
The sigma-1 receptor as key common factor in cocaine and food-seeking behaviors

By David Aguinaga, Mireia Casanovas, Rafael Rivas-Santisteban, Irene Reyes-Resina, Gemma Navarro, and Rafael Franco
Excerpt from the article published in Journal of Molecular Endocrinology, 63(4), R81-R92, November 2019, DOI: https://doi.org/10.1530/JME-19-0138
Editor’s Highlights
- Food-seeking behavior involve peripheral and central players, ghrelin being one of the most relevant.
- Ghrelin is a peptide hormone that controls food intake and energy homeostasis.
- The ‘hunger’ hormone, ghrelin, interact with σ1R in CNS neurons, both compounds arising as key players in the central control of food/energy intake.
- Cocaine is able to interact with sigma-1 and sigma-2 receptors. σ2R subtype is involved in psychomotor stimulant effects while σ1R subtype participates in the cocaine-induced convulsions’.
- The sigma-1 receptor (σ1R), is a key piece of the puzzle showing interrelationships between cocaine and food-seeking behaviors
Abstract
Addiction and eating disorders involve brain reward circuits. Binge eating predisposes to addictive behavior, while the cessation of exposure to drugs of abuse leads to reward activities, including intake of tasty foods. Cocaine use is associated with a decrease in food intake, with reversal after drug use is discontinued. Exciting new findings show that receptors for the ‘hunger’ hormone, ghrelin, directly interact with the sigma-1 receptor (σ1R), which is a target of cocaine. σ1Rs are key players in regulating dopaminergic neurotransmission and ghrelin-mediated actions. This review focuses on the σ1 receptor as a general neuroendocrine regulator by directly interacting with neuronal G-protein-coupled receptors. This review also covers the early mechanisms by which cocaine binding to σ1 blocks the food-seeking behavior triggered by ghrelin. Those findings appear as fundamental to understand common mechanisms in drug addiction and eating disorders.
Introduction
Food-seeking behavior involve peripheral and central players, ghrelin being one of the most relevant. Ghrelin is synthesized in the stomach but its endocrine action is exerted, among others, in heart, gastrointestinal (GI) tract, pancreas and central nervous system (CNS) (Fig. 1). Ghrelin, the ‘hunger’ hormone, acts via a specific receptor, GHS-R1a, which belongs to the superfamily of G-protein-coupled receptors (GPCRs). Dopamine, one of the main neurotransmitters in the brain, is a key player in the circuits in which neuronal GHS-R1a expression occurs. Also, dopamine is central to the actions of a variety of drugs of abuse, in particular cocaine. South American tribes used cocaine for endurance enhancement and appetite suppression, indicating a long history of use without fully understanding the mechanisms of action. Recent results show that one atypical receptor of unknown physiological function, the sigma-1 receptor (σ1R), is a key piece of the puzzle showing interrelationships between cocaine and food-seeking behaviors. First, this review covers the role of σ1R in cocaine addiction via GPCR-mediated regulation of dopaminergic transmission. The second part covers recent data showing how sigma receptors regulate ghrelinergic signaling. Finally, the review presents the novel and relevant evidence linking cocaine-mediated suppression of appetite with both cocaine binding to σ1R and subsequent regulation of the functionality of dopamine and ghrelin GPCRs.
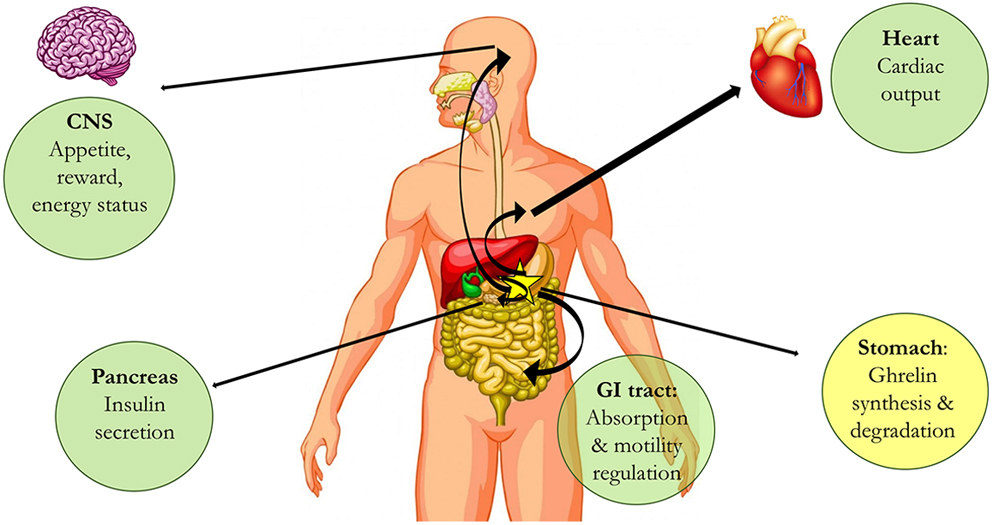
Endocrine actions of ghrelin in different body organs. Ghrelin is produced in the stomach (yellow) and exerts actions in different parts of the mammalian body. Examples of organs and actions are in green circles.
Apart from acting via inhibition of dopamine transporters in neurons, cocaine is able to interact with sigma-1 and sigma-2 receptors. The physiological role of the two receptors is unknown and, also, they are structurally unrelated despite sharing the same name (sigma). This review focuses on σ1R as a mediator of the anorexigenic effect of cocaine. The receptor, whose function remains a mystery, displays interesting features such as its capacity to (i) bind cocaine at physiologically relevant concentrations and (ii) modulate GPCR function. Early evidence indicated a blockade of stimulant effect of cocaine by targeting σ1R (Menkel et al. 1991). It should be noted that treatment with synthetic drugs acting on σ2 receptors decreases some of cocaine-induced effects (Matsumoto et al. 2007) and that antagonist of the receptor counteract induced locomotor stimulation in cocaine-administered mice (Lever et al. 2014). However, the research on σ2receptor is still too preliminary to allow establishing a solid link between the receptor and addiction mechanisms. One of the novel aspects in the review, which is based on recent results, is the advance in understanding how cocaine consumption reduces food-seeking behavior. Apparently, this is due to direct interactions with the receptor for the hunger hormone, ghrelin.
The most striking and accepted effect of cocaine in brain is an increase in inter- and extra-synaptic dopamine concentration that leads to marked activation of dopamine receptors (Fig. 2). These and many other neuronal receptors belonging to the family of GPCRs use cAMP as second messenger. Information taken from the British Society guide for pharmacology shows sigma receptors’ atypical properties: they do not use either cAMP or any other second messenger as calcium ions or inositol-3-phosphate. As σ1R are not coupled to any known signal transduction machinery, the guide indicates that ‘there is only a modest pharmacological overlap and no structural convergence with the GPCRs’ (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=785). On the other hand, structurally different compounds may bind to σ1R receptors at topologically intracellular sites. In fact, the binding of a drug seemingly acting on opioid receptors suggested that σ1R receptors belonged to the opioid receptor family. The uniqueness of the receptor was first confirmed by the lack of effect of opioid receptor antagonists and, secondly, by cloning and discovering that it was not a GPCR. Solving of 3D structure (see details below) has confirmed some of the assumptions about these receptors and has provided brand new information related to atypical membrane insertion and oligomeric (trimeric) structure.
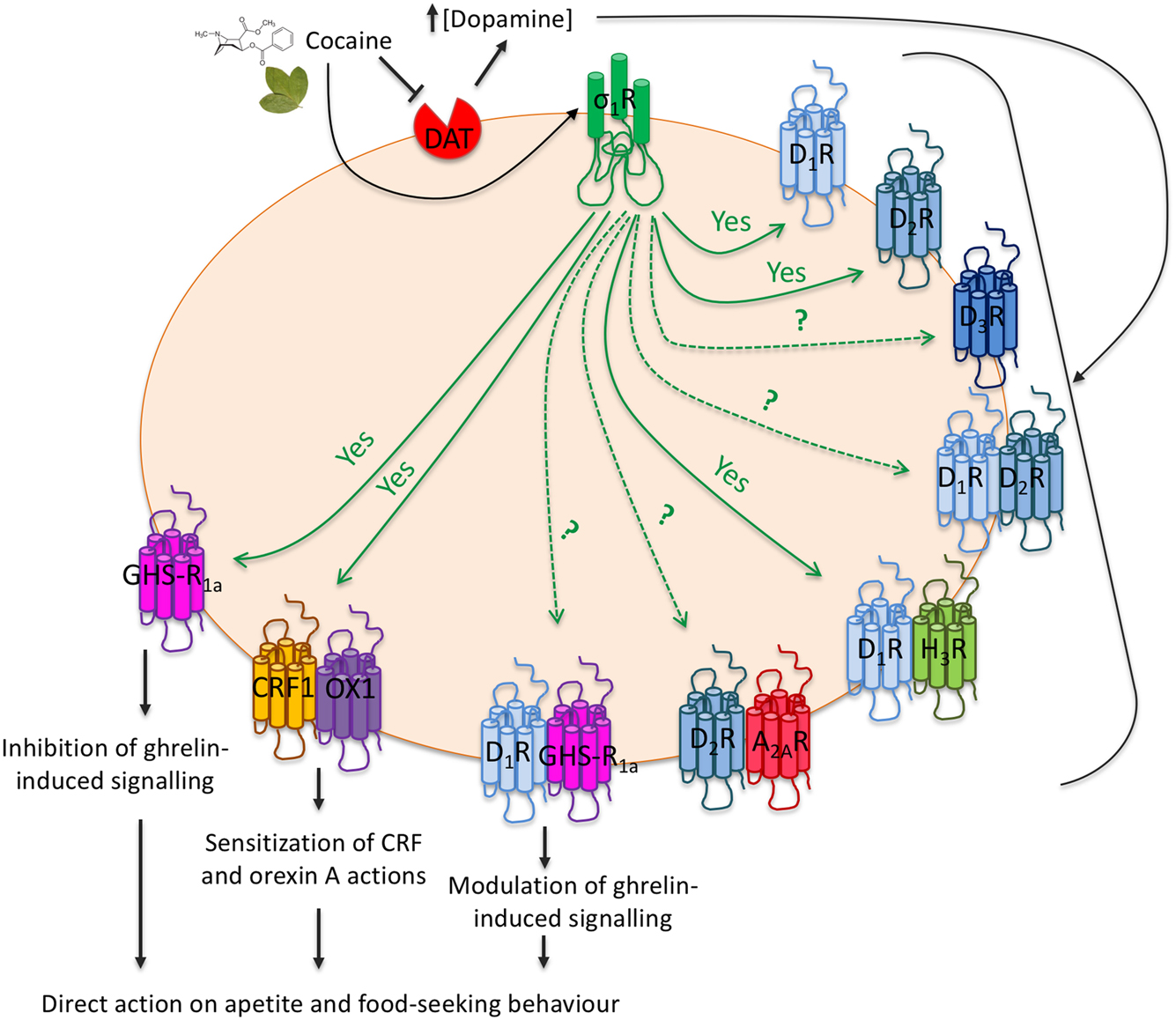
Schema of σ1 receptor-mediated actions related with food intake and mediated by neuronal GPCRs. Cocaine increases extraneuronal dopamine concentration and activates σ1 receptors. Interactions of σ1 and dopamine receptors or dopamine receptor-containing heteromers indirectly regulate food-seeking behavior. Interactions with ghrelin, orexin or CRF receptors directly regulate food consumption habits. Arrows originating at σ1 receptors indicate proven (continuous ‘yes’) or suspected/unproven interactions. Dopamine receptors types appear as D1R, D2R, or D3R. Adenosine A2A receptors appear as A2AR. Histamine H3 receptors appear as H3R. Other abbreviations are for ghrelin (GHS-R1a), CRF (CRF1) and orexin (OX1) receptors.
Evidence for a relevant role of σ1R on cocaine addiction
SKF-10,047 displays psychomimetic activity while it does not selectively bind to μ- or k-opioid receptor. Although it was firstly assumed that the compound could bind to a novel opioid receptor (Martin et al. 1976), its effect was not blocked by nonselective opioid receptor antagonists (Vaupel 1983). The compound in fact binds to σ1R whose cloning showed that it does not have the seven transmembrane domains of all GPCRs (Hanner et al. 1996). While waiting for more precise details on its physiological role, the σ1R is considered a pluripotent modulator able to interact with different specific proteins (e.g. binding immunoglobulin protein (BiP)) and/or components of signal transduction machineries (Su et al. 2016). σ1R may interact with receptors for a variety of hormones/neurotransmitters and modulate cell surface expression and/or function. Research on this protein is gaining momentum due to its potential as a target to combat neuropathic pain (Mei & Pasternak 2002, Corbera et al. 2006, Sun et al. 2016). Agonistic activity in the case of σ1R may be indirectly measured by subcellular translocation, establishment of protein-protein interactions or regulation of ion channel activity (Wu & Bowen 2008, Kim et al. 2010, Navarro et al. 2010, Su et al. 2010). Indirect evidence also shows the involvement of σ1R in neurological disorders such as depression (Su et al. 2010). Also interesting is the correlation between a mutation in the receptor and debut and progression of juvenile lateral amyotrophic sclerosis (Al-Saif et al. 2011, Mavlyutov et al. 2013). More recently, a biophysical approach assessing homomerization of σ1R and heteromerization of σ1R with BiP has allowed distinguishing molecules with differential potential on stabilizing multimerization of the receptor and/or facilitating the σ1R/BiP interaction (Yano et al. 2018). Despite the ambiguity of ‘agonist/antagonist’ words in the case of this ‘receptor’, the authors show that haloperidol and (+)-pentazocine have opposite effects, thus paving the way to effectively test or select molecules with ‘agonistic’ versus opposite (‘antagonistic’) actions.
Solving the 3D structure (Schmidt et al. 2016) provided a further unexpected finding. σ1R does not have, as previously hypothesized, two transmembrane domains and extracellular C- and N-terminal domains, but an extracellular N-terminal end, a transmembrane α–helical domain and a C-terminal tail having a cupin-like β-barrel with a buried ligand-binding site. Adjacent molecules show a homo-oligomeric arrangement consisting of three closely interacting σ1R protomers. Despite homology (relatively poor) with a fungal sterol isomerase (Cobos et al. 2008), no catalytic activity has been yet allocated to σ1R.
σ1R may interact with receptors for a variety of hormones/neurotransmitters (see below and Fig. 2), thus raising the hypothesis that the receptor could regulate the expression and/or the function of cell-surface receptors. Kruse recently reviewed the intriguing features of this protein that shows no structural resemblance with any other membrane receptor, that has an occluded ligand-binding site and that resembles a yeast enzyme, yeast sterol isomerase (Kruse 2017). Remarkably, while the physiological function remains elusive and the endogenous ligand is yet to be discovered, cocaine binds to σ1R (Skuza 1999, Shull 2002, Lever et al. 2016). The design of drugs impeding the interaction of cocaine with σ1R was proposed to reduce drug-seeking behavior (Matsumoto et al. 2001a). It is now well established that σ1R-mediated cocaine actions in the CNS are dependent on molecular interactions with dopamine and other GPCRs.
Cocaine drives many of its effects via activation of σ1R
Apart from the well-known fact that cocaine markedly enhances the extracellular levels of dopamine in several regions of the CNS, the mechanism underlying drug addiction seems to be mediated by the σ1R.
To our knowledge there are few reports addressing cocaine binding to σ1R. It is however relevant that the interaction of cocaine with σ1R was suspected several years ago (see; Hayashi & Su 2007 for review). Pioneering studies on cocaine binding to σ1R were performed in 2001 (Matsumoto et al. 2001b). Competition assays in radioligand-binding studies showed that K i of cocaine was 2.3 in mouse and 2.9 µM in rat brain. Values were similar in mice brain membranes (Lever et al. 2016) thus confirming that at ‘physiological’ concentrations occurring after cocaine exposure the drug binds to σ1Rs. Based on these results, Matsumoto proposed that σ1Rs were targets to combat cocaine addiction (Matsumoto et al. 2003). More recently, a detailed study (Lever et al. 2016) showed that cocaine administration to mice reduces the binding of a radiolabeled ligand and it does so in a similar proportion in the 20 different brain regions analyzed by quantitative autoradiography. Average reduction of specific binding after i.p. administration of 100 µmol/kg cocaine was 54%. It should be noted that the σ1R is expressed in all brain regions in a narrow range of 2–4 fmol/mg tissue.
A significant advance in addiction research has been provided by demonstrating a link between σ1R and the MAP kinase pathway. The receptor, by itself or upon cocaine binding, cannot convey signals that directly impact on such key signaling pathway. The most reasonable assumption is that cocaine binding to σ1R produces conformational changes in cell-surface GPCRs that are in direct contact with σ1R. This possibility not only affects signaling but also the overall biology of those GPCRs that have a crucial role in drug addiction and control of food intake.
The first in-depth review on the role of σ1R in the many different sides of cocaine addiction appeared in 2002 (Romieu et al. 2002). The already existing data made authors state ‘σ1R is not only necessary for acquisition of the cocaine-induced conditioned place preference, but that it is also implicated in its expression, confirming that activation of the σ1R is induced during cocaine’s early effects’. A more recent review, that summarizes the main properties of σ1R, emphasizes its role as a chaperone (see; Katz et al. 2017 and references therein). The review also presented the evidence of receptor involvement in three specific aspects of drug addiction, namely self-administration, discrimination and place conditioning. We will here focus on the molecular mechanisms that triggered by cocaine binding to σ1R are mediated by other proteins. It was proposed that there is a ‘concomitant targeting of both dopaminergic pathways and σR proteins’ (Katz et al. 2017). However, recent evidence is more consistent with a more direct effect of cocaine on dopaminergic transmission and on other signaling systems operating in neurons of reward circuits. It is quite noteworthy that in the review by Romeu the authors intuitively noticed this possibility and stated: ‘The σ1R is activated consequently to dopamine reuptake blockade and is not sufficient to induce conditioned place preference (CPP) by itself. The mechanism of the σ1R involvement in CPP and the selectivity toward the CPP-inducing drug remains however to be determined’ (Romieu et al. 2002). In fact, data obtained in the last decade suggest new possibilities that have increased the knowledge of underlying mechanisms. In what concerns dopaminergic signaling, cocaine binding to σ1R produces a direct effect on dopaminergic transmission. This is possible because the σ1R directly interacts with dopamine receptors (Fig. 2) and, accordingly, cocaine is binding to and affecting the structure and function of σ1/dopamine receptor complexes.
Lessons from drugs behaving as σ1R ligands
Pioneering studies aimed at understanding the role of σ1R in cocaine effects, showed a synthetic ligand, NPC 16377, was protective against the behavioral effects of cocaine. NPC 16377 did not show any noticeable side effect while its efficacy was quite marked; for instance, it totally prevented the development of cocaine sensitization and significantly reduced diazepam-sensitive cocaine convulsions. Its effects were quite selective; it was not a NMDA receptor ligand and was not efficacious against discriminative stimuli triggered by other drugs (Witkin et al. 1993). Similar results were obtained using compounds with different chemical structures that reduced cocaine-induced hyperlocomotion and convulsions being able to bind to σ1R but not to other receptors (McCracken et al. 1999, Shull 2002).
Skuza tested different sigma receptor ligands, rimcazole and panamesine among others, on cocaine-induced locomotor activity in rats and convulsions (Skuza 1999). Whereas panamesine reduced the two actions of the drug of abuse, rimcazole increased the total time of cocaine-evoked convulsions and locomotor activity. The author concluded: ‘σ2R subtype is involved in psychomotor stimulant effects of cocaine while σ1R subtype participates in the cocaine-induced convulsions’. Also, Romieu and colleagues showed that two novel σ1R ligands, BD1063 and BD1008, significantly increased the ED50 for the locomotor effects of cocaine attributing the effects to dopamine transporter inhibition (Romieu et al. 2000). However, testing different concentrations of analogs of rimcazole for their ability to bind to the two main targets of cocaine, dopamine transporters and σ1R, (Matsumoto et al. 2001a and 2001b) were successful in showing that drugs protective against convulsion-producing concentrations of cocaine were not acting via dopamine transporter inhibition but via binding to σ1R. In a follow-up study six analogs of BD1008 were tested against a variety of targets showing significant affinity for σ1R, moderate for σ2R and low or very low affinity for dopamine transporters and for dopamine, opioid, NMDA and 5-hydroxytryptamine receptors (Matsumoto et al. 2004). The involvement of σ1R was further confirmed using an antisense oligodeoxynucleotide approach, which was efficacious in reducing the behavioral effects of cocaine (Matsumoto et al. 2001a and 2001b). Taking into account all the evidence, antagonists of σ1R were proposed as therapeutic agents to fight against cocaine addiction (Matsumoto et al. 2001c, 2003, Maurice & Romieu 2004). It should be however noted that σ1R-based therapies may not work on acute symptoms and may be better suited to address drug sensitization. Indeed, a common phenomenon displayed by all ligands tested is the reduction in psychostimulant-induced sensitization, not only to cocaine but also to methamphetamine (Ujike et al. 1996).
σ1R impacts on signaling pathways originating at GPCRs and also on the function of ion channels; overall, these functional interactions shape the behavioral and neuroanatomical alterations leading to cocaine addiction (Kourrich et al. 2013). One relevant aspect to consider is how σ1R may affect dopaminergic signaling. As an example, the D1 receptor-mediated signaling and dopamine-induced inositol 1,4,5-trisphosphate production have been studied in dissociated neurons of the nucleus accumbens (NAc). The main finding was that the calcium mobilization produced by administration of inositol 1,4,5-trisphosphate was enhanced by cocaine in a σ1R-dependent fashion. Not only cocaine increases the effects of agonists on cAMP levels but it also alters the kinetics of the MAP kinase pathway engagement. Remarkably, D1 and σ1 receptors do interact and the cocaine effect on dopaminergic signaling was dependent on σ1R, a fact that was confirmed by using σ1R KO mice (Navarro et al. 2010).
Cocaine, dopamine, dopamine receptors, and dopamine heteroreceptor complexes
Dopamine is one of the main neurotransmitters in the brain, and its actions, mediated by dopamine receptors, are relevant for motor control and are important in the reward circuits targeted by addictive drugs. Pioneering electrophysiological studies by Uchimura and North showed that intracellular recordings in NAc neurons triggered by 5-hydroxytryptamine or by dopamine were affected by cocaine (Uchimura & North 1990). The drug at doses in the 1–30 µM range affects dopamine D1-receptor-mediated hyperpolarization and D2-receptor-mediated depolarization. Cocaine was more effective, that is, less doses were required to alter the actions of 5-hydroxytryptamine.
Nowadays, GPCR-containing heteroreceptor complexes are considered as real functional units. In cocaine addiction, the engagement of σ1R by cocaine and the interaction of these receptors with GPCRs has led to investigate how heteromers may contribute to the behavior and motor effects of the drug. As an example, complexes formed by dopamine D1 and histamine H3 receptors display particular properties: the heteromer is needed for signaling to the MAP kinase pathway (Ferrada et al. 2009). Again, cocaine alters heteromer function via the σ1R (Moreno et al. 2014). These results probably reflect the occurrence of a macromolecular complex constituted by H3, D1 and σ1 receptors, whose structure and function becomes altered in the presence of cocaine. This interpretation is supported by the finding that σ1R antagonists restore the homeostatic interplay between H3 and D1 receptors and, therefore, supports the potential of σ1R antagonists to combat drug addiction.
Cocaine affects signaling of other dopamine receptor-containing heteromers also in a σ1R-dependent fashion (Fig. 2). Adenosine A2Aand dopamine D2 receptor heteromers were among the first-identified GPCR heteromers (Hillion et al. 2002). A2AR-D2R heteromers play a relevant role in the striatum and are the target for therapeutic approaches addressed to combat Parkinson’s disease; the homeostatic mechanism consists of A2AR-mediated brake of dopamine actions on D2 receptors. On the one hand, energy transfer studies showed that cocaine altered the structure of adenosine receptor homomers and of adenosine/dopamine receptor heteromers. On the other hand, the drug affected some but not all of the signaling pathways engaged by activation of D2 receptors (Marcellino et al. 2010). Taking into account data on the molecular mechanisms of cocaine actions, Borroto-Escuela et al. (2016b) hypothesized that the drug is significantly altering the allosteric interactions occurring in homo- and heteroreceptor complexes, especially in those containing dopamine receptors. Taking also into account the differential distribution of receptors in different brain regions and the changes in receptor expression after cocaine exposure, the authors suggested that (i) anti-cocaine actions of A2A agonists do not depend on heteromerization of A2AR with D2receptors and (ii) cocaine self-administration courses with a loss in dorsal striatopallidal GABAergic neurons of the brake on D2 receptor signaling within the A2AR-D2R receptor heteroreceptor complex.
Cocaine administration to non-human primates results in brain concentration peaks appearing as soon as 5 min to go back to basal at 30 min (Bradberry et al. 2000). A key region for addiction behavior establishment is the NAc. A novel technique of heteromer detection shows that cocaine self-administration increases the expression of A2AR/D2R and D2R/σ1R complexes in the shell of the NAc, whereas it decreases the expression of those constituted by D2R and σ1R in the dorsal striatum (Borroto-Escuela et al. 2017). Thus, self-administration likely increases in a regional-selective way the expression of receptors that may establish direct interactions and display particular signaling pathways. It should be noted that σ1R may form heteromers with D2 receptors but not with other D2-like receptors such as D3 or D4 receptors. The drug inhibits dopaminergic signaling in the striatum of wild-type mice but not in the striatum of σ1R KO mice. As commented below, altering D1-receptor-mediated signaling modifies the D1-D2 balance required for proper motor control (Navarro et al. 2013). σ1R-mediated dysbalance on striatal dopaminergic signaling is probably one of the main factors underlying locomotor actions of cocaine.
A small but significant percentage of neurons in the NAc express D1 and D2 dopamine receptors. There is dispute on the possibility that D1 and D2 may form heteroreceptor complexes in motor control brain areas (Lee et al. 2004, Rashid et al. 2007, Perreault et al. 2010, George et al. 2014, Frederick et al. 2015, Rico et al. 2016). The most likely hypothesis is that 10–20% of neurons in the NAc express both receptors and that they establish heteroreceptor complexes shifting dopaminergic transmission from cAMP- to Ca2+-dependent signaling (Hasbi et al. 2009). As the increase of dopamine in this nucleus is fundamental for addiction and a significant amount of these cells express the two dopamine receptors, it is hypothesized that D1/D2 heteroreceptor complexes are important for the establishment of cocaine-seeking behavior. In line with this hypothesis, recent studies show that disruption of the heteromer has profound consequences in animals treated with cocaine. Intracerebroventricular administration of disrupting peptides, induces, sustains, accelerates and exacerbates the incentive motivational and locomotor activating effects of cocaine in a self-administration paradigm. The blocking peptides were also able to increase ∆FosB expression in the NAc. These findings suggest a model for tonic inhibition of basal and cocaine-induced reward (Perreault et al. 2016). Future experiments should address the question of whether σ1R may interact with D1/D2heteroreceptors and mediate cocaine actions in neurons expressing the two receptors.
Cocaine alters mitogen-activated protein kinase (MAP) pathway via GPCRs
One of the common factors in drug addiction is the involvement of the MAP kinase pathway. Many drugs of abuse and also natural compounds with psychoactive effects have an impact on the pathway. Structurally different drugs such as tetrahydrocannabinol (THC) or cocaine increase the phosphorylation of extracellular signal-regulated kinases (ERKs) in different brain regions thus fitting with impaired CPP upon pharmacological blockade of the pathway. Furthermore, activation of the MAP kinase pathway contributes to the plastic changes induced by drugs of abuse (see; Valjent et al. 2004 and references therein). Activation of the MAP kinase pathway is needed for establishing an association between drug consumption and CPP (Valjent et al. 2006, Du et al. 2017). Of the two predominant ERK isoforms, ERK2 seems more directly involved in remodeling produced by repeated exposure to cocaine. This conclusion arises from data in ERK1 KO mice, which display a facilitation of acquisition of cocaine CPP and of locomotor sensitization (Ferguson et al. 2006). Also interesting is the finding that dopamine D1 receptor mediates cocaine-induced long-term plasticity in the NAc (Zhang et al. 2017). On the one hand, dopamine supersensitivity, which is subsequent to cocaine consumption, alters the homeostasis of the pathway. Although the temporal course of cocaine-induced increase of dopamine occurs mainly in ventral striatum (Kalivas 1993, Kalivas & Duffy 1993), it affects other brain regions in virtue of the volume transmission mechanism defined by Agnati et al. (1986). On the other hand, whereas THC or caffeine act by direct binding to GPCRs, a direct effect of cocaine on those receptors is unlikely. Interestingly enough, cocaine binding to σ1R does impact on the signal transduction mechanisms that originate at GPCRs and regulate the MAP kinase and mTOR pathways (see; Franco et al. 2017 and references therein). In the amygdala, which is also affected in cocaine addiction, acute and chronic drug administration produced different patterns of immediate early gene expression that correlated with ERK phosphorylation (Radwanska et al. 2005). In summary, the MAP kinase pathway arises as a key mediator of the central actions produced in response to cocaine administration.
Orexin and ghrelin receptors
Orexin/hypocretin physiology and orexin receptor pharmacology data suggest that orexigenic receptors have therapeutic potential in food disorders and drug addiction (Kukkonen & Leonard 2014, Leonard & Kukkonen 2014). Such hypothesis is partly based on the fact that orexin G-protein-coupled receptor pharmacology is multifaceted, ranging from activation of G proteins to ion flux regulation.
Cocaine-seeking behavior involves the ventral tegmental area and several mediators such as orexin-A and corticotropin-releasing factor (CRF). Again, receptors related with cocaine effects have the possibility of forming heteromers whose function is affected by the drug. As a relevant example, CRF1 (CRF1R) interacts with orexin OX1 receptors, and the interaction results in a negative crosstalk between orexin-A and CRF; these endogenous transmitters regulate dendritic dopamine release in the ventral tegmental area. σ1R also interacts with those heteroreceptor complexes, and cocaine binding to σ1R sensitizes cells to excitatory effects of CRF and orexin-A; the mechanism consists of a crosstalk within protomers in CRF1-OX1 receptor heteromers (Navarro et al. 2015). These results reflect an interplay between addiction, stress and, importantly, control of food intake by orexigenic factors (Fig. 2).
Ghrelin is a peptide hormone that controls food intake and energy homeostasis (Fig. 1). Its action is mediated by specific receptors that have received a variety of denominations such as growth hormone-releasing peptide or growth hormone secretagogue receptor. Ghrelin receptors belong to the GPCR superfamily and only one type has been identified. The full-length 388 amino acid long human ghrelin receptor containing seven transmembrane domains is known as GHS-R1a, to differentiate it from the GHS-R1b splice variant, which is 289 amino-acid-long and lacks the fifth and sixth transmembrane (TM) domains. These TM domains are required for coupling to heterotrimeric G proteins and, therefore, ghrelin cannot signal via GHS-R1b receptors (Mary et al. 2013). The truncated variant seems to serve as modulator of GHS-R1a surface expression and signaling. In fact, GHS-R1b is expressed in the same cells than GHS-R1a and both receptors interact to form signaling units (Mary et al. 2013). It has been reported that GHS-R1b negatively influences ghrelin action by reducing surface expression of functional G-protein-coupled ghrelin receptors (Chow et al. 2012) and by allosteric interactions that reduce the efficacy of the hormone (Mary et al. 2013).
In a detailed review Schellekens et al. (2013a) nicely summarize how activation of specific receptors in the brain shapes the many actions of the so-called hunger hormone. The link between ghrelin and dopaminergic transmission in reward circuits is highlighted: ‘The ghrelin signaling system has recently been suggested to play a key role at the interface of homeostatic control of appetite and the hedonic aspects of food intake, as a critical role for ghrelin in dopaminergic mesolimbic circuits involved in reward signaling has emerged’. They also point out that ghrelin receptors may establish interactions with other proteins (Schellekens et al. 2013a,b); in fact, ghrelin receptors establish direct protein-protein interactions with a variety of GPCRs: dopamine, melanocortin, prostanoid, serotonin, somatostatin and neurotensin receptors (see; Borroto-Escuela et al. 2014 and references therein; see www.gpcr-hetnet.com). In a detailed study in which complexes formed by GHS-R1a-GHS-R1b and dopamine D1 receptors were detected, ghrelin receptor signaling was different in hippocampus versus striatum. In the latter, D1 receptors were involved in GHS-R1a-Gs/olf coupling. Thus, the dopamine receptor may switch from Gi/o to Gs/olf coupling but only if GHS-R1b is also expressed (Navarro et al. 2016). It then appears that anything affecting D1 receptor-mediated signaling may in turn affect ghrelin actions.
Links between drug addiction and anorexic behavior
Used today as recreational drug, cocaine was first consumed by humans in the form of Coca leaves. Indeed, indigenous peoples of South America knew that chewing coca leaves was key for keeping their life style. Coca served to cope with the harsh living conditions, for instance when people had to travel long distances and cross Andean mountains with little food. Despite such ancient knowledge, that is, the appetite suppressant action of cocaine, the molecular basis of hunger dissipation by the drug has remained elusive. Interestingly, years ago it was shown that antagonists of σ1R blocked compulsive eating behavior in rats (Cottone et al. 2012). These early results fit nicely with those of a recent report that has provided insight into the underlying molecular mechanisms.
Addiction and eating disorders (i.e. binge eating, anorexia, bulimia) share a central control that involves reward circuits in the brain. This leads to bidirectional influences: on one hand, previous history of binge eating predisposes to the addictive behavior, whereas the cessation of exposure to drugs of abuse leads to reward-providing activities, including the intake of palatable foods. This fact reflects a vicious circle in which the weight gain that follows cocaine abstinence secondarily causes a significant distress which can make a patient more prone to relapses. Many uncertainties remain about the biological substrate of these changes, particularly at the level of signaling systems involved. Thus, establishing the molecular mechanisms of such complex interactions is of immense public health relevance.
Exciting new findings substantiate the concept that receptors for the ‘hunger’ hormone, ghrelin, interact with σ1R in CNS neurons (Fig. 2). The results provide solid evidence of the anorexic effect of cocaine being mediated by ghrelin receptors, both compounds arising as key players in the central control of food/energy intake (Aguinaga et al. 2019). Importantly, the ghrelin/σ1R interaction creates qualitatively new, higher-order structures, with altered signaling properties. Unraveling the mechanisms applicable in this setting may ultimately be translatable into new approaches in endocrinology and behavioral neuroscience fields. In addition, they arise as useful to address the social impact of anorexia/bulimia/binge-eating, now particularly worrisome in young populations.
Cocaine affects ghrelin receptor traffic and function via σ1R
A first relevant piece of information concerning cocaine and ghrelin receptors concerns cell surface GHS-R1a expression. Cocaine at a physiologically relevant dose is able to increase plasma membrane expression of σ1R (Navarro et al. 2010). Molecules acting as σ1R agonists do the same effect, thus confirming specificity. Remarkably, both cocaine and σ1R agonists increase the colocalization of the two receptors at the cell surface. Accordingly, any drug interacting in an ‘agonistic’ manner with σ1R is able to concomitantly affect the coexpression of the two receptors at the cell surface. Direct interactions have been confirmed in brain sections of cocaine-treated animals and in an heterologous expression system (Aguinaga et al. 2019).
The in situ proximity ligation assay allows determining the occurrence of receptor complexes in natural sources (Borroto-Escuela et al.2016a). The technique showed occurrence of σ1/GHS-R1a receptor heteromeric complexes in rat brain sections. Eleven percent of cells in rat striatum displayed the fluorescent signal corresponding to those macromolecular complexes. The percentage of positive cells in animals chronically treated with the addictive drug increased to 61% and the amount of signal, measured as red clusters, also increased. The acute treatment led to a more marked increase in both the percentage of labeled cells (76%) and the degree of labeling (3.2-fold increase). The results are consistent with both occurrence of σ1/ghrelin receptor complexes and marked upregulation of those complexes upon acute or chronic cocaine treatment. Upregulation of σ1/GHS-R1a receptor heteromeric complexes was also obtained in primary cultures of striatal neurons treated with cocaine. These findings led to the hypothesis that cocaine binding to σ1 receptors could affect the ghrelin receptor-mediated signaling. The hypothesis was confirmed in a heterologous expression system and in primary cultures of striatal neurons (Aguinaga et al. 2019).
Consistent with the coupling of ghrelin receptor to heterotrimeric Gi proteins, its activation using endogenous ghrelin in the presence of forskolin significantly decreases intracellular cAMP levels. Signaling via Gi was blocked by both agonists of σ1R and by cocaine. Signal transduction in neurons expressing GHS-R1a leads to activation of the MAP kinase pathway. This signaling, that is, ERK phosphorylation triggered by GHS-R1a activation, was inhibited not only by ghrelin receptor antagonists but also by cocaine and σ1R agonists (Fig. 3). Therefore, both G-dependent and G-independent signaling becomes compromised by cocaine binding to σ1R and disappear when σ1R is silenced by a siRNA methodology (Aguinaga et al. 2019).
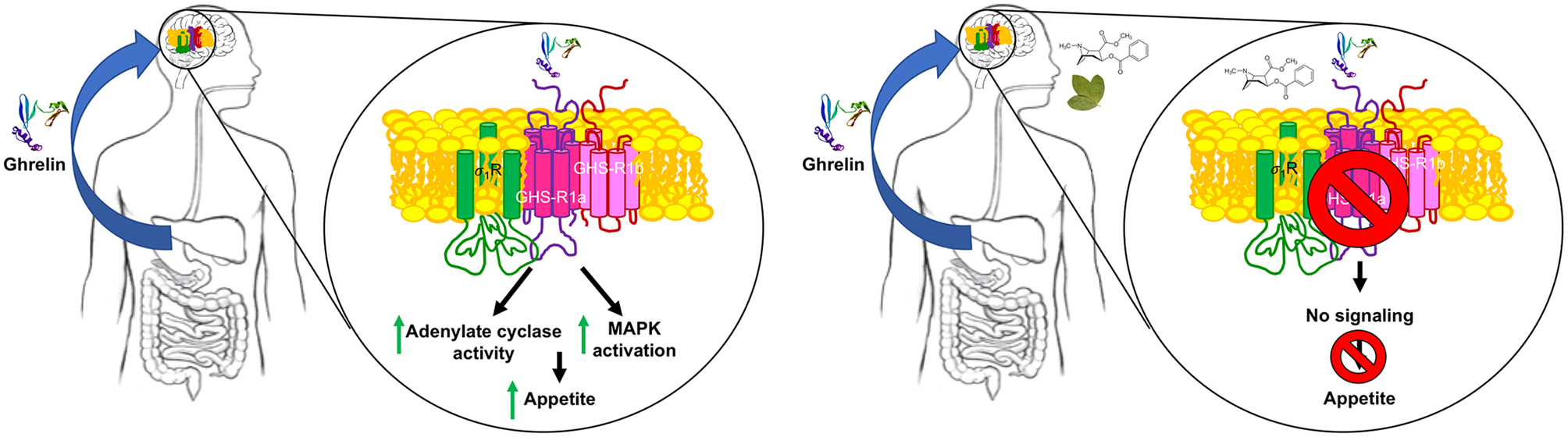
Mechanisms of ghrelin receptor-mediated cocaine-suppression of appetite. In the absence of cocaine, the peptide hormone ghrelin arrives to the CNS and activates GHS-R1a in CNS neurons to engage Gi signaling and the MAPK pathway (left). In the presence of cocaine, activation of σ1 produces conformational changes in GHS-R1a (see Aguinaga et al. 2019) that block any signaling originating at the ghrelin receptor (right). The chemical structure of cocaine is shown near the coca leaf.
A further relevant question was to know whether the cocaine effects were mediated by either σ1 containing heteroreceptor complexes or by indirect mechanisms involving second messengers or other signaling molecules. Cocaine acting as agonist of the receptor may stabilize its trimeric structure (Gromek et al. 2014). Taking advantage of the (trimeric) 3D-structure of σ1R, a model was proposed for the σ1R-GHS-R1a interaction (Schmidt et al. 2016). The model predicted that the transmembrane 1 (TM1) domain of the ghrelin receptor participates in the interaction interface, whereas TM7 does not. The issue was addressed taking advantage of interfering peptides. They have been successfully used to disrupt the structure of interactions involving GPCRs (Navarro et al. 2018). These peptides consist of receptor TM sequences followed by a short sequence of the cell-penetrating HIV transactivator of transcription (TAT) (Schwarze et al.1999). In agreement with the model provided for σ1R-GHS-R1a heteromers, the TAT-TM1 but not the TAT-TM7 blocked the effect of cocaine on GHS-R1a-mediated signaling, while the ghrelin action was still blocked by the selective ghrelin receptor antagonist. In summary, at the mechanistic level the cocaine blockade of ghrelin action occurs at a proximal level in CNS neurons by σ1Rs directly interacting with ghrelin receptors (Fig. 3).