
Proteostasis collapse is a driver of cell aging and death
By Mantu Santra, Ken A. Dill, and Adam M. R. de Graff
Excerpt from the article published in PNAS, 116(44) 22173-22178, October 16, 2019, DOI: https://doi.org/10.1073/pnas.1906592116
Editor’s Highlights
- Aging results from declining protein quality-control systems involved in protein synthesis, degradation, and chaperoning that normally protect the proteins in the cell’s proteome
- Proteostasis, the decline in protein quality control is the principal risk factor is advancing age, “probably because cell regulation and protein production and disposal becomes increasingly compromised with age”
- Chaperones are proteins that help other proteins fold. By binding to nonnative conformations and giving them a fast route to folding, Adenosine triphosphate (ATP)-dependent chaperones are responsible for maintaining a high degree of foldedness.
- Chaperones reduce the levels of unfolded, oxidation-prone conformations, thereby decreasing the rates of protein damage and aggregation.
- With age, the abundances of irreparably damaged proteins are predicted to increase, distracting the chaperones from folding the healthy proteins the cell needs.
- Chaperone Overexpression Can Extend Life Span.
SigmaR1 and SigmaR2/Tmem97 are endoplasmic reticulum (ER)-resident membrane chaperones.
Significance
Cells and organisms grow old and die. We develop a biophysical model of the mechanism. Young cells are kept healthy by the positive processes of protein synthesis, degradation, and chaperoning (the activity of keeping proteins properly folded). But, with age, negative processes increase: Oxidative damage accumulates randomly in the cell’s proteins, healthy synthesis and degradation slow down, and—like overfilled garbage cans—chaperone capacity is exceeded. The chaperones are distracted trying to fold irreversibly damaged proteins, leading to accumulating misfolded and aggregated proteins in the cell. The tipping point to death happens when the negative overwhelms the positive. The model makes several quantitative predictions of the life span of the worm Caenorhabditis elegant.
Abstract
What molecular processes drive cell aging and death? Here, we model how proteostasis—i.e., the folding, chaperoning, and maintenance of protein function—collapses with age from slowed translation and cumulative oxidative damage. Irreparably damaged proteins accumulate with age, increasingly distracting the chaperones from folding the healthy proteins the cell needs. The tipping point to death occurs when replenishing good proteins no longer keeps up with depletion from misfolding, aggregation, and damage. The model agrees with experiments in the worm Caenorhabditis elegans that show the following: Life span shortens nonlinearly with increased temperature or added oxidant concentration, and life span increases in mutants having more chaperones or proteasomes. It predicts observed increases in cellular oxidative damage with age and provides a mechanism for the Gompertz-like rise in mortality observed in humans and other organisms. Overall, the model shows how the instability of proteins sets the rate at which damage accumulates with age and upends a cell’s normal proteostasis balance.
In higher organisms, cells age and die by natural processes. What are the molecular mechanisms that drive it? It has been difficult to disentangle causes from effects because aging impacts most cellular biomolecules. Oxidative damage is known to play a key role. Much of what is known about cellular aging comes from “bottom-up” experiments, by perturbing a few genes at a time—by knockouts, knock-ins, or point mutations or by gene-to-gene comparisons using sequence databases (1). Our interest here is in the “top-down” question of the aging mechanism, which we take to be a more system-wide failure in the cell. Any single gene cannot reverse aging or abolish life span limits. Oxidative damage is indiscriminate and nonspecific in which class of biomolecule it hits or its spatial location in the cell. We take the mechanism of aging and longevity to be more about a general and stochastic destruction than a pinpoint action.
Theories of cell aging are diverse (1–5). Some theories are gene-centric, attributing primary importance to deleterious changes to DNA that accumulate throughout life (6). Such changes include mutations (7) and the progressive shortening of telomeres (8, 9). Other theories are epigenetic, focusing on how changes to DNA and DNA-binding proteins affect how genes are read. These changes include DNA methylation, histone modification, and loss of chromosomal organization (1). Further, the significant depletion of histone levels and changes in their stoichiometry (10) could alter transcription factor binding (11). Such changes may be responsible for the loss of proper messenger RNA and protein stoichiometry observed with age (12–16).
Another view is that aging results from declining protein quality-control systems involved in protein synthesis, degradation, and chaperoning that normally protect the proteins in the cell’s proteome (17–20). Central to proteostasis, the decline in protein quality control is implicated in more than 50 diseases of abnormal protein deposition (proteinopathies), for which the principal risk factor is advancing age, “probably because cell regulation and protein production and disposal becomes increasingly compromised with age” (21). Proteostasis is a natural culprit in aging because it is a front line of response to stress and because proteins are the primary repairers of the cell and sustainers of the genome (10). It has been noted that “all known gerontogenes confer resistance to stress” (22). Protein damage is a key measure of such resistance, as survival of radiation is better predicted by protein oxidation than by DNA damage (23, 24). Damage to proteins can lead to loss of their stability and function (25–27) and to impaired protein synthesis (24) and degradation (28). While protein damage is only one of a broad set of changes impacting the proteome with age (15, 29), it is an important component. We focus not on the genetics and biochemistry of any particular gene or protein, but on the cell-wide biophysics of how proteins fold (30), the impact of temperature and oxidation (25, 31), and how proteostasis declines with age.
Previous models of proteostasis have focused primarily on the mechanism of heat-shock activation (20, 32) or the means by which chaperone systems promote folding (33–35). Others have looked for broader principles determining folding yield (36) or partitioning between folding and degradation (37). While these studies have contributed to a solid understanding of protein properties upstream of damage, their downstream properties and impact remain poorly studied (20, 32). Our modeling addresses the critical impact of protein conformation on oxidizability (38–40) and the role of damaged protein in diverting the limited chaperone resources away from the folding of their undamaged counterparts (40, 41).
Premises of the Proteostasis Collapse Model
Here is our model of proteostasis collapse with age. The mathematical details, parameters, and justifications are given in Materials and Methods and SI Appendix. First, each protein in the cell has a rate of folding, unfolding, misfolding, and aggregation. Many of these rates are known (33, 43, 44). Here, we considered a single type of average protein—namely, ones that are clients of the HSP70 chaperone system, because of experiments (45) and theory (40) showing that these proteins are the dominant targets of protein oxidation. Second, we modeled the trafficking of proteins on and off of chaperones. Many of these rates, too, are known (46–49). Third, whereas these first 2 aspects—folding and chaperone trafficking—describe only normal proteostasis in young healthy cells, we included also a slow, cumulative oxidative-damage component that is known for cell growth and protein degradation (50, 51). Some of this damage will be irreversible, leading to proteins that cannot reach their functional native structures and thus are biased toward misfolding and aggregation. Of necessity, we leave out many details, such as the folding of membrane proteins, the mitochondrial generation of energy, and regulatory processes. It is not our goal to model everything. Here are the premises:
- Each protein in a cell has an intrinsic propensity to fold or misfold. Most proteins carry out their function only when in their properly folded, native conformation (17–19). Here, we model the folding process of an average HSP70 chaperone-dependent protein as it transitions between 4 conformational states: its folded, functional native state (N), unfolded state (U), misfolded state (M), and aggregated state (A) (Fig. 1). The interconversion processes are modeled by using coupled Ordinary Differential Equations (ODEs) (Materials and Methods and SI Appendix). The transition rates and equilibria between these states were previously determined (33, 40, 42), as well as their dependencies on temperature (SI Appendix).
- Each protein sustains damage at a rate determined by its foldedness. A principal cause of proteome aging is oxidative damage (27, 52). Both experimental (38, 39) and theoretical (40) studies support the idea that nonnative conformations are the major target of oxidative damage. As in previous work (40), the flux of damage 𝐽𝑑𝑎𝑚=𝑘𝑑𝑎𝑚[𝑈]Jdam=kdam[U], where 𝑘𝑑𝑎𝑚kdam is the rate constant of damage, scaling proportionately to reactive oxygen species (ROS) levels, and [U] is the concentration of unfolded protein (see SI Appendix, Table S1 for parameter values). Once oxidized, proteins are assumed to be unable to fold, as shown in Fig. 1, consistent with experiments (45).
- Chaperones modulate the foldedness and damage levels of their client proteins. Chaperones are proteins that help other proteins (their clients) fold. By binding to nonnative conformations and giving them a fast route to folding, ATP-dependent chaperones are responsible for maintaining a high degree of foldedness (53). The collective effect of these chaperones, called foldases, is modeled by taking the kinetics and concentrations of their dominant member, the HSP70 chaperones (12) (SI Appendix). Given that nonnative conformations are thought to be the major target of oxidation, chaperones play a key role in controlling damage levels (40, 45).
- Chaperones can become preoccupied handling damaged proteins that are unable to fold, preventing them from helping their undamaged client proteins that are either newly synthesized or spontaneously unfolded (54–56). Experimental support for this assumption includes the increased chaperone binding observed for proteins with disease-causing point mutations, which mirror the destabilizing effects of side-chain oxidation (25, 57, 58). Further, the presence of oxygen (and hence ROS) has been shown to significantly amplify the heat-shock response during the periods of high mistranslation, a process evoked when chaperones become titrated (45). Due to the inability of irreversibly damaged proteins to fold, their repeated binding to chaperones creates a futile cycle that can only be terminated by their degradation or aggregation.
- Proteome health also depends on the rates of synthesis and degradation of proteins. To complete the model, we therefore need to know how the rates of cell growth 𝑘𝑔𝑟(𝑡)kgr(t) and degradation 𝑘𝑑𝑒𝑔(𝑡)kdeg(t) change with age, 𝑡t. Given the highly regulated nature of these processes, it is outside the scope of the current model to predict them from first principles. Instead, since both have been measured experimentally (12, 50) (SI Appendix, Fig. S1), we take them as inputs to the model, approximating the data using exponential fits: 𝑘𝑔𝑟=𝑘𝑜𝑔𝑟𝑒−𝜆𝑔𝑟𝑡kgr=kgroe−λgrt and 𝑘𝑑𝑒𝑔=𝑘𝑜𝑑𝑒𝑔𝑒−𝜆𝑑𝑒𝑔𝑡kdeg=kdegoe−λdegt, where 𝑘𝑜𝑔𝑟kgro and 𝑘𝑜𝑑𝑒𝑔kdego are the initial values. The decay constants are determined from fitting the experimental data, giving values at 20 ○C of 𝜆𝑔𝑟=0.4d−1λgr=0.4 d−1 and 𝜆𝑑𝑒𝑔=0.14d−1λdeg=0.14 d−1, respectively. These rates are assumed to scale with temperature following Arrhenius kinetics, doubling roughly every 8 ○C, as observed for enzymatic processes (SI Appendix, Eq. S4 and Table S1).
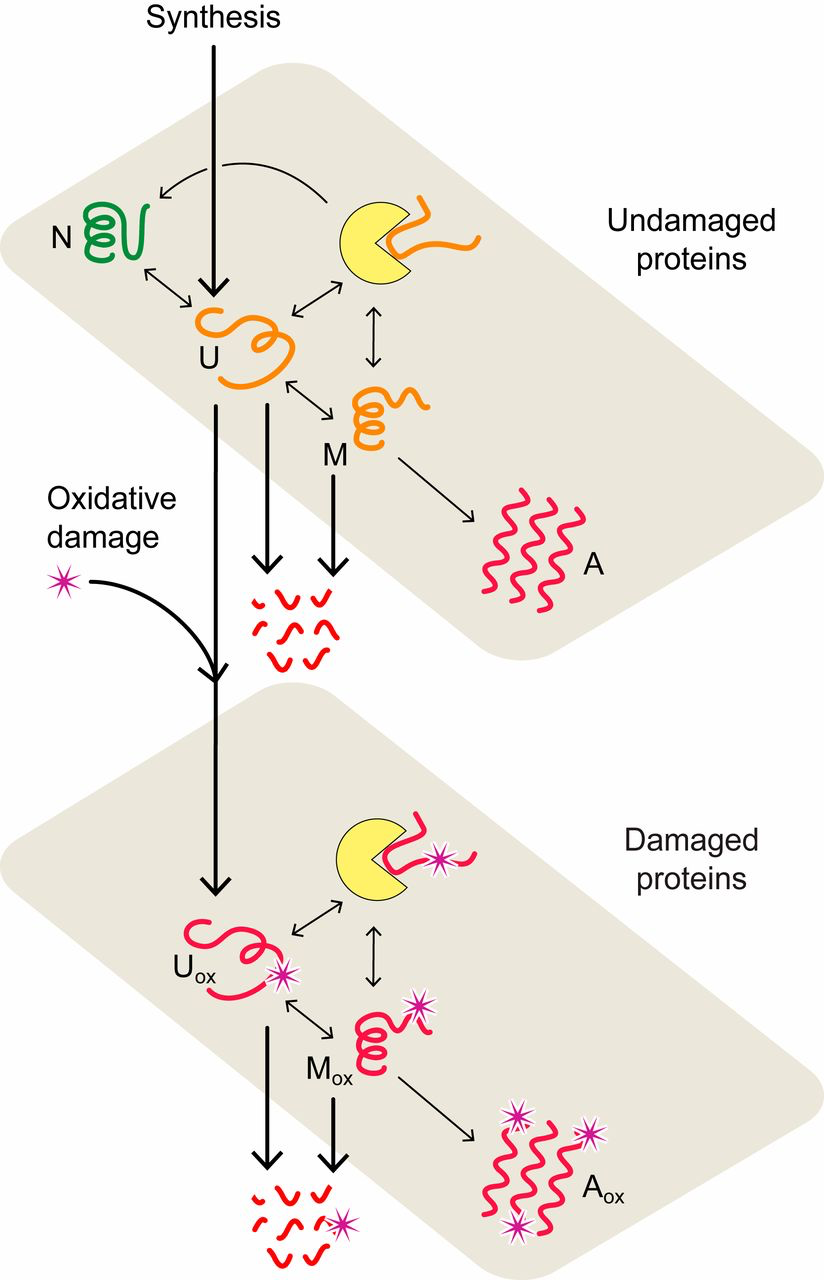
Proteostasis model of cell aging. In the model, proteins convert between native (N), unfolded (U), misfolded (M), and aggregated (A) states (upper plane) with rates estimated from experimental data (33, 42). Chaperone-dependent folding of newly synthesized or thermally unfolded proteins compete with the processes of protein oxidation, aggregation, and degradation, collectively defining the quality of the proteome and proteostasis at a given age. The lower plane describes the interconversion of oxidatively damaged unfolded (𝑈𝑜𝑥/Uox), misfolded (𝑀𝑜𝑥/Mox), and aggregated (𝐴𝑜𝑥/Aox) states. Chaperones are represented by yellow disks.
By solving the ODEs (SI Appendix and Materials and Methods) with the parameters described above, we can compute how the rates of trafficking, folding, and aggregation of an average HSP70 chaperone client protein changes over time as oxidative damage accumulates. Below, we show the resulting predictions for several properties—the survival probability 𝑆(𝑡)S(t)reflecting the fraction of the population remaining alive at time 𝑡t; the corresponding mortality rate 𝜇=−𝑑(𝑙𝑛𝑆)/𝑑𝑡μ=−d(lnS)/dt describing the death rate per individual; the distribution of protein between folded, misfolded, aggregated, and damaged states; and their dependence on temperature or added oxidizing agent—and compare them to corresponding experiments in the worm. The model gives a picture of how proteostasis collapse contributes to cell aging and death.
How Does Proteostasis Decline with Age?
Misfolding and Aggregation Grow Nonlinearly with Age.
Fig. 2A shows predicted properties as a function of age. Healthy proteostasis in young cells (the first few days in the worm) is achieved by the dilution of damaged proteins due to the high rates of growth. However, fast growth is short-lived, with protein synthesis rates declining 10-fold over the reproductive time window (12). As the synthesis of new protein slows with age, the production of healthy protein no longer keeps up with the rate of oxidative damage, causing damage levels to rise (Fig. 2 A and B). Rising damage results in increased misfolding of the proteome (Fig. 2A, orange curve), as chaperones become increasingly preoccupied in futile attempts to refold irreversibly damaged proteins. This leaves fewer chaperones to fold the healthy undamaged proteins the cell needs to function. As misfolded proteins approach their solubility limit, the concentration of aggregates increases exponentially (Fig. 2A, red curve) before reaching a plateau. This predicted sequence of events is consistent with experiments, which find: 1) an exponential rise in damage levels (Fig. 2B) (25, 27); 2) increased protein misfolding (59); and, finally, 3) the rapid onset of aggregation that appears to grow exponentially (60) before reaching a plateau (12). The model is also qualitatively consistent with the observed Gompertzian, time-dependent mortality rate and associated survival probability (Fig. 2 C and D), although it does lack late-life slowing in mortality seen experimentally (61, 62) and modeled elsewhere (62). For discussion of the mortality rate 𝜇(𝑡)μ(t) and survival probability 𝑆(𝑡)S(t), see SI Appendix.
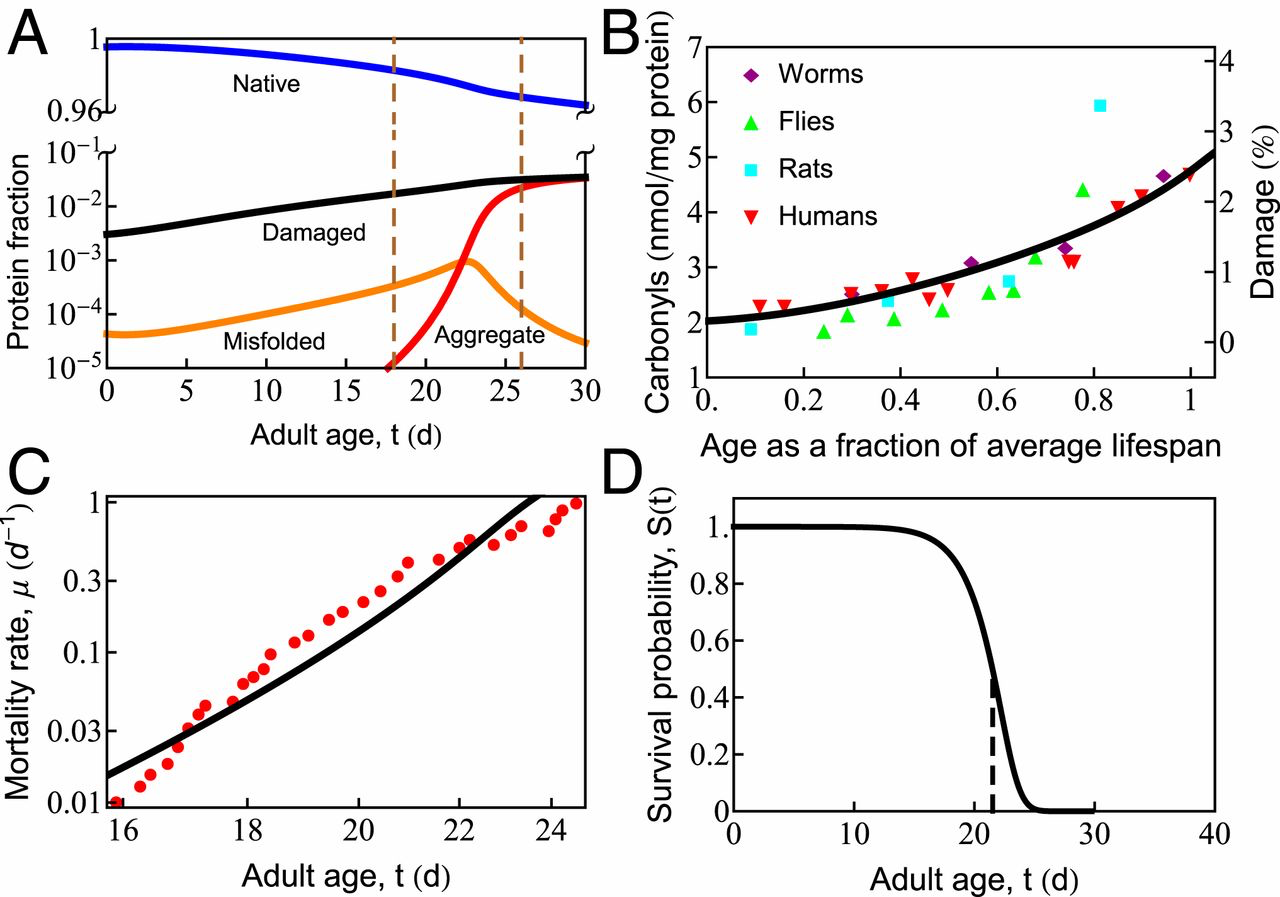
In cell aging, oxidative damage leads to increased misfolding and aggregation, which eventually causes cell death. (A) The age-dependent slowing of synthesis and degradation causes the accumulation of damaged (black line), misfolded (orange line), and aggregated (red line) states and depletion of the native (blue line) state. Progressive cell aging causes proteostasis to transition through 2 phases: 1) the progressive titration of chaperones by damaged proteins, causing the accumulation of misfolded protein, followed by 2) misfolded proteins crossing their solubility threshold, triggering late-life aggregation (the region inside the vertical dashed lines). (B) The gradual increase of protein damage with age in different organisms, as measured by carbonyl content. The black line indicates the percentage of damaged protein predicted by the current model. (C) Age-dependent mortality rate data at T = 20.1 ○C (red symbols). The black line is the theoretical fit to the data. (D) Predicted survival curve in wild-type Caenorhabditis elegansat T = 20 ○C. The vertical dashed line shows the point of 50% survival and is taken as the average life span of the organism.
How Does Life Span Depend on Environmental Properties?
Life Span Shortens with Increasing Temperature.
In simple organisms, life span has a remarkable and strong dependence on temperature (51, 61). Maintained at higher temperatures, organismal life span shortens considerably. For example, Caenorhabditis elegans grown at 20 ○C lives about 22 d, yet at 25 ○C, they live only 10 d (61). Qualitatively, such observations appear to support the Rate-of-Living Hypothesis (63)—namely, that life span is shorter at higher temperatures because of faster metabolism, which, in turn, is expected from the physical chemistry of Arrhenius kinetics dictating the temperature-dependent acceleration of biochemical reactions (64, 65). In that theory, the rate of aging is proportional to the speed of these reactions, so a plot of the logarithm of the inverse life span (which is a rate of living) vs. 1/T (T is the absolute temperature) should give a straight line with a slope equal to the activation energy of the process. However, Fig. 3C shows that life span follows a more complex behavior than the Rate-of-Living Hypothesis would predict. Our model gives the following interpretation.
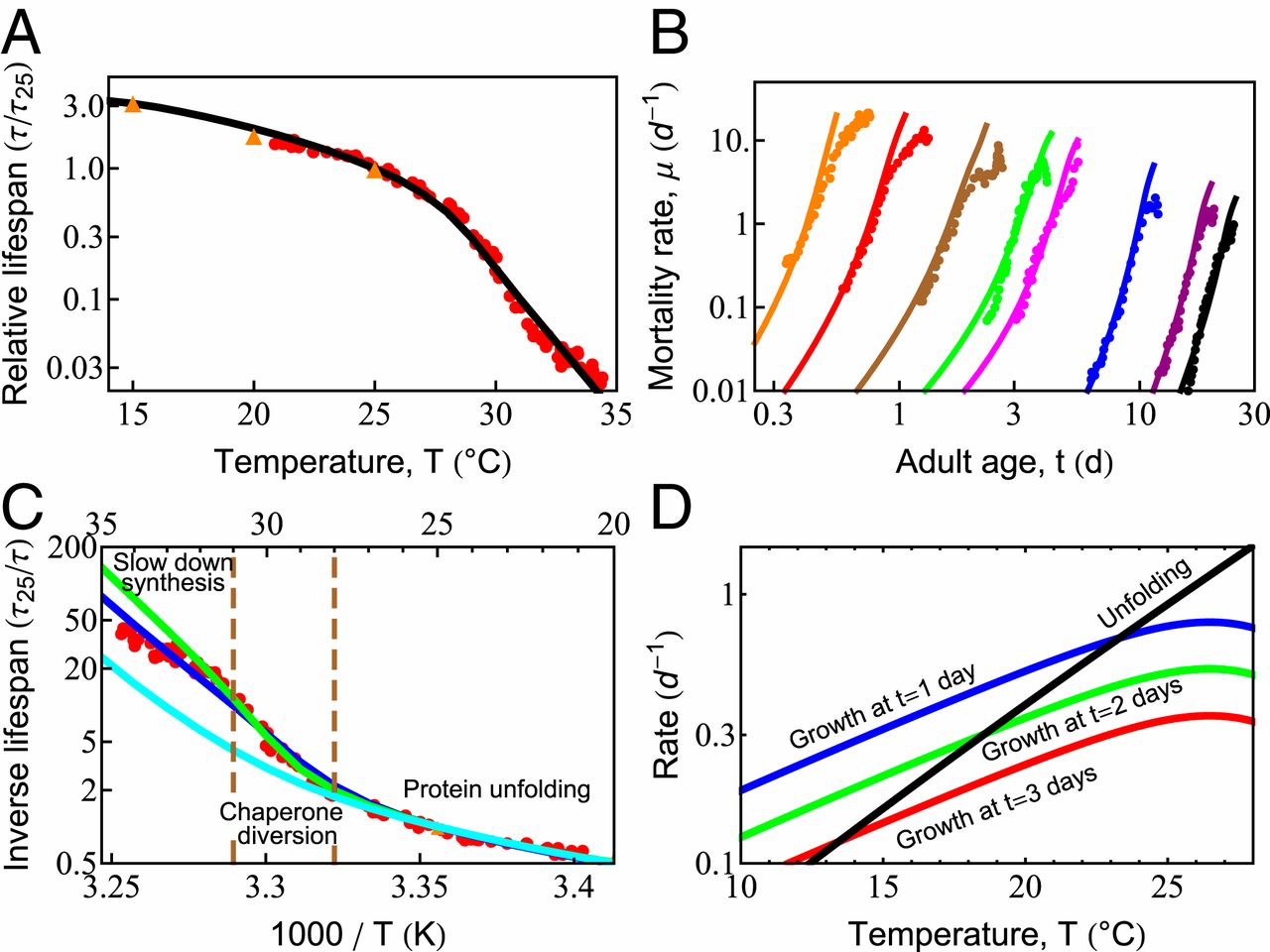
Temperature dependence of survival probability and life span is dictated by the fundamental properties of proteins. (A) Predicted decay of life span with increasing temperature (black line), compared with experimental data from Stroustrup et al. (61) (red circles) and Van Voorhies and Ward (51) (orange triangles). (B) Predicted rates of mortality at different temperatures (solid lines) compared with experimental data from Stroustrup et al. (61) (symbols). The temperatures are, from black to orange, 20 ○C (20.1 ○C), 22.1 ○C (23.7 ○C), 25.5 ○C (25.2 ○C), 28.3 ○C (29.1 ○C), 28.9 ○C (30 ○C), 30 ○C (30.9 ○C), 31.3 ○C (31.3 ○C), and 32.4 ○C (32.5 ○C); the values in the brackets are those reported in Stroustrup et al. (61). (C) The inverse life span as a function of inverse absolute temperature shows distinct regions in which different processes dominate life-span behavior. The blue line represents the prediction of the current model. The green line is the prediction in the absence of the slowed growth rate observed experimentally at high temperatures (SI Appendix, Fig. S2, orange line). The cyan curve represents the prediction if damaged proteins are not allowed to bind and effectively deplete chaperones. (D) With increasing temperature, the average unfolding rate of proteins (black line) increases faster than enzymatic processes like protein synthesis, shown at 1, 2, and 3 d of adulthood (blue, green, and red lines, respectively).
Heating at normal temperatures between 15 °C and 24 °C increases metabolism.
Across the proteome, and across species, enzymatic rates are known to roughly double for every 8 ○C rise in temperature, following Arrhenius kinetics (SI Appendix, Eq. S4). Around room temperature, the dominant proteostasis processes—synthesis, folding, degradation, and their rates of decay—all scale similarly with temperature, causing life span to scale inversely with metabolic rate. Thus, over this narrow temperature range, life span scales consistently with the Rate-of-Living Hypothesis (63).
At Temperatures from 24 °C to 28 °C, Protein Unfolding Rates Begin to Exceed Synthesis Rates.
Protein unfolding rates have a steeper temperature dependence than other proteostasis processes, roughly doubling every 4 ○C (66, 67). By using kinetics inferred previously for HSP70 clients (33), unfolding rates are predicted to catch up to the rates of protein synthesis in early adulthood within this temperature range (Fig. 3D). This rapid acceleration of spontaneous unfolding of proteins is a central reason why greater chaperone levels are beneficial at higher temperatures, regulated through the heat-shock response (68). Yet, oddly, this stress response is turned off very early in C. elegans adulthood (69). Other stress-response pathways such as the mitochondrial unfolded protein response and the endoplasmic reticulum unfolded protein response are also strongly down-regulated (59) by a mechanism that can be modulated by gonadal signaling (70–72) and dietary restriction (73). Given that a single temperature was used to raise the worms (61), our model therefore uses a single, constant chaperone level for all adult temperature conditions and ages (12).
Heating at Temperatures Above 28 ○C Rapidly Fills Available Chaperone Capacity, Forcing Protein Synthesis to Slow.
Life span is predicted to shorten much more rapidly at temperatures above 28 ○C, as unfolding loads begin to overshadow synthesis (SI Appendix, Effective Parameters). The temperature dependence of the total unfolding burden faced by the cell thus transitions from a synthesis-dominated regime, where the burden doubles every 8 ○C, to an unfolding-dominated regime, where the burden doubles every 4 ○C. At such high unfolding loads, we hypothesized that life span might be particularly sensitive to the burden from damaged proteins. To test this, we modeled aging with (blue) and without (cyan) the ability for damaged proteins to bind chaperones. Consistent with our hypothesis, chaperone distraction by damaged proteins begins decreasing life span between T = 28 °C and 31 °C (Fig. 3C). This sensitivity is further compounded by the rising levels of unfolded, oxidation-prone proteins at these temperatures. Together, these processes provide a dangerous feedback loop that may explain why the heat-shock response is highly sensitive to the presence of damaging free radicals (41).
The increasing diversion of chaperones away from the folding of newly synthesized proteins may also be why, above 28 ○C, an additional behavior is observed experimentally: Growth begins to slow (SI Appendix, Fig. S2). In this extreme heat, slowing growth may be one of the few mechanisms the cell has to diminish the folding burden and delay proteostasis collapse. To test this, we modeled aging with (blue) and without (green) slowed growth (Fig. 3C). The difference between the 2 curves suggests that slower synthesis provides a modest benefit to life span, predicted to make a 2-fold difference by T = 35 ○C. It reiterates a key result observed throughout our modeling—namely, that proteostasis capacity is tightly coupled to the rate of protein synthesis.
In short, while our model does show a component of life span that is attributable to the rate-of-living via metabolic speed-up, it also shows an additional component based on cell stresses. As the cell is stressed by heat, proteins unfold, misfold, and aggregate. Chaperones are recruited, but with age, the synthesis of “good protein” and the chaperoning of those spontaneously unfolding ultimately succumb to damage levels, at which bad protein becomes overwhelming.
Life Span Shortens with Added Oxidant Concentration.
Since oxidation is a well-known aging agent, adding an oxidant to cells should decrease their life span, as shown in recent experiments (61). To explore the behavior predicted from the proteostasis model, we assumed that damage to the proteome occurs in linear proportion to the amount of oxidant, consisting of the basal level of ROS made by the cell as well as that from the added oxidant tert-butyl hydroperoxide (tBuOOH). The damage rate can be expressed as 𝑘𝑑𝑎𝑚=𝑘𝑜𝑑𝑎𝑚(1+[𝑡𝐵𝑢𝑂𝑂𝐻]𝑐𝑜)kdam=kdamo(1+[tBuOOH]co), where 𝑘𝑜𝑑𝑎𝑚kdamo is the rate of damage in the absence of tBuOOH and 𝑐𝑜co is a constant with units of concentration, determined by the best fit to experimental data. Fig. 4A shows that the predicted life span (black curve) agrees well with the experimental measurements [red points; Stroustrup et al. (61)]. Life span changes very little at low levels of tBuOOH, as it makes only a small contribution to the overall damage rate. However, beyond a critical value, life span drops sharply. The age-dependent rise in protein damage is shown in Fig. 4B for 3 values of [tBuOOH] (shown by vertical dashed lines in Fig. 4A). Consistent with the experimental observation, the extent of damage at death (Fig. 4B, vertical dashed lines) increases with shortening life span (74, 75).
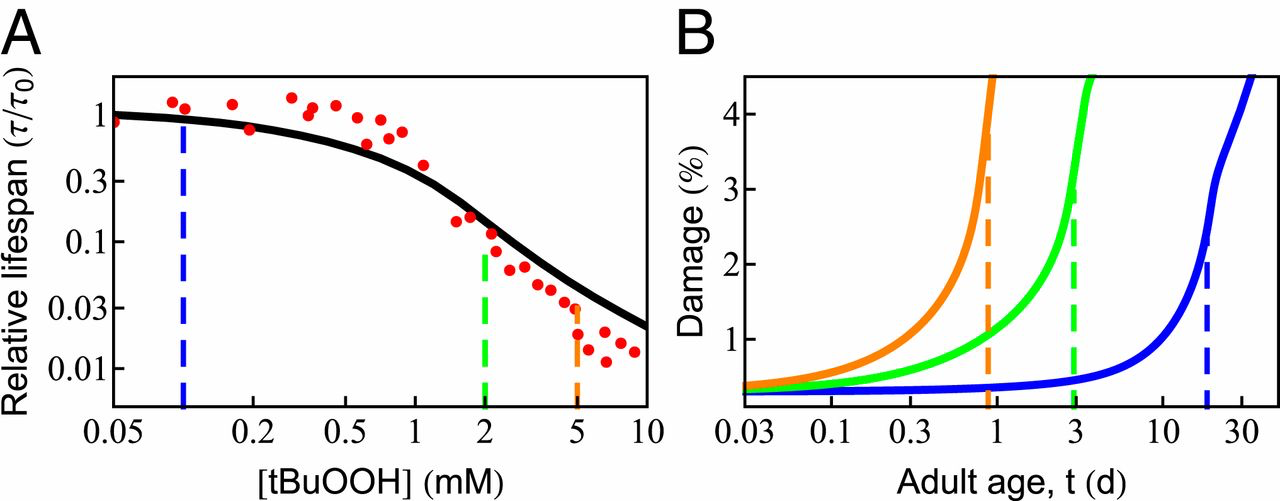
Effect of oxidative damage on survival probability and life span. (A) The predicted dependence of life span on the concentration of external oxidant, tBuOOH (black solid line), compared with experimental data [red circles; Stroustrup et al. (61)]. Beyond a critical concentration (0.5 mM), life span drops sharply in an approximately power-law fashion. (B) The age-dependent rise in protein damage is shown for 3 concentrations of tBuOOH (indicated by vertical dashed lines in A). The vertical dashed lines in B indicate the average life span.
How Does Life Span Depend on Cellular Properties?
Chaperone Overexpression Can Extend Life Span.
The model gives an accounting for how chaperone levels can affect a cell’s aging and longevity. Chaperones reduce the levels of unfolded, oxidation-prone conformations, thereby decreasing the rates of protein damage and aggregation (76). Proteins are susceptible to damage when they are synthesized, as well as during each thermal unfolding event, which occurs in our model at a rate 𝑘𝑢≈0.4d−1ku≈0.4 d−1 at T = 20 ○C (SI Appendix, Table S1). To explore this dependence more precisely, we predicted life span for chaperone levels ranging from 80 to 300% of the wild-type value at 3 different temperatures, as shown in Fig. 5A. The model predicts that at T = 25 ○C, an 80% increase in chaperone level can increase life span by ≈≈45%. This is in quantitative agreement with the experimental observation (77). Extension of life span by increasing chaperone levels has also been observed in other studies on C. elegans (78, 79), fruit flies (80), and mice (81). Although there are apparent exceptions, such as the acceleration of neurodegeneration upon overexpression of HSP90 in mice (82), it should be noted that HSP90 can be a negative regulator of the HSP70 chaperones studied here. Interestingly, life-span extension from increased chaperone availability is predicted to have a strong nonlinear temperature dependence, rising sharply above 28 ○C (Fig. 5B, solid line), coincident with the sharp rise in chaperone diversion and the inhibition of protein synthesis observed experimentally (SI Appendix, Fig. S2). To test if damaged proteins play a central role in this model behavior, we turned off the ability of damaged proteins to bind chaperones. When we did, the strong nonlinear behavior largely disappeared (Fig. 5B, dashed line). As chaperones cannot be strongly up-regulated in adult worms (69), Fig. 5B shows the importance of freeing up chaperone capacity by decreasing synthesis at high temperatures.
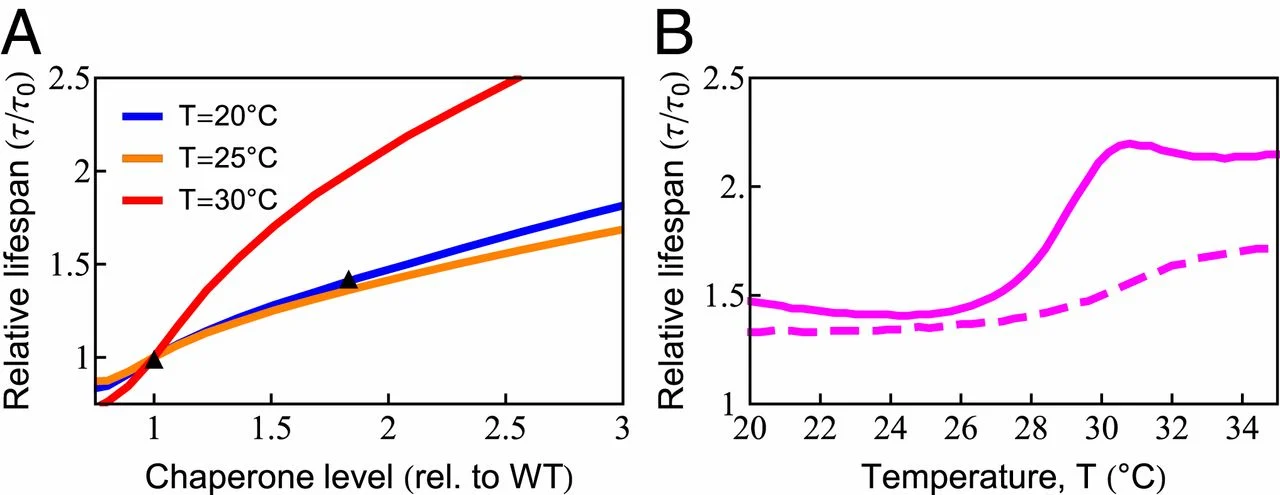
Increased chaperone capacity extends life span. (A) Chaperone dependence of relative (rel.) life span (𝜏/𝜏𝑜), where 𝜏𝑜
is the life span with wild-type [WT] chaperone level) at T = 20 ○C (blue line), T = 25 ○C (orange line), and 𝑇=30○C (red line). The predicted increase of life span (orange line) quantitatively captures the experimentally observed life span extension at 𝑇=25○C
(black triangles) (77). (B) The temperature dependence of life-span extension due to a 2-fold increase in chaperone level with (solid line) and without (dashed line) the ability of chaperones to bind, and thus be titrated by, damaged protein.
Overexpressing the Degradation Machinery Extends Life Span.
The model also predicts the dependence of cellular life span on degradation rates. Proteasome overexpression has been shown to extend life span by directly ridding the cell of protein damage and indirectly by controlling the levels of regulatory proteins (83). By up-regulating degradation capacity in the model, we found—similar to chaperones—that life span can be extended significantly (83) (Fig. 6A). The amount of benefit also showed nonmonotonic behavior due to chaperone diversion by damaged proteins (Fig. 6B). Consistent with these predictions, up-regulating 26S proteasomal activity showed negligible benefits at 20 ○C, yet increased life span by 25% at 25 ○C (71). However, experiments showed continued life-span benefit at 34 ○C, where death normally occurs in less than a day, suggesting that our model may not be adequately capturing certain processes under such severe stress conditions.
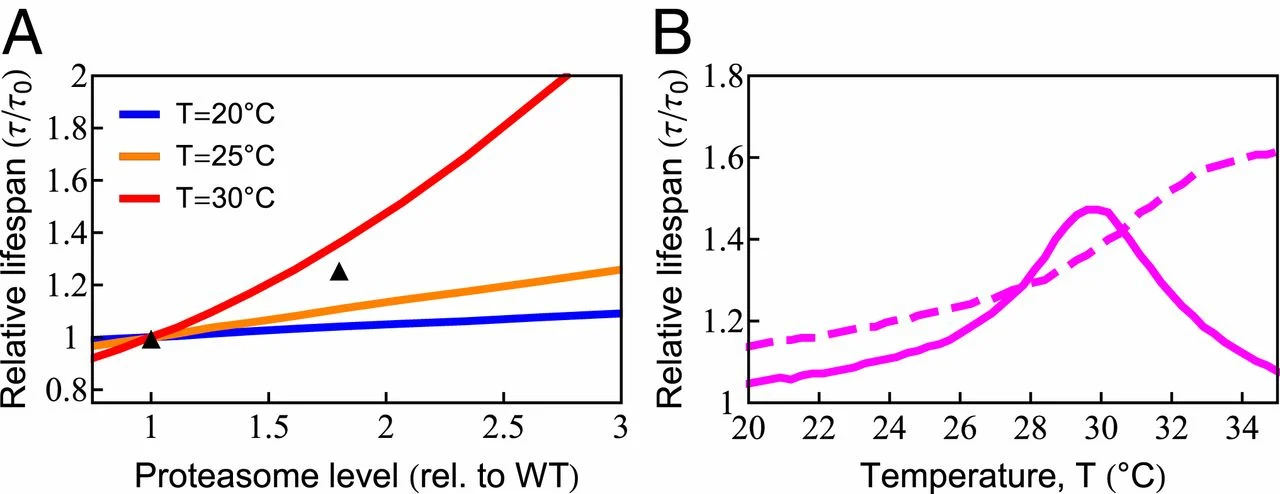
Increased degradation capacity extends life span. (A) Increasing degradation capacity is predicted to increase life span at different temperatures. The black symbols are experimental data at 𝑇=20○C (83). rel., relative; WT, wild type. (B) The benefit of increased degradation capacity depends highly on temperature (solid line). The dashed line is the model prediction in the absence of chaperone diversion.
Conclusions.
We describe how proteostasis declines with age in the worm C. elegans. The model explains 2 remarkable experimental results from ref. 61—namely, that worms’ life span decreases by 2 orders of magnitude when they are grown in heat or added oxidant. Most importantly, the model gives an underlying mechanistic explanation for how this dependence emerges from well-known properties of protein-folding kinetics and the targeting of nonnative protein conformations by oxidants. This, in turn, provides a mechanism for one of the most prominent features of aging—namely, the nonlinear increase in death rate with age, called Gompertz Law. With age, the abundances of irreparably damaged proteins are predicted to increase, distracting the chaperones from folding the healthy proteins the cell needs. The tipping point to death occurs when replenishing good proteins no longer keeps up with losses from damage and aggregation. The progressive aging of the proteome is seen to be a direct consequence of slowed protein synthesis and turnover, relative to the relentless onslaught of damage.