly binding to the orthosteric Sig1R binding site, fenfluramine should act as a full agonist, partial agonist, full antagonist or inverse agonist of Sig1R. For example, it has already been demonstrated that fenfluramine can block i.c.v. administered NMDA-induced seizures in mice, thus resembling the activity of Sig1R antagonists (Table 1) [199]. Fenfluramine was shown to disrupt the association between Sig1R and the NR1 subunit of NMDA receptors [199], which is a well-defined mechanism for the activity of Sig1R antagonists [32], [35]. Acting as a Sig1R antagonist was also suggested as a possible anti-seizure mechanism of fenfluramine in a zebrafish model of Dravet syndrome (Table 1) [197]. Therefore, the classification of fenfluramine as a positive allosteric modulator should be considered critically.
However, the difficulty with classifying fenfluramine raises an important question of what the allosteric modulators of Sig1R are, with emphasis on the need to elucidate possible direct mechanisms. In line with this topic of study, a dual synergistic activity has been demonstrated between dextromethorphan and the first known positive allosteric modulator, phenytoin. In an in vivo preclinical study, the Sig1R agonist dextromethorphan potentiated the anti-seizure activity of the allosteric Sig1R modulator and anti-seizure drug phenytoin [114]. In addition, it was shown that other high affinity Sig1R agonists, carbetapentane and caramiphen, exhibit similar properties [105], [114]. Facilitation of the anti-seizure effects of antiepileptic drugs, including phenytoin, has also been demonstrated for fluoxetine in several reports [287], [288], [289], [290]. Since dual synergistic activity occurs between several Sig1R agonists and the prototypic allosteric modulator phenytoin in vivo, it is not possible to directly describe this interaction based on a purely classical interpretation of allosteric mechanisms. Additionally, the positive allosteric modulatory effect of phenytoin in vitro is observed only at high concentrations [266]. Detailed knowledge of the direct molecular mechanisms underlying the activity of allosteric modulators is inevitably needed.
The idea that some positive allosteric modulators may achieve their pharmacological effects due to direct activity at several other molecular targets cannot be excluded. This so-called “off-target” activity may indirectly trigger an activated state of Sig1R by inducing Sig1R-dependent chaperoning activity to “off-target” proteins. Taking this into account, all positive allosteric modulators could be viewed as Sig1R sensitizers. Nevertheless, Sig1R modulators, which achieve their in vivo effects by either direct Sig1R modulation or modulation of molecular mechanisms that are actuated by Sig1R, are promising antiseizure drugs not only for the treatment of seizures but also for disease-associated comorbidities. Allosteric modulators have been suggested to offer several distinct potential advantages, such as high level of selectivity by binding to sites that are less preserved across receptor subtype families, potential to retain spatial and temporal aspects of endogenous receptor signaling, and, so called, “ceiling” level to their effect, which may lead to an improved therapeutic window [291], [292]. Due to enriched expression of Sig1R together with its wide functional roles in the brain, allosteric modulators may provide a better approach in developing novel anti-seizure drugs.
3.2. Proconvulsive and convulsive effects of Sig1R ligands
Sig1R ligands have been shown to have a significant biphasic dose-response in several preclinical and clinical studies [293]. The biphasic dose-response of Sig1R ligands has also been clearly observed in preclinical models of seizures [120], [183], [294]. Therefore, this hormesis phenomenon [295] could explain why some Sig1R ligands have demonstrated both anti-convulsive (Table 1) and pro-convulsive (Table 2) effects.
| Compounds | Administration (dose, route) | Observations | Affinity to Sig1R, Ki (nM) | Additional targets, Ki (nM) | References |
| Sig1R agonists | |||||
| ( ± )Alazocine (( ± )SKF-10,047) | 1800–4657 | Kappa opioid R(0.4) Mu opioid R (1.2) Delta opioid R(IC50 = 184) NMDA(2554) | |||
| Increasing cumulative i.v. doses of compound were given at 30-min intervals over a range of 0.1 – 51.2 mg/kg | Convulsive activity:induced convulsions in female Sprague-Dawley rats (ED50 = 25.6 mg/kg, i.v.) | [90] | |||
| 0.2 µmol or higher concentration, i.c.v. | Convulsive activity: induced diffuse clonic seizures in DBA/2 mice | [92] | |||
| Citalopram | 167–404 | SERT (0.5–33.0) 5-HT2C(156–617) H1 (283–371) Alpha1 (711–1820) Ca2+channels (996–1588) 5-HT2B (1171) M1 (1430) Na+channels (2409) NET (2178–6190) TMEM97 (5410) K+channels (IC50 = 2600–3981) DAT (9270) 5-HT2A (9500) | |||
| 15 mg/kg i.p. 30 min before timed intravenous infusion of pilocarpine (24 mg/ml) at the constant infusion rate of 150 µl/min | Pro-convulsive activity: facilitated limbic seizures induced by pilocarpine in male NMRI mice | [117] | |||
| 25 and 50 mg/kg i.p. 30 min before PTZ (0.5%, i.v.) infusion (1 ml/min) | Pro-convulsive activity: significantly decreased seizure threshold in male NMRI mice | [119] | |||
| 25 and 50 mg/kg i.p. 30 min before PTZ (0.5%) i.v. infusion (1 ml/min) | Pro-convulsive activity: significantly decreased PTZ-induced seizure threshold in male NMRI mice | [120] | |||
| 82 and 122 mg/kg i.p. | Convulsive activity: induces seizures in Sprague-Dawley rats | [296] | |||
| 10 mg/kg/day s.c. delivered with osmotic minipumps for 4 weeks during amygdala kindling | Pro-convulsive activity: increased susceptibility to limbic epileptogenesis in male Wistar rats | [297] | |||
| Dextromethorphan((+)methorphan) | 10–652 | SERT (1.4–40.0) NET (240–6000) nAChR (IC50 = 700–8900) Alpha1 (830–3000) Mu opioid R (1280) Na+channels (4800) NMDAR (780–8945) M1 (5070) Kappa opioid R (7000) | |||
| More than 20 mg/kg bolus, or more than 10 mg/kg/h i.v. | Convulsive activity: male New Zealand white rabbits developed muscular tremor, focal motor seizures, hypotension, opisthotonic posturing and apnea | [298] | |||
| 50 mg/kg p.o. 15 min before i.v. infusion of theophylline until the onset of maximal seizures | Pro-convulsive activity: after treatment with dextromethorphan a reduced convulsant dose and concentration of theophylline was necessary to produce seizures in female Levis rats | [299] | |||
| 50 mg/kg p.o. 15 min before i.v. infusion of PTZ until the onset of maximal seizures | Pro-convulsive activity: significantly lower dose of the convulsant to produce maximal seizures in female Levis rats | [299] | |||
| 60 mg/kg i.p. 30 min before each daily stimulation in amygdaloid kindled seizure model | Pro-convulsive activity: potentiated development of amygdaloid kindling in male Sprague-Dawley rats | [136] | |||
| 60 mg/kg i.p. | Convulsive activity: produced spontaneous seizure activity in male Sprague-Dawley rats | [136] | |||
| 50 mg/kg i.p. 15 min before each daily stimulation in amygdaloid kindled seizure model | Pro-convulsive activity: accelerated the expression of kindled seizures in rats. Combination with phenytoin resulted in progressive seizure buildup | [300] | |||
| 50 and 75 mg/kg | Pro-convulsive activity: decreased NMDA-induced seizure threshold in rats | [129] | |||
| 30 mg/kg i.p. 30 min before stimulation in amygdaloid kindled seizure model | Convulsive activity: induced seizures in kindled female Wistar rats | [128] | |||
| ED50 = 19 mg/kg i.v. | Convulsive activity: induced seizures in rats | [301] | |||
| 56 mg/kg i.p. | Pro-convulsive activity: increased sound-induced wild running in Wistar rat pups | [144] | |||
| 212 µmol/kg i.v. infusion | Convulsive activity: induced seizures in male Sprague-Dawley rats | [302] | |||
| 80 mg/kg i.p. | Convulsive activity: Sprague-Dawley rats showed increased seizure occurrence and intensity | [303] | |||
| ( ± )Fluoxetine | 191–240 | SERT (0.3–270.0) Alpha2(6–5012) DAT (11–6670) 5-HT2C(50–398) 5-HT2A(55–1820) Na+channel(82–90) NET (85–6670) M2 (512–2700) Alpha1 (605–5260) M1 (702–1030) Ca2+channels (736–1095) M3 (762–3100) 5-HT6 (771–1770) H1 (933–5400) M5 (976–2070) 5-HT7 (1000) TMEM97 (1610) 5-HT1A (1820–8310) M4 (2090) K+channels (5012) 5-HT2B (5030) 5-HT1D (4270) 5-HT1B (6170) H3 (7300) | |||
| 100 µM | Pro-convulsive activity (in vitro): increased seizure-like event frequency in preictal phase was observed in a low Mg2+/high K+ -induced seizure model in hippocampal slices which were isolated from male Wistar rat pups | [158] | |||
| 10 mg/kg i.p. for 21 days (the last administration was 1 h before the PTZ) | Pro-convulsive activity: reduced PTZ-induced (30 mg/kg, i.p.) seizure threshold and increased seizure severity in male Wistar rats. | [304] | |||
| 10 and 20 mg/kg i.p. 30 min prior to pilocarpine (400 mg/kg, s.c.) | Pro-convulsive activity: decreased the latency to first seizures, increased the number of seizures and decreased the survival in male rats | [305] | |||
| 10 mg/kg/day s.c. delivered with osmotic minipumps for 4 weeks during amygdala kindling | Pro-convulsive activity: increased susceptibility to limbic epileptogenesis in male Wistar rats | [297] | |||
| 30 mg/kg/day i.p. for 7 weeks in WAG/Rij rat model of absence epilepsy | Pro-convulsive activity: pro-absence effect was observed after treatment of fluoxetine in 6-month old rats with established absence seizures | [171] | |||
| 15 mg/kg p.o. for 17 days | Pro-convulsive activity: increased seizure behavior and sudden death in male APPswe/PS1dE9 mice | [306] | |||
| 10 mg/kg/day s.c. for 30 days during amygdala kindling | Pro-convulsive activity: accelerated kindling epileptogenesis in male C57BL/6J (wild-type) and homozygous 5-HT2A knockout mice | [307] | |||
| 20 mg/kg i.p. 30 min after penicillin (500 IU, 2.5 µl) i.c. injection into the left sensorimotor cortex | Pro-convulsive activity: increased the frequency and amplitude of penicillin-induced epileptiform activity in male Wistar rats | [173] | |||
| Fluvoxamine | 17–36 | DAT (1.5–9200) SERT (1.5–540) NET (299–5000) Alpha1 (1290–4800) Alpha2 (1900) 5-HT2C (5790–6700) TMEM97 (8439) | |||
| 40 mg/kg i.v. | Convulsive activity: caused some small spikes (EEG recording) in 2 out of 10 Wistar male rats | [308] | |||
| Ifenprodil (erythro diastereomer) | 2–125 | TMEM97(5–98) EBP (1) NMDAR(8–94) Alpha1 (12–140) 5-HT1A (46) 5-HT2B (66–95) DAT (363) 5-HT2A (421–510) 5-HT7 (513) 5-HT2C (748) Alpha2 (1600–2300) | |||
| 3.2, 10 and 32 µg, i.c.v. | Pro-convulsive activity: enhanced kindling in male Sprague-Dawley rats, biphasic dose-dependent effect was observed | [183] | |||
| 3 and 10 mg/kg i.p. 30 min before the priming of audiogenic seizures | Pro-convulsive activity: increased the incidence of wild-running in male and female Wistar rat pups (ED50 = 5.3 mg/kg, i.p.) | [144] | |||
| Opipramol | 0.2–50.0 | H1 (6) EBP (13) ERG2 (17) TMEM97 (110) 5-HT2A (120) D2 (120–300) Alpha1 (200) D1 (900) H2 (4470) Alpha2 (6100) | |||
| 5 and 10 mg/kg i.p. | Pro-convulsive activity: enhanced PTZ (30 mg/kg, i.p.) induced kindling in male Balb/c mice | [309] | |||
| ( ± )Pentazocine | 15–135 | Kappa opioid R(2.2–75.0) Mu opioid R (2.7–6.9) Delta opioid R(49) NMDAR (25–2820) TMEM97 (1500–1900) D2 (3500) | |||
| 60 mg/kg i.p. | Convulsive activity: induced seizures in female Sprague-Dawley rats | [310] | |||
| 3 mg/kg i.m. | Pro-convulsive activity: increased incidence of clonic-tonic seizures after the end of the PTZ (10 mg/kg per min, i.v.) infusion in dogs | [311] | |||
| 9 and 12 mg/kg i.v. | Convulsive activity: a characteristic pattern of unambiguous EEG and clinical signs of epilepsy in male Wistar rats were noticed 30 s after administration, responses were augmented by naloxone | [312] | |||
| (+)Pentazocine | 1.5–42 | TMEM97 (224–2060) M1 (225) M2 (525) Mu opioid R (1700) NMDAR (4190) | |||
| 12.5, 25 and 50 mg/kg s.c. | Pro-convulsive activity: by 20–40% reduced threshold for flurothyl-induced seizures in male Sprague-Dawley rats | [5] | |||
| (+)3-PPP | 6–75 | D2 (9–776) Mu opioid R (79) TMEM97 (IC50 = 76–940) D4 (130) D3 (132–217) | |||
| 60–100 mg/kg i.p. | Convulsive activity: dose-dependently produced behavioral clonic and electrical (EEG) tonic-clonic seizures in CD-1 male mice | [313] | |||
| 30 mg/kg i.p. given 30 min before the test | Pro-convulsive activity: significantly decreased the electroconvulsive threshold (auricular MES, 50 Hz, 0.2 s) in female Swiss mice, the effect was reversed by haloperidol | [314] | |||
| Sig1R antagonists | |||||
| NE-100 | 1.0–2.4 | TMEM97 (85–212) DAT (3600) 5-HT1A(IC50 = 6500) | |||
| 25 mg/kg i.p. 30 min before the i.v. infusion of PTZ | Pro-convulsive activity: significantly decreased PTZ-induced clonic seizure threshold in Swiss-Webster mice | [210] | |||
| 50 and 75 mg/kg i.p. | Convulsive activity: induced seizures in male Swiss-Webster mice | [210] | |||
| 75 mg/kg i.p. | Convulsive activity:induced seizures in CD-1 background WT and Sig1R KO mice | [18] | |||
| Rimcazole (BW-234 U) | 97–2380 | DAT (98–224) TMEM97(302) SERT (825–1710) Kappa opioid R (896) NET (2160) 5-HT2A (5010) D2 (7980–8700) | |||
| 5 and 10 mg/kg i.p. 30 min before the MES test | Pro-convulsive activity: significantly lowered the threshold for electroconvulsions in female Swiss mice, enhanced the protective activity of phenobarbital and valproate against MES | [315] |
Sig1R ligands with reported pro-convulsive and/or convulsive effects in preclinical animal models.
Affinities of compounds were obtained from databases PubChem and Binding Database (accessed from May 25, 2022 till June 28, 2022). Table demonstrates the binding affinities (Ki) to targets of compounds at concentrations below 10 µM (< 1 ×10-5). NE-100: 4-methoxy-3-(2-phenylethoxy)-N,N-dipropylbenzeneethanamine; 3-PPP: 3-(1-propylpiperidin-3-yl)phenol. MES – maximal electroshock-induced seizures; PTZ – pentylenetetrazole. Targets: Sig2R – sigma-2 receptor (known as TMEM97); EBP – emopamil binding protein; M1-M5 – muscarinic recepotrs; nAChRs – nicotinic actehylcholine receptors; Mu, Kappa, Delta – opioid receptor subtypes; NMDAR – N-methyl-D-aspartate receptors; NET – norepinephrine transporter; alpha-1 and alpha-2: adrenergic receptors; SERT – serotonin transporter; 5-HT1-7 – serotonin receptor subtypes; DAT – dopamine transporter; D1 and D2 – dopamine receptors; H1-3 – histamine receptors; Na+, K+ and Ca2+ – corresponding ion channels.
Elucidating the exact mechanism that generates pro-convulsive and convulsive effects of Sig1R ligands is challenging. For several high-affinity Sig1R ligands, Sig1R is not considered the primary target due to high affinities to other proteins (Table 2). Therefore, it is hard to identify pathways that specifically account for only Sig1R-related effects. It should be noted that the biphasic dose-dependent effects on the modulation of seizure thresholds are commonly observed, e.g., for opioid receptor ligands, SSRIs and compounds with dopaminergic activity [295], [316]. Indeed, high dose of administered Sig1R ligands demonstrating high affinity to SERT, TMEM97, dopamine and opioid receptors have been reported to induce seizures in vivo (Fig. 6).
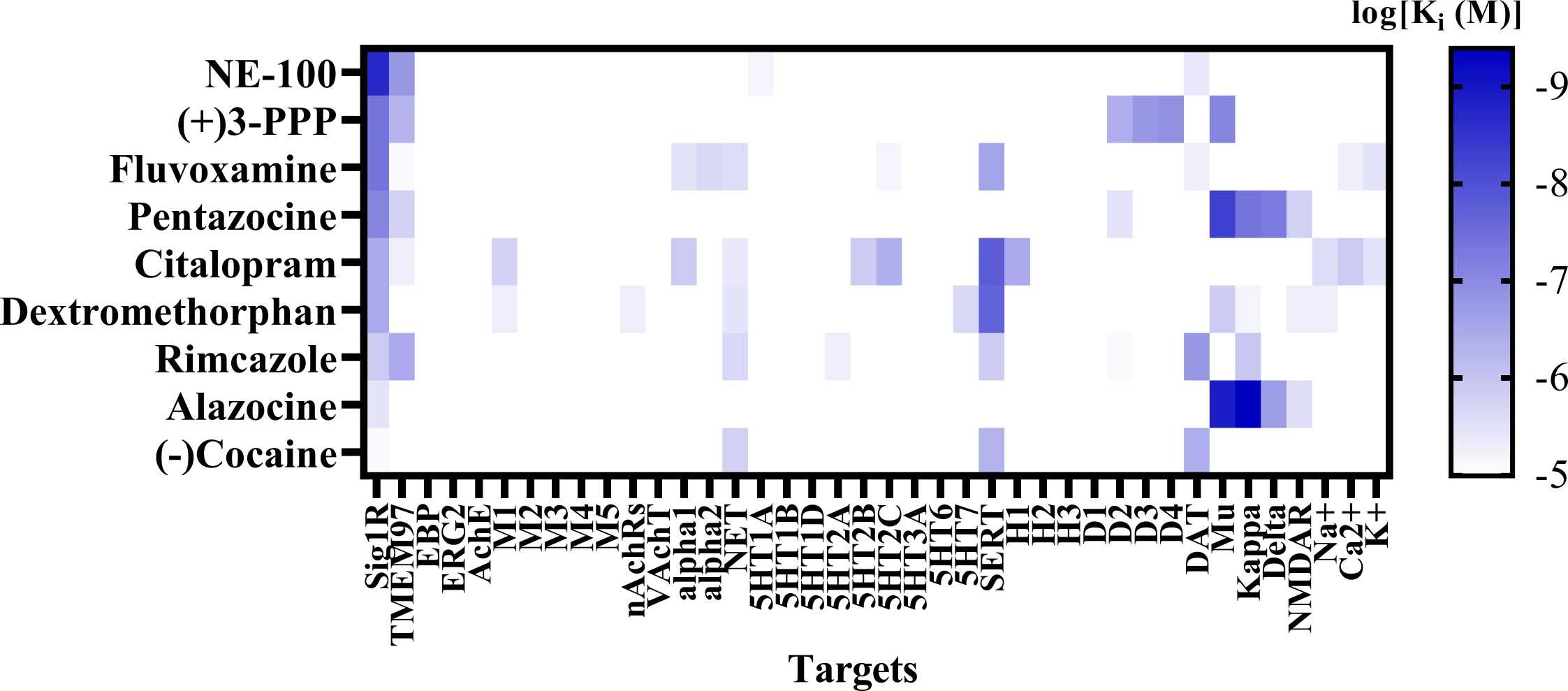
Sig1R ligands with demonstrated convulsive effects and their molecular targets.
Heatmap demonstrates the binding affinities (Ki) to targets of compounds at concentrations below 10 µM (< 1 ×10-5). Affinities of compounds were obtained from the PubChem and Binding Databases (accessed from May 25, 2022, to June 28, 2022). TMEM97 – transmembrane protein 97; EBP – emopamil binding protein; ERG2 – C-8 sterol isomerase; AchE – acetylcholine esterase; M1-M5 – muscarinic receptors; nAChRs – nicotinic acetylcholine receptors; VAchT – vesicular acetylcholine transporter; alpha-1 and alpha-2: adrenergic receptors; NET – norepinephrine transporter; 5-HT1–7 – serotonin receptor subtypes; SERT – serotonin transporter; H1-H3 – histamine receptors; D1-D4 – dopamine receptors; DAT – dopamine transporter; Mu, Kappa, Delta – opioid receptor subtypes; NMDAR – N-methyl-D-aspartate receptors; Na+, K+ and Ca2+ –corresponding ion channels. Graph was generated using GraphPad Prism software. For more details, see Table 1.
Seizures induced by Sig1R ligands are usually observed after administration of a dose greater than therapeutic doses or after overdose. Data obtained from animal studies are consistent with clinical reports showing that high doses of some Sig1R agonists could be pro-convulsive and induce seizures. For example, proconvulsive effects and seizures have been reported after overdoses of dextromethorphan [300], several SSRIs with high affinity for Sig1R [317], [318], [319], [320], [321], and pentazocine [322], [323], [324]. Therefore, the dose and route and time of administration together with the pharmacokinetics and pharmacodynamics of the compound all play significant roles in the hormesis phenomenon.
Sig1R antagonists seem to be promising compounds to block the seizures induced by psychotomimetic drugs. However, proconvulsive and convulsive effects of Sig1R antagonists have been demonstrated. Rimcazole, which reached clinical trials for the treatment of schizophrenia, was discontinued due to seizure induction in patients [325], [326]. The highly potent Sig1R antagonist NE-100 also reached phase 2 clinical trials for the treatment of schizophrenia but was discontinued in 2007 and no detailed information available. At high doses, NE-100 has demonstrated both proconvulsive (25 mg/kg, i.p.) and convulsive (50 and 75 mg/kg, i.p.) effects in mice (Table 2) [18], [210]. In addition, it was shown that the Sig1R antagonist NE-100 (75 mg/kg, i.p.) can induce seizures in Sig1R KO mice [18], thus indicating that proconvulsive/convulsive effects can be induced through mechanisms that probably involve some off-target activity. Studies have described the modulation of the release of dopamine, glutamate and GABA from nerve terminals by NE-100 [327], [328]. In addition, NE-100 and rimcazole demonstrate reasonably high binding affinity to TMEM97 and DAT (Table 2), which might be involved in the proconvulsive/convulsive effects of these compounds, especially when administered at high doses. Although NE-100 induced seizures in both WT and Sig1R KO animals, the severity of seizures was significantly less in Sig1R KO mice, with no generalized seizures, fewer clonic seizures, and a significantly shorter duration of seizures [18], which confirm the ability of Sig1R to modulate the severity of seizures.
It should be kept in mind that hormesis is an indicator of biological plasticity and the overall capacity of a biological system [295]. Seizure control aims to reach and maintain the balance between excitation and inhibition. However, the window in which excitation and inhibition are balanced is sometimes very narrow. In addition, pro-convulsive effects have also been observed for approved anti-seizure drugs, e.g., phenytoin, and for some specific conditions and several types of seizures, anti-seizure drugs are contraindicated [329]. Likely, more than one pathophysiological mechanism could be involved in the pro-convulsive/convulsive effects of Sig1R ligands.
4. Evidence from clinical studies
There are several ongoing clinical trials involving Sig1R ligands that are testing therapies for the treatment of Alzheimer’s, Parkinson’s and Huntington’s diseases, amyotrophic lateral sclerosis, depression and anxiety, chronic pain, COVID-19 and seizures (Fig. 7).
Number of current clinical trials involving Sig1R ligands.
Data were obtained from ClinicalTrials.gov (January 2023). DDE – developmental and epileptic encephalopathies.
Several clinical trials involving Sig1R ligands that are testing therapies for the treatment of developmental and epileptic encephalopathies, e.g., CDKL5 deficiency disorder (NCT05064878), Dravet (NCT03936777), Lennox-Gastaut (NCT02655198, NCT03936777, NCT03355209) and Rett (NCT04304482) syndromes, and photosensitive epilepsy(NCT03790137), are currently underway. Previous and ongoing clinical trials with Sig1R ligands for the treatment of seizures include dextromethorphan (brand name: Delsym®), safinamide (brand name: Xadago®), blarcamesine (brand name: ANAVEX®2–73), fluoxetine (brand name: Prozak®) and fenfluramine (brand name: Fintepla®).
Dextromethorphan (affinity to Sig1R Ki = 10–652 nM) is used for the treatment of seizures in infantile nonketotic hyperglycinemia [330]. However, the efficacy of dextromethorphan (5–35 mg/kg/day) is reported to be dependent on disease stage and severity, and the justification for long-term treatment has been questioned [330]. Reports of the efficacy of dextromethorphan in Phase 1 [331] and Phase 2 [332] clinical trials are available. A phase 1 study aiming to evaluate the safety of dextromethorphan (160 and 200 mg/day) in patients with intractable partial epilepsy showed that treatment with dextromethorphan for 16 weeks was safe, and seizure control in patients was improved. However, two patients experienced increased seizure frequency and were excluded from the study [331]. The phase 2 study was a randomized open-label trial for the evaluation of dextromethorphan in 35 Rett syndrome patients. Although no significant improvement was observed in global clinical severity as measured by the Rett Syndrome Severity Scale, patients who were administered dextromethorphan showed significant dose-dependent improvements in clinical seizures, receptive language, and behavioral hyperactivity [332]. Over a period of 6 months, some of the patients experienced an increase in seizure frequency after using dextromethorphan at oral doses of 2.5 mg/kg/day and 5 mg/kg/day [332]. Nevertheless, it was concluded that dextromethorphan is safe for use in young girls with Rett syndrome. To date, no further development of dextromethorphan has been reported.
Safinamide is a third-generation reversible MAO-B inhibitor that also blocks voltage-gated Na+ channels and modulates the stimulated release of glutamate [333]. Safinamide shows a high affinity for Sig1R (Ki = 19 nM) [334]. Following initial screening as a possible anti-seizure drug, safinamide was selected for its potency, broad spectrum of action, and good safety margin, which allowed its efficacy to be evaluated in a pilot phase 2 clinical study [334]. Thirty-eight refractory epilepsy patients received 12 weeks of safinamide treatment (starting at a dose of 50 mg/day that increased until it reached 300 mg/day) [334]. The results demonstrated an improvement in 41% of subjects who showed a reduction in their number of seizures by at least half [334]. However, since safinamide was mainly developed for the treatment of Parkinson’s disease, no further studies have been performed to pursue its development for clinical use in the treatment of seizures. It was approved by the FDA in 2017 for the treatment of Parkinson’s disease.
Blarcamesine (ANAVEX2–73) is a mixed Sig1R (IC50 = 0.9 µM) and muscarinic receptor(IC50 = 3.3 µM) agonist. It entered clinical trials for the treatment of Alzheimer’s disease in 2014. Currently, blarcamesine is still being actively studied in phase 2 and 3 clinical trials for the treatment of Alzheimer’s disease, Parkinson’s disease dementia and Rett syndrome. Therefore, information about its clinical efficacy against seizures in the academic literature is scarce. However, it has been reported that blarcamesine given orally at doses of up to 30 mg/day was well tolerated in Rett syndrome patients [335]. Primary and secondary efficacy endpoints have demonstrated statistically significant and clinically meaningful reductions in Rett syndrome symptoms together with reductions in concentrations of potential biomarkers (GABA and L-alpha-aminoadipic acid) of disease pathology, as well as dose-related significant improvements in overall quality of life (measured with CHQ-PF50) [335]. Overall, more details of its efficacy in clinical trials are needed.
Fenfluramine (affinity to Sig1R Ki = 0.3 µM) was initially approved by the FDA in 1973 as an appetite suppressive drug to treat obesity. However, it was withdrawn from the market in 1997 due to cardiac side effects, which included heart valve disease, pulmonary hypertension and cardiac fibrosis [336]. Later, considerably high anti-seizure effects were demonstrated in a small group of patients with Dravet syndrome who received a low dose of fenfluramine as an add-on therapy [337]. After this finding was reported, a repurposing of fenfluramine began, and fenfluramine has been studied in randomized controlled trials and open-label studies for the treatment of seizures in rare epileptic encephalopathies. Fenfluramine, as an orphan drug, has been approved by the FDA for the treatment of seizures associated with Dravet syndrome (2020) and very recently Lennox-Gastaut syndrome (2022). Due to its promising efficacy, fenfluramine has gained interest and attention in the field. Therefore, there are several recent reviews available summarizing its clinical efficacy [336], [338], [339], [340], [341], [342].
Cannabidiol (Epidiolex®) was approved by the FDA in 2018 as an orphan drug and is indicated for the treatment of seizures associated with Lennox-Gastaut syndrome, Dravet syndrome, and tuberous sclerosis complex. Although cannabidiol has not been shown to bind to Sig1R, a Sig1R-related anti-seizure effect of cannabidiol has been demonstrated [39]. Cannabidiol can act as a Sig1R antagonist, diminishing NMDA-induced seizures in mice [39]. Therefore, additional studies are needed to confirm the involvement of Sig1R in the anti-seizure activity of cannabidiol. Cannabidiol is also being investigated in clinical trials for the treatment of electrical status epilepticus during slow-wave sleep and other types of seizures.
Based on the high affinity to Sig1R of compounds that have reached clinical trials and are still in active investigation for the treatment of seizures, Sig1R has gained high recognition as a valid target in epilepsy.
5. Future challenges and perspectives
Several recent findings in Sig1R KO mice [17], [18], [51] suggest possible future directions that may elucidate specific details of molecular mechanisms and pathways that could be used for the treatment of seizures. A very recently developed conditional Sig1R KO mouse model using CRISPR-Cas9 gene targeting [343] will allow researchers to study cell type and brain region specificity and will help to elucidate the involvement of Sig1R-dependent pre- and postsynaptic mechanisms in greater detail. In addition to Sig1R KO mice, a Sig1R-deficient zebrafish line has been developed and described [344]. Zebrafish models could be very useful in studying gene-mediated seizure mechanisms, e.g., double KO animals, and performing high-throughput in vivo drug screening [345]and would allow us to better understand and classify the involvement of Sig1R in the pharmacodynamics of Sig1R ligands demonstrating anti-seizure effects.
Currently, one of the greatest issues in the field of Sig1R remains the inconsistent ligand classification methods that do not always allow us to directly translate observations of ligand activities and their agonist-antagonist interactions to specific properties of Sig1R. The biphasic dose-dependent response of Sig1R ligands is an important issue to consider, especially with regard to seizure management. The dose-dependent effects of Sig1R ligands must be extremely well characterized to avoid undesired side effects in clinical settings. The accumulation of knowledge based on a number of studies performed with selective Sig1R ligands would allow us to better distinguish between very specific Sig1R-independent and -dependent activities regarding modulation of the seizure threshold. The most challenging aspect of Sig1R ligands and their possible clinical application is understanding which Sig1R-protein interactions (both direct and indirect) and whether activation or inhibition of such complexes and pathways will be useful for the treatment of some specific type of seizures.
6. Conclusions
Studies of Sig1R KO animals have begun to provide basic knowledge in the understanding of Sig1R-mediated pathways involved in mechanisms of seizure control. Current knowledge allows us to propose some possible directions of Sig1R-targeted drugs for the treatment of seizures. While agonists and positive allosteric modulators seem to act as a promising tool for the management of seizures and seizure-related comorbidities by reducing undesired side effects such as significant CNS depression, Sig1R antagonists seem to be useful for the acute treatment of intoxication-induced seizures due to overdoses of various drugs, especially psychostimulants and drugs of abuse. Treatment strategies, especially for epilepsy, are mainly based on a combination of several drugs due to comorbidities appearing with this devastating disease. Identification of new drug targets is needed to find novel anti-seizure drugs, especially for the one-third of people for whom existing therapies are ineffective. As a chaperone protein, Sig1R has already demonstrated its potential as a multidimensional drug target and has been recognized as a valid target for the management of seizures.