
Therapeutics in the Pipeline Targeting α-Synuclein for Parkinson’s Disease
By Hilary Grosso Jasutkar, Stephanie E. Oh, and M. Maral Mouradian
Excerpt from the article published in Pharmacological Reviews January 1, 2022, 74 (1) 207-237; DOI: https://doi.org/10.1124/pharmrev.120.000133
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disorder and the fastest growing neurologic disease in the world, yet no disease-modifying therapy is available for this disabling condition. Multiple lines of evidence implicate the protein α-synuclein (α-Syn) in the pathogenesis of PD, and as such, there is intense interest in targeting α-Syn for potential disease modification. α-Syn is also a key pathogenic protein in other synucleionpathies, most commonly dementia with Lewy bodies. Thus, therapeutics targeting this protein will have utility in these disorders as well. Here we discuss the various approaches that are being investigated to prevent and mitigate α-Syn toxicity in PD, including clearing its pathologic aggregates from the brain using immunization strategies, inhibiting its misfolding and aggregation, reducing its expression level, enhancing cellular clearance mechanisms, preventing its cell-to-cell transmission within the brain and perhaps from the periphery, and targeting other proteins associated with or implicated in PD that contribute to α-Syn toxicity. We also discuss the therapeutics in the pipeline that harness these strategies. Finally, we discuss the challenges and opportunities for the field in the discovery and development of therapeutics for disease modification in PD.
I. Introduction
I. A. The Link between α-Synuclein and Neurodegenerative Diseases
The protein α-synuclein (α-Syn) was first linked to Parkinson’s disease (PD) in 1997 when a missense mutation (A53T) was found in its encoding gene (SNCA) (also known as PARK1) in a large Italian family known as the Contursi kindred and in three unrelated families of Greek origin with autosomal dominant early onset PD (Polymeropoulos et al., 1997). Soon after, α-Syn was shown by immunostaining to be a major component of Lewy bodies (LBs), the intraneuronal cytoplasmic inclusions that have long been considered the hallmark pathologic feature of PD, and in dystrophic processes known as Lewy neurites (Spillantini et al., 1997). Notably, this postmortem evidence of α-Syn aggregates was detected in the substantia nigra of individuals with nonfamilial PD and in the cingulate cortex and substantia nigra of individuals with dementia with Lewy bodies (DLB), another neurodegenerative disorder among what are now known as synucleinopathies, indicating the critical link between α-Syn and neurodegenerative diseases beyond those with rare genetic mutations. In fact, pathologic α-Syn aggregates are present in the vast majority of individuals with sporadic PD (Goedert et al., 2017; Riederer et al., 2019). Since those initial groundbreaking discoveries in the late 1990s, several additional missense mutations (Fig. 1) (Polymeropoulos et al., 1997; Kruger et al., 1998; Zarranz et al., 2004; Markopoulou et al., 2008; Appel-Cresswell et al., 2013; Kiely et al., 2013; Lesage et al., 2013; Pasanen et al., 2014) as well as multiplication of the SNCA gene locus have been linked to early-onset, autosomal dominant PD and dementia (Singleton et al., 2003; Chartier-Harlin et al., 2004; Farrer et al., 2004; Ibanez et al., 2004; Nishioka et al., 2006; Ahn et al., 2008; Ikeuchi et al., 2008; Obi et al., 2008; Uchiyama et al., 2008; Ibanez et al., 2009; Nishioka et al., 2009; Sironi et al., 2010). Interestingly, duplication and triplication of SNCA exhibit gene dosage effect, with each extra copy of the SNCA gene reducing the onset age of the clinical disease by about a decade (Trinh et al., 2018), and the resulting increase in α-Syn protein levels correlates directly with the severity of Lewy pathology and PD symptoms (Singleton et al., 2003; Kasten and Klein, 2013; Trinh et al., 2018). Additionally, reduced epigenetic silencing of SNCA has been detected in the brains of patients with sporadic PD (Jowaed et al., 2010), suggesting, again, that the level of α-Syn expression is an important determinant of PD pathogenesis. Furthermore, numerous single-nucleotide polymorphisms in SNCA have been identified as PD susceptibility variants in several independent genome-wide association studies and meta-analyses (Simon-Sanchez et al., 2009; Houlden and Singleton, 2012; Nalls et al., 2014; Blauwendraat et al., 2019). This demonstrates that common variants of SNCA and its noncoding regulatory regions that control its expression may influence the development of sporadic PD, strengthening the link between the disease and α-Syn (Fuchs et al., 2008; Campelo and Silva, 2017). Consistent with this, α-Syn levels are altered in the cerebrospinal fluid (CSF) of patients with PD, such that total α-Syn is decreased, but oligomeric, phosphorylated, and aggregated forms are increased (Parnetti et al., 2019).
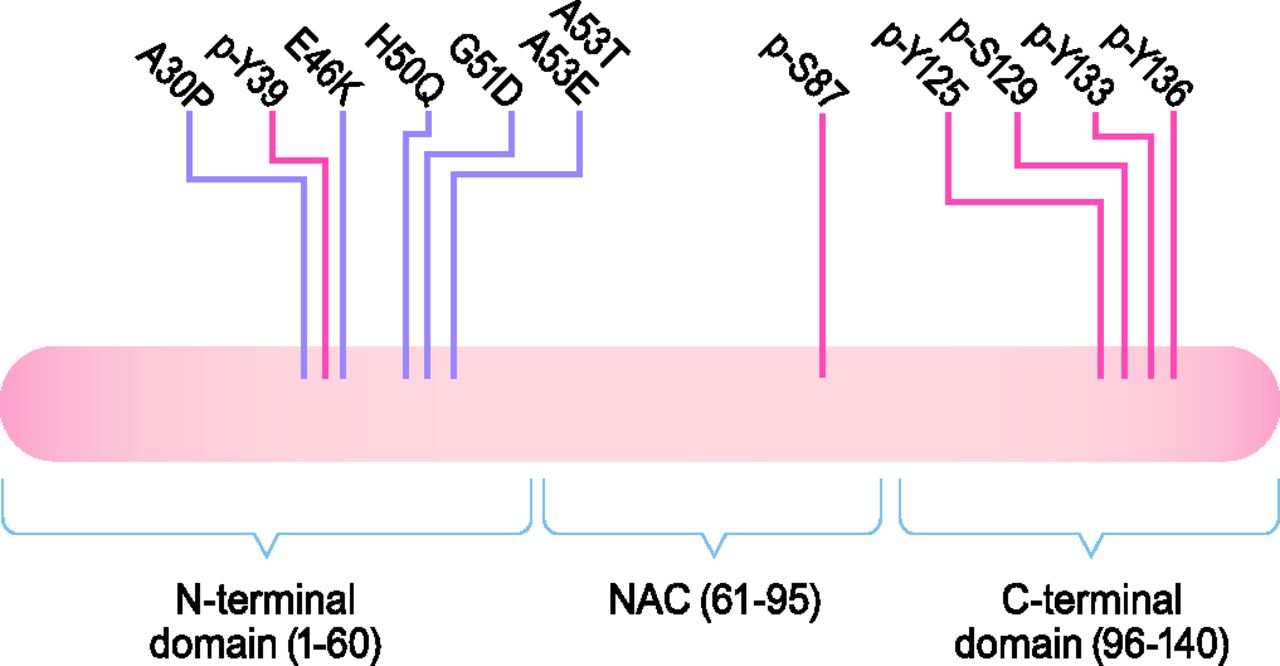
The structure of α-Syn. α-Syn is a 140-amino-acid (aa) protein composed of three domains: an N-terminal domain (aa 1–60), the NAC domain (aa 61–95), and a C-terminal domain (aa 96–140). There is a sequence of 11 residues that are repeated seven times found within the NAC and N-terminal domains. These repeats take on an amphipathic α helix secondary structure upon binding to lipids, which mediates the interaction of α-Syn with lipid membranes. All known disease-associated mutations are found in the N-terminal domain. There is also a confluence of glutamate residues and residues that are targets for phosphorylation in the C-terminal domain, which gives this domain a negative charge and may affect the propensity of α-Syn to misfold. Disease-associated mutations are marked in purple, and phosphorylation sites are marked in red.
In addition to PD and DLB, aggregates of misfolded α-Syn are also seen in multiple system atrophy (MSA), another synucleinopathy (Goedert et al., 2017). DLB and PD exhibit overlapping pathology, with α-Syn aggregates within the neuronal cytoplasm (Lewy bodies) and neuronal processes (Lewy neurites), albeit in different brain regions (Goedert et al., 2013). However, the clinical manifestations and progression differ, whereby cognitive symptoms develop in later stages in PD, whereas motor symptoms develop either simultaneously with or within a year of the cognitive decline in DLB (Irwin et al., 2017; McKeith et al., 2017). On the other hand, α-Syn deposits in MSA occur in glial cytoplasmic inclusions with relatively little deposition in neurons, indicating a process distinct from PD pathogenesis but linked through α-Syn in a neuropathological spectrum. In MSA, the disease is marked by atypical parkinsonism, autonomic dysfunction, and cerebellar abnormalities (Gilman et al., 2008; Peeraully, 2014).
α-Syn neuronal inclusions and alterations in CSF α-Syn levels are also found in several other neurodegenerative disorders, including Alzheimer disease (AD) (Twohig and Nielsen, 2019). A substantial number of patients with AD harbor Lewy bodies alongside amyloid plaques and neurofibrillary tau deposits, although they are more often restricted to the amygdala than in PD, suggesting that α-Syn may play a role in the pathophysiology of AD in a subset of patients (Lippa et al., 1998; Hamilton, 2000; Toledo et al., 2016).
Because of the strong genetic and pathologic links between α-Syn and neurodegenerative disease, it is considered a major therapeutic target for disease modification. Accordingly, elucidating its structure and biologic properties has been helpful to formulate therapeutic strategies to mitigate its toxicity.
I. B. α-Syn Structure
α-Syn is a small, 140-amino-acid protein containing three domains: N-terminal domain (1–60), central nonamyloid component (NAC) domain (61–95), and C-terminal domain (96–140) (Fig. 1) (Bendor et al., 2013). All known point mutations in SNCA associated with PD are in the N-terminal domain, which is predicted to form an amphipathic α-helix; has a lysine-rich, highly conserved motif similar to lipid-binding motifs; and is responsible for the ability of α-Syn to associate with vesicles and membranes (Rochet et al., 2012; Bendor et al., 2013). The central NAC domain of α-Syn is responsible for its propensity to misfold into β-sheet–rich amyloid aggregates (Spillantini et al., 1997; Giasson et al., 2001). The polar C-terminal domain contains a number of negatively charged residues as well as a serine and three tyrosine residues that can be phosphorylated, which may impact α-Syn structure, membrane binding, aggregation, and toxicity (Uéda et al., 1993; Burre et al., 2018).
The native monomeric form of α-Syn is intrinsically disordered in solution (Weinreb et al., 1996; Bertoncini et al., 2005; Fauvet et al., 2012). However, it polymerizes into oligomers that then progress to form higher-order species and fibrils (Karpowicz et al., 2019; Gracia et al., 2020) that accumulate in Lewy bodies and Lewy neurites (Spillantini et al., 1997). Although native α-Syn may serve to maintain presynaptic function and regulation of transmitter release (Burre et al., 2018), misfolded α-Syn oligomers and aggregates have been shown to impair neuronal homeostasis through several mechanisms, including by causing oxidative and endoplasmic reticulum stress, vesicular trafficking dysfunction, autophagy-lysosomal pathway dysfunction, and mitochondrial dysfunction (Dehay et al., 2015).
I. C. α-Syn Function
The normal physiologic function of α-Syn is yet to be fully elucidated, but it is enriched in presynaptic compartments where it can associate with vesicles and membranes (Iwai et al., 1995; Kahle et al., 2000). In addition to its direct interaction with synaptic vesicles at presynaptic neuronal terminals, α-Syn interacts with synaptic proteins, such as phospholipase D2 (Payton et al., 2004) and Rab Small GTPases (Dalfo and Ferrer, 2005).
A number of studies in cellular and animal models that examined synaptic homeostasis after overexpressing or decreasing α-Syn levels have implicated it in regulating neurotransmitter release, synaptic activity, and plasticity (Lashuel et al., 2013). Although SNCA knockout animals are viable, they exhibit impairments in the trafficking of synaptic vesicles (Cabin et al., 2002) similar to overexpression models (Nemani et al., 2010; Lundblad et al., 2012).
I. D. α-Syn Seeding, Aggregation, and Propagation: How the Spread of Lewy Pathology May Explain Clinical Progression of PD
Systematic neuropathological studies of α-Syn aggregates in PD brains have suggested a pattern of progression and spread of pathology that corresponds with the clinical progression of disease. The Braak staging system (Braak and Braak, 2000) supports the notion that in the early stages of PD, deposition of α-Syn is seen in the anterior olfactory nucleus, the dorsal motor nucleus of the vagus, peripheral autonomic ganglia, and unmyelinated lamina-1 spinal-cord neurons. Over time, these aggregates spread to the brainstem, cortex, and limbic areas (Braak et al., 2003, 2007). Increasing evidence also points to α-Syn deposition in the gut and the possibility of transmission from the peripheral nervous system (PNS) to the central nervous system (CNS), a paradigm that aligns with the temporal sequence of symptom development in PD. Although the brain region prominently affected by the neurodegenerative process in PD and that accounts for much of the classic motor manifestations of the disease is the substantia nigra pars compacta, where dopaminergic neurons become depleted (Massano and Bhatia, 2012), many patients experience a host of nonmotor symptoms that appear years before the onset of motor symptoms. These nonmotor manifestations include REM sleep behavior disorder, hyposmia, constipation, and anxiety and depression, reflecting areas of the PNS and CNS found impacted by α-Syn deposition, such as the olfactory cortex and myenteric plexus (Savica et al., 2010; Postuma et al., 2012). Thus, it is presumed that therapeutic interventions during the prodromal period may prevent or delay the progression of PD to disabling motor symptoms and eventually cognitive impairment (Kalia and Lang, 2015). The spreading pathology from the periphery to the brain also opens up the possibility for therapeutics that might act in the periphery to prevent or slow down the propagation to the brain and emergence of Parkinsonian motor symptoms.
In addition to histopathological studies that suggest the spread of α-Syn pathology through different PNS and CNS regions (Braak et al., 2003; Parkkinen et al., 2008), early studies detecting α-Syn in CSF and plasma suggested the ability of these pathogenic α-Syn species to be released from neurons (El-Agnaf et al., 2003), although this study could not rule out the possibility that this α-Syn may originate from outside the CNS, such as from blood cells. But evidence for cell-to-cell transmission in the human brain was first detected in 2008, when healthy human embryonic mesencephalic grafts that were transplanted into PD patients’ striatum later exhibited α-Syn–positive LB-like pathology in postmortem studies from 4 to 24 years post-transplant (Kordower et al., 2008; Li et al., 2008; Mendez et al., 2008; Li et al., 2016). This host-to-graft propagation of α-Syn pathology was validated in vitro and in vivo in α-Syn transgenic mice engrafted with mouse cortical neuronal stem cells soon after (Desplats et al., 2009). The mechanism underlying α-Syn cell to cell transmission is still under investigation. It has been suggested that synaptic activity, particularly synaptic vesicle exocytosis, results in the release of α-Syn from neurons (Yamada and Iwatsubo, 2018). Binding of extracellular α-Syn to the transmembrane protein lymphocyte-activation gene 3 (LAG3) (Mao et al., 2016) or to complex N-linked glycans, particularly neurexin 1 (Birol et al., 2019), has been implicated in mediating the uptake of extracellular α-Syn into receiving neurons.
From these observations, a transmission hypothesis for synucleinopathies has emerged involving pathologic α-Syn seeding and nucleation, cell-to-cell transmission, and subsequent amplification and aggregation of toxic protein species in a prion-like manner (Peng et al., 2020). Multiple studies have shown that α-Syn pathology can be generated and propagated in in vitro and in vivo models through the introduction of pathologic α-Syn seeds in the form of preformed fibrils (PFF) generated from recombinant α-Syn monomers (Luk et al., 2012), α-Syn derived from brain homogenates of transgenic mice or human disease-affected brains (Masuda-Suzukake et al., 2013; Recasens et al., 2014), or α-Syn aggregates produced via viral vector-mediated overexpression of wild-type α-Syn (Ulusoy et al., 2013).
One factor complicating our understanding of the cell-to-cell transmission of α-Syn is that it is unclear which forms of α-Syn are pathogenic. It has been suggested that there may be distinct “strains” of α-Syn fibrils that may be particularly pathogenic and more likely to spread (Uemura et al., 2020). However, the toxic species of α-Syn capable of seeding and acting as prion-like templates for endogenous α-Syn may actually be soluble, prefibrillar intermediates (oligomers or protofibrils) (Danzer et al., 2007; Karpinar et al., 2009; Winner et al., 2011; Cremades et al., 2012) rather than the amyloid-like, insoluble fibrils. Experimental evidence has also suggested that LBs may be protective and present a form of aggresomes (Conway et al., 2000; Tanaka et al., 2004). Evidence for an alternative view has been developed as well, suggesting that LB formation itself can be a major driver of neurodegeneration involving a complex interplay between α-Syn fibrillization and post-translational modifications as well as interactions with mitochondria, autophagosomes, and endolysosomes (Mahul-Mellier et al., 2020).
Insights into factors responsible for the formation of toxic α-Syn species and the demonstration of cell-to-cell transmission have opened the way to a wide range of therapeutic approaches to slow down or stop disease progression. Broadly, these approaches involve 1) immunization, 2) reducing α-Syn expression, 3) inhibiting intracellular α-Syn aggregation, 4) enhancing α-Syn clearance and degradation, 5) targeting other genes/proteins that influence α-Syn aggregation or processing, and 6) decreasing synaptic activity (Fig. 2).
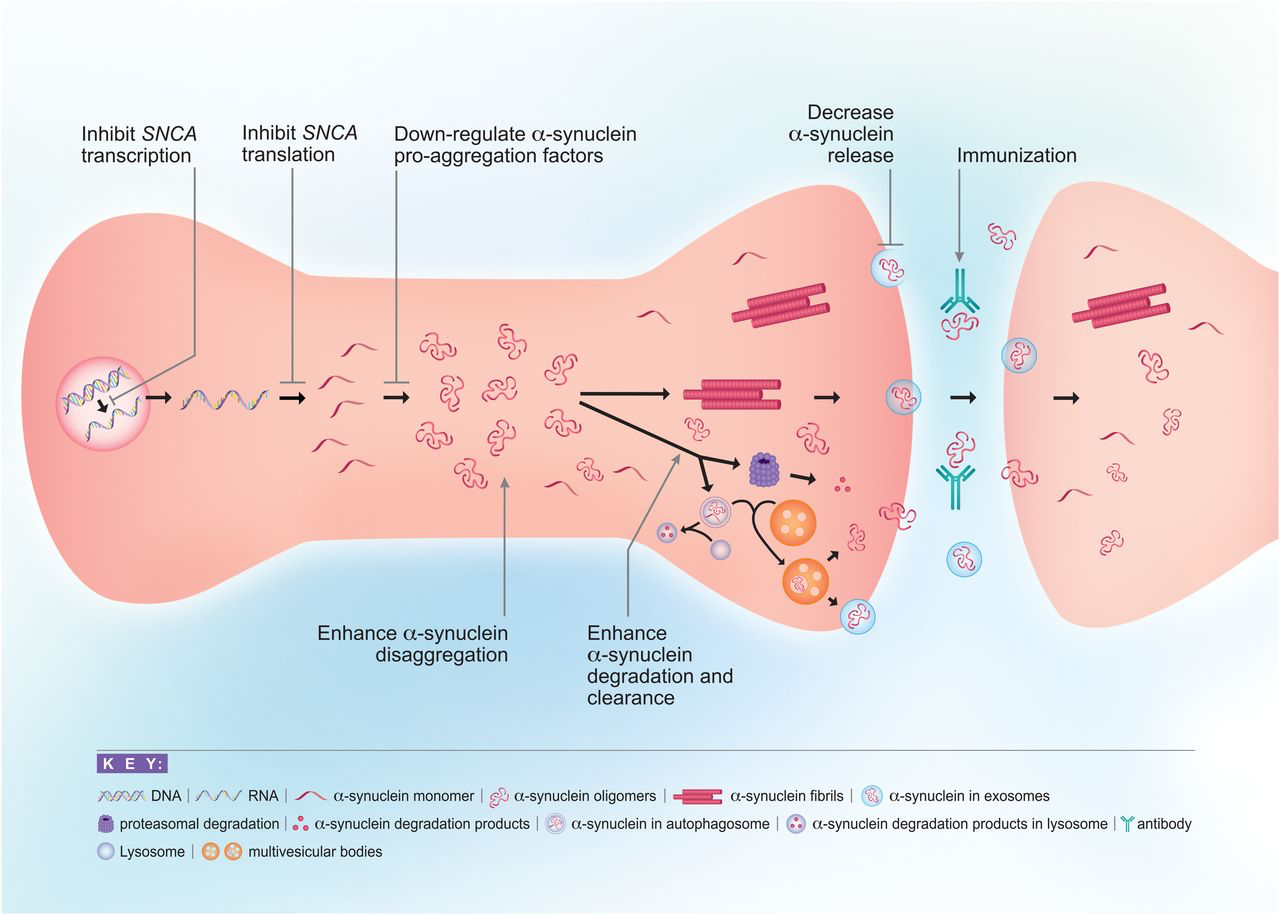
Approaches to reducing α-Syn toxicity. There are multiple points along the life cycle of α-Syn at which therapeutics may be used to intervene with its toxicity. These include reducing α-Syn transcription or translation, inhibiting α-Syn aggregation through the use of disaggregators, or interfering with proaggregation factors, enhancing α-Syn clearance and degradation through autophagy or the ubiquitin-proteasome system, decreasing release of α-Syn into the extracellular space, and targeting pathologic α-Syn through immunization. Another approach is to target other genes/proteins that influence α-Syn aggregation or processing, such as GBA, LRRK2, and PARP-1 (not pictured).
II. Immunization
A major therapeutic approach being pursued against α-Syn is immunization (Table 1). Mechanistically, antibodies are believed to bind α-Syn aggregates extracellularly during transmission, but with technological advances to facilitate intracellular trafficking of antibodies, immunization has the added benefit that it may also be able to clear intracellular α-Syn aggregates (Bhatt et al., 2013; Spencer et al., 2016; Messer and Butler, 2020). Immunogenic epitopes within the C- and N-termini of α-Syn have been well characterized, particularly within amino acids 89–140. Therapeutics based on active immunization using these epitopes and passive immunization using monoclonal antibodies against these epitopes are being developed and tested in clinical trials (Valera et al., 2016; Wang et al., 2019; Kwon et al., 2020). Both passive and active immunization have shown neuroprotection in transgenic mouse models of PD, DLB, and MSA (Masliah et al., 2005, 2011; Bae et al., 2012; Games et al., 2014; Mandler et al., 2014, 2015; Tran et al., 2014; Spencer et al., 2017; Henderson et al., 2020; Lemos et al., 2020). Together, these studies demonstrate that immunization approaches targeting various α-Syn epitopes can lead to reduced accumulation of α-Syn oligomers; diminished neurodegeneration, including in tyrosine hydroxylase (TH)-positive dopaminergic neurons; and improvements in the motor phenotype of various mouse models of α-synucleinopathies. Some concerns regarding immunization have been raised. For example, the animal models in which they have been studied are typically transgenic mice with human α-Syn overexpressed in the brain, which may not be fully representative of human disease. There is also the concern that antibodies against α-Syn could have other off-target effects, such as triggering nonselective autophagy (Masliah et al., 2011). Furthermore, given that the physiologic function of α-Syn is still not fully understood, the potential detrimental effects of targeting normal α-Syn are difficult to predict. In addition, there are logistical challenges related to antibodies that are either made or introduced in the periphery crossing the blood-brain barrier (BBB), although recent evidence suggests that the brain may not be as immune-privileged as once thought (Wang et al., 2019), which would somewhat mitigate this challenge. Despite these concerns, both active and passive immunization against α-Syn are being tested in clinical trials.
II. A. Active Immunization
Clinical trials using α-Syn antigens include the vaccines AFFITOPE PD01A, AFFITOPE PD03A, and UB312. AFFITOPE PD01A is a synthetic peptide based on C-terminal amino acids 110–130 of α-Syn (Mandler et al., 2014). Mice treated with this immunogen produced antibodies that reacted with both C terminus truncated and full-length α-Syn and both oligomeric and monomeric forms and had a preference for α-Syn as opposed to β-Syn. Furthermore, mThy1-α-syn transgenic mice treated with AFFITOPE PD01A showed reduced accumulation of α-Syn in the substantia nigra and TH-positive fibers in the striatum and protection against TH fiber loss and motor deficits as measured by the body suspension test (Mandler et al., 2014). Platelet-derived growth factor (PDGF)–α-Syn transgenic mice treated with AFFITOPE PD01A also showed reduced α-Syn aggregation, protection against synapse loss, decreased inflammation, and protection against learning and memory deficits. A follow-up study of AFFITOPE PD01A in the MBP–α-Syn mouse model of MSA showed similar protection against α-Syn aggregation and TH fiber loss as well as reduced demyelination and motor protection as measured by errors on the round-beam test (Mandler et al., 2015). Based on these preclinical findings, multiple phase I clinical trials of AFFITOPE PD01A in both PD and MSA patients have been completed (NCT02618941, NCT02216188, NCT02270489, NCT01568099, NCT01885494). Dosing regimens investigated ranged from a single dose to four doses administered every 4 weeks of 15 ug or 75 ug and in some studies including a booster immunization 36 weeks after the first injection. These dosing regimens resulted in a measurable humoral immune response and demonstrated good safety and tolerability, with the most common adverse events reported being injection-site reaction, fatigue, and headache (Meissner et al., 2020; Volc et al., 2020). Phase 2 trials are intended, but as of this writing, they have not been registered on clinicaltrials.gov.
A second AFFITOPE antibody, AFFITOPE PD03A, which was designed using the same technology used to develop AFFITOPE PD01A, has also reached the clinical-trial stage in patients with early PD and with MSA (NCT02267434 and NCT02270489). A single phase 1 trial in each of the PD and MSA patients has been completed. In the PD trial, patients received a low- (15 ug) or high- (75 ug) dose injection once per week for 4 weeks followed by a booster immunization 36 weeks after the first injection. The dosing paradigm in the trial for patients with MSA was the same but lacked the low dose arm. Results have only been published for the MSA trial but, as with AFFITOPE PD01A, showed a measurable immune response and good safety and tolerability (Meissner et al., 2020). No follow-up trials are currently registered with clinicaltrials.gov.
UB-312 is another synthetic peptide-based vaccine, which targets a 12-amino-acid sequence in the C terminus of α-Syn (Nimmo et al., 2020). Antibodies generated in guinea pigs using UB-312 were found to bind α-Syn oligomers and fibrils in post-mortem brain tissue from PD, MSA, and DLB patients. However, preclinical studies of UB-312 did not evaluate for protection in animal models of any of these diseases. Despite this, a phase 1 clinical trial for this vaccine in PD is currently ongoing. This trial has a dose escalation period, part A, during which the optimal dose is identified followed by an evaluation for safety and tolerability period, part B. The expected completion date is June 2022.
II. B. Passive Immunization
For passive immunization, patients are treated with antibodies against α-Syn that are believed to bind extracellular α-Syn, preventing it from spreading to other cells (Brundin et al., 2017; George and Brundin, 2017). A number of monoclonal antibodies are being investigated for this purpose. One antibody currently in a relatively advanced stage of testing is prasinezumab (PRX002/RO7046015). This is a humanized IgG1 monoclonal antibody directed against epitopes near the C terminus of α-Syn based on murine monoclonal antibody 9E4, which recognizes amino acids 118–126 of mouse α-Syn (Masliah et al., 2011). Preclinical data evaluating the effect of antibody 9E4 in PDGF–α-Syn transgenic mice demonstrated a protective effect with regard to α-Syn accumulation, synapse loss, and performance on the rotarod, pole test, and water maze (Masliah et al., 2011). In human trials of prasinezumab, a phase 1, single ascending-dose study showed a good safety profile, serum half-life of 18.2 days, and dose-dependent lowering of “free” α-Syn in the serum, with a maximal effect at a dose of 30 mg/kg (Schenk et al., 2017). This safety profile was replicated in another phase 1, multiple ascending-dose study in patients with PD, but no alteration in the level of free α-Syn in the CSF could be detected (Jankovic et al., 2018). This was hypothesized to be due to low CSF concentration of the antibody, a CSF/serum ratio of 0.3%, coupled with relatively lower affinity for monomeric α-Syn as opposed to aggregated α-Syn. Phase 2 trials are currently underway to investigate the efficacy of prasinezumab (NCT03100149, NCT04777331).
Cinpanemab (BIIB054) is a fully human IgG1 monoclonal antibody directed against an epitope near the N terminus of α-Syn that is also being testing for passive immunization in PD. This antibody was identified in serum associated with isolated B-cell lines derived from healthy individuals using direct ELISA (Weihofen et al., 2019). It binds an epitope within the first 15 amino acids of the N terminus of α-Syn and is highly selective for aggregated α-Syn. In α-Syn A53T transgenic mice that had been inoculated with α-Syn preformed fibrils injected into the striatum, treatment with BIIB054 delayed the onset of paralysis and was protective against striatal dopamine transporter loss (Weihofen et al., 2019). A phase 1, single ascending-dose study showed it was well tolerated at doses up to 45 mg/kg, although it achieved CSF concentrations of only 0.13%–0.56% of that in plasma (Brys et al., 2019). However, recently a phase 2 trial (NCT03318523) missed its primary and secondary endpoints, did not achieve proof of concept, and was, therefore, discontinued from further development.
A third monoclonal antibody being investigated is MEDI1341, which binds to the C terminus of α-Syn, likely an epitope within amino acids 103–129 based on analysis of binding various mutated forms of α-Syn (Schofield et al., 2019). It does not bind β– or γ-synuclein but does bind aggregated α-Syn, including Lewy bodies and Lewy neurites from post-mortem patient samples. Intravenous treatment of rats with MEDI1341 reduced CSF α-Syn levels by as much as 81.5% at peak, with reduction still at 45% by 4 days later and reduced brain interstitial fluid α-Syn levels by 75% at 4 hours post-treatment (Schofield et al., 2019). Additionally, intravenous treatment of cynomolgus monkeys with MEDI1341 led to detectable levels in the CNS, although only 0.2% of that found in serum. Free α-Syn in the CSF decreased by up to 88%. Furthermore, in a mouse model of PD in which α-Syn was delivered unilaterally to the mouse hippocampus via lentiviral injection, intraperitoneal treatment with MEDI1341 was protective against spread of α-Syn to the contralateral hippocampus (Schofield et al., 2019). There are currently two phase 1 trials underway for MED1341: a single ascending-dose study in healthy volunteers (NCT03272165) and a multiple ascending-dose study in patients with PD (NCT04449484).
A phase 1 trial investigating the monoclonal antibody Lu AF82422 is currently recruiting participants. No preclinical data have been published, but per news releases by the sponsor, this is a humanized monoclonal IgG1 antibody against the C terminus of α-Syn. The initial phase 1 clinical trial of Lu AF82422 has an expected completion date of August 2021 (NCT03611569).
BAN0805/ABBV-0805 is another humanized monoclonal antibody targeting α-Syn oligomers and protofibrils. Although preclinical data for this specific antibody are not available, studies have been done using mAb47, a murine antibody reportedly analogous to ABBV-0805 (Lindström et al., 2014). In this study, (Thy-1)-h[A30P] α-synuclein transgenic mice treated with mAb47 exhibited decreased levels of α-Syn protofibrils in spinal cord tissue compared with untreated mice. However, despite this change in levels of α-Syn protofibrils, levels of monomeric and fibrillar forms of α-Syn in brain and spinal cord tissue did not differ between the groups. A phase 1 trial of ABBV-0805 was registered but was withdrawn before participants were enrolled (NCT04127695). Additional monoclonal antibodies targeting α-Syn are in the pipeline at the preclinical stage for PD, including one by Denali Therapeutics known as ATV: α-Syn and by AC Immune.
Overall, early trials have suggested good tolerability to immunotherapy, which is promising. However, these biologics reach much lower levels in the CSF than in the plasma, which may limit their effectiveness (Sardi et al., 2018). Further investigations are needed to determine whether these experimental therapeutic agents will have a disease-altering effect in PD.
III. Reduction of α-Syn Expression
Another potential strategy for developing disease-modifying treatments for α-synucleinopathies is to reduce the expression of the pathogenic protein (Table 2). Approaches to decrease α-Syn expression include reducing transcription of the SNCA gene through epigenetic modifications or transcription factors using small molecules or biologics and reducing translation of SNCA mRNA using nucleic acid–based therapeutics or small molecules.
III. A. Reducing SNCA Transcription
The β2-adrenergic receptor agonists clenbuterol and salbutamol may reduce α-Syn levels by altering histone acetylation at the promoter and enhancer regions of SNCA. This class of drugs was identified in a high-throughput screening of 1126 compounds that included US Food and Drug Administration (FDA)-approved drugs and nonregulated supplements by measuring SNCA mRNA levels after treatment of SK-N-MC cells (Mittal et al., 2017). In accordance with this, in rat primary cortical neurons, treatment with the β2-adrenergic receptor agonist metaproterenol reduced α-Syn mRNA and protein levels, and intraperitoneal treatment of mice with the β2-adrenergic receptor agonist clenbuterol led to decreased α-Syn mRNA and protein levels in the substantia nigra (Mittal et al., 2017). Interestingly, follow-up of four million Norwegians over 11 years demonstrated that salbutamol, which is a brain-penetrant β2-adrenergic receptor agonist used to treat asthma, was associated with reduced risk of developing PD (Mittal et al., 2017). A similar study following 1.7 million adults in Israel found that use of β2 agonists significantly reduced the risk of developing PD (Gronich et al., 2018). However, a study in the United States following 100,000 adults found no association between the β2 agonist salbutamol and risk of developing PD after controlling for demographics, smoking status, and overall use of medical care and suggested that the association found in the earlier studies may have been driven by cigarette use leading to asthma protecting against PD rather than the β2 agonist itself (Searles Nielsen et al., 2018). Further investigation is needed to clarify the effect of β2 agonists on PD pathophysiology.
Two biologics have also been studied as potential therapeutics for PD that act by decreasing transcription of the SNCA gene: SLS-004 and ST-502. The former, SLS-004, is a lentiviral vector containing DNA-methyltransferase 3A, which targets DNA-methylation editing within intron 1 of the SNCA gene to downregulate expression (Kantor et al., 2018). SLS-004 treatment of human induced-pluripotent stem cells from a patient with PD with SNCA triplication led to decreased levels of α-Syn mRNA and protein levels as well as reduced reactive oxygen species (ROS) and increased cell viability. On the other hand, ST-502 is a zinc-finger transcription factor from Sangamo therapeutics, which is delivered by an adeno-associated virus (AAV) vector and functions at the DNA level to selectively repress the expression of SNCA. Releases from the company report that ST-502 can decrease SNCA mRNA, but this work is not yet published in the scientific literature. Both of these biologics are at the preclinical stage.
III. B. Reducing SNCA Translation
III.B.1. Nucleic Acid–Based Therapeutics
To date, no nucleic acid–based therapeutics targeting α-Syn have reached the stage of clinical trials, but testing in animal models has provided proof of concept for this approach. Early work focused on generating siRNA against human α-Syn mRNA and found that siRNA against nucleotides 288–309 reduced expression of α-Syn in HeLa cells transfected with human α-Syn, whereas short hairpin RNA (shRNA) against this sequence reduced expression of endogenous α-Syn in SH-SY5Y cells by 90%; this shRNA also eliminated expression of human α-Syn in the rat striatum after coinjection of a human α-Syn expressing lentivirus (Sapru et al., 2006). Similarly, continuous infusion of naked siRNA against murine α-Syn into the mouse CA1 region of the hippocampus led to a significant reduction in SNCA mRNA levels (Lewis et al., 2008). Meanwhile, infusion of a 21-basepair siRNA duplex against the SNCA transcript into the substantia nigra of the squirrel monkey led to decreased α-Syn mRNA and protein expression, with no adverse effects in terms of inflammation, number of dopaminergic cells, or levels of dopamine or its metabolites (McCormack et al., 2010). In the rat rotenone model of PD, AAV-mediated delivery of shRNA against the SNCA transcript to the substantia nigra led to a 35% reduction in α-Syn protein levels in substantia nigra dopamine neurons and was associated with protection against loss of dopaminergic neurons, presynaptic terminals, and dendritic processes as well as protection against motor deficits, as measured by the postural instability and spontaneous cylinder exploration tests (Zharikov et al., 2015). In another study, siRNA against SNCA mRNA normalized α-Syn levels in fibroblasts from patients with SNCA triplication and shRNA against SNCA rescued motor impairments in a Drosophila model of PD as measured by the climbing assay (Takahashi et al., 2015). In mice transgenic for S129D α-Syn, peripheral injection of exosomes expressing rabies virus glycoprotein containing siRNA against α-Syn reduced α-Syn mRNA expression in the midbrain, striatum, and cortex; decreased protein levels in the midbrain, striatum, cortex, cerebellum, brainstem, and thalamus; and lessened aggregates in the midbrain (Cooper et al., 2014). In mice transgenic for WT α-Syn, intraventricular injection of AAV expressing siRNA against α-Syn reduced SNCA mRNA and α-Syn protein expression in the striatum (Helmschrodt et al., 2017). Some studies have shown that in vivo RNAi-mediated α-Syn silencing induces nigrostriatal degeneration and inflammation in rodent models and in nonhuman primates (Gorbatyuk et al., 2010; Khodr et al., 2011, 2014; Collier et al., 2016), although evidence for this is mixed. Initially there was concern that use of RNAi technology would be limited by poor penetration of many viral vectors across the BBB, but the development of AAV-based vectors with good ability to cross the BBB, such as AAV9 and AAV-PHP.eB, has mitigated this concern (Chan et al., 2017; Liu et al., 2021). In addition, alternate delivery methods, such as intranasal delivery, have shown some preclinical success. The nasal BBB is semipermeable, allowing for easier transit into the brain. Using this delivery approach, intranasal treatment of mice with conjugated RNAi, IND-499-siRNA, which is targeted to monoamine neurons, reduced α-Syn levels in monoaminergic neurons to 70% that of controls 1 day after treatment (Alarcón-Arís et al., 2018).
The use of antisense oligonucleotides (ASOs) to target mRNAs of interest has gained attention in recent years with the approval of such treatments for spinal muscular atrophy, Duchenne muscular dystrophy, and polyneuropathy of hereditary transthyretin-mediated amyloidosis by the FDA and ongoing clinical trials in other neurologic indications (Scoles and Pulst, 2019; Bennett et al., 2021). For example, antisense oligonucleotides promoting ribonuclease H-mediated degradation of SNCA mRNA leading to reduction of α-Syn protein levels were shown to protect nigral dopaminergic neurons in rodent PFF models of PD (Cole et al., 2016; Cole et al., 2021). Interestingly, intraventricular delivery of ASO against SNCA mRNA led to a decrease in the number of established α-Syn aggregates in the substantia nigra in rats that had previously been challenged with PFF injection into the striatum (Cole et al., 2021). Similarly, intraventricular injection of ASO against SNCA mRNA in mice transgenic for A53T human α-Syn decreased α-Syn mRNA and protein levels in the cerebral hemisphere and in WT human α-Syn transgenic mice was protective against behavioral deficits (Uehara et al., 2019). Recently, ASO against SNCA mRNA targeted to monoaminergic neurons led to significant decreases of α-Syn mRNA and protein in AAV-h-α-Syn mice and was also protective against accumulation of phosphorylated α-Syn and reductions in dopamine neurotransmission seen in these animals (Alarcón-Arís et al., 2020). Use of ASOs to decrease α-Syn mRNA and protein has also been demonstrated in nonhuman primates (Alarcón-Arís et al., 2020; Cole et al., 2021) but has not yet reached human trials.
Another approach for knocking down α-Syn is the use of microRNA (miRNA) (Mouradian, 2012). miRNAs are abundant, endogenous, short, noncoding RNAs that act as post-transcriptional regulators of gene expression by base-pairing with their target mRNA, reducing protein translation, and degrading mRNA (Bartel, 2004; Junn and Mouradian, 2010; 2012). For example, miRNA-7 targets the 3′-UTR of SNCA mRNA to decrease expression of α-Syn (Junn et al., 2009). Additionally, miRNA-7 facilitates the clearance of preformed α-Syn aggregates by promoting autophagy (Choi et al., 2018). Like miRNA-7, miRNA-153 binds the 3′-UTR of SNCA mRNA, leading to decreased α-Syn expression in a way that is additive to that of miRNA-7 (Doxakis, 2010). In addition, miRNA-34b and miRNA-34c, which are downregulated in PD, have also been shown to downregulate α-Syn in cellular model systems (Kabaria et al., 2015).
Ribozymes, which are antisense RNAs that have enzymatic activity, are another potential technology for downregulating α-Syn expression (Ohkawa et al., 1995). Intranigral injection of ribozyme against α-Syn mRNA using an AAV vector led to decreased α-Syn protein expression and was protective in a rat model of PD created by 1-methyl-4-phenylpyridinium (MPP+) iodine injection into the medial forebrain bundle (Hayashita-Kinoh et al., 2006).
III.B.2. Small Molecules that Reduce α-Syn Levels by Inhibiting Translation
Although nucleic acid based therapeutic approaches are gaining momentum, targeting RNA using small molecules to modulate protein expression is increasingly being recognized as a new frontier in therapeutic development (Disney et al., 2020; Meyer et al., 2020). This is particularly useful for intrinsically disordered proteins, such as α-Syn, that are undruggable. The SNCA mRNA has an iron-responsive element (IRE) in its 5′-UTR that assumes a hairpin structure. Approximately 50% of SNCA mRNA isoforms in SH-SY5Y cells, which is a human dopaminergic cell line, contain the IRE, meaning that targeting this element would reduce α-Syn expression without eliminating expression entirely (Zhang et al., 2020b). Using sequence-based design, screening of small molecules against the SNCA IRE identified Synucleozid to bind to a specific site in this sequence, selectively lowering α-Syn protein levels and protecting SH-SY5Y cells from the toxicity of α-Syn PFF (Zhang et al., 2020b).
Interestingly, posiphen, which is the (+) enantiomer of the cholinesterase inhibitor phenserine, has also been shown to reduce α-Syn protein translation (Rogers et al., 2011; Mikkilineni et al., 2012), although its exact mechanism of action is unknown. Posiphen is better known for its action of decreasing levels of amyloid precursor protein, one of the disease-associated proteins in Alzheimer disease (Lahiri et al., 2007), and, as such, has been found to reduce CSF amyloid precursor protein levels in healthy volunteers (Maccecchini et al., 2012). More recent work demonstrating that posiphen rescues colonic motility in the A53T and A30P α-Syn transgenic mouse models of PD (Kuo et al., 2019) suggests that posiphen may also be protective in PD. Currently, there is a phase 1 and 2 safety, tolerability, and dose-finding study of posiphen in AD and PD patients (NCT04524351).
IV. Inhibition of α-Syn Aggregation
IV. A. α-Syn Disaggregators
Although α-Syn is a natively unfolded protein, it can misfold into aggregation-prone secondary structures. As such, rational drug design has aimed to target this pathway of misfolding leading to the formation of higher order structures to interfere with aggregation (Table 3). This class of treatments is known as disaggregators. For example, Anle138b, which was identified using high-throughput screening for compounds that could inhibit prion protein aggregation, was found to inhibit α-Syn oligomer formation in vitro. It also protects against the rotenone and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse models as well as against the motor deficits and pathologic changes in A30P α-Syn transgenic mice (Wagner et al., 2013). Further testing demonstrated that treatment with Anle138b slowed disease progression in the A30P α-Syn transgenic mouse model of PD even when treatment was initiated after disease onset (Levin et al., 2014). The protective effect of Anle138b extends to the MI2 mouse model of PD, which expresses human, aggregation-prone truncated 1–120 α-Syn under the control of the tyrosine hydroxylase promoter (Wegrzynowicz et al., 2019) as well as to the motor deficits in models of MSA (Fellner et al., 2016; Heras‐Garvin et al., 2019; Lemos et al., 2020). Anle138 completed a first-in-human, placebo-controlled, double-blind, randomized trial to evaluate safety, tolerability, and pharmacokinetics in healthy volunteers in August of 2020 (NCT04208152). Although the results of this trial are not yet published, the company has reported excellent safety and tolerability and that the drug reached adequate plasma levels for efficacy. Another phase 1 dose-finding study in patients with PD is currently recruiting (NCT04685265).
Other small molecules have been designed to interfere with α-Syn interaction with membranes, which is a step that may promote oligomerization (Miraglia et al., 2018). The small molecule NPT100-18a was found to interact specifically with membrane-bound α-Syn, leading to displacement of α-Syn from the membrane, reduced formation of α-Syn oligomers in vitro, reduced α-Syn aggregation, and protection against neurite loss in primary neuronal cultures overexpressing α-Syn. In α-Syn transgenic mice, NPT100-18a protected against α-Syn accumulation, neurodegeneration, and motor impairment (Wrasidlo et al., 2016). A similar compound with improved pharmacokinetic properties, NPT200-11, has been developed, which also prevents α-Syn aggregation and protects against pathologic changes and motor deficits in α-Syn transgenic mice (Price et al., 2018). A single ascending-dose, phase 1 clinical trial using NPT200-11 was completed in early 2016 (NCT02606682), but results of this study have not yet been published, and no further human trials for this compound are currently registered with clinicaltrials.gov. Squalamine is another disaggregator that functions by displacing α-Syn from lipid membranes (Perni et al., 2017). In Caenorhabditis elegans–overexpressing α-Syn, squalamine protects against α-Syn aggregation and paralysis (Perni et al., 2017). This compound is being investigated for symptomatic treatment of PD, as its oral administration to A53T α-Syn transgenic mice restored gut motility (West et al., 2020). Based on this preclinical work, the synthetic salt of squalamine ENT-01 has been investigated in patients with PD (NCT03047629). In this phase 2 trial, patients were treated with a single escalating dose of ENT-01 every 3–7 days from 25 mg to 200 mg and demonstrated negligible systemic absorption but improved constipation (Hauser et al., 2019). Two follow-up phase 2 studies of ENT-01 for constipation in PD are still recruiting (NCT04483479 and NCT03781791). Additionally, a phase 1 study in patients with PD dementia to investigate the effect of ENT-01 on cognitive symptoms is currently active but not recruiting (NCT03938922). The related compound trodusquemine also inhibits α-Syn aggregation but has improved penetration of the BBB relative to ENT-01 (Perni et al., 2018). Treatment of c. elegans overexpressing α-Syn with oral trodusquemine was protective against α-Syn aggregation and paralysis (Perni et al., 2018).
Another molecule using the mechanism of preventing α-Syn aggregation is the fusion protein NPT088. This is a human immunoglobulin fused to a phage-derived capsid protein known as g3p, which has a general amyloid-association motif that binds to aggregated forms of multiple misfolded proteins, including α-Syn, and disrupts the amyloid conformation (Krishnan et al., 2014; Levenson et al., 2016). NPT088 reportedly reduces proteinase K resistant α-Syn aggregates in transgenic mice, although these results have not yet been reported in the scientific literature. A multiple-dose safety study of NPT088 delivered intravenously has been completed in patients with Alzheimer disease (NCT03008161). This trial demonstrated good safety and tolerability, although there was no effect on markers of disease progression, such as amyloid plaques, τ aggregates, or clinical symptoms (Michelson et al., 2019). Studies of this molecule in patients with PD are still pending.
Intrabodies/nanobodies are small antibody fragments that can enter cells. These can be engineered to bind α-Syn and inhibit oligomerization as well as fused to motifs that target them to the proteosome, as discussed below in section V.B. Targeting the Ubiquitin-Proteasome System. Such engineered intrabodies/nanobodies have been shown to reduce α-Syn aggregates and nigral neurodegeneration in rodents induced by viral vector mediated α-Syn overexpression (Bhatt et al., 2013; Butler et al., 2016; Chatterjee et al., 2018). One example of this is VH14, an intrabody that targets the NAC domain of α-Syn and inhibits aggregation (Chatterjee et al., 2018).
A family of peptides known as SLS-007 that aim to prevent α-Syn aggregation by targeting the NAC core have been developed. These compounds were designed using computational modeling based on the NAC core of α-Syn, and two members of this class, S62 and S71, were found to prevent α-Syn aggregation in vitro and slow seeding of α-Syn aggregates in HEK293 cells expressing human WT and A53T α-Syn (Sangwan et al., 2020). These peptides are being tested in preclinical models using AAV vectors for delivery.
IV. B. Targeting α-Syn Proaggregation Factors
Multiple cell biologic factors, such as post-translational modifications, exposure to certain elements such as iron, disruption of lipid homeostasis, interaction with other proteins, and oxidative stress, have been identified to promote α-Syn oligomerization, fibrillization, and aggregation (Rochet and Liu, 2009; Breydo et al., 2012; Ghosh et al., 2017). These findings provide additional potential targets for disease modification through inhibiting α-Syn aggregation (Table 3).
Post-translational modifications that are believed to promote α-Syn aggregation and misfolding include phosphorylation, ubiquitination, tyrosine nitration, acetylation, and oxidation (Schmid et al., 2013). Targeting phosphorylation seems particularly promising. α-Syn is hyperphosphorylated at serine 129 in the brains of patients with PD and DLB, and animal models of synucleinopathy, including transgenic mice and PFF-injected animals, exhibit aggregates that are phosphorylated at S129 (Paleologou et al., 2008b; Lee et al., 2011; Luk et al., 2012; Oueslati, 2016; Yan et al., 2018). Evidence has been presented that the S129 phosphorylated form of the protein fibrillizes in vitro to a greater extent than its nonphosphorylated counterpart (Fujiwara et al., 2002; Smith et al., 2005), although a subsequent report showed that this post-translational modification increases the conformational flexibility of α-Syn and inhibits its fibrilization (Paleologou et al., 2008a). Ser87 is another site of phosphorylation found in the brains of patients with PD and in models of PD (Okochi et al., 2000; Fujiwara et al., 2002; Anderson et al., 2006). Despite some evidence that phosphorylation may inhibit α-Syn aggregation in vitro, pharmacological manipulation of a specific isoform of the protein phosphatase protein phosphatase 2A (PP2A) that is responsible for dephosphorylating S129-p-synuclein mitigates the phenotype of α-Syn transgenic mice and PFF-injected mice (Lee et al., 2013; Yan et al., 2018). This can be accomplished by inhibiting protein phosphatase methylesterase 1 (PME1) using eicosanoyl-5-hydroxytryptamide (EHT), allowing PP2A to be maintained as a methylated active holoenzyme to dephosphorylate S129-p-synuclein (Lee et al., 2011, 2013; Yan et al., 2018). Furthermore, this activity of EHT, which is a component of coffee, is synergized when combined with caffeine, both of which effect an increase in activity of PP2A (Lee et al., 2013; Yan et al., 2018). Reducing α-Syn phosphorylation pharmacologically through activating PP2A is particularly pertinent mechanistically considering that in PD and DLB brains, PP2A is demethylated, PME1 is upregulated, and the methylating enzyme leucine carboxyl methyltransferase is downregulated (Park et al., 2016).
Increased iron in the substantia nigra of patients with PD compared with controls is another factor suspected to contribute to the neuropathology of the disease (Acevedo et al., 2019), and in vitro studies have indicated that iron exposure may contribute to α-Syn aggregation (Uversky et al., 2001). To this end, deferiprone, an iron chelator that crosses the BBB, has been investigated. Deferiprone is unique among iron chelators in that it captures intracellular iron and releases it extracellularly (Devos et al., 2020). It has been shown to be protective in multiple models of PD, including in SH-SY5Y cells challenged with MPP+ (Molina-Holgado et al., 2008), rats lesioned with 6-hydroxydopamine (6-OHDA) (Dexter et al., 2011), mice lesioned with MPTP (Devos et al., 2014), and human mesencephalic dopaminergic cells treated with MPP+ (Devos et al., 2014). In a pilot study, patients with PD within 3 years of disease onset were treated with 30 mg/kg/day of deferiprone for 6 months (delayed start) or 12 months (early start) and had reduced levels of iron in the nigrostriatal system based on MRI imaging and improvement on motor performance as measured by the Unified Parkinson’s Disease Rating Scale (UPDRS) (Devos et al., 2014). Similar results on iron levels and motor score were found in a follow-up study, in which patients received deferiprone for 12 months (delayed start) or 18 months (early start); in addition, deferiprone had a greater effect in patients with a gene polymorphism that is associated with low ceruloplasmin-ferroxidase activity (NCT00943748) (Grolez et al., 2015). A smaller study using either 20 mg/kg/day or 30 mg/kg/day of deferiprone for 6 months demonstrated good safety and tolerability as well as reduced iron levels in the caudate, as measured by MRI; there was a trend toward improvement in motor scores on the UPDRS, but this did not reach significance in this small study (NCT01539837) (Martin-Bastida et al., 2017). Two further clinical trials of deferiprone have been initiated. One is a phase 2, multicenter, placebo-controlled, randomized clinical trial of deferiprone at 30mg/kg/day for 9 months, which has finished recruiting but is still in progress (NCT02655315). The other was also a placebo-controlled, randomized clinical trial of deferiprone at doses ranging from 600 mg to 2400 mg per day for 9 months; this study is completed, but results have not yet been published (NCT02728843).
An iron attenuator, PBT434, has been shown to inhibit α-Syn aggregation in vitro and protect against neuropathologic changes and decline in motor activity in the 6-OHDA, MPTP, and A53T α-Syn transgenic mouse models of PD (Finkelstein et al., 2017). A phase 1 clinical trial has been undertaken to investigate the safety, tolerability, and pharmacokinetics of PBT434 in healthy adults in either a single ascending dose of up to 600 mg or multiple ascending doses of up to 250 mg twice per day for 8 days; although the results of this trial have not yet been published, they have been presented at scientific conferences and suggest good oral bioavailability, safety, and tolerability (Stamler et al., 2019, 2020).
Disruption of lipid homeostasis may also promote α-Syn misfolding and aggregation, as interaction of α-Syn with lipid membranes promotes its oligomerization (Miraglia et al., 2018; Fanning et al., 2019). Unbiased phenotypic screening in yeast identified inhibitors of Ole1, the yeast homolog of human stearoyl-CoA desaturase (SCD), as protective against growth inhibition after activation of α-Syn expression (Vincent et al., 2018). The protective effect may stem from reduced formation of oleic acid, a monounsaturated fatty acid that is generated by SCD, as excess oleic acid led to an increase in α-Syn aggregation in human neuronal cultures (Fanning et al., 2019). Accordingly, the SCD inhibitor 5b was protective against the formation of α-Syn aggregates and motor impairment in mice transgenic for human WT α-Syn or triple-mutated α-Syn harboring the human disease-associated mutation E46K along with the amplifying mutations E35K and E61K (Nuber et al., 2021). YTX-7739, a small-molecule inhibitor of SCD, has reportedly been found to be safe and well tolerated in healthy volunteers, and a phase 1b trial in people with PD is now underway in the Netherlands.
Finally, targeting enzymes that contribute to crosslinking of α-Syn into high-molecular-weight aggregates is another potential approach for blocking its aggregation. Transglutaminase 2 (TG2) is one such enzyme. TG2 is a crosslinking enzyme that has been shown to promote the formation of high-molecular-weight aggregates of α-Syn in vitro (Junn et al., 2003) and in vivo (Grosso et al., 2014). Its activity is regulated by a number of factors that become disrupted in the neurodegenerative cascade, including loss of calcium homeostasis, energy depletion, and the formation of reactive oxygen species (Grosso and Mouradian, 2012), suggesting a pathway by which an initial insult leads to upregulation of TG2 activity, which then contributes to α-Syn crosslinking and aggregation. Pathologic data in patients with PD support this hypothesis; there is an increase in TG2 mRNA and protein expression in the substantia nigra of patients with PD (Citron et al., 2002; Andringa et al., 2004; Wilhelmus et al., 2011). In α-Syn transgenic mice expressing wild-type human SNCA under the control of the Thy-1 promoter, overexpression of TG2 exacerbates α-Syn aggregation, neuritic damage, and behavioral deficits (Grosso et al., 2014), whereas deletion of TG2 is protective in both the MPTP and α-Syn transgenic mouse models (Battaglia et al., 2007; Zhang et al., 2020a). Additionally, the TG2 inhibitor cystamine and its reduced, active form, cysteamine, are protective in MPTP and 6-OHDA lesioned mouse models of PD (Tremblay et al., 2006; Stack et al., 2008; Sun et al., 2010). Taken together, this body of work suggests that TG2 can be a therapeutic target for blocking α-Syn aggregation and neuroprotection.
V. Enhancing Clearance and Degradation of α-Syn
Enhancing the clearance and degradation of α-Syn is another potential approach toward disease-modifying therapy for PD (Table 4). α-Syn is degraded by both the ubiquitin-proteasome system and the autophagy/lysosomal pathway (ALP) (Bennett et al., 1999; Webb et al., 2003; Cuervo et al., 2004). It is suspected that normal, soluble α-Syn may be primarily degraded by the ubiquitin-proteasome system, whereas aggregates may be handled by the ALP (Ebrahimi-Fakhari et al., 2011). Pathogenic depletion of proteasome components and lysosomes has been seen in sporadic PD brains and in rodent models (Chu et al., 2009; Dehay et al., 2010). In addition, several other proteins implicated in the pathogenesis of PD that are mutated in familial forms of the disease are important for the ALP. These include leucine-rich repeat kinase 2 (LRRK2) (Alegre-Abarrategui et al., 2009; Ramonet et al., 2011; Bravo-San Pedro et al., 2013; Soukup et al., 2016); glucocerebrosidase, which is a degradative enzyme within the lysosome and the most common genetic risk factor for PD (Neumann et al., 2009); PTEN-induced kinase 1 (PINK1) and Parkin, which work together to target damaged mitochondria to the lysosome (Narendra et al., 2008; Matsuda et al., 2010; Stamatakou et al., 2020); and DJ-1, which contributes to activation of chaperone-mediated autophagy (Nash et al., 2017; Xu et al., 2017). In addition, α-Syn itself may compromise autophagy function. In in vitro studies and PC12 cell cultures, A30P and A53T α-Syn interfere with the uptake and degradation of substrates into lysosomes by chaperone-mediated autophagy (Cuervo et al., 2004). Interestingly, in PC12 cells, expression of A30P or A53T α-Syn also leads to an upregulation of macroautophagy, and this is thought to be compensatory for the blockage of chaperone-mediated autophagy (Cuervo et al., 2004). However, experimental support for α-Syn interfering with macroautophagy has also been reported, as overexpression of WT but not A30P or A53T α-Syn in SK-N-SH human neuroblastoma cells led to decreased levels of the autophagosome marker LC3-II (Winslow et al., 2010), and α-Syn preformed fibril treatment of HEK293 cells stably expressing α-Syn led to decreased levels of LC3-II as well as accumulation of the macroautophagy substrate p62 (Tanik et al., 2013). Given that autophagy is implicated in the degradation of α-Syn (Webb et al., 2003; Ebrahimi-Fakhari et al., 2011), the importance of multiple familial PD-associated proteins in the ALP suggests a model by which autophagic activity is decreased because of abnormalities in the function of these proteins, precluding it from degrading α-Syn. In addition, autophagy is a complex pathway that interacts with the endosomal system at multiple points, and thus abnormalities in autophagy may also contribute to increased endosomal escape of misfolded α-Syn into the cytosol and exocytosis of α-Syn to the extracellular space, contributing to its spread to other neurons (Fig. 2) (Hyttinen et al., 2013; Fox and Yamamoto, 2015; Brock et al., 2019; Zhang et al., 2019; Fakhree et al., 2021).
V. A. Targeting Autophagy
Based on the importance of autophagy in clearing misfolded proteins and in the neurodegenerative process of PD, autophagy enhancers have been studied as potential disease-modifying agents. Early work investigated rapamycin, which enhances autophagy by inhibiting mechanistic target of rapamycin (mTOR). For example, intraperitoneal administration of rapamycin protected against cell death in the MPTP mouse model of PD (Malagelada et al., 2010); intracerebral infusion of rapamycin in PDGFβ promoter-driven WT α-Syn transgenic mice resulted in decreased accumulation of α-Syn and reduced synaptic pathology (Crews et al., 2010; Dehay et al., 2010); and intraperitoneal delivery of the rapamycin derivate temsirolimus in the AAV–α-Syn injected mouse model of PD was protective against pathologic changes and motor deficits (Decressac et al., 2013). However, the utility of rapamycin is limited because the mTOR pathway is involved in a multitude of cellular processes, and as such rapamycin has been found to induce severe adverse effects, including oral ulcers, hypertriglyceridemia, and impaired wound healing (Jahrling and Laberge, 2015). The compound MSDC-0160, which is an inhibitor of mitochondrial pyruvate carrier, also inhibits mTOR through alteration of cellular metabolism and has been shown in C. elegans to reduce α-Syn toxicity (Ghosh et al., 2016). It has not been tested in patients with PD yet, but a very small, 3-month clinical trial in patients with AD demonstrated increased nonserious adverse effects, such as diarrhea, compared with placebo (NCT01374438) (Shah et al., 2014). Additionally, resTORbio has developed the compound RTB-101, which works downstream of mTOR as a target of rapamycin kinase complex 1 inhibitor. Phase 1/2 trials were initiated, but completion was delayed and, as of this writing, no trials are registered on clinicaltrials.gov.
Tyrosine kinase inhibitors have been investigated as autophagy enhancers as well. These compounds activate autophagy by decreasing activation of the phosphatidylinositol 3-kinase-AKT (PI3K-AKT)-mTOR pathway and have been extensively studied for use in cancer (Tanaka et al., 2020) but may also have utility in neurodegenerative diseases. For example, c-Abl is a tyrosine kinase that has been implicated as a mediator of PD through multiple pathways, including increasing phosphorylation of α-Syn and interfering with Parkin activity (Brahmachari et al., 2017). Several c-Abl inhibitors are under investigation as therapeutics for PD. The c-Abl inhibitor nilotinib has been shown to enhance autophagic clearance of α-Syn (Hebron et al., 2013) and protect dopaminergic neurons in the MPTP mouse model of PD (Karuppagounder et al., 2014). Small, nonblinded safety trials (NCT02954978 and NCT02281474) of nilotinib in patients with DLB and PD dementia reported some improvement in motor function within 6 months of starting treatment (Pagan et al., 2016, 2019; Wyse et al., 2016). However, a subsequent 6-month, randomized parallel-group, double-blind, placebo-controlled efficacy trial in patients with moderately advanced PD demonstrated poor CSF penetration and no improvement relative to placebo (NCT03205488) (Simuni et al., 2020). Along similar lines, another c-ABL1 tyrosine kinase inhibitor, K0706, also known as SCC-138, is being evaluated for disease modification in PD and DLB. According to the sponsor’s website, K0706 protected against dopaminergic cell loss on the substantia nigra in the MPTP mouse model of PD. Safety and tolerability studies in healthy adults (NCT03316820, NCT03445338) and patients with PD (NCT02970019) have shown adequate safety, and trials in PD and DLB are currently in phase 2 (NCT03996460, NCT03655236). An additional c-Abl tyrosine kinase inhibitor under investigation is radotinib, which, when administered orally, was protective against dopaminergic cell loss, neuroinflammation, and motor deficits in mice injected with α-Syn PFF in the striatum (Lee et al., 2018). A phase 2, dose-escalation study of radotinib in patients with PD is registered with clinicaltrials.gov but not yet recruiting (NCT04691661). The c-Abl tyrosine kinase inhibitor FB-101 is being investigated as well. A phase 1 trial with a single ascending dose and multiple ascending dose arm is currently recruiting (NCT04165837), but preclinical data specific to this compound are not currently available. Finally, ikT-148009, another c-Abl tyrosine kinase inhibitor, is also under investigation for disease modification in PD. Preclinical data specific to this compound are not currently available, but a phase 1 single and multiple ascending-dose trial in healthy volunteers is enrolling (NCT04350177).
Bosutinib is another tyrosine kinase inhibitor being investigated in PD. In mice challenged with lentiviral injection of α-Syn into the substantia nigra and in A53T α-Syn transgenic mice, intraperitoneal injection of bosutinib decreased α-Syn protein levels and markers of inflammation (Lonskaya et al., 2013; Hebron et al., 2014). Accordingly, a phase 2 trial of bosutinib at 100 mg daily for 90 days in patients with DLB is currently active and not recruiting (NCT03888222). Trehalose is another compound that has been shown to enhance autophagy, likely through activation of transcription factor EB (TFEB) (Rusmini et al., 2019). Oral trehalose provided in the drinking water has been found to be protective on multiple outcome measures in a variety of animal models of PD, including in rats injected in the striatum with A53T α-Syn in an AAV vector, rats lesioned with rotenone or 6-OHDA, and mice lesioned with MPTP (Sarkar et al., 2014; Ferguson et al., 2015; Wu et al., 2015; He et al., 2016; Darabi et al., 2019; Howson et al., 2019). Human trials of trehalose for PD have not been undertaken, but phase 1 trials have been initiated in AD and spinocerebellar ataxia 3.
Another upstream regulator of autophagy is Beclin 1. Overexpression of Beclin-1 via lentivirus vector prevented α-Syn accumulation and synapse loss in the PDGF promoter driven α-Syn transgenic mouse model (Spencer et al., 2009). MicroRNA-7 also activates autophagy (Choi et al., 2018) and may, therefore, be a potential agent for disease modification in PD through this mechanism in addition to reducing α-Syn protein levels (Junn et al., 2009). Both overexpression of Beclin-1 and treatment with miRNA-7 are at the preclinical stage.
One of the challenges in targeting autophagy in PD is that the autophagic pathway is complex, and, therefore, different steps in the pathway likely have different impacts on the pathophysiology of PD. Additionally, although inadequate autophagy can lead to the pathologic accumulation of substrates, excessive autophagy may be detrimental as well. This may explain conflicting results in the literature, demonstrating that in some cases, such as those described above, upregulating autophagy is protective, whereas in others (Moors et al., 2017), autophagy activation can lead to cellular dysfunction and death. These opposing findings underline the need to better understand the function of autophagy in both healthy cells and in disease to target this pathway for neuroprotection in a more refined way.
V. B. Targeting the Ubiquitin-Proteosome System
α-Syn is also degraded by the proteosome (Bennett et al., 1999; Webb et al., 2003), and as a result, enhancing proteasomal function or enhancing targeting of α-Syn to the proteosome may be protective in PD. The small molecule IU1 has been shown to enhance proteasomal function by inhibiting the deubiquinating activity of human ubiquitin-specific peptidase 14 (USP14) (Lee et al., 2010). Inhibition of USP14 has been shown to be protective in Drosophila mutant for PTEN-induced kinase 1 (PINK1) and Parkin (Chakraborty et al., 2018). Additionally, enhanced proteasomal function through inhibition of USP14 was associated with increased clearance of tau; however, there is some suggestion that IU1 may be neurotoxic (Kiprowska et al., 2017) and, therefore, requires further study before it can be considered for human trials. Alternatively, mechanisms to enhance targeting of α-Syn to the proteasome have also been investigated. For example, as discussed above, constructs have been developed in which an intrabody/nanobody that targets α-Syn is fused to a proteasome-targeting proline, aspartate or glutamate, serine, and threonine (PEST) motif. In addition to stalling α-Syn aggregation, these constructs mediate its proteasomal clearance (Butler et al., 2016; Chatterjee et al., 2018). Two examples of this are VH14PEST, which binds to the NAC region of α-Syn, and NbSyn87PEST, which targets the C-terminal region. Both nanobodies demonstrate protection against cell death in ST14A rat striatal progenitor cell line transfected with α-Syn (Butler et al., 2016) and against α-Syn aggregation in the intrastriatal AAV-α-Syn injected rat model of PD (Chatterjee et al., 2018). Notably, VH14PEST is also protective against synaptic loss and decreased dopamine transporter expression in the striatum seen in this model, whereas NbSyn87PEST is associated with an increase in microglial density in the substantia nigra (Chatterjee et al., 2018).
VI. Targeting Other Genes/Proteins Involved in Synucleinopathies
VI. A. Glucocerebrosidase
A number of proteins and genes are implicated as contributing to PD pathogenesis in addition to α-Syn. Notable among these is glucocerebrosidase (GCase), a lysosomal hydrolase that converts glucosylceramide into ceramide and glucose. Homozygous mutations in the GBA gene leading to deficiency of GCase cause Gaucher disease, which increases one’s risk for PD (Bultron et al., 2010). Additionally, heterozygosity for Gaucher disease-linked mutations of GBA is associated with an increased risk of synucleinopathies (Sidransky et al., 2009; Migdalska-Richards and Schapira, 2016). Patients with PD carrying GBA mutations have a younger age of symptom onset than those with sporadic PD and are less likely to have typical features, such as asymmetric onset, resting tremor, and rigidity (Gan-Or et al., 2008; Sidransky et al., 2009; Bultron et al., 2010; Ryan et al., 2019) but have more postural instability with gait difficulty and dementia as the disease progresses (Davis et al., 2016; Ryan et al., 2019). Interestingly, GCase activity has also been shown to be decreased in the brains of patients with PD not associated with GBA mutations, including both idiopathic PD and LRRK2-associated PD (Alcalay et al., 2015). Although the precise mechanism by which GBA mutations increase the risk of synucleinopathies is not fully understood, it has been suggested that these mutations contribute to the accumulation of misfolded α-Syn. For example, decreased GCase activity may lead to the accumulation of proteins processed through the ALP, including α-Syn, in neurons (Schondorf et al., 2014; Mazzulli et al., 2016a; Sardi et al., 2017; Taguchi et al., 2017). Alternatively, alterations in glycosphingolipid homeostasis that affect membrane composition may impair vesicular transport and lysosomal/endosomal function in a way that promotes α-Syn aggregation (Kim et al., 2018; Zunke et al., 2018).
A number of strategies are currently being employed to target GCase as a potential PD therapy (Table 5). For example, small molecules that can cross the BBB and increase lysosomal GCase activity are being investigated. The mucolytic agent Ambroxol, which binds the active site of GCase and exhibits chaperone activity, is one such agent. It increases enzyme activity in the brainstem, midbrain, cortex, and striatum of mice (Migdalska-Richards et al., 2016) and in the midbrain, cortex, striatum, and cerebellum of a single nonhuman primate (Migdalska-Richards et al., 2017). Ambroxol is also protective against motor dysfunction in Drosophila-expressing disease-associated mutation in GBA (Sanchez-Martinez et al., 2016). Results of AIM-PD, a phase 2A clinical trial investigating safety and tolerability of Ambroxol in patients with PD, were recently published (Mullin et al., 2020). This open-label, noncontrolled trial analyzed data from 17 patients with PD, 8 of whom had a GBA1 mutation and 9 of whom did not. Ambroxol was given for 186 days, and the dose was titrated up to 420 mg three times per day. This regimen demonstrated good safety and tolerability with no serious adverse effects. CSF α-Syn concentration increased by 13%, and GCase protein levels increased by 35%. An improvement in motor rating scores was also reported. A randomized phase 2 trial of up to 1050 mg/day in 75 patients with PD dementia is currently recruiting (NCT02914366), and two further phase 2, randomized trials for DLB are planned but not yet recruiting (NCT04588285 and NCT04405596).
VI. Targeting Other Genes/Proteins Involved in Synucleinopathies
VI. A. Glucocerebrosidase
A number of proteins and genes are implicated as contributing to PD pathogenesis in addition to α-Syn. Notable among these is glucocerebrosidase (GCase), a lysosomal hydrolase that converts glucosylceramide into ceramide and glucose. Homozygous mutations in the GBA gene leading to deficiency of GCase cause Gaucher disease, which increases one’s risk for PD (Bultron et al., 2010). Additionally, heterozygosity for Gaucher disease-linked mutations of GBA is associated with an increased risk of synucleinopathies (Sidransky et al., 2009; Migdalska-Richards and Schapira, 2016). Patients with PD carrying GBA mutations have a younger age of symptom onset than those with sporadic PD and are less likely to have typical features, such as asymmetric onset, resting tremor, and rigidity (Gan-Or et al., 2008; Sidransky et al., 2009; Bultron et al., 2010; Ryan et al., 2019) but have more postural instability with gait difficulty and dementia as the disease progresses (Davis et al., 2016; Ryan et al., 2019). Interestingly, GCase activity has also been shown to be decreased in the brains of patients with PD not associated with GBA mutations, including both idiopathic PD and LRRK2-associated PD (Alcalay et al., 2015). Although the precise mechanism by which GBA mutations increase the risk of synucleinopathies is not fully understood, it has been suggested that these mutations contribute to the accumulation of misfolded α-Syn. For example, decreased GCase activity may lead to the accumulation of proteins processed through the ALP, including α-Syn, in neurons (Schondorf et al., 2014; Mazzulli et al., 2016a; Sardi et al., 2017; Taguchi et al., 2017). Alternatively, alterations in glycosphingolipid homeostasis that affect membrane composition may impair vesicular transport and lysosomal/endosomal function in a way that promotes α-Syn aggregation (Kim et al., 2018; Zunke et al., 2018).
A number of strategies are currently being employed to target GCase as a potential PD therapy (Table 5). For example, small molecules that can cross the BBB and increase lysosomal GCase activity are being investigated. The mucolytic agent Ambroxol, which binds the active site of GCase and exhibits chaperone activity, is one such agent. It increases enzyme activity in the brainstem, midbrain, cortex, and striatum of mice (Migdalska-Richards et al., 2016) and in the midbrain, cortex, striatum, and cerebellum of a single nonhuman primate (Migdalska-Richards et al., 2017). Ambroxol is also protective against motor dysfunction in Drosophila-expressing disease-associated mutation in GBA (Sanchez-Martinez et al., 2016). Results of AIM-PD, a phase 2A clinical trial investigating safety and tolerability of Ambroxol in patients with PD, were recently published (Mullin et al., 2020). This open-label, noncontrolled trial analyzed data from 17 patients with PD, 8 of whom had a GBA1 mutation and 9 of whom did not. Ambroxol was given for 186 days, and the dose was titrated up to 420 mg three times per day. This regimen demonstrated good safety and tolerability with no serious adverse effects. CSF α-Syn concentration increased by 13%, and GCase protein levels increased by 35%. An improvement in motor rating scores was also reported. A randomized phase 2 trial of up to 1050 mg/day in 75 patients with PD dementia is currently recruiting (NCT02914366), and two further phase 2, randomized trials for DLB are planned but not yet recruiting (NCT04588285 and NCT04405596).
Other small molecules that enhance GCase activity include LTI-291, which has undergone a small phase 1 clinical trial showing that single doses up to 90 mg and 14 consecutive daily doses up to 60 mg were safe and well tolerated (Heijer et al., 2021). Further studies of this molecule are still pending. Another small molecule, S-181, has been shown to increase lysosomal function in induced pluripotent stem cell (iPSC)-derived dopaminergic neurons from patients with PD and decrease insoluble α-Syn in D409V GBA1 mutant mice (Burbulla et al., 2019). Clinical trials have not yet been initiated for this compound.
Another strategy for treating PD by targeting GCase is to stabilize the enzyme using chaperones that interact with its active site, such as isofagomine (also known as afegostate-tartrate, t2101) (Steet et al., 2006). Isofagomine has been shown to protect against the decline in motor function in Drosophila expressing disease-associated mutations in GBA (Sanchez-Martinez et al., 2016) and to rescue the phenotype of human wild-type α-Syn transgenic mice (Richter et al., 2014). This drug has not been studied in patients with PD, but safety and tolerability trials have been completed in patients with Gaucher disease (NCT00446550 and NCT00433147). Although full results are not yet published, early reports from these trials demonstrate good safety and tolerability at doses of 225 mg daily for up to 7 days at a time, suggesting that it would also be safe to trial isofagomine in patients with PD. Other GCase chaperones that lead to increased enzyme activity or half-life include NCGC758 and NCGC607 (Patnaik et al., 2012; Aflaki et al., 2014; Aflaki et al., 2016). These have been shown to reverse α-Syn accumulation in dopaminergic neurons derived from iPSCs from patients with PD (Aflaki et al., 2016; Mazzulli et al., 2016b) but have not yet reached the stage of clinical trials.
Gene therapy is another strategy to increase GCase levels. AAV1-mediated GBA delivery in the striatum has been demonstrated to decrease α-Syn protein levels in the A53T α-Syn transgenic mouse model of PD (Sardi et al., 2013). An open-label phase 1/2a trial of PR001, which is an AAV9-mediated delivery of GBA, in patients with PD with at least one GBA mutation is recruiting (NCT04127578). This is a one-time delivery of the vector intracisternally.
An alternative strategy to prevent the accumulation of lipid substrates in GBA linked disease is to inhibit their biosynthesis. To this end, the novel glucosylceramide synthase inhibitors GZ/SAR402671, also known as venglustat, and GZ667161 have been developed. GZ667161 decreased the accumulation of toxic α-Syn and was protective against cognitive deficits in both the GbaD409V/D409V and A53T–α-Syn overexpressing mouse models of synucleinopathies (Sardi et al., 2017). A phase 2 trial of venglustat in 270 patients with PD known as MOVES-PD (NCT02906020) showed good safety and tolerability (Judith Peterschmitt et al., 2019) but recently failed to meet its primary endpoint of improving MDS-UPDRS parts II and III.
Another small molecule that is being developed specifically for GBA-PD is ESB-1609. This therapeutic is a sphingosine 1-phosphate receptor 5 (S1P5) agonist, which, according to the company website, upregulates lipid transporters with the intent to rescue lysosomal deficits in patients with a mutated copy of GBA. This would, theoretically, lead to increased degradation of α-Syn, and thus neuroprotection in PD. ESB-1609 is reportedly undergoing a phase 1 investigation, although this trial is not registered on clinicaltrials.gov. Another sphingosine 1-phosphate receptor agonist is Fingolimod, which is approved by the FDA for multiple sclerosis (MS). Fingolimod is less selective than ESB-1609, as it agonizes S1P1, S1P3, S1P4, and S1P5 and in MS is actually believed to be protective by acting as a functional S1P1 antagonist, leading to reduced lymphocyte release from lymph nodes (Vogt and Stark, 2017). However, given the preliminary findings that ESB-1609 may rescue lysosomal deficits, it is possible that Fingolimod could be protective in PD as well. Indeed, treatment of SH-SY5Y cells with exogenous S1P is protective against cell death and the generation of reactive oxygen species induced by MPP+ (Pyszko and Strosznajder, 2014; Alessenko and Albi, 2020). The protective effect seen in this study was mediated by S1P1 receptors, which, taken together with the findings with ESB-1609, suggests that agonism of the S1P pathway may be a potential route for neuroprotection via more than one receptor. Indeed, studies in mouse models have demonstrated a protective effect of Fingolimod in the A53T transgenic mouse, GM2 synthase transgenic mouse (GM2+/−), and 6-OHDA challenged mouse models of PD on various outcome measures, including α-Syn aggregation, motor symptoms, and inflammatory markers (Vidal-Martínez et al., 2016, 2019; Ren et al., 2017). A role for the S1P pathway was not investigated in these papers, but a more recent article demonstrated that treatment with either Fingolimod or a selective S1P1 receptor agonist, SEW2871, protects against markers of inflammation in the striatum, neuronal loss in the substantia nigra, and motor deficits in the MPTP-challenged mouse (Pépin et al., 2020), supporting the role of the S1P pathway in neuroprotection using these compounds.
VI. B. LRRK2
Another important protein in the pathogenesis of PD and α-Syn toxicity is LRRK2 (Hur and Lee, 2021). Mutations in LRRK2 are found in 1% of patients with sporadic PD and 4% of familial cases worldwide (Healy et al., 2008). In patients with PD of Ashkenazi Jewish descent, these figures are 13.3% for sporadic disease and 29.7% of familial disease (Ozelius et al., 2006). This is a large protein with a kinase and GTPase domain, and most pathologic mutations in LRRK2 are within these domains (Daher, 2017). LRRK2 has been implicated in autophagy, vesicle trafficking, and retromer complex modulation; however, studies investigating pathologic mutations of LRRK2 suggest that these induce degeneration through a gain of function mechanism (Daher, 2017). This may occur in part because of its effect on α-Syn. PD-associated mutations in LRRK2 contribute to α-Syn aggregation, release from the cell, and propagation to other cells (Bae et al., 2018; Schapansky et al., 2018; Bieri et al., 2019; Nam et al., 2021). Therefore, therapies targeting LRRK2 aim to inhibit this protein and in particular its kinase activity (Table 6). A number of efforts are underway to achieve this goal, with the small molecules DNL201 and DNL151 and the ASO ION859 in the most advanced stages. Phase 1 studies in healthy participants and in patients with PD have been completed for DNL201 and in patients with PD for DNL151, but the full results of those trials are pending (NCT03710707, NCT04551534, and NCT04056689). Additionally, a phase 1 trial in healthy volunteers is currently underway for DNL151 (NCT04557800). Early unpublished reports suggested that these compounds may have induced pulmonary toxicity, and indeed, LRRK2 inhibition in nonhuman primates caused reversible lung morphologic changes (Fuji et al., 2015). However, more recent press releases from the sponsor suggest that the pulmonary side effects may be mild, and studies in nonhuman primates demonstrated that treatment with one of three different LRRK2 inhibitors, GNE-7915, MLi-2, and PFE-360, lead to only mild pathologic changes in the lungs and no change in pulmonary function (Baptista et al., 2020).
Preclinical data have shown that treatment of primary hippocampal cell cultures with the LRRK2 ASO ION859, also known as BIIB094, protects against the formation of α-Syn aggregates seen in response to challenge with PFF. In mice, intraventricular treatment with ION859 protects against the accumulation of phosphorylated α-Syn, dopaminergic cell loss, and motor deficits in the PFF model of PD (Zhao et al., 2017). A phase 1 clinical trial to determine the safety and tolerability of ION859 is currently underway (NCT03976349).
Many other potential therapeutics that inhibit LRRK2 activity or expression have been tested in preclinical models: the compound MLi-2 decreased LRRK2 kinase activity, although it was not protective in the MitoPark mouse model of PD with respect to motor behavior or striatal levels of dopamine and its metabolites (Fell et al., 2015; Scott et al., 2017); LRRK2-IN-1 blocked LRRK2 kinase activity in HEK293 cells (Deng et al., 2011); PF-06447475 blocked LRRK2 kinase activity and was protective against dopaminergic cell loss in the AAV–α-Syn rat model of PD (Daher et al., 2015); JH-II-127 inhibited LRRK2 kinase activity and was found to have good oral bioavailability and brain penetration in mice (Hatcher et al., 2015); and micro-RNA-205 downregulated LRRK2 expression and protected against neurite outgrowth defects induced by expression of LRRK2 R1441G in primary neuronal culture (Cho et al., 2013). None of these therapeutics have yet been advanced to clinical trials.
VI. C. Poly(adenosine 5′-diphosphate–ribose) Polymerase-1
Poly(adenosine 5′-diphosphate–ribose) polymerase (PARP)-1 is a protein that may contribute to cell death in PD in response to α-Syn aggregation (Kam et al., 2018). Accordingly, blocking PARP-1 with any of the three PARP inhibitors veliparib, rucaparib, or talazoparib has been shown to be protective against dopaminergic cell loss and rescued the motor phenotype in the PFF mouse model of PD (Kam et al., 2018). Further studies of compounds that can block PARP-1 are needed.
VII. Decreasing Synaptic Activity
Based on the transmission hypothesis of advancing α-Syn toxicity and disease progression, preventing this process can also be a disease-modifying strategy in PD. Experimental evidence has shown that synaptic activity contributes to the transcellular spread of α-Syn and that decreasing synaptic activity in mouse primary cortical neurons using the N-methyl-d-aspartate (NMDA) receptor antagonist AP5 prevents the release of α-Syn into the extracellular space induced by glutamate treatment (Table 7) (Yamada and Iwatsubo, 2018). The NMDA receptor antagonist memantine is already FDA-approved for AD and is therefore a readily available drug to target this mechanism of α-Syn spread. To this end, a phase 3 trial of memantine in patients with PD is currently recruiting (NCT03858270). Patients will receive 10 mg of memantine twice a day for 51 weeks and be evaluated on measures of cognitive function and by brain MRI. The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonist perampanel, which is FDA-approved for the treatment of epilepsy, is also being investigated for its ability to block α-Syn transmission. Perampanel was found to protect against the formation of phosphorylated α-Syn aggregates in the anterior olfactory nucleus and piriform cortex in mice challenged with α-Syn PFF in the olfactory bulb (Ueda et al., 2021).
VIII. Challenges and Opportunities
As detailed above, multiple approaches are being pursued to develop disease-modifying therapies for PD that either directly or indirectly impact α-Syn, and substantial progress has been made in recent years. And yet, there are still no proven safe and effective treatments to slow down the progression of this disabling condition. Many challenges can help explain why. First, the pathogenesis of PD is complex and not yet fully understood. This makes it difficult to predict the ideal point in the pathogenetic pathway(s) to target. Further contributing to this complexity is the heterogeneity of the disease with multiple genetic causes and risks, variable penetrance of certain genes, and different clinical courses and pathologic variations. Additionally, no animal models that are currently available faithfully recapitulate the pathogenesis of the human disease. As a result, effects of treatments seen in preclinical trials may not be representative of how a drug will perform in patients. Another challenge in the development and testing of PD therapeutics is the lack of reliable biomarkers of prodromal PD and disease progression. The variability in the progression of symptoms among individuals (Cedarbaum et al., 2015) increases the challenge of choosing candidates for clinical trials and to assess drug efficacy. In addition, it is often difficult to identify and recruit patients in the prodromal stage, which would be necessary to initiate trials prior to the onset of symptoms when treatments may be more effective. To address this gap in the field, multiple initiatives are underway to develop PD biomarkers. These include the Parkinson’s Progression Markers Initiative (PPMI) by the Michael J. Fox Foundation for Parkinson’s Research, which seeks to collect prospective datasets with detailed clinical, imaging, genetic, and fluid biomarker data from patients with PD (Marek et al., 2018); the National Institute of Neurological Disorders and Stroke PD Biomarkers Program; and the Critical Path for Parkinson’s initiative (Parkinson’s UK and Critical Path Institute of UK). Additionally, dopamine transporter (DAT) imaging is available to aid in diagnosing PD clinically and may be of use as a biomarker in clinical trials (Conrado et al., 2018). But it is recognized that a critical need exists for the ability to image α-Syn aggregates in the brain, and substantial efforts are being made toward this objective. In addition, a combination of CSF α-Syn aggregates, serum exosomal α-Syn levels, and blood neurofilament light chains are being investigated for use in PD diagnosis, prognostication, and theragnosis (Parnetti et al., 2019), but further validation in large, independent cohorts is needed. Having validated biomarkers of disease and specifically pathologic α-Syn deposition will greatly enhance our ability to identify at risk individuals in the premotor stage of the disease and intervene with efficacious and safe therapeutics to prevent progression to clinically manifest disease. Biomarkers and genetic studies will also allow delivery of personalized medicine.
The necessity for any compound to cross the BBB to be effective also limits the pool of potentially useful compounds. However, the possibility that α-Syn aggregates begin in the gut and propagate to the central nervous system through the vagus nerve (Kim et al., 2019) may open the possibility of treating peripheral α-Syn disease as a way to prevent the brain pathology. Additionally, it has been suggested that focused ultrasound may be used to temporarily open the BBB (Xhima et al., 2018; Karakatsani et al., 2019; LeWitt et al., 2019), which would broaden the pool of potentially useful therapeutics. A recent small clinical trial on 5 patients with PD demonstrated good safety and tolerability of using MRI-guided focused ultrasound to reversibly open the BBB (Gasca-Salas et al., 2021).
In addition to the advances in biomarker identification and drug delivery methods discussed above, the PD field has become well positioned to developing personalized medicine through a greater understanding of the genetic risk factors for PD. Because of this, the wide range of approaches being tested in PD raises the prospect of combination therapies in the future to maximize benefit, a practice that is commonly employed in other medical fields, including cancer and human immunodeficiency virus infection. Although this review focused on therapeutics directed toward α-Syn toxicity, there has also been progress in development of therapeutics aimed at other aspects of the pathologic cascade, such as glucagon-like peptide-1 receptor (GLP1R) agonists, which are thought to address “brain insulin resistance” and may target neuroinflammation (Yun et al., 2018; Ntetsika et al., 2021) and neurotrophic factors, which have neuroprotective functions (Gash et al., 1996; Pascual et al., 2011). Some of these therapeutics that have primary mechanisms of action not directly linked to α-Syn misfolding and toxicity may still impact the α-Syn pathway. For example, reactive oxygen species may increase post-translational modifications that promote α-Syn aggregation (Rochet and Liu, 2009), and thus, antioxidants, such as Coenzyme Q10, glutathione, and N-acetyl-cysteine, may have an impact on α-Syn misfolding in addition to their effects of decreasing the toxicity of ROS. Conversely, α-Syn overexpression itself leads to increased ROS production and cellular susceptibility to dopamine (Junn and Mouradian, 2002; Dias et al., 2013). Many of these classes of therapeutics have been evaluated clinically (Heiss et al., 2019; Whone et al., 2019; Brauer et al., 2020; Chang and Chen, 2020; Mulvaney et al., 2020; Duarte-Jurado et al., 2021), and although those clinical trials have often had negative results, some of these therapeutics may eventually add to the repertoire of drugs that can be used in combination with those discussed in this review to better attack multiple points in the cascade of PD pathophysiology.
Despite the challenges, the prospect for finding disease-modifying treatments for PD is more realistic today than ever before. Our improved understanding of the disease, the innovative ideas for intervention, advances in biotechnology, the multitude of approaches being tested, and new developments in imaging and other biomarkers will make this goal possible.