
Understanding the Role of Glia-Neuron Communication in the Pathophysiology of Epilepsy: A Review
By Peng Chen, Fuchao Chen, and Benhong Zhou
Excerpt from the article published in Journal of Integrative Neuroscience (JIN) 2022, 21(4), 102. Published: 30 May 2022. DOI: https://doi.org/10.31083/j.jin2104102
Editor’s Highlights
- Epilepsy is a prevalent, chronic, and severe neurological disorder characterized by abnormal hypersynchrony of a large population of neurons.
- It is widely assumed that there is an imbalance between inhibitory and excitatory neurotransmission in the central nervous system (CNS) due to the resulting relative hyperactivity of neurons in epilepsy.
- TSP has been observed in reactive astrocytes, which interact with the auxiliary calcium channel subunit α2δ1, to promote abnormal synapses that change the neuronal circuit and amplify epileptic activity.
- The binding of TSP to α2δ1 was inhibited by the antiseizure medication gabapentin and the epileptic-like activity following epileptic injury which was effectively alleviated, suggesting that abnormal synapses, triggered by reactive astrocytes, may be a potential network hyperexcitation mechanism in neurodevelopmental disorder-related epilepsy.
Abstract
Epilepsy is a chronic brain disorder that causes repeated seizures. It affects 65 million people worldwide and is a major burden on individuals and health systems. It has been reported that factors leading to ion channel disfuntion, neuronal damage and are all involved in the pathogenesis of epilepsy. The exact etipathogenic mechanism is unknown and appropriate therapeutic targets remain elusive. Recent studies point to a significant contribution by non-neuronal cells, the glia—especially astrocytes and microglia—in the pathophysiology of epilepsy. This review critically evaluates the role of glia-induced hyperexcitability in the pathogenesis of epilepsy to provide a better understanding of the contribution of glia to epilepsy.
1. Introduction
Epilepsy is a prevalent, chronic, and severe neurological disorder characterized by abnormal hypersynchrony of a large population of neurons [1]. The latest statistics state that 2% of the population worldwide (approximately 55–65 million people), including children and the elderly, suffer from epilepsy due to diverse known and unknown etiologie [2]. Epilepsy also occurs more often in patients with other cerebral diseases, such as prolonged febrile seizures, Parkinson’s disease, traumatic brain injury and stroke [3]. Patients with epilepsy exhibit cognitive impairments, due to damage in the hippocampus, particularly in learning and memory, resulting from the damaged reorganization of neural networks [4]. Patients with epilepsy have been reported to exhibit increased mortality occurring 2–10 years earlier than that of the general population [5]. Epilepsy continues to be a major public health problem of increasing global concern. There is a great need to develop more effective treatment methods to improve the therapeutic effects and prevent the recurrence of seizures.
There is ongoing debate regarding the precise mechanisms that underlie epilepsy. It is widely assumed that there is an imbalance between inhibitory and excitatory neurotransmission in the central nervous system (CNS) due to the resulting relative hyperactivity of neurons in epilepsy [6]. Researchers have suggested that the seizure mechanism of epilepsy is closely related to channelopathy, metabolic, acid-base, and neuroinflammation [7]. Although all these mechanisms have been widely known and recognized for many years, therapeutic antiseizure strategies targeting these mechanisms and the clinical development status of these drugs are still not promising in a substantial proportion of patients [8]. Increasing attention is being paid to the role of glia in modulating synaptic connectivity [9].
Glial cells are specialized cells and outnumber neurons in the cerebral cortex by more than 3:1 by some estimates, with oligodendrocytes comprising approximately 75% of cortical glia, followed by astrocytes (17%) and microglia (6.5%) [10]. Glia exert diverse neuronal functions in the brain and are intimately involved in the active control of neuronal activity and plasticity, support homeostasis of the extracellular microenvironment through ion buffering and neurotransmitter recycling, maintain the integrity and function of the blood-brain barrier (BBB) permeability, control the inflammation and promote immune response and neural function restoration of brain cells [11, 12]. Dysregulation of glial functions may cause epilepsy or promote the triggering of seizures, and although the disease process has been illuminated, an additional examination is warranted [13]. This review will highlight studies that focus on how glial-mediated changes contribute to epilepsy development. More extensive discussion could be performed regarding the extracellular matrix (ECM) produced by astrocytes, microglia, blood vessels, immune cells, and even neuronal and non-neuronal cells as important participants in epilepsy and that may serve as novel therapeutic targets for epilepsy treatment [14, 15].
2. Gliosis in Epilepsy
Prominent gliotic scar formation was found to be a common hallmark in a model of chronic focal epilepsy. Gliosis is probably omnipresent in all forms of epilepsy [16]. Gliosis refers to a nonspecific reactive change of glial cells, especially microglia and astrocytes, in response to various types of damage and repair of the CNS [17]. The nature and degree of these changes may vary depending on the damage severity and injury types. Reactive gliosis is characterized by some obvious features, including hypertrophy of the cell bodies and processes. The expression of some proteins, such as glial fibrillary acidic protein and vimentin, is upregulated in these processes. These were considered specific biomarkers for astrocytes and ionized calcium-binding adaptor molecule 1, which was a novel calcium-binding protein and specifically expressed in microglia in the brain. Cell proliferation occurs, and the formation of ECM molecules, such as chondroitin sulphate proteoglycans, are produced by reactive astrocytes in the tenacious glial scar [18, 19]. Gliosis may result in beneficial or detrimental effects for nearby cells, depending on the context [20]. The brain cellular damage can be mitigated or prevented via reactive glia-releasing neurotrophins such as nerve growth factor and brain-derived neurotrophic factor. These molecules help improve the inflammation status, remove debris and promote synaptic plasticity strengthening tissue remodeling [21, 22]. Exorbitant reactive gliosis could result in excessive uncontrolled signal tissue and cell damage. The latest evidence has confirmed that reactive astrocytes can assume neurotoxic or neuroprotective priming states accounting for the possible role of reactive glia in the pathophysiology of the CNS [19, 23].
There has been a heated debate, in academia, about whether gliosis is a cause or consequence of epilepsy. It has been suggested that gliosis precedes the development of epilepsy, and it is supposed to be causative under the circumstance of trauma-associated epilepsy [24]. The best evidence for gliosis being causative of epilepsy comes from studies in which gliosis was induced by the conditional astrocyte-specific deletion of the ββ1 integrin gene Itgb1 [25]. It was reported that widespread gliosis was the initial pathological feature and that the animals finally developed epilepsy with spontaneously recurrent seizures, indicating that astrogliosis alone is almost sufficient to induce epilepsy [26]. In an unrelated study, the researchers found that astrogliosis in the hippocampus induced by virus exposure is enough to produce neuropathic excitability [27]. Dysplastic neurons, abnormal cortical organization, and astrogliosis are due to an astrocyte-specific deletion of the TSC1gene (which can cause tuberous sclerosis complex (TSC)) and is accompanied by epileptic seizures in severe cases [28]. Seizures are one of the most common groups of neurological conditions caused by a heterozygous mutation in the coding region of the glial fibrillary acidic protein, an astrocyte-specific intermediate filament protein. Insufficient glutamate transporters were expressed in astrocytes of affected individuals, and seizures and excitotoxic neuronal death can be documented in the brain following status epilepticus (SE) [29]. These results exhibited solid evidence of a direct causal relationship between associated dysfunctions of astrocytes and the development of recurrent seizures. A growing body of evidence has confirmed that reactive astrogliosis seems to be found in most acquired epilepsy animal models, as well as in the tissues of patients with epilepsy, further supporting that reactive astrogliosis is generally related to epilepsy (Fig. 1) [25, 30].
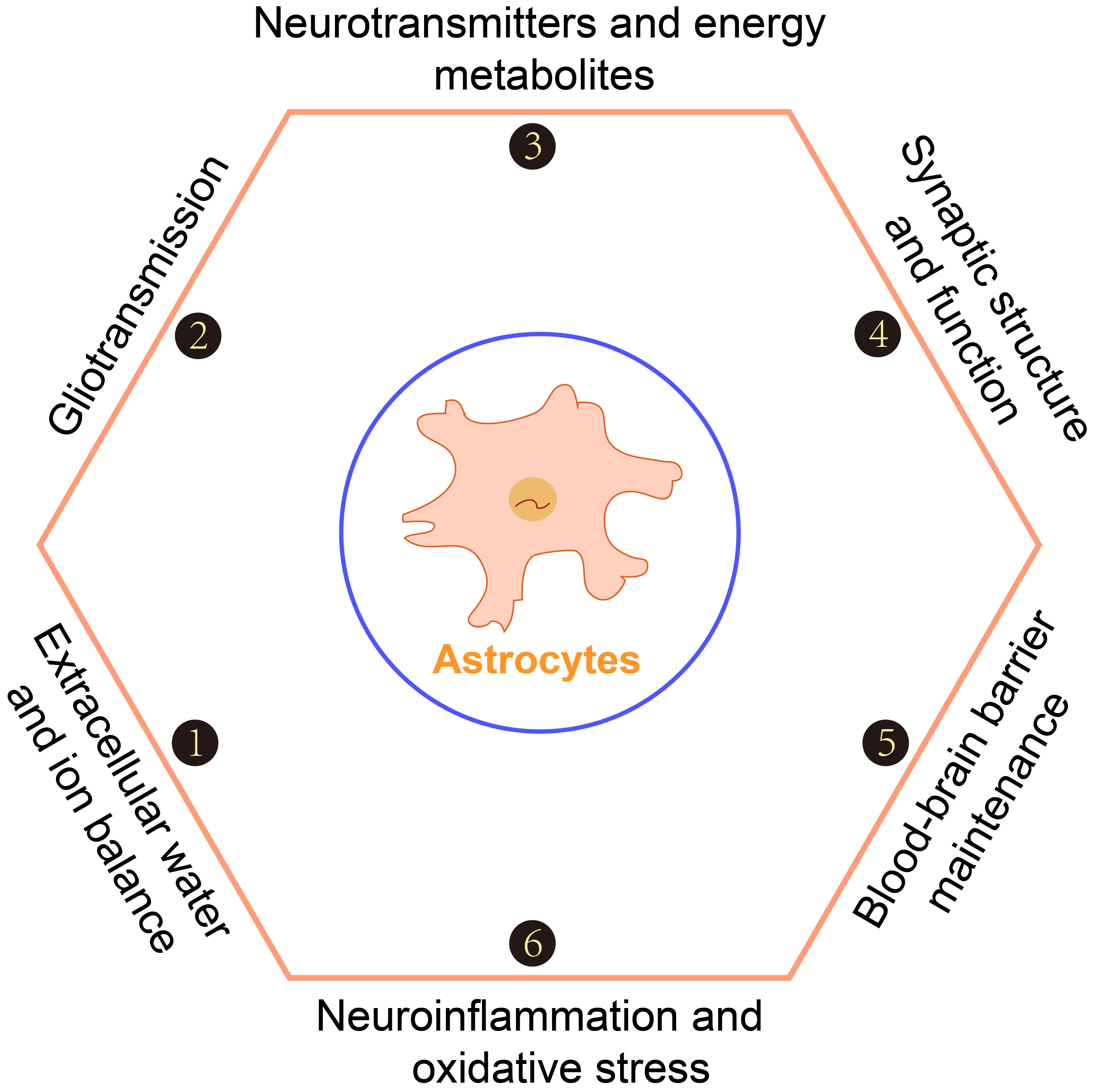
Processes within epilepsy in which astrocytes are involved.
Astrocytes are involved in important processes such as extracellular water and ion balance, gliotransmission, neurotransmitters and energy metabolites, synaptic structure and function, blood-brain barrier maintenance, neuroinflammation, and oxidative stress.
Some studies have reported that the degree of interdependence between seizures and astrogliosis is very slight, and there is no connection between these events. One study resected brain tissue of focal cortical dysplasia in patients and observed that there was no gliosis in the nonlesional epileptogenic cortex [31]. A study reported that, soon after administration with pilocarpine in epileptic mice, the seizure frequency tended to increase and was found to correlate highly with a loss of gamma-aminobutyric acid (GABA) ergic interneurons in the dentate gyrus but not with reactive gliosis [32]. The above results suggest that gliosis is an important part of epilepsy histopathology and plays a crucial role in diverse types of epileptogenesis but not all forms of epilepsy. Elucidating the causes and processes of gliosis leading to seizure is a topic of much research and the possible mechanisms are widely discussed in the light of various experiments and detailed in Fig. 2.
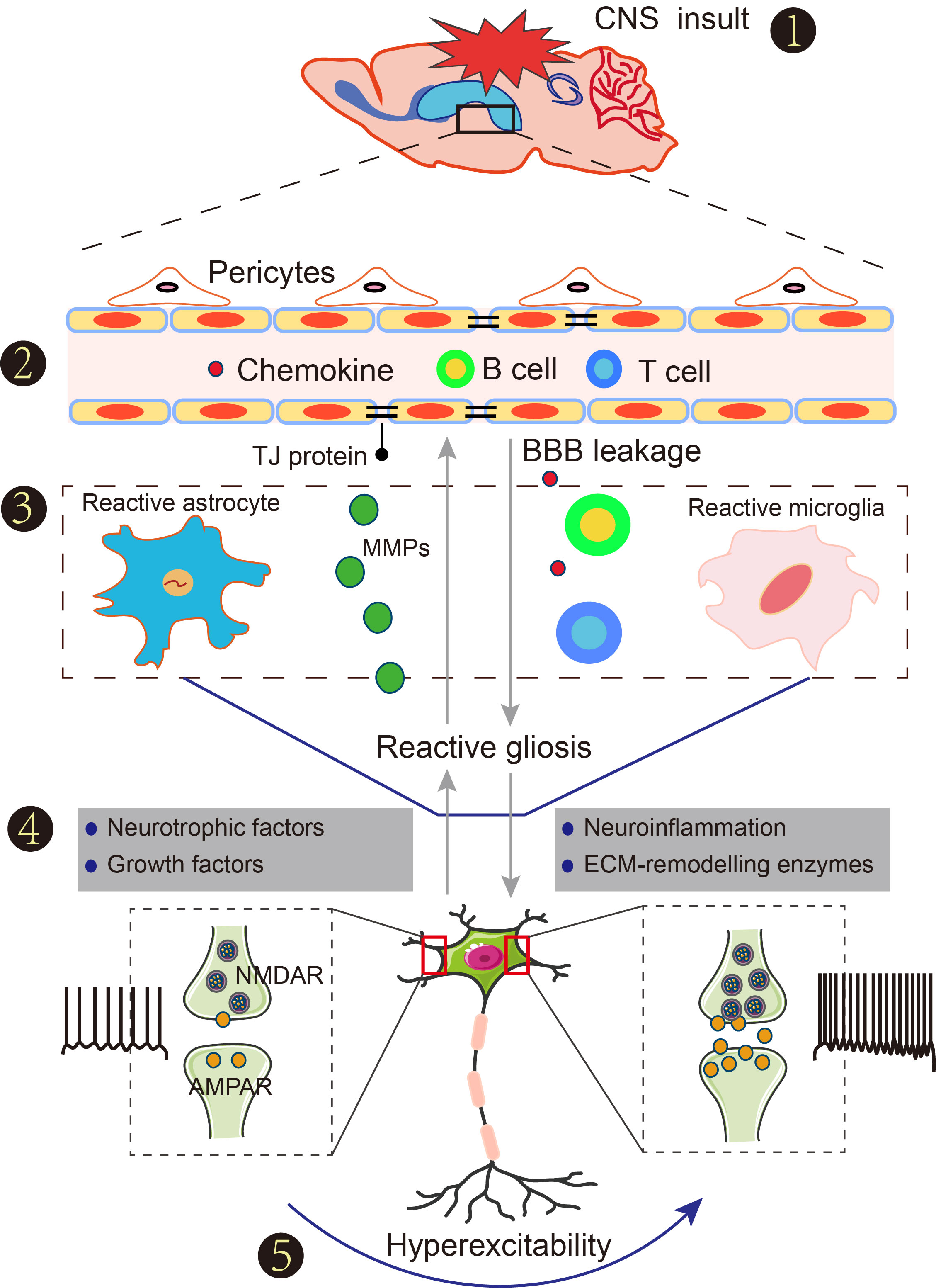
Connection of reactive gliosis and seizures after insults to the central nervous system (CNS).
(1) Blood-brain barrier (BBB) damage following acute insults to the CNS such as stroke, traumatic brain injury, infection, and hyperpyrexia. (2) Increased BBB permeability contributes to the extravasation of serum albumin, peripheral immune cells and chemokines into the peripheral vasculature of the brain, accompanied by reactive gliosis. (3) Reactive gliosis is a common term that refers to a range of molecular, morphological and functional changes, such as the nuclei becoming enlarged, the chromatin becoming less dense and nucleoli becoming more prominent after suffering any insult to the CNS. (4) Neurotrophic factors, growth factors, neuroinflammatory factors and extracellular matrix remodeling enzymes are produced from reactive gliosis to regulate immune and nerve function. (5) The consequences induce neuronal hyperexcitability and cause seizures.
3. Neurotransmitters and Energy Metabolites
The main neuropathologic hallmark of epilepsy, in humans and non-human model animals is increased extracellular glutamate, which plays a key role in increasing the neuronal excitability of seizures [33, 34]. The key physiological function of astrocytes in the normal brain is the clearance of neuronally released K++ and glutamate from the extracellular space [35]. There are two other amino acid transport systems named excitatory amino acid transporter 1 (EAAT1) and excitatory amino acid transporter 2 (EAAT2). By coupling the uptake of one glutamate to the cotransport of three Na++ and release of one K++, they accomplish an electrogenic glutamate transportation [36]. Early expression of EAAT1 primarily occurs during brain development, whereas EAAT2 is primarily responsible for the transportation of astrocyte-specific glutamate and is mostly expressed in adult brains. In the hippocampus, glutamate aspartate transporter and glutamate transporter 1 (GLT1) (rodent analogs of EAAT1 and EAAT2, respectively) are expressed as glial glutamate transporters in astrocytes. Synaptic transmission can be terminated via transporters mediating a rapid clearance rate of synaptic glutamate in astrocytes to control the changes in excitatory glutamatergic neurotransmission and protect the extrasynaptic N-methyl-D-aspartate receptors that are coupled to cell-death pathways from accidental activation [37]. Due to the bidirectional properties of glutamate transporters, the neuronal glutamate (GLU) must be sequestered and metabolized, once taken up by astrocytes via GLU transporters, and the amount of glutamate released into the synapse was promoted by increased concentration of intracellular glutamate in astrocytes. Many intracellular glutamates can be converted into αα-ketoglutarate within the catalyzation of glutamate dehydrogenase, which then enters into the tricarboxylic acid (TCA) cycle [38]. Glutamine activates a key role as a major energy source in the brain. If the remaining portion of intracellular levels of glutamate is decreased, by the conversion to glutamine via glutamine synthase catalysis, the glutamine is exported into the interstitial space, where it can be acquired by neurons for chemoenzymatic synthesis of glutamate via a phosphate-activated glutaminase (a synthesizing enzyme for glutamate). This is called the glutamate-glutamine cycle. In excitatory neurons of the nervous system, glutamate is packed into the specialized secretory through several vesicles vesicular transporter, and when incoming information reaches the synapse, glutamate is released into the gap. Glutamate in inhibitory GABAergic neurons can be reconverted into GABA by the enzyme glutamic. Astrocytes play a significant role in the transformation of vesicular glutamate and GABA. It would be expected that interfering with the cycle at any stage would rapidly impact neurotransmitter supply and synaptic function. Astroglial reactivity may be associated with epileptogenesis.
A growing number of studies have reported that GLT1 null mice show lethal spontaneous seizures. Glial GLT1 overexpression in mice is not susceptible to pentylenetetrazol and pilocarpine-induced seizures [39, 40]. These findings support the importance of glial glutamate transporter GLT1 in the maintenance of extracellular glutamate homeostasis in epileptogenesis. Treatment with ceftriaxone, a ββ-lactam antibiotic that acts as a transcriptional activator to modulate the functional expression of GLT1, was found to reverse the glutamate uptake deficit [41]. Expression of GLT1 in astrocytes was upregulated, and seizure frequency was reduced via early administration of ceftriaxone in mouse models of TSC before the onset of epilepsy. A new treatment strategy highlighted that heat shock protein 90ββ (HSP90ββ)-mediated proteasomal degradation down-regulated the expression of GLT1 in reactive astrocytes [42]. Kainic-acid-induced seizures were suppressed by chronic treatment with novel small molecule HSP90 inhibitors [42].
Examination of the expression of EAATs in human brain tissue, showed that glutamate secretion was increased in pathophysiological states and could be assessed in patients before and during seizures, indicating that the clearance of neuronally released glutamate astrocytes failed in astrocytes [33]. During the interictal and ictal periods in epilepsy, extracellular glutamate levels are abnormally elevated, and the expression and function of EAAT2 in astrocytes are changed, accompanied by impairment of glutamine synthetase (GS) function. The enzymatic activity and expression of GS were significantly reduced in the hippocampus of individuals with mesial temporal lobe epilepsy (MTLE) and astrocytosis was particularly seen in areas in which GS activity was lost [43, 44]. When methionine sulfoximine (MSO), a selective GS inhibitor, was continuously infused into areas of the hippocampus, in adult laboratory rats, it led to an evident reduction in GS activity, neuropathological features and changes strikingly similar to that reported in the majority (>>95%) of MSO-treated MTLE and epilepsy model animals, supporting a role for GS downregulation in the pathogenesis of MTLE seizure generation [45].
Many of the hallmarks of reactivity reactive gliosis were observed in vivo adeno-associated virus-induced epilepsy models; virally infected astrocytes showed an increased expression of the glial fibrillary acidic protein, vimentin, and downregulation in GS expression. There were no alterations of anatomy and intrinsic properties in neighboring neurons. The regions with virally transduced circuits showed epileptiform activity in the CA1 neurons of the hippocampus, specifically evident as cerebral cortical hyperactivation directly inputs to the distal dendrites of hippocampal CA1 pyramidal neurons [46]. Previous studies have shown that this pathway was regulated by local circuit inhibition in the cerebral cortex and extensively triggered isocortex hyperactive- and prone-to-epilepsy in animals [47]. All of these astrogliosis-mediated consequences could be mimicked by inhibiting the glutamate-glutamine cycle and were reversed by a supply of exogenous glutamine (Fig. 3). Similar results have also been investigated in an earlier study via co-culturing astrocyte neurons and rat spinal dorsal horn neurons, suggesting that a disturbed astrocyte-neuron metabolic crosstalk could explain the neuronal excitability underlying epilepsy.
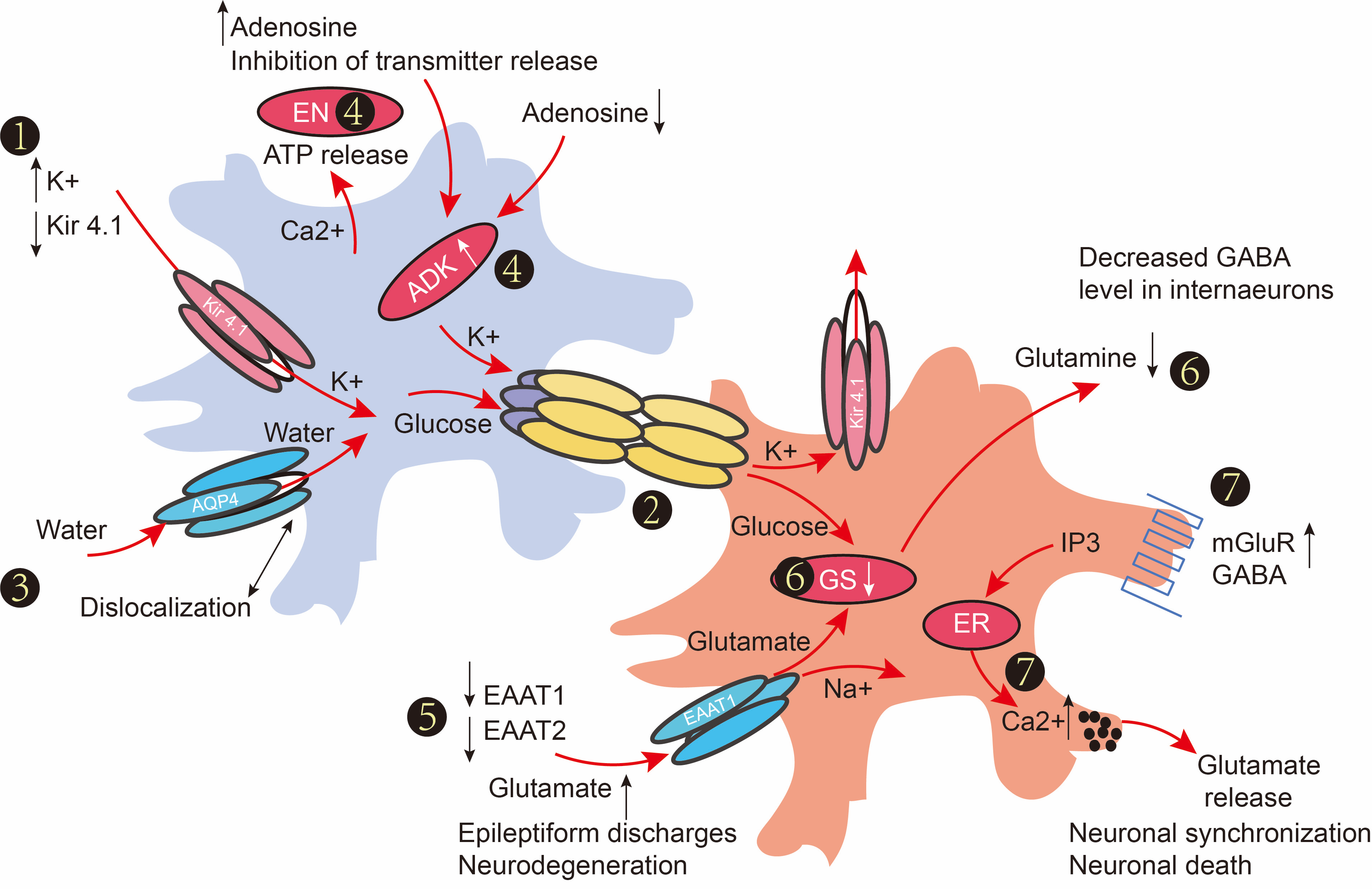
Astroglial dysfunction in epilepsy.
(1) Seizure-induced upregulation of astrocytic [K] buffering and hyperactivity intensified the dysfunction or ablation of gap junctions in epilepsy. (3) Dislocation of water channels contributes to impaired K1 buffering. (4) Seizure susceptibility increased by a reduction of ambient adenosine (controlled by ATP release) and upregulation of adenosine kinase (ADK) concentration; thus, genetic knockdown of ADK attenuated seizure activity. (5) Astrocytes are the major cell type responsible for glutamate uptake. The expression of EAAT1 and EAAT2 is decreased during the epilepsy process. Excessive extracellular glutamate can lead to subsequent neuronal death after seizure induction. (6) Glutamine converts into glutamine, which is then transported into extracellular space through GS. Loss of GD can make excessive GABA release from interneurons to impair the inhibition. (7) Activation of mGluRs or GABABRs leads to intracellular Ca
increases. Glutamate release from astrocytes was amplified after the activation of mGluR.
Astrocytes fulfill key functions in the regulation of extracellular ion homeostasis essential for modulating synaptic transmission [48]. The process of astrocytic glucose-derived glutamine and extracellular glutamate clearance could contribute to synaptic hyperactivity. Following the release of glutamate from excitatory synapses, EAATs sequester this glutamate driven by an electrochemical sodium gradient with a stoichiometry of this process for one glutamate molecule taken up with three Na++ ions. Na++ gradient across the photoreceptor membrane is then re-established by the activity of the enzyme Na++/K++-ATPase. The conversion of glutamate to glutamine, catalyzed by glutamine synthetase, also depends on ATP. The glutamate-glutamine cycle places a huge burden on the astrocytes that trigger the taking up of glucose from the blood through GLUT1 transporters in their end feet. Due to the cell-specific expression of glucose metabolism mechanisms, the glucose processing pathways between astrocytes and neurons are different. Glucose is mainly converted into pyruvate by the glycolysis in astrocytes and this process is predominant in astrocytes and can be upregulated. The utilization of pyruvate, through the TCA cycle, is limited. Neurons have limited capacity to obtain energy through increasing glycolysis, due to the lack of activity of glycolysis, but the TCA cycle and oxidative phosphorylation were maintained actively [49]. Glucose metabolism in neurons is directed mainly to the pentose phosphate pathway to allow the cell to maintain its redox status to strike a balance between promoting and suppressing oxidative stress. The lactate is proposed to be generated by glycolysis in astrocytes and then converted into lactate via lactate dehydrogenase (LDH) 5. The lactate, released by astrocytes, is taken up by neurons through specific monocarboxylate transporters (MCT), where it is employed as an energy source via the TCA cycle after being metabolized into pyruvate under the action of LDH 1. This metabolic pathway is called the astrocyte-neuronal lactate-shuttle (ANLS), and the expression levels positive correlate with neuronal activity. Brain glycogen stored in astrocytes provides lactate as an energy source to neurons during sustained neuronal activity via the ANLS pathway and enhances glycolysis. Any perturbations to this pathway may modulate some aspects of intrinsic neuronal excitability. Established therapies, such as a high-fat, low-carbohydrate “ketogenic diet”, that shift the energy source from glucose to ketones, such as ββ-hydroxybutyrate and acetoacetate, have been confirmed as a successful therapy for patients with difficult-to-treat epilepsies [50]. The stabilizing effect of glucose restriction on neuronal excitability was eliminated by exogenous lactic acid, pyruvate and their downstream energy metabolites. There is a decrease in excitability and excitatory neurotransmission in the mouse hippocampus upon hypothalamic LDH inhibition in astrocytes indicating that lactate produced and released from astrocytes is a potential source of energy for neurons. Although the underlying downstream molecular mechanisms remain largely unknown, the author suggested that neuronal hyperpolarization mediated by the activation of ATP-sensitive potassium channel (KATP) channels is due to LDH inhibition being abolished in the presence of KATP channel inhibition. Stiripentol, an LDH inhibitor, is a new-generation anti-seizure medication that potently ameliorates the mice seizures induced by kainic acid injection.
These findings show that astrocytes play an important role in supplying energetic compounds to neurons for meeting the energy requirements involved in brain homeostasis and the synthesis of neurotransmitters and amino acids. Astrocytes exert a crucial effect in controlling the physiological excitation/inhibition balance. A mechanistic understanding of their role in providing structural and metabolic support to neurons may lead to the development of novel anti-seizure interventions.
4. Regulation of Synaptic Structure and Function Through Gliotransmitters
A growing body of evidence indicates that epilepsy is a chronic neurological disorder in which neural circuits are gradually dysregulated. As these synapses are formed by gap junctions between neurons, it makes sense to consider that epilepsy remains a functional disorder resulting from a synaptic impairment or changes in the synaptic structure. Glial cells maintain the synaptic structure and function and promote the development of the neurons via regulating the long-term processes of synapse formation, synapse pruning, synaptic maturation and releasing gliotransmitters.
Although glia has been identified as critical to brain function and the development of the nervous system for many years, several reviews have reported that astroglia and microglia are identified as key players in synaptogenesis of the normal brain and that several molecular players participate in synapse maturation and activity-dependent plasticity [51]. These molecular players include thrombospondins (TSPs), integrins, transforming growth factor-ββ (TGFββ), brain-derived neurotrophic factor, estradiol, glypicans, and hevin. Some of these molecules, such as TSPs, have been shown to take part in the process of brain maturation, after which their expression is subsequently downregulated [52]. When the CNS is subjected to external stimulation or injury, excess TSP has been observed in reactive astrocytes, which interact with the auxiliary calcium channel subunit α2δ1, to promote abnormal synapses that change the neuronal circuit and amplify epileptic activity [53]. The binding of TSP to α2δ1 was inhibited by the antiseizure medication gabapentin and the epileptic-like activity following epileptic injury which was effectively alleviated, suggesting that abnormal synapses, triggered by reactive astrocytes, may be a potential network hyperexcitation mechanism in neurodevelopmental disorder-related epilepsy [54].
Glial cells have been reported to be involved in regulating some of the molecular pathways shared by synaptic pruning. During the process of the optic thalamic circuit, numerous studies have demonstrated that eye-specific dissociation events mainly occur in axons from retinal ganglion cells through the elimination of bioelectrical sources overlapped by microglia and astrocytes. Astrocytes induce CNS synapse formation via the overexpression of the C1q-dependent protein gene and promote the activation of the complement signaling pathway, enhancing the level of phagocytosis synapses in microglia [55]. Complement protein C1q knockout mice show reduced pruning excitatory cortical and enhanced synaptic connections to epileptic and normal neurons [56]. Astrocytes trigger phagocytosis by microglia, exhibit some phagocytic activity in physiological and pathological conditions, and trigger phagocytosis through microglia [57]. Synaptic pruning can be enhanced via upregulating complement components in rodent models of epilepsy, which may be due to the rescue synaptic functions in the microglia during puberty or adulthood.
Molecules released from astrocytes are also emerging as important players in synaptic function. Cholesterol enhances quantal neurotransmitter release synaptic vesicles within presynaptic terminals, glial-derived tumor necrosis factor regulates glutamate and GABAAARS transportation onto postsynaptic membranes and glypicans 4 and 6 promote the expression of amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors at the postsynaptic sites [58, 59]. Astrocyte-released molecules can also regulate proper synaptic differentiation and synaptic transmission. These molecular characteristics and mechanisms are still unclear.
Many in vivo experiments, on selective silence of microglial proteins via genetic ablation strategies, revealed that the role of microglia in the regulation of neuronal synaptic plasticity depends critically on the interaction between microglia and neurons. These biochemical and molecular genetic studies were conducted on neuron-to-glia signal transmission activated by fractalkine (FKN) secreted by neurons in mice that bind FKN receptors expressed in the microglia of CNS parenchyma [60]. There was a severe synaptic and neuronal loss in FKN receptor knockout mice [61]. Mice lacking FKN signaling display decreased the proliferation of microglia in the CNS and inhibited the conversion of N-methyl-D-aspartate receptor subunit Glun2B to Glun2A during synapse development. These deficits also exhibited the changes with the loss of synaptic connections and perhaps neurons, reduced neurogenesis, a poorer ability of animals to sustain long-term potentiation and psychiatric disorders and cognitive dysfunctions. Depletion of the colony-stimulating factor-1 receptor expressed by embryonic microglia caused activation of synaptic plasticity, hyperactivity and anxiolytic-like behavior, probably due to a decrease in the number of pro-opiomelanocortin neurons, suggesting that microglia could be important for the development of cell types key to hypothalamic satiety centers [62].
Although there is little direct empirical evidence to support that glial-mediated synaptogenesis and pruning regulatory dysfunction is responsible for seizures, a few studies have revealed that synaptic pruning and aberrant synaptogenesis in neurodevelopmental disorders may be associated with epilepsy. Reactive astrocytes can accelerate the decrease of synaptic density and affect dendritic morphological development [63]. These phenomena are usually observed in fragile X syndrome (a disease associated with intellectual disability) which is closely related to epileptic seizures [64]. The impaired synaptic pruning function of microglia can cause neural circuit disruption, inducing epileptic seizures in Rett syndrome model mice. Dysfunctional synaptic pruning significantly increases the frequency of temporal lobe epilepsy in individuals with mutations in the glioma inactivated 1 (LGI1) gene encoding leucine-rich glioma inactivating protein 1.
5. Gliotransmission and Epilepsy
An expanding potential range of glial transmitters released from astrocytes, has been proposed, including glutamate, ATP and adenosine, and these molecules act as important neurotransmitters in the communication between astrocytes and neurons. Since these neuromodulators effectively induce neuronal excitability, evidence that gliotransmission contributes to epilepsy is growing, although it is seen that these processes keep their role still a controversial issue [65]. Glutamic acid, secreted by astrocytes, has various regulatory effects on the physiological functions of neurons, including enhancing neuronal excitability and activity synchronization reactivity and improving synaptic plasticity [66]. Glutamic acid, released from astrocytes, is usually induced by the photodecomposition of inositol 1, 4, 5-triphosphate. Although these stimuli cause a slow inward current, leading to transient depolarization, which is associated with paroxysmal depolarization, the physiological correlation of this astrocyte stimulation has been questioned [67]. Since the paroxysmal depolarization of these neurons is entirely a response to calcium signals from astrocytes and precedes neuronal overactivity, to some extent, astrocyte glutamic acid release can induce neuronal overexcitability.
There is new and stronger evidence to show that glutamate is a driving factor in epilepsy. Astrocyte-derived tumors which are referred to commonly as gliomas are notorious for causing seizures which occur in more than 70% of people before diagnosis. About one-third of people present with epilepsy (most of them belong to drug-resistant epilepsy), which is related to tumor diseases. Glutamate released from the tumor has been observed in tumor-bearing mice and humans due to overexpression of the system Xc encoded by the SLC7A11gene (an elevated expression in patients with glioma) [68]. The main function of this transporter is to synthesize glutathione, an intracellular antioxidant. The mandatory release of glutamic acid will cause the concentration of extracellular glutamic acid to be significantly higher than the normal level, causing excessive excitation which will aggravate the excitotoxicity of peripheral neurons [69]. GABAergic neurons also suffered damage after the protective coating of peripheral neural networks was decomposed and destroyed by matrix metalloproteinases released from gliomas [70]. The resulting imbalance, between excitement and inhibition, eventually leads to spontaneous and behavioral convulsive seizures.
Astrocytes can release ATP and are widely expressed in ion channels, G protein-coupled receptors activated by extracellular nucleotides and some products cleaved by these exonucleotidases [71]. Astrocyte astrogliosis, the release of glutamic acid and pro-inflammatory factors, remodeling of the ECM and synaptic formation are the main mechanisms for purinergic signaling to promote excitation and epilepsy. Adenosine (a lysate of ATP) and a subset of ATP receptors mainly mediate the occurrence of synaptic activity inhibition, and the reduction of excitatory drive can be realized by enhancing or inhibiting excitatory neurotransmission [72]. Presynaptic and postsynaptic Gi/o protein (which was coupled with adenosine A1 receptors [A1Rs]) were activated by adenosine, and the downregulation of A1Rs has been reported in rodent models of epilepsy in several recent studies. In presynaptic terminals, A1Rs can reduce calcium influx by inhibiting N-type calcium channels after activation, weakening excitatory synaptic transmission. Excitatory synaptic transmission can also be inhibited by reducing the sensitivity of presynaptic A1R-mediated neurotransmitter release elements [73]. Activated A1R can effectively reduce neuronal excitability at postsynaptic terminals, mainly by enhancing the outflow of potassium from the G protein-coupled Kir channel.
Evidence of the endogenous anticonvulsant role of adenosine has been confirmed, and it has been proposed to have neuroprotective effects through activation of its A1Rs. The physiological correlation of adenosine has been reported, with increased adenosine levels caused by epileptic activity. Studies of adenosine treatment, in acquired epilepsy models, have revealed the importance of astrocyte-based adenosine circulation in the disease process. Adenosine mainly comes from the rapid degradation of astrocytes-released ATP in extracellular space (ECS) and is subsequently removed by astrocyte nucleoside transporters and phosphorylated by ADK which is expressed in astrocytes [74]. Since the level of extracellular adenosine mainly depends on ADK, any change in ADK expression can limit the adenosine level, reducing its anticonvulsant and neuroprotective functions. ADK dysfunction has been found in both animal and human epileptic models, and strong overexpression of ADK was observed in reactive astrocytes, indicating that adenosine deficiency is at least a contributing factor to seizure generation [75]. Even in the absence of astrogliosis, or any other epileptic event, ADK overexpression is sufficient to cause epileptic seizures [74]. Data are sufficient to support that A1R agonists or ADK inhibitors can be used to increase adenosine levels to treat epilepsy, and a large number of epilepsy model studies have confirmed the effectiveness and reliability of this method for reducing epileptic seizures [74].
Glial cells can release a variety of substances and neurotransmitters. These small molecular substances can effectively improve the shaping of the synaptic structure and function caused by epilepsy and other diseases. Neurodegenerative diseases, such as epilepsy, are closely related to glial cell dysfunction.
6. Blood-Brain Barrier
The BBB, a dynamic biological interface, which actively controls the passage of substances between the CNS and peripheral circulation is an essential requisite for the maintenance of homeostasis and the physiological environment of the neural circuits [76]. A life-threatening condition, SE, has been confirmed to be closely linked to a rapid and robust increase in permeability of the BBB, through extensive experimental animal models of SE induction [77]. In this model, quantification of BBB leaks could reveal the extent of BBB destruction and suggest that 48 h BBB dysfunction (BBBD) could be a highly sensitive and specific predictor of epileptic progression after four to eight weeks [78, 79]. These experimental results are consistent with other previously reported animal models of SE induced by organophosphates, pilocarpine or kainic acid and electrical stimulation that have shown robust inflammatory responses with dysfunction of the BBB [80].
Albumin may play a critical role in the compromise of the BBB that occurs immediately following epilepsy [81]. The activation of the TGF-ββ pathway has emerged as a central mechanism underlying epileptogenesis development [82]. Similarities in outcomes, such as seizure-like activity, were observed in the activation of the TGF-ββ signaling pathway and after exposure to BBB disruptors or albumin [83]. Co-immunoprecipitation experiments confirmed that albumin activates the canonical TGF-ββ receptor-Smad signaling pathway, and the pathways downstream of TGF-ββ signaling are involved in epilepsy [84, 85]. Transcriptome analysis revealed similar expression patterns, following BBB breakdown and albumin and TGFββ1 exposure, including the up or downregulation of genes related to inflammation, synaptic transmission and early astrocytic activation. A new drug, named losartan (an angiotensin II type 1 receptor blocker), approved by the Food and Drug Administration, has been confirmed to exert a protective effect in rats following SE via blocking TGF-ββsignaling in peripheral tissue [86]. When the animals were in an epileptic state or after the BBB was disrupted by a pathological agent, losartan could downregulate the signal transduction of brain TGF-ββ mediated by albumin and inhibit reactive astrocytes to alleviate the BBBD, preventing the occurrence of epilepsy [87]. These results are consistent with other previously reported animal data and support the antiepileptic and neuroprotective effects of losartan on epilepsy and brain injury.
Vascular endothelial growth factor (VEGF) is another molecule associated with BBB dysfunction in the brain [88]. Among VEGF proteins, VEGF-C and its receptor VEGFR-3, are well-recognized regulators of lymphangiogenesis [89]. In the CNS, the expression of VEGFR-3 mRNA which is widely distributed throughout the entire brain region is known to localize mostly within neurons and some astrocytes and its receptor provides an auto-/paracrine growth and chemotactic system for glial precursors in the developing brain [90]. Under neuropathological conditions, the induction of VEGFR-3 occurs in reactive astrocytes and activated microglial cells in animal models of multiple sclerosis and stroke [91]. VEGFR-3 is highly expressed in reactive astrocytes in patients with TSC and MTLE [92]. VEGFR-3 expression was predominantly increased in reactive astrocytes after pilocarpine-induced SE [93]. This evidence suggests that changes in VEGFR-3 expression in astrocytes may play a key role in the pathogenesis of epilepsy. More research will be necessary to fully evaluate the potential of the VEGF signaling pathway to be therapeutic in epilepsy, including the precise mechanisms by which it works to maintain a normal astrocyte phenotype.
Pericytes have been proposed to play a vital role in the formation and maturation for the maintenance of BBB integrity during embryogenesis [94]. The proliferation of endothelial cells and pericyte rearrangement are all observed in the progression of epileptogenesis and after pilocarpine-induced SE [95]. Pericytes are actively participated in the “seizure network” through in vitro experiments using patch-clamp techniques to perform whole-cell recordings for pericytes derived from cortical organotypic slice cultures. It was speculated that the prolongation of inward currents, in pericytes, might promote acute epileptic seizures. Recurrent seizures develop downstream of pericytic damage, progressive loss of vascular cells and BBBD. Both in vitro slice cultures and in vivo animal model experiments confirmed that the sensitivity of capillaries and arterioles to vasodilation response induced by recurrent epilepsy was reduced [82].
7. Glial Activation Mediated Neuronal Ferroptosis is a Hot Spot in Epilepsy Research
Ferroptosis is an iron-dependent form of regulated cell death that is initiated by abnormal iron metabolism and severe lipid peroxidation, leading to oxidative stress and cell death [96]. Ferroptosis has been extensively reported to be involved in various neurological disorders, including epilepsy [97]. The evidence on ferroptosis in epilepsy, from glia-neuron crosstalk, is still lacking. Glial activation is involved in the onset and progression of epilepsy through various pathways. Large numbers of activated microglia were found around the degenerated neurons in epilepsy patients’ brains. Reactive astrocytes were also detected in autopsies of epilepsy patients’ brains, accompanied by the formation of neuron discharge [98]. Activated glia can induce neuronal death in vitro by releasing proinflammatory factors, nitric oxide, ROS, and glutamate. Different types of glia may contribute to epilepsy pathogenesis via different pathways such as demyelination and cytokine release [99]. Upon activation, they may also induce neuronal ferroptosis by disrupting iron homeostasis, altering amino acid metabolism and increasing oxidative stress [100]. Ferroptosis may play a role in the interaction between glia and neurons and thus modulate the pathogenetic process of epilepsy (Fig. 4). Researchers need to address current limitations to elucidate the pathogenesis of epilepsy and develop corresponding effective treatment measures.
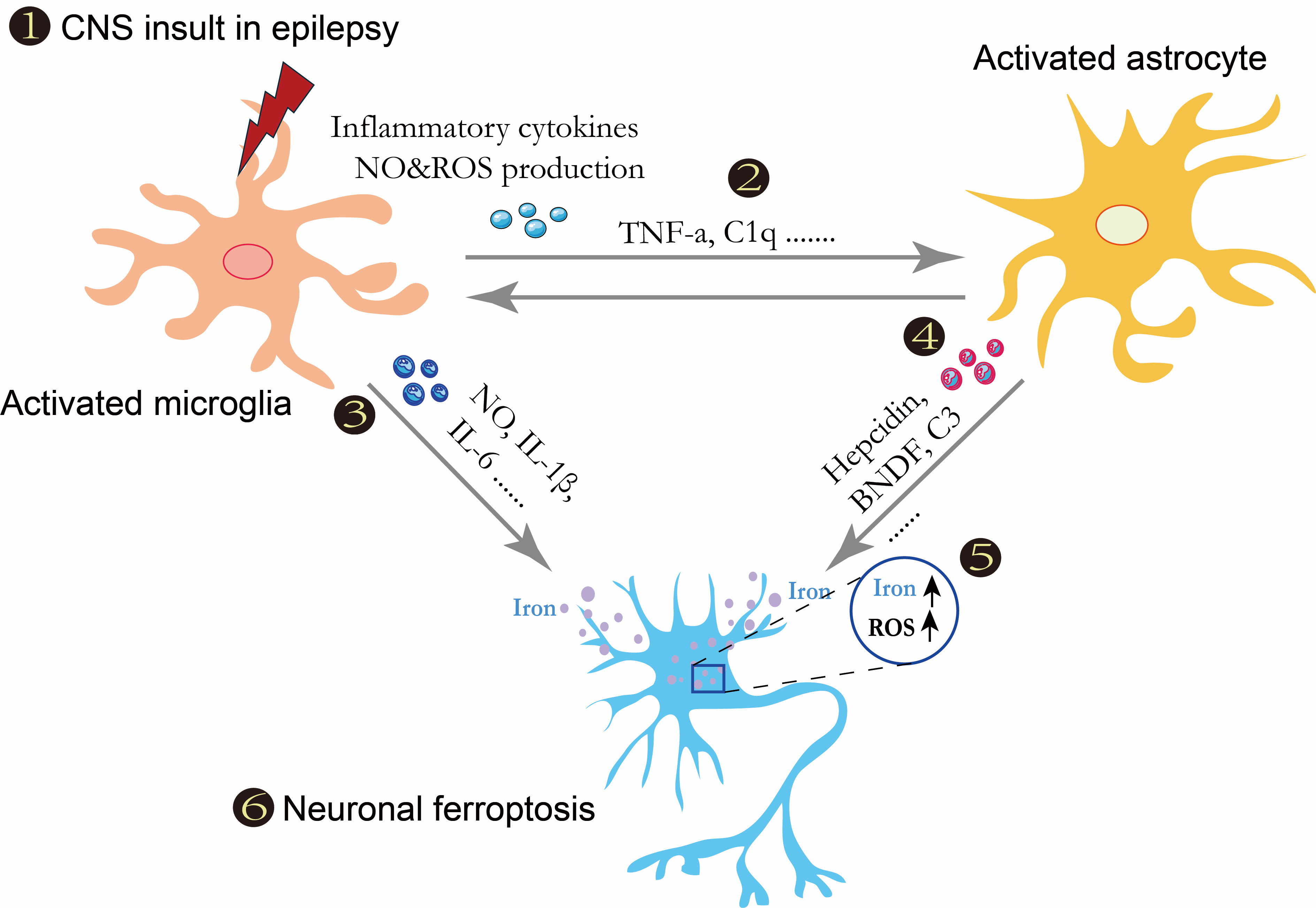
Hypothesis for ferroptosis in epilepsy implying crosstalk between glia and neurons.
(1) Central nervous system insult in epilepsy-induced microglial activation. (2) Activated microglia release tumor necrosis factor-
and C1q to induce astrocyte activation. (3–5) Under activation conditions, different phenotypic microglia could release nitric oxide, IL-1
8. Conclusions
The above contribute to an increased scientific understanding of epilepsy and leads one to suggest that the neuronal microenvironment plays a significant role in the etiology of epilepsy. This microenvironment is envisaged as dynamic and largely influenced by the contact between the neuron and glial cells. There seems little doubt that glia function alterations might critically contribute to epileptogenesis and ictogenesis. Although the neural mechanism of epilepsy seems to have been explained by glial-mediated neural network regulation, there is ongoing discussion regarding whether the glial cells have major homeostasis functions or directly regulate neuronal signals, in normal and diseased brains, including epilepsy.
The evidence provided does not agree with the concept of strict separation between “passive” homeostasis regulation and “active” neuron-glial cell communication. Disruption of glia-mediated changes in extracellular ionic and neurotransmitters, ECM, vascular tone and metabolite supply destroy the maintenance of lasting equilibrium. It will also have direct or indirect effects on the excitability of neurons. Because glial cells and neurons have almost the same set of ion channels, structural morphology and receptors, some important questions and challenges need to be resolved, including confirming that alterations in glial cells are causative to some forms of epilepsy and identifying glial targets as a basis for the development of more specific, an anti-seizure medication.