
Thrombospondin receptor α2δ-1 promotes synaptogenesis and spinogenesis via postsynaptic Rac1
By W. Christopher Risher, Namsoo Kim, Sehwon Koh, Ji-Eun Choi, Petar Mitev, Erin F. Spence, Louis-Jan Pilaz, Dongqing Wang, Guoping Feng, Debra L. Silver, Scott H. Soderling, Henry H. Yin, and Cagla Eroglu
Excerpt from the article published in Journal of Cell Biology (2018) 217 (10): 3747–3765.
https://doi.org/10.1083/jcb.201802057
Editor’s Highlights
- The thrombospondin (TSP) family of extracellular matrix proteins exert synaptogenic effects via binding to their neuronal receptor, the calcium channel subunit α2δ-1
- α2δ-1 is a drug target for epilepsy and neuropathic pain; thus the TSP–α2δ-1 interaction is implicated in both synaptic development and disease pathogenesis.
- α2δ-1 is crucial for the establishment of proper cortical synapse connectivity. In the absence of α2δ-1, intracortical excitatory synaptogenesis, synaptic function, and spinogenesis are greatly diminished.
- The small Rho GTPase, Ras-related C3 botulinum toxin substrate 1 (Rac1), is a key component of the synaptogenic signaling cascade downstream of TSP–α2δ-1.
- But Rac1 activation alone was unable to cluster pre- and postsynaptic machinery in the absence of the α2δ-1.
- Rac1 impairments have been suggested as a mechanism underlying structural abnormalities in fragile X syndrome as well as aberrant synaptic transmission in schizophrenia.
- Cortical development and synaptic growth wherein astrocytes can control neuronal Rac1 signaling via TSP–α2δ-1.
Abstract
Astrocytes control excitatory synaptogenesis by secreting thrombospondins (TSPs), which function via their neuronal receptor, the calcium channel subunit α2δ-1. α2δ-1 is a drug target for epilepsy and neuropathic pain; thus the TSP–α2δ-1 interaction is implicated in both synaptic development and disease pathogenesis. However, the mechanism by which this interaction promotes synaptogenesis and the requirement for α2δ-1 for connectivity of the developing mammalian brain are unknown. In this study, we show that global or cell-specific loss of α2δ-1 yields profound deficits in excitatory synapse numbers, ultrastructure, and activity and severely stunts spinogenesis in the mouse cortex. Postsynaptic but not presynaptic α2δ-1 is required and sufficient for TSP-induced synaptogenesis in vitro and spine formation in vivo, but an α2δ-1 mutant linked to autism cannot rescue these synaptogenesis defects. Finally, we reveal that TSP–α2δ-1 interactions control synaptogenesis postsynaptically via Rac1, suggesting potential molecular mechanisms that underlie both synaptic development and pathology.
Introduction
The controlled development of neuronal networks is crucial for proper function of the central nervous system (CNS). Thus, the formation of CNS synapses, the smallest cell biological units of neural circuits, is tightly regulated (Scheiffele, 2003; West and Greenberg, 2011), but the manner in which this occurs is poorly understood. Research in the last two decades revealed that astrocytes, the most abundant glial cell in the brain, promote the establishment of synaptic connectivity (Clarke and Barres, 2013). Astrocyte-secreted factors strongly enhance excitatory synapse formation in vitro and in vivo (Baldwin and Eroglu, 2017; Bosworth and Allen, 2017). The thrombospondin (TSP) family of extracellular matrix proteins were the first astrocyte-secreted synaptogenic factors to be identified via purified retinal ganglion cell (RGC) neuron cultures. Transgenic mice lacking two of the TSP isoforms (TSP1 and TSP2), which are expressed by astrocytes during early postnatal development, show decreased excitatory synaptic density in the cortex (Christopherson et al., 2005).
TSPs exert their synaptogenic effects via binding to their neuronal receptor, the calcium channel subunit α2δ-1 (Eroglu et al., 2009), also known as the gabapentin receptor (Gee et al., 1996). α2δ-1 (Cacna2d1) is highly expressed by neurons throughout the CNS (Cole et al., 2005), with enrichment in cortical and hippocampal pyramidal neurons. It is predicted to be a type I membrane protein consisting of an entirely extracellular α2 portion and a δ part that spans extracellular (ECD), transmembrane (TM), and cytosolic regions (Davies et al., 2007). Both α2 and δ are translated from the same transcript but are proteolytically cleaved into two mature polypeptides (De Jongh et al., 1990; Jay et al., 1991; Douglas et al., 2006). α2δ-1, via its Von Willebrand factor A (VWF-A) protein–protein interaction domain, interacts with the synaptogenic fragment of TSP2 containing epidermal growth factor–like repeats to promote synapse formation (Eroglu et al., 2009).
α2δ proteins are involved in the trafficking and function of voltage-gated calcium channels (VGCCs; particularly the high voltage L-type channels) and influence presynaptic release probability and homeostasis (Geisler et al., 2015). However, despite the importance of VGCCs to synaptic transmission and plasticity (Catterall and Few, 2008; Li et al., 2016), manipulation of channel expression or function does not affect TSP-induced synaptogenesis (Eroglu et al., 2009). Furthermore, α2δ proteins were shown to have functions independent of their roles in regulation of Ca2+ channels (Kurshan et al., 2009). Therefore, we previously proposed that α2δ-1 acts as a surface receptor for TSP and, after TSP binding to the VWF-A domain of α2δ-1, triggers a downstream synaptogenic signaling cascade that promotes the formation of excitatory synapses (Risher and Eroglu, 2012). α2δ-1 is also the receptor for the drug gabapentin, which is used for the treatment of epilepsy and neuropathic pain (Gee et al., 1996; Field et al., 2006). Gabapentin interferes with the binding of TSP to α2δ-1 and thereby inhibits synaptogenesis (Eroglu et al., 2009), suggesting that impairments in astrocyte signaling may underlie aberrant connectivity that drives neurological disease.
In this study, we have determined that dendritic α2δ-1 is crucial for the establishment of proper cortical synapse connectivity. In the absence of α2δ-1, intracortical excitatory synaptogenesis, synaptic function, and spinogenesis are greatly diminished. Furthermore, we found the small Rho GTPase, Ras-related C3 botulinum toxin substrate 1 (Rac1), to be a key component of the synaptogenic signaling cascade downstream of TSP–α2δ-1.
Results
TSP receptor α2δ-1 is critical for cortical synaptic development
We observed via Western blot from mouse cortical and hippocampal lysates that α2δ-1 expression increases rapidly between postnatal days 5 (P5) and P10 (Fig. 1 A), corresponding with a period in which excitatory synapse formation is initiated (Semple et al., 2013). To determine whether α2δ-1 is required for proper synaptic connectivity in the cortex, we used a mouse line lacking α2δ-1 (i.e., α2δ-1 knockout [KO]; Fig. S1, A and B; Park et al., 2016). No α2δ-1 protein is detected in synaptosome lysates isolated from the α2δ-1 KO mice (Fig. S1 C). We focused our analysis on the Layer I (L1) synaptic zone (S/Z) of primary visual cortex (V1; Fig. 1 B), which receives dendritic projections from excitatory pyramidal neurons in cortical layers II and III (Fig. 1 B), and which has the highest α2δ-1 expression in the CNS (Cole et al., 2005). We immunostained brain sections with pre- and postsynaptic marker pairs that label the two main classes of excitatory synapses in the cortex: (1) intracortical connections positive for presynaptic vesicular glutamate transporter 1 (VGluT1) and (2) sensory pathway VGluT2+ inputs from thalamus (Fig. 1 C). Colocalization of VGluT1 with postsynaptic PSD95 is severely decreased (−58.1%) in α2δ-1 KO mice compared with littermate WT mice at P21 (Fig. 1 D), indicating that loss of α2δ-1 disrupts intracortical synaptic connectivity. By contrast, thalamocortical synapses, quantified as the colocalization of VGluT2/PSD95, do not differ between WT and α2δ-1 KO at P21 (Fig. 1 E). The colocalization of VGluT1/PSD95 puncta is not merely by chance, since randomization of puncta by rotating the channels out of alignment by 90° nearly abolished occurrence of colocalized puncta in both the WT and KO (Fig. S1, D and E). These results suggest that α2δ-1 is specifically required for the formation of intracortical synapses in V1.
Area S1 somatosensory cortex also showed a strong decrease in the number of intracortical synapses, with no change in thalamocortical synapses (Fig. S1, F and G). The deficit in intracortical synapses becomes apparent between postnatal weeks 2 and 3, coinciding with peak α2δ-1 expression (Fig. 1 A): we found no significant differences between genotypes at P14 V1, but the synaptic growth that typically takes place between P14 and P21 failed to occur in the α2δ-1 KO mice (Fig. S1 H). We also observed a significant deficit in intracortical synapses in the KO at P40, indicating that the synaptic phenotype observed in α2δ-1 KOs is not merely a developmental delay (Fig. 1 F).
The striking decrease in the number of intracortical synapses was not caused by a decrease in neurons (determined by NeuN+ cell density and cortical thickness of V1 at P21; Fig. S1, I–K). However, overall neuronal complexity was significantly decreased in the KOs as quantified by Sholl analysis of Golgi-Cox–stained LII/III pyramidal neurons (Fig. 1, G and H). There was also a trend toward a decrease in total dendrite length (Fig. 1 I). Taken together, these results show that α2δ-1 is also required for dendritic elaboration, which is tightly linked with excitatory synaptogenesis during development (Cline and Haas, 2008).
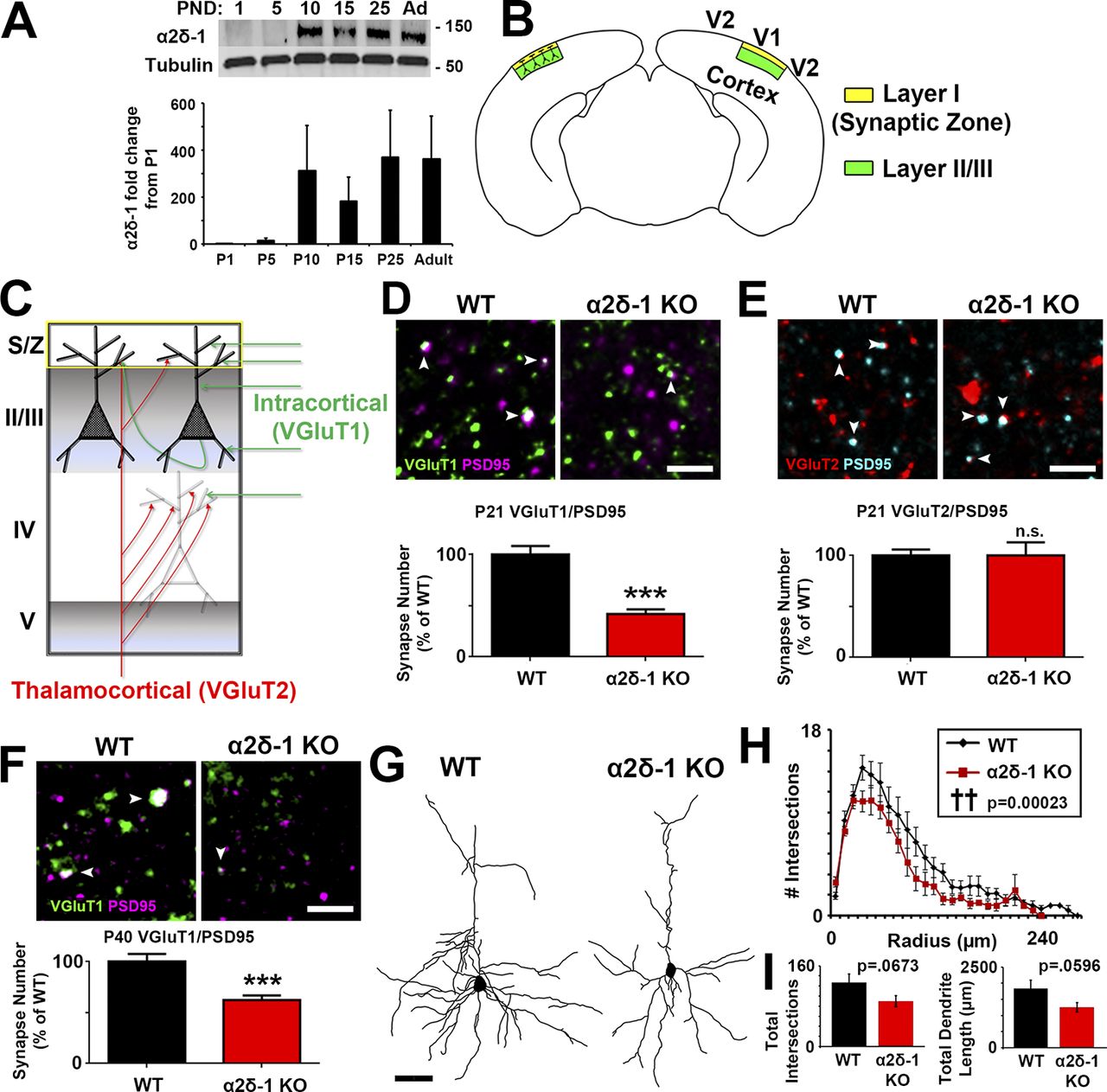
Impaired synaptic connectivity in α2δ-1–deficient cortex. (A) Top: Western blot of α2δ-1 expression from WT cortex and hippocampus from postnatal day (PND) 1 to adult. Tubulin: loading control. Bottom: α2δ-1 expression as fold change from P1 (n = 3 mice per age). (B) Diagram of a mouse coronal brain slice including area V1. Layer I is the S/Z where IHC analyses were performed. Dendrites from this region are primarily from neurons whose cell bodies reside in LII/III. (C) Schematic of excitatory synaptic input to area V1. (D) Top: IHC images of pre- (VGluT1), post- (PSD95), and colocalized (white arrowheads) synaptic puncta from V1 of P21 WT and α2δ-1 KO mice. Bottom: Intracortical synapse quantification as percentage of WT (n= 3 mice per genotype). One-way ANOVA with Tukey’s multiple comparisons post hoc test. (E) Top: Thalamocortical synapse staining shown with pre- (VGluT2), post- (PSD95), and colocalized (white arrowheads) synaptic puncta from V1 of P21 WT and α2δ-1 KO mice. Bottom: Quantification of thalamocortical synapses as percentage of WT (n = 3 mice per genotype). One-way ANOVA. Error bars represent SEM. (F) IHC staining from area V1 at P40 showing VGluT1/PSD95 intracortical synapses in WT and α2δ-1 KO (n = 3 mice per genotype). Nested ANOVA. (G) Representative camera lucida drawings from LII/III pyramidal neurons in P21 WT and α2δ-1 KO. (H) Sholl analysis results show morphological complexity in P21 WT and α2δ-1 KO neurons (n = 4 neurons/mouse; three mice/genotype). ††, P = 0.00023; ANCOVA. (I) Total number of intersections measured via Sholl analysis (left) and total length of the dendritic arbor (right) compared between P21 WT and α2δ-1 KO neurons. Unpaired two-tailed t test. Error bars represent SEM. Bars: (D, E, F) 2 µm; (G) 50 µm. ***, P < 0.0001.
We next tested whether the impaired synaptic connectivity in the α2δ-1 KO resulted in disrupted synaptic function. We performed electrophysiological recordings of miniature excitatory postsynaptic currents (EPSCs; mEPSCs) from LII/III pyramidal neurons in P21 V1 of littermate α2δ-1 WT and KO mice (Fig. 2 A). In accordance with our histological findings, mEPSCs from α2δ-1 KO cortices showed a strong reduction in frequency and a small but significant decrease in amplitude, reflecting highly decreased synaptic density and mildly decreased synaptic strength, respectively (frequency: WT, 2.534 ± 0.3843 Hz; KO, 1.046 ± 0.1668 Hz [−58.72%]; amplitude: WT, 16.02 ± 0.8360 pA; KO, 14.63 ± 0.5270 pA [−8.68%]; Fig. 2, B–D). On the contrary, miniature inhibitory postsynaptic currents (mIPSCs) showed no differences between genotypes (Fig. 2, E–G). We next recorded evoked EPSCs (eEPSCs) to determine whether loss of α2δ-1 affected the glutamate receptor subtype composition of postsynapses. We found a significant decrease in the N-methyl-D-aspartic acid (NMDA)/α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) ratio (Fig. 2, H and I) in line with significantly decreased rise and decay times observed in the mEPSCs (Fig. S2, A and B) as NMDA receptors contribute to the slow component of synaptic waveforms (Lester et al., 1990). Paired-pulse ratio (PPR) recordings showed no difference between the WT and α2δ-1 KO, indicating that α2δ-1 is not required for the control of presynaptic release properties in the cortex (Fig. 2, J and K). Taken together, these results show that α2δ-1 is required for proper excitatory synapse function but dispensable for the function of inhibitory synapses in the developing cortex.
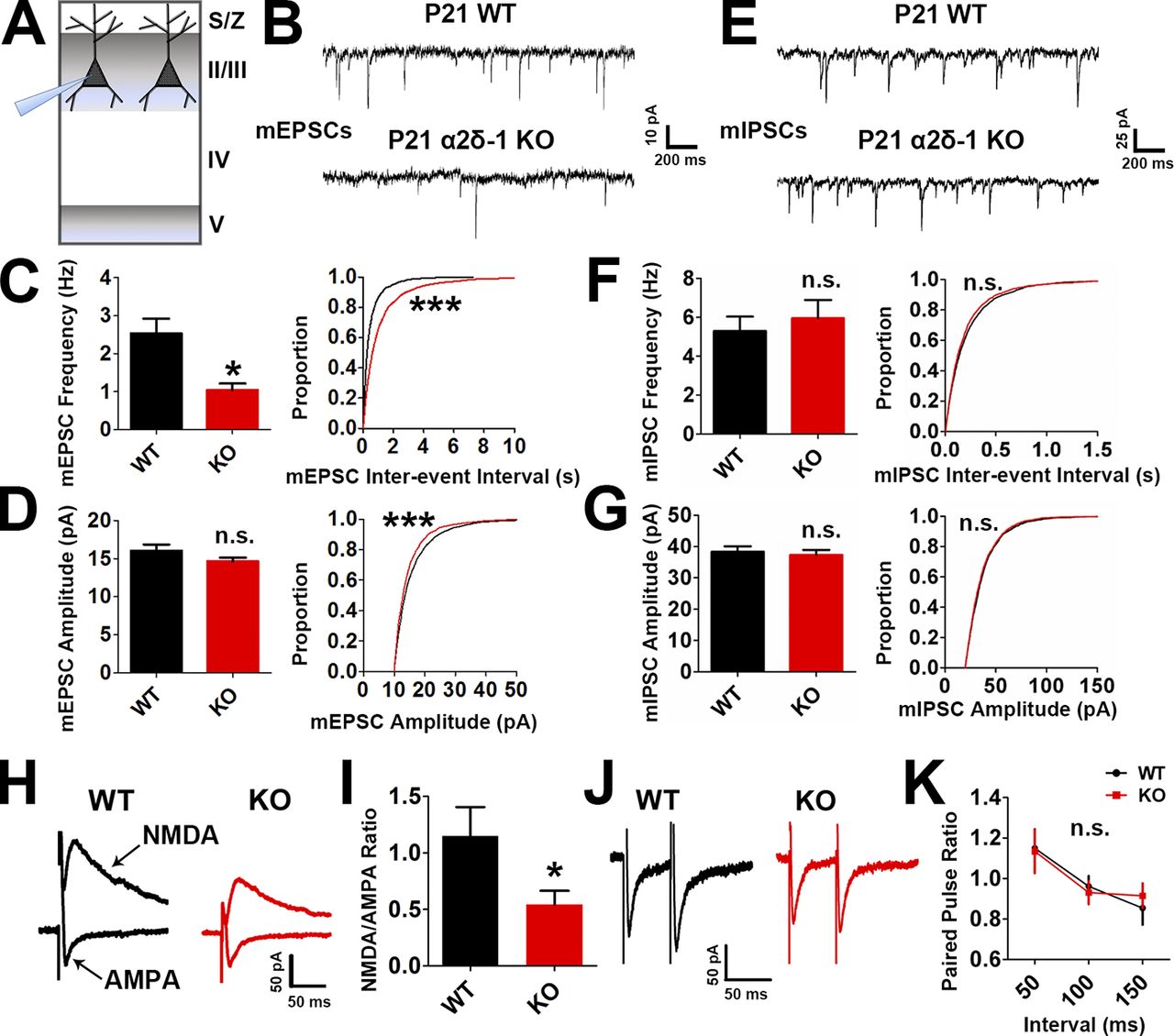
Lack of α2δ-1 results in decreased excitatory synaptic function. (A) Recordings were made from LII/III pyramidal neurons in V1 at P21. (B) mEPSC traces from WT and α2δ-1 KO pyramidal neurons. (C and D) Frequency (C, left), interevent interval (C, right), and amplitude (D) of mEPSCs from WT and α2δ-1 KO neurons (n = total of 12 cells from three animals/genotype). Left: Two-tailed t test. Right: Kolmogorov–Smirnov test. (E) mIPSC traces from WT and α2δ-1 KO pyramidal neurons. (F and G) Frequency (F, left), interevent interval (F, right), and amplitude (G) of mIPSCs from WT and α2δ-1 KO neurons (n = total of 10 cells from three animals/genotype). Left: Two-tailed t test. Right: Kolmogorov–Smirnov test. (H) Traces of NMDA-only and AMPA-only evoked currents from WT and α2δ-1 KO pyramidal neurons. (I) Quantification of NMDA/AMPA ratio between WT and α2δ-1 KO (n = total of 12 cells from three to four animals/genotype). Two-tailed t test. Error bars represent SEM. (J) Traces from paired pulse recordings from WT and α2δ-1 KO neurons. (K) Comparison of PPR between WT and α2δ-1 KO (n = total of 12 cells from three to four animals/genotype). One-way ANCOVA. Error bars represent SEM. *, P < 0.05; ***, P < 0.0001.
Our immunohistochemical and electrophysiological experiments revealed major changes in excitatory synaptic connectivity of α2δ-1 KO cortex. Based on its up-regulation at P10 (Fig. 1 A), we hypothesized that α2δ-1 is required for formation and/or maturation of dendritic spines. To investigate synaptic connectivity, we used serial section transmission EM (ssEM) to visualize synaptic components including dendrites, filopodia, spines, and postsynaptic densities in 3D (Harris et al., 2006). We focused on secondary and tertiary dendrites within the S/Z of V1 from P21 mice. EM analysis further confirmed the decreased excitatory synaptic density per spine length in the α2δ-1 KOs compared with WT (Fig. 3, A–C). α2δ-1 KO dendrites have far fewer protrusions than WT (Fig. 3, B and C) and regularly display “bulged” regions separated by thin constrictions of the shaft (Fig. 3 B), with the KO having increased frequencies of unusually small and large shaft measurements compared with WT (Fig. S3 A).
Dendritic protrusions receive the majority of excitatory synaptic inputs in the CNS, presenting in a variety of morphologies that reflect different stages of synaptic maturation (Harris and Kater, 1994; Irwin et al., 2001). In particular, as synapses mature, dendritic protrusions decrease in length and increase in width, switching from a filopodial morphology into mushroom spines (Grutzendler et al., 2002). Protrusion density was severely reduced in α2δ-1 KO dendrites compared with WT (Fig. 3 D), primarily driven by a decrease in mature mushroom spines as well as intermediate thin spines (Fig. 3, E and F). The majority of protrusions in the α2δ-1 KO tended to be thin and filopodia-like; occasionally harboring excitatory postsynaptic densities but often barren of synaptic contacts (Fig. 3 B, red arrowheads, and Fig. 3 G). Accordingly, overall protrusion length was increased in the α2δ-1 KO (Fig. S3 B). Comparison of spine versus filopodia protrusions revealed a failure of spinogenesis in the KOs, whereas filopodia numbers were unaffected between WT and KO (Fig. 3 H). α2δ-1 KO dendritic protrusions do not undergo the stereotypical filopodia-to-spine transition during excitatory synaptic development, thus revealing a crucial role for α2δ-1 in this process.
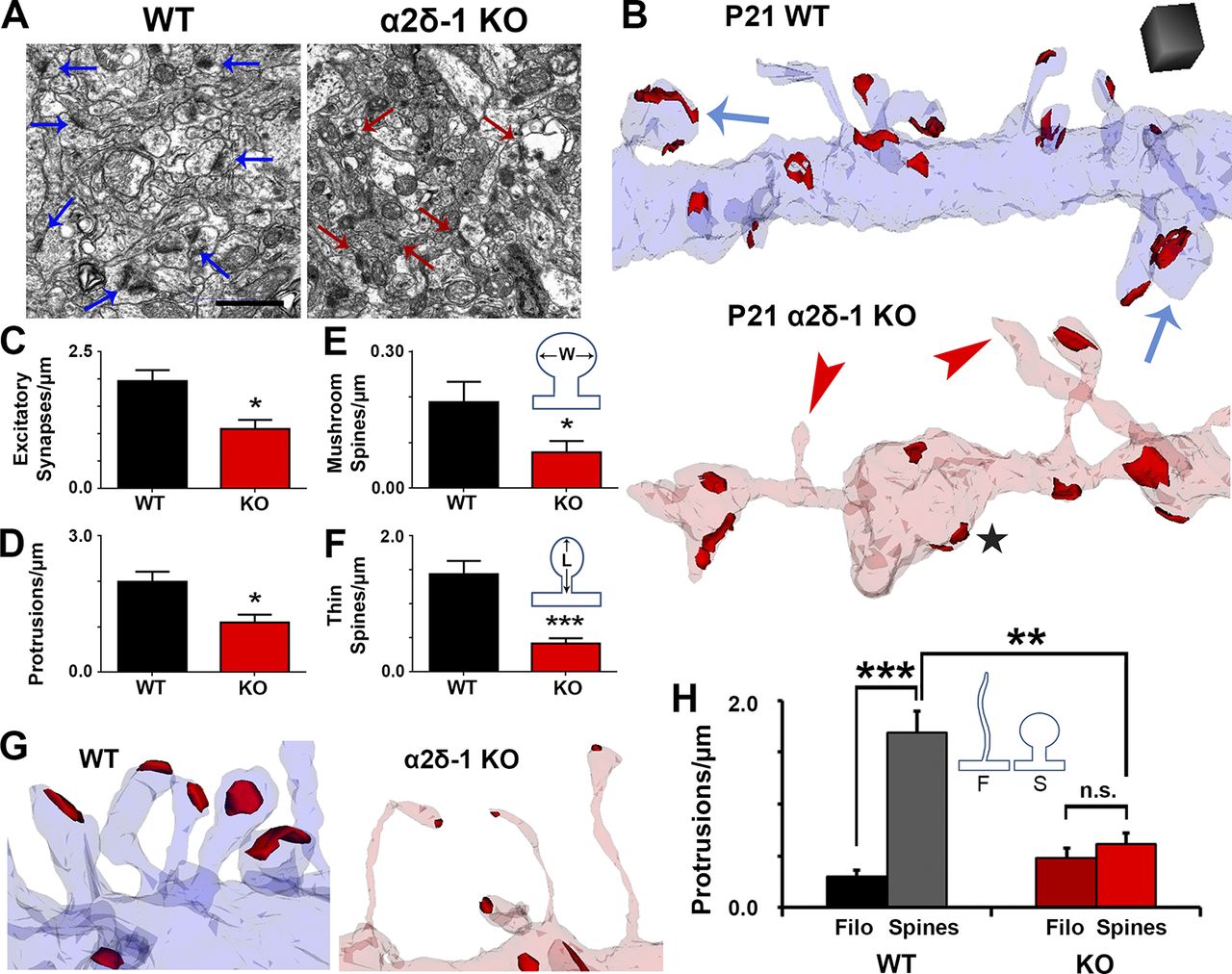
Ultrastructural analysis reveals that α2δ-1 promotes synapse and spine maturation. (A) Electron micrographs from P21 V1 WT and α2δ-1 KO brains. Arrows, excitatory synapses. Bar, 1 µm. (B) ssEM 3D reconstructions of LI dendrites from P21 WT and α2δ-1 KO V1. Red, excitatory postsynaptic densities; blue arrows, mushroom spines; red arrowheads, nonsynaptic filopodia; black star, bulged region of dendritic shaft. Cube, 0.5 µm3. (C–F) Densities calculated from reconstructions of excitatory synapses (C), protrusions (D), mushroom spines (E), and thin spines (F). E and F show the width (W) and length (L) measurements used in spine identification (n = 4 dendrites/animal; three animals per genotype). Two-tailed t test. Error bars represent SEM. (G) Spines in the WT and filopodia in the α2δ-1 KO. (H) Comparison of filopodia (F) and spine (S) densities between the WT and α2δ-1 KO (n = 4 dendrites/animal; three animals per genotype). One-way ANOVA with Tukey’s multiple comparisons post hoc test. *, P < 0.05; **, P < 0.001; ***, P < 0.0001.
α2δ-1 is required cell autonomously for the formation and maturation of dendritic spine synapses
α2δ-1 is highly expressed in regions outside the brain, particularly in skeletal and cardiac muscles (Gong et al., 2001). To investigate whether the synaptic abnormalities in the global KO are caused by cell-autonomous loss of α2δ-1 in neurons and not secondary to its effects elsewhere, we used a transgenic mouse strain with floxed alleles of α2δ-1 (α2δ-1f/f; Park et al., 2016) to delete α2δ-1 by Cre-dependent recombination. These mice also carry Rosa(STOP)loxP-tdTomato alleles (a.k.a. Ai14, hereafter referred to as RTm), which express tdTomato under control of the Rosa locus after Cre-mediated recombination (Fig. 4 A). Cre recombinase was expressed in a sparse population of LII/III cortical pyramidal neurons by in utero electroporation of α2δ-1f/f–RTmf/f pups at embryonic day 15.5 (E15.5; Fig. 4 A). With this approach, we could preferentially knock out α2δ-1 in a small fraction of LII/III neurons that would be RTm+ (Fig. 4 B). Brains were harvested at P21, and RTm+dendrites were imaged along with their associated pre- and postsynaptic puncta (Fig. 4 C). After reconstructing dendritic structures and synaptic puncta in 3D via Imaris, we observed similar impairments between this cell-specific loss of α2δ-1 (i.e., α2δ-1 conditional KO; cKO) and the global α2δ-1 KO. Total spine density was decreased in cKO dendrites compared with age-matched control (α2δ-1+/+-RTmf/f) dendrites driven largely by a reduction in mature spines (Fig. 4, D and E). Furthermore, both pre- (VGluT1) and postsynaptic (PSD95) markers associated with α2δ-1 cKO dendrites were decreased as was their colocalization (Fig. 4, D and E). Density of the NMDAR subunit NR1 as well as the juxtaposing of NR1 puncta with presynaptic VGluT1+ terminals were also significantly decreased in α2δ-1 cKO dendrites (Fig. 4, F and G). These findings show that α2δ-1 is required cell autonomously for intracortical synapse formation and spinogenesis.
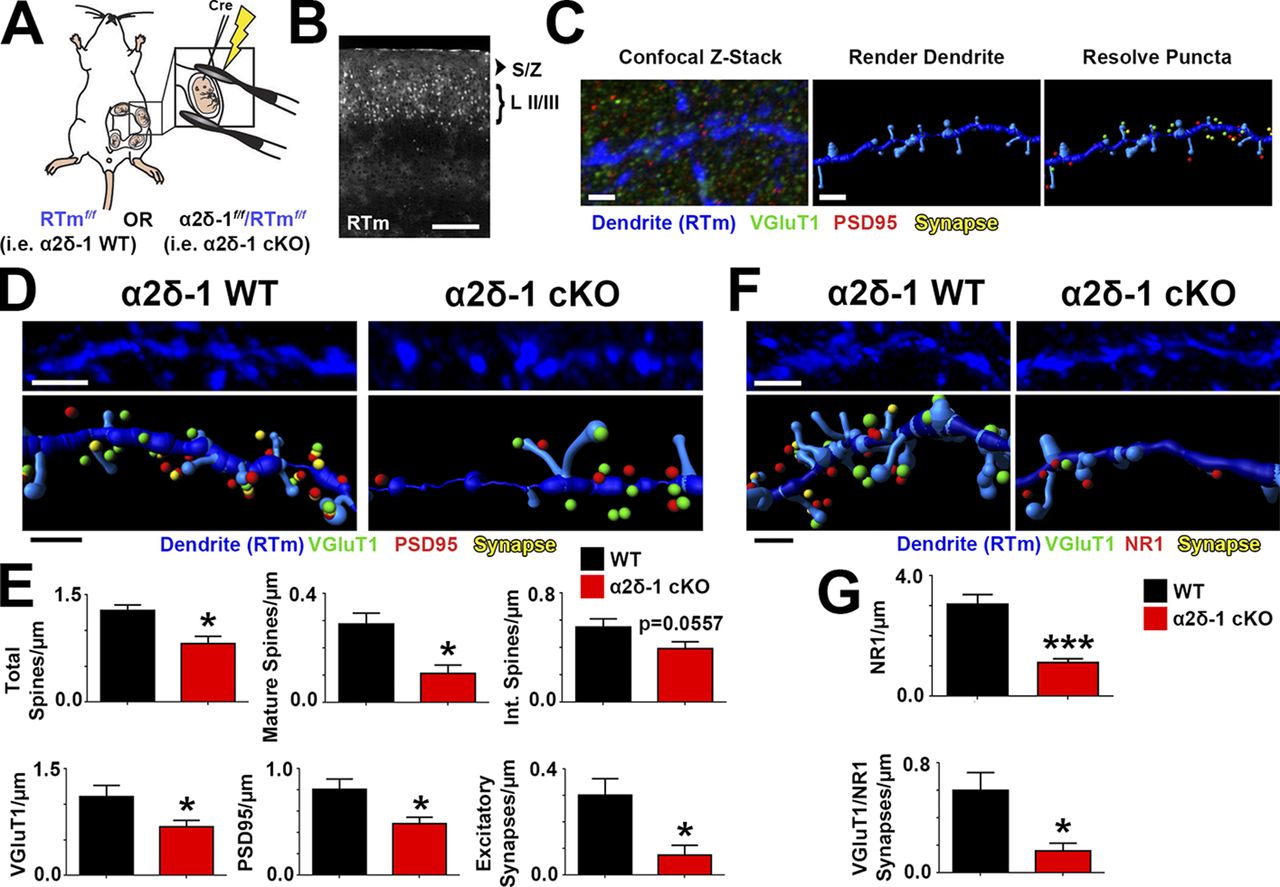
α2δ-1 promotes synapse and spine development cell autonomously. (A) Schematic for in utero electroporation. (B) Confocal image of P21 V1. Cre+ neurons in LII/III express RTm. Dendrites were imaged in the S/Z. (C) Workflow for analysis with Imaris. (D) 3D confocal images (top) and Imaris reconstructions (bottom) of RTm+ dendrites from WT and α2δ-1 cKO brains. Pre- (VGluT1), post- (PSD95), and colocalized synaptic puncta in close proximity to the dendrite are shown. (E) Density of total spines, mature and intermediate spines, VGluT1, PSD95, and excitatory synapses between WT and α2δ-1 cKO dendrites (four dendrites/animal; three animals per condition). Two-tailed t test. (F) 3D confocal images (top) and Imaris reconstructions (bottom) of RTm+ dendrites from WT and α2δ-1 cKO brains. Pre- (VGluT1), post- (NR1), and colocalized synaptic puncta in close proximity to the dendrite are shown. (G)Density of NR1 and colocalized VGluT1/NR1 synapses of WT and α2δ-1 cKO dendrites (six dendrites/animal; three animals per condition). Two-tailed t test. Error bars represent SEM. Bars: (B) 200 µm; (C) 1 µm; (D and F, top) 2 µm; (D and F, bottom) 1 µm. *, P < 0.05; ***, P < 0.0001.
Post- but not presynaptic α2δ-1 is required for TSP-induced synaptogenesis
To determine whether TSP promotes cortical synapse formation through α2δ-1 in a manner similar to that we previously showed in RGCs (Eroglu et al., 2009), we isolated cortical neurons with >95% purity from P1 littermate α2δ-1 heterozygous (Het) and KO pups (Fig. 5 A). After 7 d in vitro (DIV7), neurons were treated with TSP2 for an additional 6 d. In DIV13 cultures from α2δ-1 Het mice, TSP2 promoted a significant increase in excitatory synapses, determined as a twofold increase in colocalization of presynaptic VGluT1 and postsynaptic Homer (Fig. 5, B and C). As expected, the synaptogenic effect of TSP2 was blocked by cotreatment with gabapentin (32 µM), which interferes with the binding of TSP to α2δ-1 (Fig. 5, B and C; Eroglu et al., 2009). The same TSP2 treatment did not induce a synaptogenic response in cortical neurons from α2δ-1 KO mice, which did not differ in synapse density from α2δ-1 Het neurons in the absence of TSP2 (Fig. 5, B and C). These results confirm that TSP2 induces intracortical excitatory synapse formation in vitro in an α2δ-1–dependent manner.
α2δ-1 is present both pre- and postsynaptically (Taylor and Garrido, 2008; Bauer et al., 2009). To determine whether pre- or postsynaptic α2δ-1 is critical for excitatory synaptogenesis, α2δ-1 Het and KO mice were crossed with a transgenic mouse line expressing GFP in all cells (Hadjantonakis et al., 1998), generating the following littermates: α2δ-1 Het/GFP (tg/0); α2δ-1 Het/GFP (0/0); α2δ-1 KO/GFP (tg/0); and α2δ-1 KO/GFP (0/0; Fig. 5 D). Cortical neurons from P1 pups were plated such that GFP+ neurons comprised 5% of the cells per condition (Fig. 5 E). In this way, when imaging synapses on GFP+ cells, the majority of the presynaptic inputs would come from surrounding GFP− neurons (Fig. 5 E). We tested three different scenarios: (1) control, where both the “presynaptic” (GFP−) and the “postsynaptic” (GFP+) neurons were α2δ-1 Het; (2) postsynaptic necessity, where a GFP+ α2δ-1 KO cell was surrounded by GFP− α2δ-1 Het neurons; and (3) presynaptic necessity, where GFP−α2δ-1 KO neurons synapse onto an GFP+ α2δ-1 Het neuron (Fig. 5 E). After TSP2 treatment (Fig. 5 A), we observed increased excitatory synapses in the control scenario, where all neurons expressed α2δ-1 (Fig. 5, F and G). Postsynaptic necessity was confirmed when TSP2 treatment had no effect on synapse number even though the presynaptic neurons still expressed α2δ-1. Unexpectedly, when α2δ-1 was present only in the postsynaptic cell, TSP-induced synaptogenesis was significantly enhanced even above control (Fig. 5, F and G), showing that presynaptic α2δ-1 is not required and potentially may have an inhibitory role for the synaptogenic function of TSP. These results confirm that postsynaptic α2δ-1 is necessary and sufficient for TSP-induced cortical synapse formation.
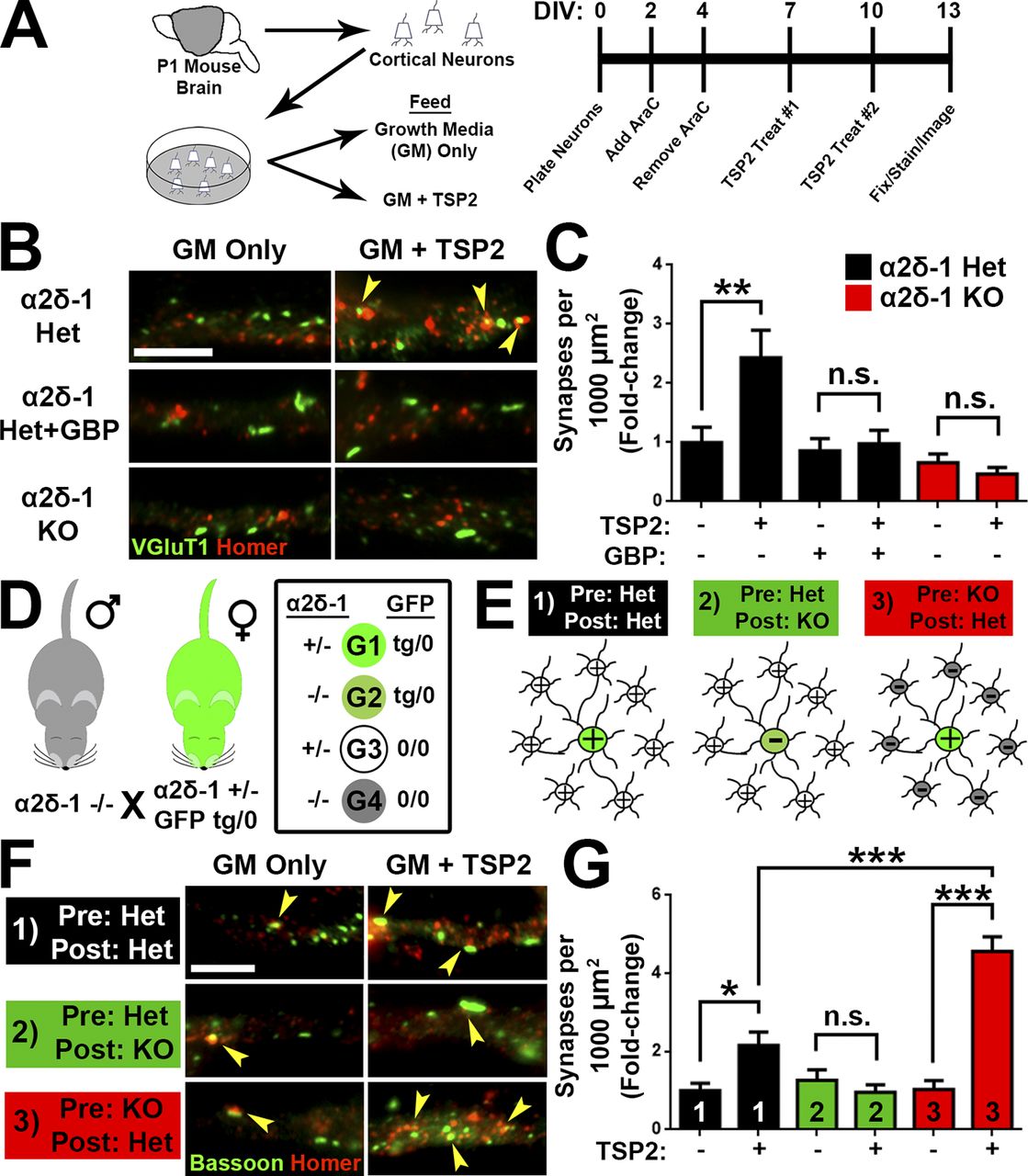
TSP stimulates synaptogenesis via postsynaptic α2δ-1. (A) Mouse cortical neuron purification and TSP2 treatment timeline. (B) Cortical dendrites from α2δ-1 Het or KO mouse pups. Cells were treated with TSP2-containing or deficient growth media as well as the α2δ-1 ligand gabapentin. Colocalized pre- (VGluT1) and postsynaptic (Homer) puncta reveal sites of excitatory synapses (yellow arrowheads). (C) Density of excitatory synapses shown as fold change compared with α2δ-1 Het GM only (n = 30 cells per condition; two independent experiments). (D) Mating strategy to generate four genotypes (G1–G4) needed to determine site of action of α2δ-1 for synaptogenesis. (E) Cortical neuron plating scheme for α2δ-1/GFP experiments. (F) GFP+ cortical neuron dendrites from α2δ-1 Het or KO mouse pups. Cells were treated with TSP2-containing or -deficient growth media. Colocalized pre- (Bassoon) and postsynaptic (Homer) puncta reveal sites of excitatory synapses (yellow arrowheads). (G) Density of excitatory synapses shown as fold change compared with α2δ-1 Het/Het GM only (n= 30 cells per condition; two independent experiments). One-way ANOVA with Tukey’s multiple comparisons post hoc test. Bars, 5 µm. Error bars represent SEM. *, P < 0.05; **, P < 0.001; ***, P < 0.0001.
Rac1 is required for induction of excitatory synaptogenesis and spinogenesis by TSP–α2δ-1
Previously, we hypothesized that the TSP–α2δ-1 interaction activates a synaptogenic signaling event in neurons (Risher and Eroglu, 2012), but the downstream components of this process were unknown. Our results from knocking out α2δ-1 cell autonomously showed reduced synaptic localization of NR1 (Fig. 4, F and G), and the global α2δ-1 KO had a significantly diminished NMDA/AMPA ratio (Fig. 2, H and I). Therefore, we next used the CRISPR/Cas9 gene silencing approach to test whether deletion of NR1 impairs TSP-induced synapse formation. Small guide RNAs (sgRNAs) targeting α2δ-1 (Cacna2d1) and NR1 (Grin1) were cloned into the pX601 vector expressing an HA-tagged SaCas9, with gene silencing verified with the T7E1 assay, sequencing, and protein expression (Fig. S4, A–D). Rat cortical neurons were purified, transfected with CRISPR constructs, and treated with TSP2 before immunostaining for pre- and postsynaptic markers (Fig. 6 A). As expected, CRISPR-mediated deletion of Cacna2d1 completely abolished TSP2-induced synapse formation. Deletion of Grin1 did likewise (Fig. 6, B and C), revealing a role for NMDAR-mediated signaling in TSP-induced synapse formation.
Our analysis of the synaptic connectivity in α2δ-1 KOs (Figs. 1, 2, and 3) showed that α2δ-1 regulates developmental processes in dendrites known to be highly dependent on the actin cytoskeleton (Ethell and Pasquale, 2005; Spence and Soderling, 2015), suggesting the activity of the Rho family of small GTPases. Among these GTPases, Ras-related C3 botulinum toxin substrate 1 (Rac1) and cell division control protein 42 (Cdc42) are involved in various stages of synaptic development including dendrite elaboration, filopodia formation, and spine maturation (Tashiro et al., 2000; Scott et al., 2003). Because these processes are all impaired in α2δ-1 KOs (Figs. 1, 2, and 3), we hypothesized that Rac1 and/or Cdc42 may be required downstream of TSP–α2δ-1. We used small hairpin RNAs (shRNAs) to diminish expression of Rac1 or Cdc42 in rat cortical neurons (Fig. S4, E and F). After verifying that these shRNAs are effective in down-regulating Rac1 or Cdc42 expression (Fig. S4, G and H), we transfected cortical neurons with either an shScr (scramble) control, shRac1, or shCdc42, followed by TSP2 treatment (Fig. 6 A). shRNA against Rac1 but not Cdc42 completely prevented the synaptogenic effect of TSP2 (Fig. 6, D and E). In spines, Rac1 is downstream of guanine nucleotide exchange factors (GEFs), which facilitate the GTP binding that converts Rac1 to its active state (Okamoto et al., 2009). We next used shRNA to determine whether the GEFs Kalirin-7, Tiam1, or β-Pix/Cool-1 participate in TSP-induced synapse formation (Fig. S4, E and F; verifications in Fig. S4, I–K). Interfering with the expression of either Kalirin-7 or β-Pix but not Tiam1 prevented TSP2-induced synaptogenesis (Fig. 6, F and G). Taken together, these results show that Rac1 and its upstream GEFs Kalirin-7 and β-Pix are required for TSP-induced synapse formation.
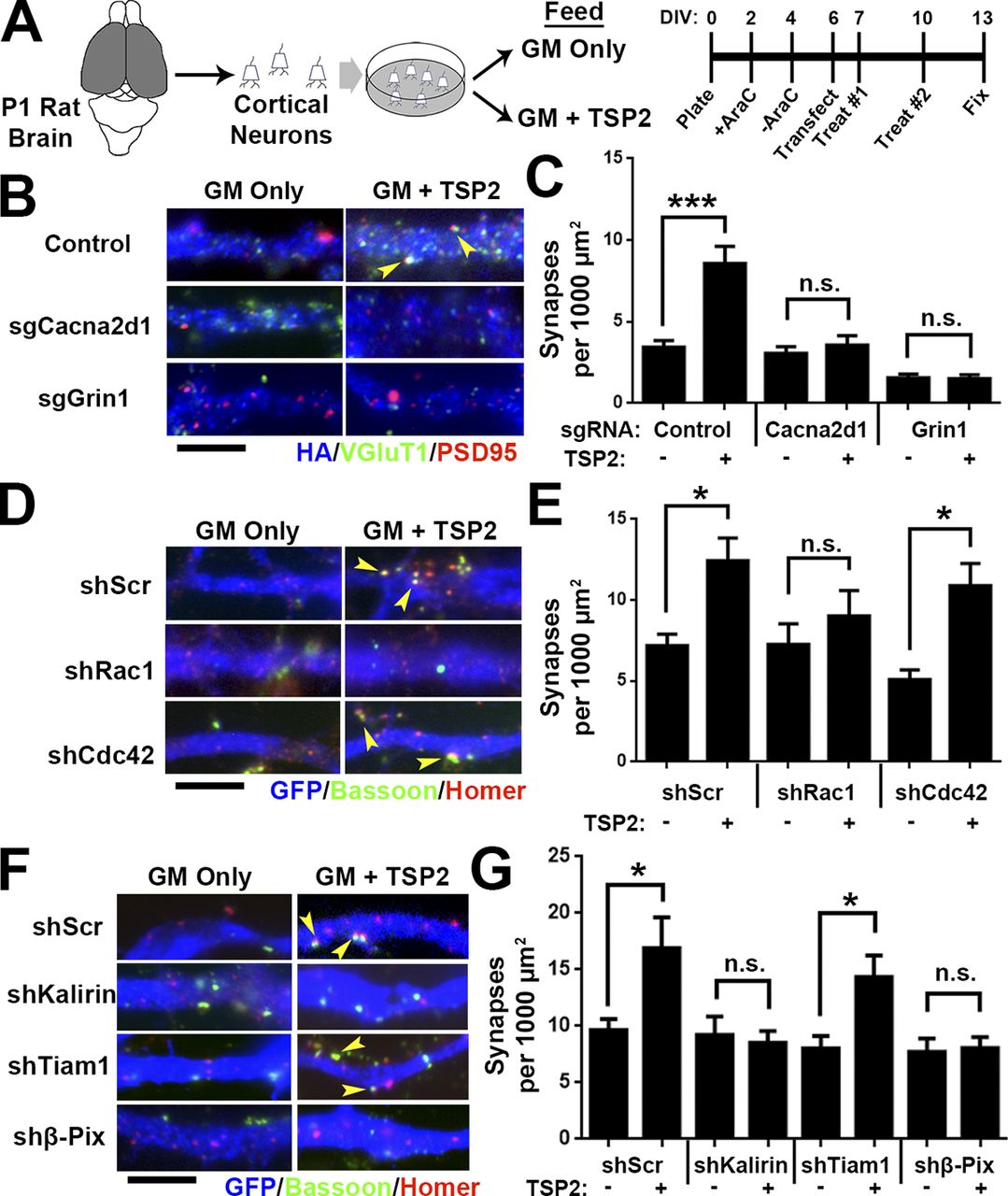
TSP-induced synapse formation requires Rho GTPase Rac1. (A) Rat cortical neuron purification and TSP2 treatment timeline. (B) Rat cortical neurons transfected with a pX601 vector containing saCas9 and sgRNA identified by HA expression. The pX601-only control (top) did not include an sgRNA sequence. Neurons were treated with TSP2-containing or deficient growth media. Colocalized pre- (VGluT1) and postsynaptic (PSD95) puncta reveal sites of excitatory synapses (yellow arrowheads). (C) Excitatory synapse density on HA+ neurons (n = 30 cells per condition; two independent experiments). (D) GFP+ rat cortical neuron dendrites. After transfection with an shRNA-expressing vector (with a scrambled shRNA sequence [shScr] as a control), neurons were treated with TSP2-containing or -deficient growth media. Colocalized pre (Bassoon) and postsynaptic (Homer) puncta reveal sites of excitatory synapses (yellow arrowheads). (E) Excitatory synapse density on GFP+ neurons (n = 30 cells per condition; two independent experiments). Error bars represent SEM. (F and G) Same scheme as D and E except using shRNA against the Rho GEFs Kalirin-7, Tiam1, and β-Pix. One-way ANOVA with Tukey’s multiple comparisons post hoc test. Bars, 5 µm. *, P < 0.05; ***, P < 0.0001.
We next asked whether postsynaptic deletion of Rac1 would result in synapse and spine defects similar to the conditional α2δ-1 KO neurons (Fig. 4) using the Rac1f/fmice for Cre-mediated deletion of Rac1 (Glogauer et al., 2003). However, we could not use the in utero electroporation (IUE) approach as we did with α2δ-1 (Fig. 4) because this would inactivate Rac1 in radial glia and newborn neurons and affect neuronal differentiation and migration (Chen et al., 2009; Shoval and Kalcheim, 2012). To circumvent this limitation, we used an ex vivo organotypic slice culture model to delete Rac1 sparsely in cortical slices at a later developmental stage (corresponding with P5) via biolistic transfection with Cre recombinase (Fig. 7 A). Conditional Rac1 deletion resulted in severely decreased spine and synapse density (Fig. 7, B–D) at 19 d ex vivo (DEV; corresponding with P21), reminiscent of the α2δ-1 cKO (Fig. 4, A and B). To test whether α2δ-1 and Rac1 work together to promote excitatory synapse and spine formation, we overexpressed α2δ-1 in Rac1f/f slices and observed significantly increased spine and excitatory synapse densities (Fig. 7, B–D). By contrast, in Rac1f/f neurons cotransfected with Cre, α2δ-1 overexpression had no synaptogenic effect (Fig. 7, B–D). These results show that Rac1 is required downstream of α2δ-1 to promote synaptogenesis and spinogenesis.
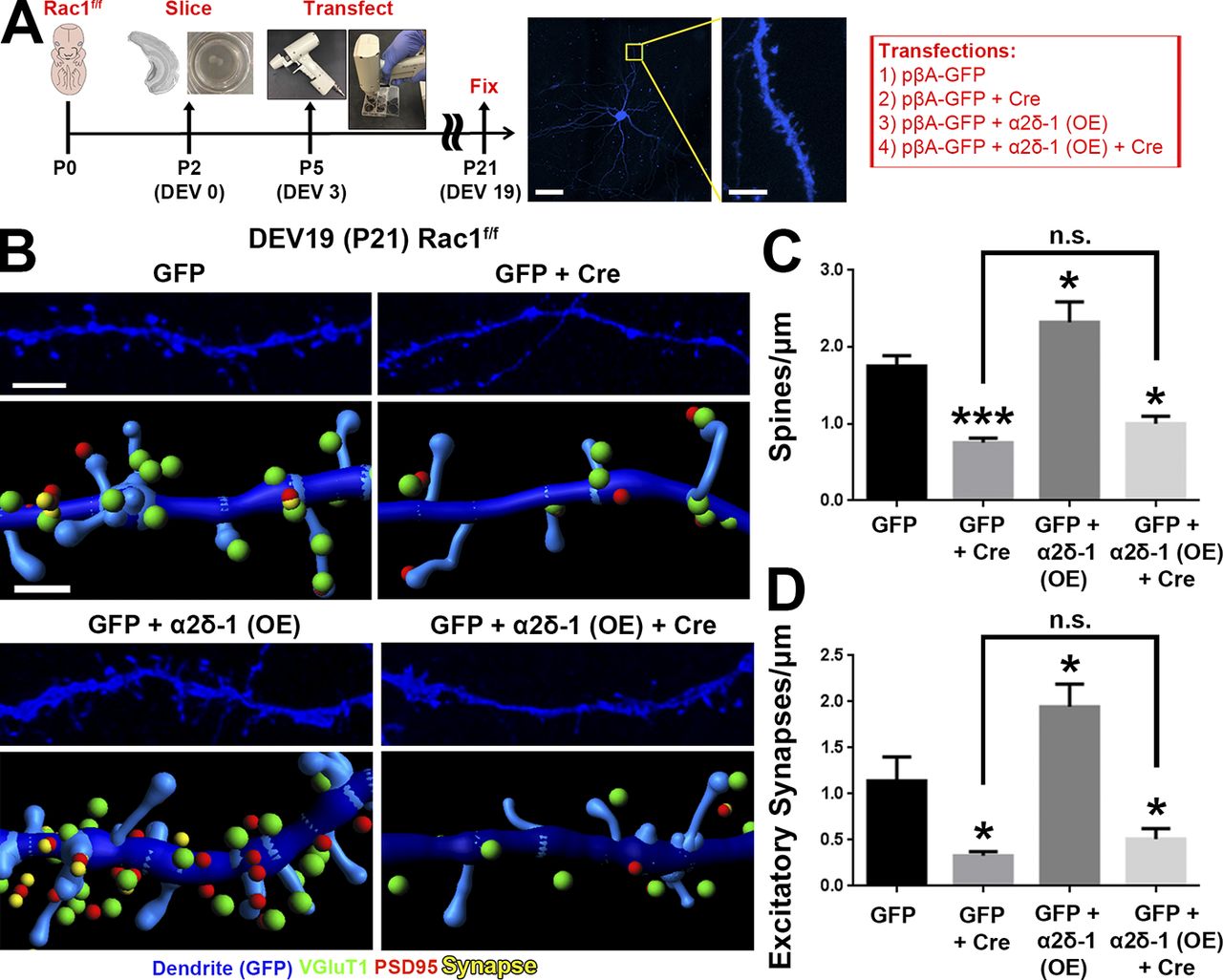
Rac1 promotes synaptic development and spinogenesis downstream of α2δ-1. (A) Schematic for organotypic slice culture/biolistic transfection. GFP+ dendrites (blue) are imaged at high magnification by confocal microscopy to capture spine morphology. (B) 3D confocal images (top) and Imaris reconstructions (bottom) of GFP+ dendrites in the S/Z of organotypic slices at DEV19 (i.e., P21) from Rac1f/f mice. Slices were transfected with cDNAs expressing GFP only or GFP plus Cre, α2δ-1 overexpression (OE), or α2δ-1 overexpression and Cre. Pre- (VGluT1), post- (PSD95), and colocalized (yellow) synaptic puncta in close proximity to the dendrite are shown. (C and D) Spine (C) and excitatory synapse (D) density from Rac1f/fdendrites (n = 12–18 dendrites per construct compiled from two independent experiments). One-way ANOVA with Dunnett’s multiple comparisons post hoc test (using GFP as control). Error bars represent SEM. Bars: (A, main) 50 µm; (A, inset) 5 µm; (B, top) 3 µm; (B, bottom) 1 µm. *, P < 0.05; ***, P < 0.0001.
Dendritic α2δ-1 bridges extracellular synaptogenic signals with intracellular Rac1 activation
Loss of α2δ-1 results in synapse and spine defects in a cell-autonomous manner (Fig. 4), raising the question whether restoring α2δ-1 expression can reverse this phenotype. To test this, we first confirmed that α2δ-1 KO dendrites in organotypic cortical slices developed significantly fewer spines and excitatory synapses than those from littermate α2δ-1 Het slices (Fig. 8, A and D). Biolistic introduction of α2δ-1 into KO neurons completely rescued spine and synapse density (Fig. 8, B–D), confirming the postsynaptic sufficiency of α2δ-1 for excitatory synapse formation and spinogenesis. To investigate the roles of the extracellular and intracellular regions of α2δ-1, we next tested the sufficiency of two α2δ-1 mutants for rescuing synaptic deficits. The first mutant contains a point mutation in the VWF-A domain of the CACNA2D1 gene of a human patient with autism (Iossifov et al., 2014), changing a highly-conserved arginine 351 residue to threonine (R351T; Fig. 8 B). We verified by heterologous expression in HEK293 cells that α2δ-1 R351T mutant protein expresses efficiently and migrates in SDS-PAGE at the same size as native α2δ-1 (Fig. S5, A and B). However, surface staining in HEK293 cells transfected with α2δ-1 R351T revealed that this mutant cannot reach the plasma membrane surface and/or cannot be stabilized there but is rather localized to a membranous organelle just beneath the outer plasma membrane (Fig. S5 C). Thus, this mutation precludes the ability of α2δ-1 to interact with extracellular ligands such as astrocyte-secreted TSPs or other secreted or cell surface molecules. In the second α2δ-1 mutant, we replaced the TM portion of α2δ-1 with a glycosylphosphotidylinositol (GPI) anchor to generate a protein (α2δ-1 ΔTM), which is effectively produced and transported to the cell surface yet incapable of interacting through its TM and cytoplasmic regions (Figs. 8 B and S5, A–C). When expressed in cortical neurons, the ΔTM mutant localized primarily to dendritic spines, whereas the full-length α2δ-1 was found both on spines and dendritic shafts; α2δ-1 R351T showed little dendritic expression (Fig. S5 D). Neither α2δ-1 mutant was capable of rescuing spinogenesis or synaptogenesis phenotypes observed in organotypic slices from the α2δ-1 KOs (Fig. 8, C and D). These results show that extracellular interactions of α2δ-1, such as its binding to TSP, must be coupled with critical interactions established with the TM and intracellular regions of α2δ-1 to control synapse and spine formation.
We next tested whether we could bypass the requirement for α2δ-1 for synaptogenesis by enhancing Rac1 activation. We used a fast-cycling Rac1 mutant (FC Rac1) that allows Rac1 to quickly cycle between its active and inactive states without the need of upstream activation via GEFs (Lin et al., 1999). Biolistic transfection of FC Rac1 rescued the spinogenesis phenotype in α2δ-1 KO cortical slices, but these spines did not fully recover excitatory contacts (Fig. 8, E and F). We speculated that the inability of Rac1 activation to rescue excitatory synapses was caused by the lack of critical interactions that α2δ-1 establishes through its extracellular regions. To test this, we coexpressed the α2δ-1 ΔTM mutant with FC Rac1 and fully restored both spine and synapse numbers in α2δ-1 KO dendrites (Fig. 8, E and F), indicating that extracellular interactions established by α2δ-1 coupled with Rac1 activation are necessary and sufficient for excitatory synapse formation. Collectively, these results show that the binding of astrocytic TSP to neuronal α2δ-1 triggers the activation of Rac1, promoting synapse formation and maturation in the developing cortex.
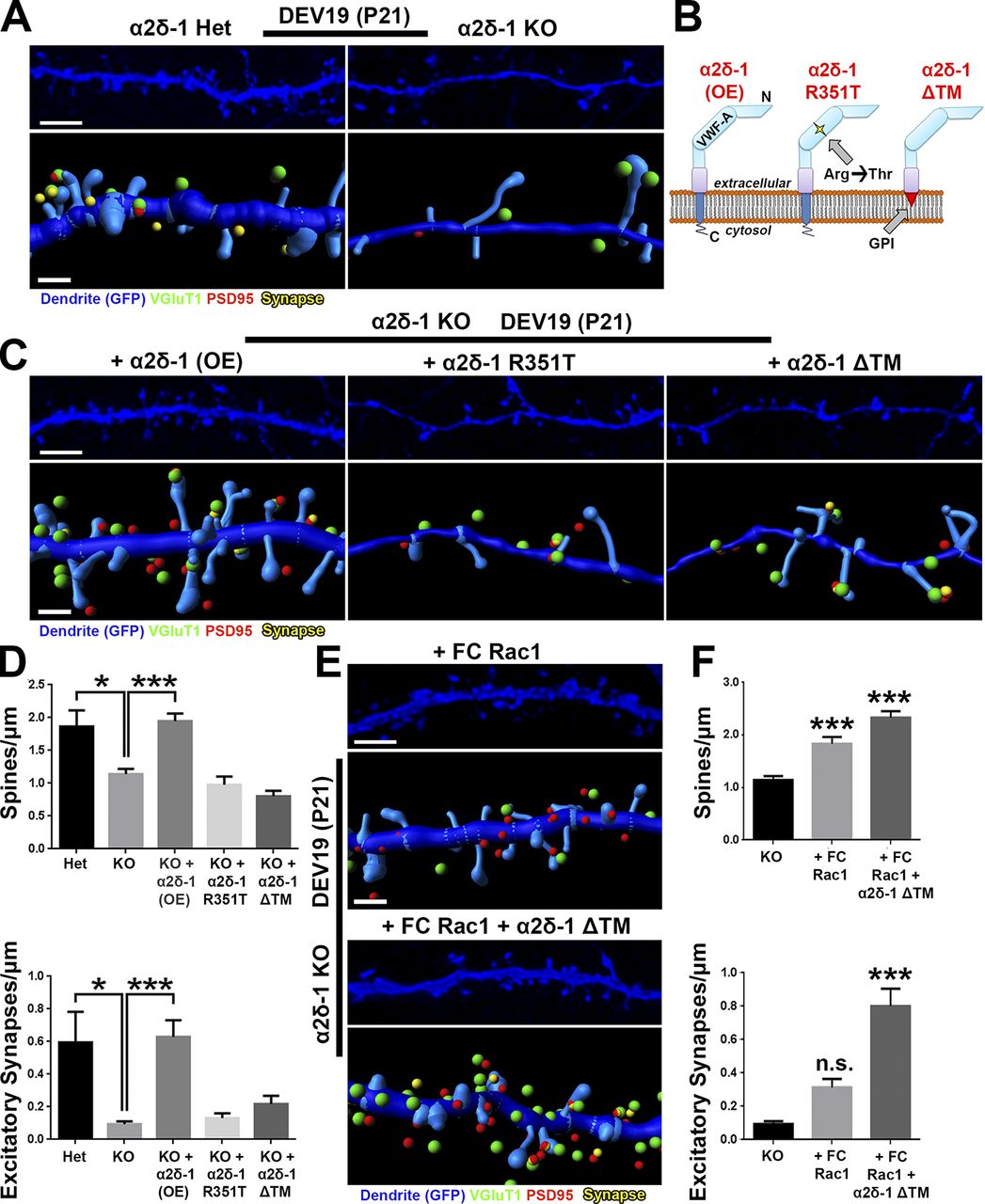
α2δ-1 and Rac1 work in concert to rescue synapses and spines in α2δ-1–null cortex. (A) 3D confocal images (top) and Imaris reconstructions (bottom) of GFP+ dendrites in the S/Z of DEV19 (i.e., P21) organotypic slices from α2δ-1 Het or KO mice. Slices were transfected with GFP to visualize dendritic morphology. Pre- (VGluT1), post- (PSD95), and colocalized (yellow) synaptic puncta in close proximity to the dendrite are shown. (B) α2δ-1–variant constructs used in the rescue experiment. (C) Same format as A but with all images taken from α2δ-1 KO mice. Slices were transfected with GFP plus either α2δ-1 (overexpression construct; OE), α2δ-1 R351T, or α2δ-1 ΔTM. (D) Spine (top) and excitatory synapse (bottom) density from α2δ-1 Het or KO dendrites. n = 12–24 dendrites per construct compiled from two independent experiments. (E) Same format as C, but slices were transfected with either fast-cycling (FC) Rac1 or FC Rac1 plus α2δ-1 ΔTM. (F) Spine (top) and excitatory synapse (bottom) density from α2δ-1 KO dendrites. n = 12–24 dendrites per construct compiled from two independent experiments. One-way ANOVA with Dunnett’s multiple comparisons post hoc test (using KO as control). Error bars represent SEM. Bars: (A, C, and E, top) 3 µm; (A, C, and E, bottom) 1 µm. *, P < 0.05; ***, P < 0.0001.
Discussion
Most excitatory synapses in the cortex are made onto dendritic spines, which undergo a complex morphological transformation that occurs in parallel with synaptic development (Harris and Kater, 1994). During this transition, immature dendritic filopodia, which are proposed to seek out appropriate axonal partners, give way to dendritic spines. Dendritic spines are more stable protrusions, containing an abundance of synaptic scaffolding proteins, neurotransmitter receptors, and an elaborate actin cytoskeleton (Grutzendler et al., 2002; Korobova and Svitkina, 2010). In this study, we show that α2δ-1, the neuronal receptor for astrocyte-secreted TSPs, is critical for the formation and maturation of cortical spine synapses. We found that these functions of α2δ-1 are cell autonomous to neurons, requiring the postsynaptic expression of α2δ-1.
Establishment of cortical connectivity by α2δ-1
In the cortical S/Z, glutamatergic excitatory synapses fall under two main categories, intracortical and thalamocortical (Nakamura et al., 2005). Previously we found that astrocyte-secreted hevin controls the formation and maturation of thalamocortical synapses by bridging presynaptic neurexin-1α and postsynaptic neuroligin-1 (Risher et al., 2014; Singh et al., 2016). In this study, we show that α2δ-1, the receptor for astrocyte-secreted synaptogenic TSPs, plays a predominant role in the formation of intracortical synapses and their localization onto dendritic spines. In agreement with our findings, TSP1/2 double null mice were shown to have a ∼30% reduction in their excitatory cortical synapses, determined by the colocalization of presynaptic protein bassoon and postsynaptic SAP102 (Christopherson et al., 2005). In the same study, the numbers of SV2+ axonal terminals were found to be significantly reduced in TSP1/2 KO mice as early as P8, whereas we found no significant changes in the numbers of excitatory synapses at ages earlier than P21. These results may indicate distinct roles of TSPs in axonal branching and elaboration versus their roles in synaptogenesis through signaling via postsynaptic α2δ-1.
Although α2δ-1 is not required for thalamocortical synaptogenesis, we previously found that overexpression of α2δ-1 under the Thy1 promoter in transgenic mice in a subset of cortical neurons dramatically increases VGluT2+ thalamocortical synapses at P21 (Eroglu et al., 2009). Moreover, in RGC cultures, TSPs induce the formation of excitatory synapses between these neurons, which exclusively express VGluT2 (Fujiyama et al., 2003; Christopherson et al., 2005). Taken together with our results, these findings indicate that an increase in α2δ-1 abundance and its subsequent activation by TSPs is sufficient to increase the overall excitatory synapse numbers regardless of the presynaptic axonal partner identity.
Mechanisms of TSP–α2δ-1–induced synapse and spine growth
Based on our findings, we propose in this study a two-step model for the synaptogenic capabilities of α2δ-1 (Fig. 9): (1) TSP binding to the extracellular domain of postsynaptic α2δ-1 on probing filopodia facilitates clustering of pre- and postsynaptic proteins to nascent synaptic sites, bringing these compartments in close apposition; and (2) the TM region and the C-terminal tail of α2δ-1 trigger intracellular signaling events such as the recruitment of GEFs Kalirin-7 and β-Pix to nascent synaptic sites within filopodia, inducing activation of Rac1 and reorganization of the actin cytoskeleton to promote spine stabilization and growth.
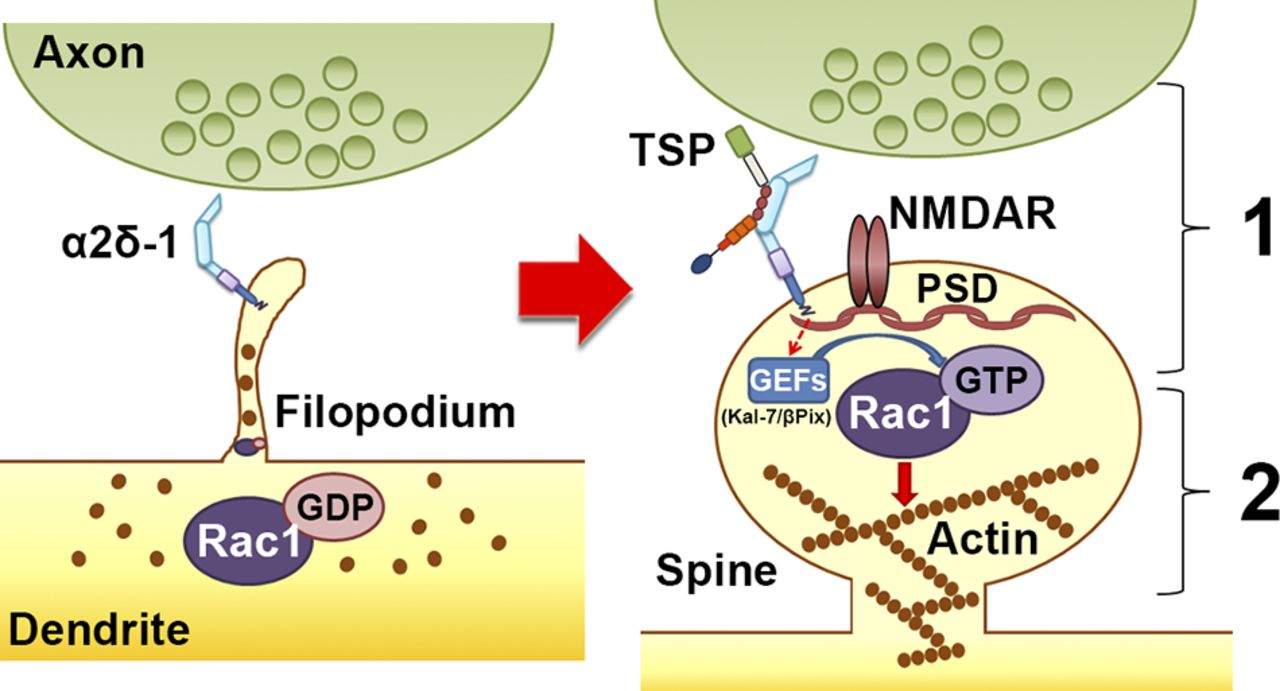
Model for α2δ-1’s dual role in promoting synapse and spine development. Early on, α2δ-1 is present on filopodia seeking contact with axonal partners. Rac1 is predominantly bound to GDP, rendering it inactive. After TSP binding, (1) α2δ-1 at the postsynaptic surface brings together pre- and postsynaptic components to form synapses; and (2) the C terminus of α2δ-1 triggers intracellular signaling via GEFs to stimulate GTP binding to Rac1, promoting actin reorganization to facilitate spine maturation.
In this study, we showed that overexpression of α2δ-1 fails to increase spine or synapse density when Rac1 function is abolished. Intriguingly, we were able to bypass the need for α2δ-1 in spinogenesis by directly stimulating Rac1, but Rac1 activation alone was unable to cluster pre- and postsynaptic machinery in the absence of the α2δ-1 ECD. Similarly, α2δ-1 ECD by itself could not induce synapse formation even though it was capable of localizing to dendritic spines. These results show that for synapse-containing spines to emerge, Rac1 activation should coincide with axon–dendrite matching and adhesion events. By virtue of its extracellular TSP-binding domain, its position within the plasma membrane, and its short C-terminal cytoplasmic tail, α2δ-1 is ideally situated to coordinate the occurrence of synaptogenesis and spinogenesis by recruiting important components of the synaptogenic signaling complex.
What comprises this synaptogenic signaling complex? We previously found that blocking or overexpressing the α1-subunits of the postsynaptic L-type calcium channels Cav1.2 and Cav1.3, the canonical interaction partners for α2δ-1 (Cantí et al., 2005), did not affect TSP-induced synaptogenesis (Eroglu et al., 2009). In this study, we instead show that disruption of the obligatory NMDA receptor subunit, NR1, abolishes the synaptogenic ability of TSP. We also observed cell autonomous loss of synaptic NR1 in the α2δ-1 cKO dendrites as well as a decreased NMDA/AMPA ratio in the α2δ-1 global KOs. These results are in line with previous findings that TSP induces the formation of silent synapses containing NMDA but not AMPA receptors (Christopherson et al., 2005). The binding of TSP to α2δ-1 may serve as an initiating event to recruit and stabilize NMDA receptors on the postsynaptic surface, which then coordinates the downstream signaling events that lead to spinogenesis. α2δ-1 may recruit NMDA receptors (NMDARs) to the synapses by a direct interaction through its C-terminal tail (Chen et al., 2018), which we found to be required for the synaptogenic activity of α2δ-1. Alternatively, TSP–α2δ-1 interaction could recruit NMDARs through an intermediary such as the neuroligin class of cell adhesion molecules, known interactors of NR1 (Budreck et al., 2013). Interestingly, TSPs have previously also been shown to interact with neuroligin family proteins (Xu et al., 2010). Furthermore, α2δ family members have recently been found to bind directly to neurexins, the primary presynaptic interaction partners of neuroligins (Tsetsenis et al., 2014; Tong et al., 2017). Possible cis-interactions between presynaptic α2δ-1 and neurexins would also provide an explanation for the enhanced TSP-induced synaptogenesis we observed when α2δ-1 was absent presynaptically. Presynaptic α2δ-1 may bind neurexins and prevent them from coupling transynaptically with neuroligins, which is important to stabilize newly formed synapses (Graf et al., 2004).
Although it is well established that dendritic protrusions undergo a maturational shift from highly motile filopodia to more stable spines, many of the molecular players underlying this transition have yet to be elucidated. The fact that both filopodia and spines are actin-rich structures means that cytoskeletal regulation is a critical mechanism underlying this morphological shift (Bilousova et al., 2006; Johnson and Ouimet, 2006). Small Rho GTPase proteins such as Rac1, RhoA, and Cdc42 differentially promote or inhibit cytoskeletal dynamics (Hall, 1998). Interestingly, TSP1 was previously shown to induce cytoskeletal reorganization in nonneuronal cells in a Rac1/Cdc42-dependent manner (Adams and Schwartz, 2000). One study showed that WT neurons cultured with TSP1-null astrocytes fail to develop mature spines (Garcia et al., 2010), indicative of problems with actin regulation. GEFs interact with NMDARs and other signaling molecules triggered by synaptic activity (Xie et al., 2007); in particular, Kalirin-7 interacts with NR1 at excitatory synapses and promotes spine enlargement via Rac1 (Xie et al., 2007; Ma et al., 2008), providing a potential link between α2δ-1 and Rac1 activation. Another GEF located in spines is β-Pix (Cool-1), which acts downstream of NMDAR to promote Rac1 activation (Saneyoshi et al., 2008). In this study, shRNA-facilitated knockdown of either Kalirin-7 or β-Pix was sufficient to abolish TSP-induced synaptogenesis in a similar manner to knocking down Rac1 function directly.
Contributions of α2δ-1 dysfunction to disease
α2δ-1 is linked to neurological disorders including epilepsy, neuropathic pain, intellectual disability, and autism spectrum disorder (Newton et al., 2001; Iossifov et al., 2014; Vergult et al., 2015). The antiepilepsy, antineuropathic pain drug gabapentin (Neurontin) binds to α2δ-1, and by doing so, it prevents TSP-induced synaptogenesis (Gee et al., 1996; Field et al., 2006; Eroglu et al., 2009). These findings had significant implications for the role of α2δ-1 in the development of maladaptive circuitry, likely by facilitating the wiring of inappropriate synaptic connections (Li et al., 2006, 2014). As we show in this study, the opposite situation is problematic as well, with too few synapses being formed and/or maintained as lack of α2δ-1 leads to a severe decrease in intracortical synapses. Reduced intracortical connectivity has been found in some patients with autism and was proposed to underlie the cognitive challenges faced by those individuals (Just et al., 2007). Although the mechanism of cortical synaptic impairment in those specific patients is unclear, we found that a previously identified autism-linked point mutation in α2δ-1 (Iossifov et al., 2014) diminished the effects of α2δ-1 on synaptic development and spine maturation. Our findings indicate that this single amino acid change in the extracellular VWF-A domain of α2δ-1 is sufficient to disrupt the localization of α2δ-1 to the cell surface, rendering it unavailable for binding TSP. Taken together, these findings underscore the importance of α2δ-1 for proper cortical development.
Our finding that Rac1 is downstream of TSP–α2δ-1 in mediating synapse and spine growth also provides a novel potential mechanistic insight into psychiatric illness and intellectual disability, where aberrant Rac1 signaling have been strongly implicated (Golden et al., 2013; Zeidán-Chuliá et al., 2013; Tejada-Simon, 2015). Rac1 impairments have been suggested as a mechanism underlying structural abnormalities in fragile X syndrome (Bongmba et al., 2011) as well as aberrant synaptic transmission in schizophrenia (Hayashi-Takagi et al., 2010). We have now elucidated a series of events in cortical development and synaptic growth wherein astrocytes can control neuronal Rac1 signaling via TSP–α2δ-1. Going forward, it will be essential to understand whether modulation of the TSP–α2δ-1–Rac1 pathway could provide a novel avenue for therapeutic approaches that aim to correct aberrant wiring of cortical circuits.