Post-Translational Modification of Cav1.2 and its Role in Neurodegenerative Diseases

By Yun Li, Hong Yang, Tianhan He, Liang Zhang, and Chao Liu
Excerpt from the article published in Frontiers in Pharmacology, volume 12, 17 January 2022 , DOI: https://doi.org/10.3389/fphar.2021.775087
Editor’s Highlights
- Cav1.2, encoded by the CACNA1C gene, is a high-voltage-activated (HVA), long-lasting (L-type), and dihydropyridine (DHP)-sensitive calcium channel.
- Cav1.2 plays important roles in the cardiovascular system, the CNS, and endocrine glands. In the brain, it mediates learning and memory, drug addiction, and neuronal development.
- Cav1.2 is a multi-protein complex that generally consists of three subunits: a pore-forming subunit α1, a β subunit, and an α2δ subunit; in skeletal muscle, a γ subunit is also found.
- The α1 subunit contains about 2000 amino acid residues, which forms four homologous domains (DI–DIV) connected by intracellular loops.
- Each domain consists of six transmembrane segments: S1 to S6. Of these, S5 and S6 form the pore; and the S4 segment serves as a voltage sensor.
- The α2δ subunit, an integral component of Cav1.2, is highly N-glycosylated by a 30-kDa oligosaccharide.
- Post-translational modulation (PTM) is a process that converts synthesized proteins to mature proteins through covalent or enzymatic modifications.
- Cav1.2 undergoes a series of PTMs before it becomes a mature and functional Ca2+ channel on the cell surface.
- Mutation of only 6/16 asparagine glycosylation sites was sufficient to decrease cell surface expression and protein stability of α2δ1 subunit, as well as α2δ1-mediated peak current density and voltage-dependent gating of the α1C subunit.
- Single mutation N663Q and double mutations N348Q/N468Q, N348Q/N812Q, and N468Q/N812Q decreased protein stability and abolished cell surface expression of α2δ1 as well as the α2δ1-induced up-regulation of Cav1.2 currents.
- The relative lack of selectivity of DHPs is a big concern for their use in the CNS; unwanted effects may arise from antagonism of Cav1.3 channels.
Abstract
Cav1.2 plays an essential role in learning and memory, drug addiction, and neuronal development. Intracellular calcium homeostasis is disrupted in neurodegenerative diseases because of abnormal Cav1.2 channel activity and modification of downstream Ca2+ signaling pathways. Multiple post-translational modifications of Cav1.2 have been observed and seem to be closely related to the pathogenesis of neurodegenerative diseases. The specific molecular mechanisms by which Cav1.2 channel activity is regulated remain incompletely understood. Dihydropyridines (DHPs), which are commonly used for hypertension and myocardial ischemia, have been repurposed to treat PD and AD and show protective effects. However, further studies are needed to improve delivery strategies and drug selectivity. Better knowledge of channel modulation and more specific methods for altering Cav1.2 channel function may lead to better therapeutic strategies for neurodegenerative diseases.
Introduction
Cav1.2, encoded by the CACNA1C gene, is a high-voltage-activated (HVA), long-lasting (L-type), and dihydropyridine (DHP)-sensitive calcium channel. Cav1.2 mediates depolarization of the cell membrane potential, calcium (Ca2+) influx, and activation of intracellular Ca2+ signaling cascades that alter gene expression, protein phosphorylation, and neurotransmitter release. Cellular excitability and signal transduction are affected by factors that modulate Cav1.2 activity. Cav1.2 channels are located in the cardiovascular system, the nervous system, and endocrine glands (Mikami et al., 1989), where they serve important physiopathological functions; for example, gain-of-function mutations in the CACNA1C gene cause Timothy Syndrome (Splawski et al., 2004; Moon et al., 2018). In neurons, two different L-type calcium channels (LTCCs) are expressed: Cav1.2 and Cav1.3 (Hell et al., 1993; Ertel et al., 2000). Cav1.2 is the major calcium channel isoform in neurons, constituting about 80% of neuronal LTCCs (Hell et al., 1993). Cav1.2 participates in learning and memory, drug addiction, and neuronal development (Striessnig et al., 2014). Large-scale genome-wide association studies have shown a strong association between susceptibility to psychiatric disorders and single nucleotide polymorphisms (SNPs) in the CACNA1C gene (Bhat et al., 2012). Yet, understanding of Cav1.2 function in the brain and its role in neurodegenerative disease remains limited.
The genetic regulation and channel modulation of Cav1.2 have been studied intensively. At the post-transcriptional level, alternative splicing of Cav1.2 increases protein diversity. Different splice variants have distinct channel properties, with tissue- and disease-specific variability (Wang et al., 2006). At the post-translational level, Cav1.2 is altered by a variety of modifications, which will be further discussed below.
Cav1.2 is an important drug target in the cardiovascular system. DHPs form a class of LTCC blockers and are the most widely prescribed drugs for hypertension and myocardial ischemia (Zamponi et al., 2015). In this review, we summarize the post-translational modifications of Cav1.2 and its role in neurodegenerative diseases, and further discuss the potential of Cav1.2 as a drug target for Alzheimer’s disease (AD) and Parkinson’s disease (PD).
Structure and Function of Cav1.2 in the CNS
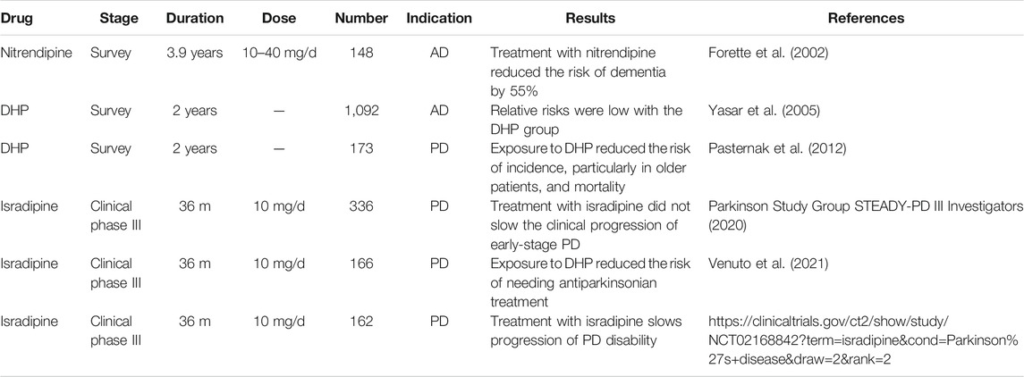
Voltage-gated calcium channels play an important role in neuronal function (Goonasekera et al., 2012). Cav1.2 is a multi-protein complex. It generally consists of three subunits: a pore-forming subunit α1, a β subunit, and an α2δ subunit; in skeletal muscle, a γ subunit is also found (Goonasekera et al., 2012). The α1 subunit contains about 2000 amino acid residues, which forms four homologous domains (DI–DIV) connected by intracellular loops (Dai et al., 2009; Alves et al., 2019). Each domain consists of six transmembrane segments: S1 to S6 (Dai et al., 2009; Alves et al., 2019). Of these, S5 and S6 form the pore; and the S4 segment serves as a voltage sensor. The gating mechanism is shown in Figure 1. At rest, the S4 segments stay inward (“down”) under the influence of the electrical field and lock the channel in its closed state. In this state, the S6 helices converge on the intracellular side, preventing ion penetration. When the membrane is depolarized, the S4 segments are released and move outward. The pore will be unlocked when all four S4 segments leave the “down” position. During continuous depolarization, the S6 gate disengages. When all the four S6 segments disengage and are in the “up” position, the pore opens. When returned to the resting potential, the deactivated voltage-sensing segment moved toward a “down” position while the pore is still open. Subsequently, the channel returns to its closed conformation at a rest state (Beyl et al., 2009; Hering et al., 2018).
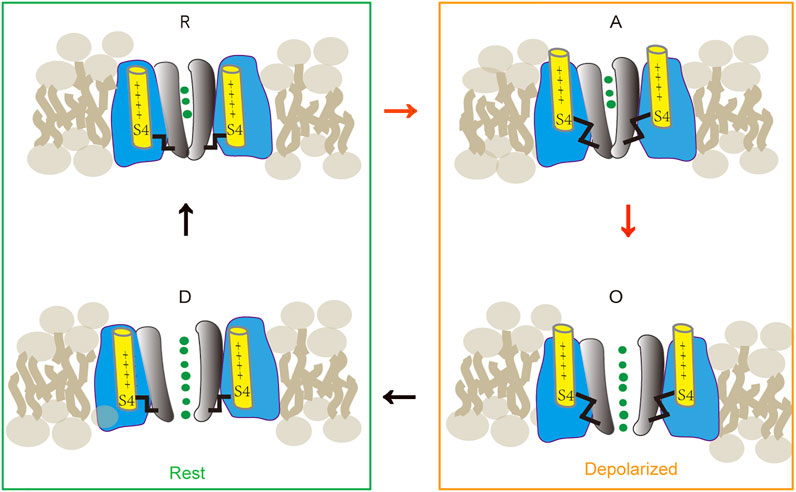
Cav1.2 state transitions during activation [modified after (Beyl et al., 2009) (Hering et al., 2018)]. The channel gating is determined by two functionally distinct processes: a voltage-sensing mechanism and a conducting pore. These two processes defined 4 states: R, at rest, pore is closed and S4 segments in the “down” position lock the pore. A, when depolarized, voltage-sensing mechanism is activated and S4 segments move to the “up” position and release the pore; but the pore remains closed. O, during continuous depolarization, all four S4 segments are in the “up” position; the pore is open. D, when returned to the resting potential, the deactivated voltage-sensing segment moved toward a “down” position while the pore is still open. Subsequently, the pore will transit to its closed conformation and at a resting state.
The α1 subunit is the binding site of most regulators and drugs that act on the channel (Zamponi et al., 2015), whereas the main functions of the other subunits are transportation, anchoring, and regulation (Hofmann et al., 2014). Cav1.2 channels usually require intense depolarization to activate and have long-lasting activity (Hofmann et al., 2014). Ca2+ entering through Cav1.2 participates in a series of physiological processes as an important second messenger.
Cav1.2 is distributed universally in the brain. In humans, moderate-to-high mRNA level is detected in the cerebral cortex, the pituitary gland, the amygdala, the basal ganglia, and the cerebellum (Splawski et al., 2004). In mice, the olfactory region, the basal ganglia, the hippocampal formation, the amygdala, and the thalamus show moderate-to-high mRNA level of Cav1.2 (Hell et al., 1993; Splawski et al., 2004; Hetzenauer et al., 2006). At the protein level, the hippocampal formation, the thalamus, and the hypothalamus have moderate-to-strong signal intensity. At the subcellular level in neurons, Cav1.2 is in the soma and at the synapses (Alves et al., 2019).
Cav1.2 plays an important role in the regulation of synaptic plasticity. Researchers found that mice with an inactivated form of the CACNA1C gene in the hippocampus and neocortex display severely impaired hippocampus-dependent spatial memory (Moosmang et al., 2005). Cav1.2 is involved in the formation of long-lasting long-term potentiation (LTP) in the hippocampus (Moosmang et al., 2005; Moon et al., 2018; Nanou and Catterall, 2018). Long-lasting LTP needs activation of gene expression and protein synthesis (Malenka and Bear, 2004). The calcium entry from Cav1.2 activates Calmodulin-dependent protein kinase II (CamKII), which binds the C-terminus of Cav1.2; and downstream CamKIV, which phosphorylate CREB and activate downstream gene expression (Cohen et al., 2015). In another pathway, the calcium-regulated phosphatase calcineurin that binds to the C-terminal domain of Cav1.2 is also activated and dephosphorylates the transcription factor NFAT (nuclear factor of activated T-cells), allowing it to translocate into the nucleus and activate gene expression (Murphy et al., 2014). The above signaling cascade increases the synthesis of mRNA encoding synaptic proteins, causing long-lasting changes in synaptic function (Nanou and Catterall, 2018). Moreover, recent studies have found a β2-adrenergic receptor and Cav1.2 signaling complex that regulates synaptic plasticity. β2-adrenergic receptors affect calcium channel activity and long-term postsynaptic plasticity through their interactions with the C-terminus of Cav1.2 channels (Qian et al., 2017).
During aging, the viability of Cav1.2 channels increases, leading to high intracellular calcium (Navakkode et al., 2018) that may modulate the processing of amyloid precursor protein (APP) and promote AD pathogenesis (Anekonda and Quinn, 2011). The calcium hypothesis of AD holds that disturbing the intracellular Ca2+ balance affects intracellular signal transmission, leading to the formation of Aβ plaques and neurofibrillary tangles, which alter the plasticity of synapses and ultimately lead to the death of neurons (Khachaturian, 1989). Furthermore, Ca2+ imbalance promotes the phosphorylation of tau and leads to disordered autophagy in neurons (Anekonda and Quinn, 2011). Endoplasmic reticulum stress (ER stress) and subsequent tau hyperphosphorylation are increased in human chronic traumatic encephalopathy. Administration of docosahexaenoic acid, an endoplasmic reticulum stress inhibitor, lowers intracellular calcium concentration, which results in the decrease of tau hyperphosphorylation and improves cognitive performance (Begum et al., 2014; Lucke-Wold et al., 2016). Separately, salubrinal, a modulator of cellular stress, can reduce neuroinflammation in mice via decreasing ER stress and oxidative stress (Logsdon et al., 2016).
Post-Translational Modulation of Cav1.2 and its Role in Neurodegenerative Diseases
Post-translational modulation (PTM) is a process that converts synthesized proteins to mature proteins through covalent or enzymatic modifications. These modifications range from the enzymatic hydrolysis of peptide bonds to the covalent addition of specific chemical groups, lipids, carbohydrates, and even entire proteins and amino acid side chains. These chemical modifications after polypeptide chain biosynthesis expand the scope of the amino acid structure and properties, thereby diversifying the structure and function of proteins. PTM can occur at any point and regulates protein activity, localization, and interactions with other molecules (Knorre et al., 2009; Walker and Nestler, 2018).
Cav1.2 undergoes a series of PTMs before it becomes a mature and functional Ca2+ channel on the cell surface. These modifications influence the channel properties, trafficking, and location and hence significantly alter the channel function. Cav1.2 modification is dramatically changed in neurodegenerative disease and may be an important component of the pathology (summarized in Figure 2).
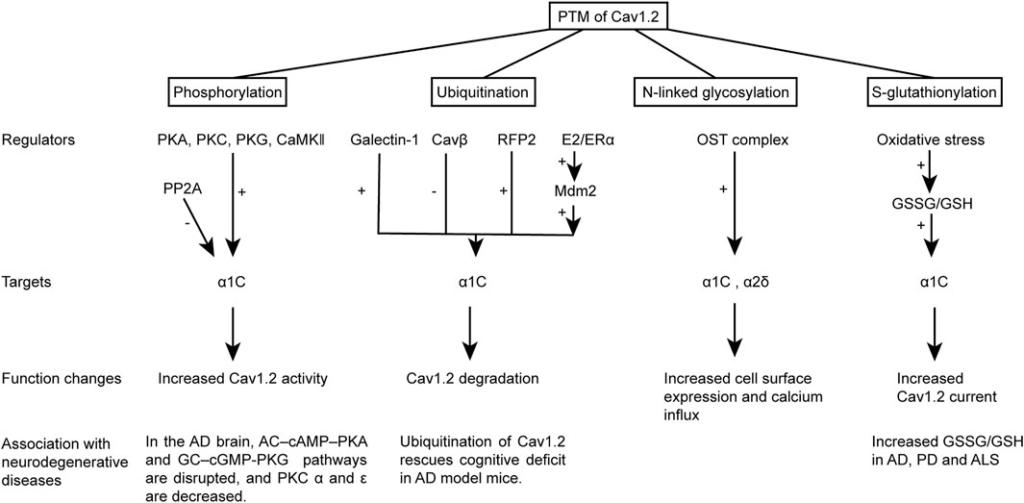
Schematic representation of the PTM of Cav1.2 and its correlation with neurodegenerative diseases. The PTM changes the channel activity, degradation, and cell surface expression of Cav1.2. The PTM of Cav1.2 or the disruption of their regulating pathways was also observed in neurodegenerative diseases.
Phosphorylation
Phosphorylation of a molecule is the attachment of a phosphoryl group. Protein phosphorylation is the most abundant post-translational modification in eukaryotes. Phosphorylation can occur on serine, threonine, and tyrosine side chains (often called “residues”) through phosphoester bond formation. Neural cells contain a plethora of protein kinases, protein phosphatases, and phosphorylated proteins, and many of these are essential for the regulation of neuronal morphology and for cell functions as diverse as membrane excitability, secretory processes, cytoskeletal organization, and cellular metabolism.
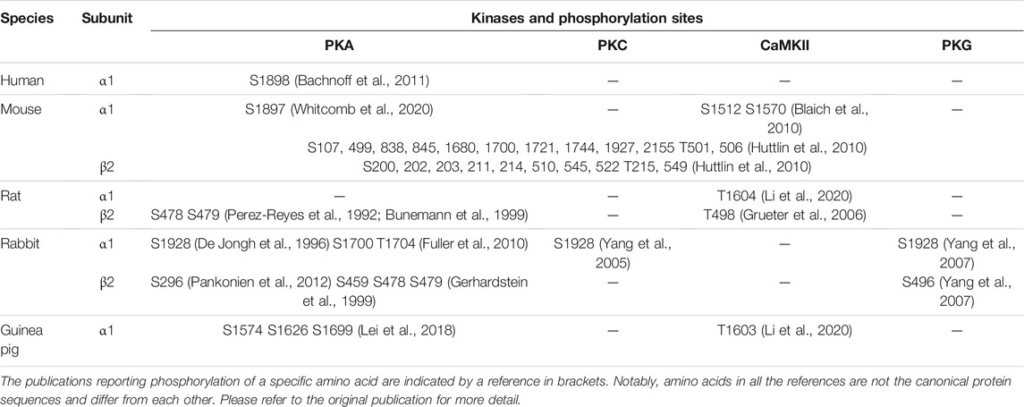
Phosphorylation of Cav1.2 channels can enhance Ca2+ influx four- to six-fold (Sculptoreanu et al., 1993; Kavalali et al., 1997). Cav1.2 channels can be phosphorylated by many protein kinases (PKA, PKC, PKG, and CAMKII) but, in most cases, the sites regulated by these kinases remain uncertain. The identified phosphorylation sites in Cav1.2 are summarized in Table 1 (Perez-Reyes et al., 1992; De Jongh et al., 1996; Gerhardstein et al., 1999; Yang et al., 2005; Grueter et al., 2006; Gui et al., 2006; Yang et al., 2007; Blaich et al., 2010; Fuller et al., 2010; Huttlin et al., 2010; Bachnoff et al., 2011; Brandmayr et al., 2012; Pankonien et al., 2012; Lei et al., 2018; Li et al., 2020; Whitcomb et al., 2020). The central subunit of Cav1.2, α1C, is the major subunit involved in the PKA-mediated increase in channel activity. The α1C subunit is phosphorylated by PKA in intact hippocampal neurons, and a two-fold increase in Ca2+ influx has been observed in hippocampal neurons in old rats compared with adult rats, suggestive of increased PKA phosphorylation of Cav1.2 with aging. S1700 phosphorylation plays a greater modulatory role than S1928 phosphorylation in the heart, which is crucial for calcium homeostasis in cardiomyocytes and prevention of heart failure (Yang et al., 2016).
However, only S1928 has been shown to increase with normal aging in the hippocampus (Davare and Hell, 2003) and S1928 is important for the upregulation of channel activity by PKA. Protein phosphatase 2A (PP2A) constitutively bound to Cav1.2 is required for dephosphorylation of S1928 and subsequent down-regulation of Cav1.2 channel activity (Xu et al., 2010). Similar to PKA, PKC can also phosphorylate α1C at the same site (Weiss et al., 2012). The channel activity of Cav1.2 increases because of the convergence of the two kinases. PKC α and ε expression is decreased with aging in the prefrontal cortex and hippocampus (Perovic et al., 2013), and they are downregulated by Aβ in AD brains (Govoni et al., 1993; Lucke-Wold et al., 2015). PKC signal cascades along with altered calcium homeostasis contribute to the development of NFTs (neurofibrillary tangles) (Lucke-Wold et al., 2014).
The S1928 site is close to the C-terminus of α1C, present only in full-length α1C. With normal aging, there is a clear increase in S1928 phosphorylation in the hippocampus but the general levels of cyclic adenosine monophosphate (cAMP), PP2A, and protein phosphatase 1 (PP1) inhibitors remain unchanged (Davare and Hell, 2003). The dentate gyrus is the major region in the hippocampus where S1928 phosphorylation occurs; no significant changes are observed in other areas of the hippocampus (Núñez-Santana et al., 2014). S1928 phosphorylation by A-kinase-anchoring protein (AKAP)-anchored PKA plays an essential role in enhancing Cav1.2 channel activity and vasoconstriction under conditions of high glucose or in diabetes (Nystoriak et al., 2017). The level of cAMP is upregulated in cerebral vessels in AD hippocampus and is associated with vascular β-amyloid peptide (Aβ) (Martínez et al., 2001). It is well established that patients with type 2 diabetes have a higher incidence of cognitive decline and morbidity of AD than the general population (Surguchov, 2020), suggestive of a link with changes in Cav1.2 activity. In the AD brain, preclinical and neuropathological data suggest that both adenyl cyclase (AC)–cAMP–PKA and guanylate cyclase (GC)–cGMP–PKG signaling are disrupted. Overall PKA activity and nuclear PKA activity appear to be suppressed in AD (Sanders and Rajagopal, 2020), which may lead to abnormal changes in Cav1.2 phosphorylation state. Furthermore, the mechanism by which the β2-adrenergic receptor (β2AR) stimulates Cav1.2 channel activity depends on S1928 phosphorylation and constitutes a critical component of the molecular mechanism underlying stable and prolonged theta-tetanus-induced LTP (Qian et al., 2017). Multiple phosphorylation sites have been found in the C-terminal domain of the Cav1.2 β subunit in vitro. However, C-terminal knock-out mice survive with no apparent physiological deficits and, most importantly, show normal function of Cav1.2 in ventricular myocytes. Thus, the phosphorylation sites on the Cav1.2 β subunit may not have essential functional roles in vivo.
Ubiquitination
Ubiquitin (UB) is a highly conserved small protein that is found in all eukaryotic cells, from single-celled yeast to humans. Its main function is to mark proteins to be degraded by 26S proteasome (Lam et al., 2000; Bennett et al., 2005; Swatek and Komander, 2016). UB binds covalently to the lysine residue of the substrate protein and the ubiquitin-labeled protein is identified and rapidly degraded. Briefly, this process requires the sequential action of three enzymes (Swatek and Komander, 2016). The C-terminal glycine residue of ubiquitin is activated by E1. Next, activated ubiquitin is transferred to an active cysteine residue of E2. Finally, ubiquitin links its C-terminus to an ε-amino group of the substrate protein’s lysine residues (Hershko and Ciechanover, 1998). In a nutshell, ubiquitination is a dynamic, multifaceted post-translational modification that is involved in nearly all physiological processes (Swatek and Komander, 2016). An abnormal UB signal is closely related to neurodegeneration.
Neurodegenerative diseases are characterized by the loss of neurons in the brain or spinal cord. Most samples from patients with neurodegenerative diseases are immunoreactive for anti-UB antibodies (Popovic et al., 2014) and abnormalities of the UB-dependent degradation systems and aggregation formation are associated with neurodegeneration (Hershko and Ciechanover, 1998; Lam et al., 2000; Bennett et al., 2005; Hara et al., 2006). In PD, α-synuclein in Lewy bodies (a diagnostic marker of PD) is modified by ubiquitin at lysines 77 and 78 (Popovic et al., 2014). Ubiquitination likely increases the aggregation and neurotoxicity of α-synuclein in cultured human dopaminergic cells (Popovic et al., 2014). In AD, a typical aggregate is ubiquitinated tau protein (Popovic et al., 2014). Thus, UB-dependent degradation systems, such as the UB-proteasome system and autophagy, likely play a role in the pathogenesis of these neurodegenerative diseases (Bennett et al., 2005; Popovic et al., 2014).
The ubiquitin–proteasome system (UPS) is closely linked to Cav1.2 degradation (Felix and Weiss, 2017). UB protein has seven lysine residues at positions 6, 11, 27, 29, 33, 48, and 63 (Ikeda and Dikic, 2008; Chen and Sun, 2009). Among these, K6/K29 take part in Cav1.2 degradation (Lai et al., 2019). E3 specifically recognizes the target proteins’ lysine residue and tags it for degradation by the proteasome (Hershko and Ciechanover, 1998). Recent studies have shown that the Cavβ subunit may serve as a molecular switch that prevents the Cav1.2 α subunit from ubiquitination by the RFP2 ubiquitin ligase and subsequent transfer of Cav1.2 channels to the endoplasmic reticulum associated protein degradation (ERAD) complex; thus, the Cavβ subunit protects Cav1.2 channels from proteasomal degradation (Felix and Weiss, 2017). Separately, Galectin-1 acts as a negative Cav1.2 channel regulator by binding to the Cav1.2 I–II loop and exposing the lysine residues inside the loop to polyubiquitination and ERAD degradation, ultimately inhibiting channel function (Hu et al., 2018; Loh et al., 2020). Furthermore, in ovariectomized APP/PS1 mice (an AD animal model), systemic administration of E2 (17β-estradiol) or the estrogen receptor α (ERα) agonist propylpyrazoletriol (PPT) increased ubiquitination of Cav1.2 in the brain, reversed elevated levels of Cav1.2 protein, and improved cognitive functioning. The binding of the E3 ligase Mdm2 with Cav1.2 is promoted by activating ERα. In Mdm2-overexpressing neurons, the intensity of Cav1.2 decreased significantly. These results suggest that Mdm2-related ubiquitination is critical for ERα regulation of Cav1.2 protein levels and that a reduction in Cav1.2 protein levels may contribute to ERα-induced cognitive improvements (Lai et al., 2019).
N-Linked Glycosylation
N-linked glycosylation is a co-translational or post-translational modification of new peptide chains in which oligosaccharides are connected to the amide of asparagine residues. N-linked glycosylation can be divided into high mannose, compound, and heterozygous types. N-linked glycosylation consists of three main steps: synthesis, transfer, and modification. Synthesis and transfer of N-linked glycosylation are carried out in the endoplasmic reticulum, whereas modification occurs in both the endoplasmic reticulum and the Golgi matrix. This progress is necessary for membrane trafficking and protein expression on the cell surface. Recent studies showed that external glucose level alters N-glycosylation (Liu et al., 2014; Villacrés et al., 2015). There are four potential N-glycosylation sites in the rabbit Cav1.2: N124, N299, N1359, and N1410. The double mutant (N124, 299Q) showed a positive shift in the voltage-dependent gating curve; and the quadruple mutant (QM; N124, 299, 1,359, 1410Q) showed a positive shift in the voltage-dependent gating curve as well as a reduction of peak current. The weaker surface fluorescence intensity of QM suggested its lower surface expression than wild-type Cav1.2 (Park et al., 2015).
The α2δ subunit, an integral component of Cav1.2, is highly N-glycosylated by a 30-kDa oligosaccharide (Marais et al., 2001). Mutation of only 6/16 asparagine glycosylation sites was sufficient to decrease cell surface expression and protein stability of α2δ1 subunit, as well as α2δ1-mediated peak current density and voltage-dependent gating of the α1C subunit. Single mutation N663Q and double mutations N348Q/N468Q, N348Q/N812Q, and N468Q/N812Q decreased protein stability and abolished cell surface expression of α2δ1 as well as the α2δ1-induced up-regulation of Cav1.2 currents (Tétreault et al., 2016). However, it is still not clear whether N-glycosylation of Cav1.2 contributes to the mechanism of Ca2+ interruption in neurodegenerative diseases.
S-Glutathionylation
S-glutathionylation is a process in which glutathione forms a disulfide bond with cysteine residues of the target protein, and is a major redox-mediated thiol modulation. Oxidative stress facilitates S-glutathionylation. The ratio of reduced and oxidized glutathione (GSH/GSSG) is important for S-glutathionylation. Glutathionylation is a reversible redox modification: it directly changes the redox state of Cav1.2 and increases calcium influx (Tang et al., 2011). However, this process is considered an oxidant-mediated reaction with low specificity for target proteins. C543 in the cytoplasmic I-II loop is the major glutathiolation target in hCav1.2. C543S mutation alters post-translational folding and shifts the channel open probability, which may lead to the onset of disease pathology (Muralidharan et al., 2016). Inflammation and ROS are known to be critical pathological manifestations of neurodegenerative diseases. Moreover, imbalance of glutathione homeostasis and dysregulation in glutathione-dependent enzyme activities are implicated in the induction and progression of neurodegenerative diseases, including AD, PD, and ALS. Therefore, impaired S-glutathionylation of Cav1.2 may contribute to the pathology of neurodegenerative diseases.
Cav1.2 as a Potential Drug Target in Neurodegenerative Diseases
Cav1.2 is a classical drug target for cardiovascular disease. Members of the dihydropyridine family of calcium channel blockers (DHPs) have been used as first-line drugs for hypertension and myocardial ischemia for decades, including amlodipine, felodipine, and nifedipine (Zamponi et al., 2015). The sensitivity of LTCCs to DHPs varies in different tissues. Cav1.2 is more sensitive to DHPs than Cav1.3 and Cav1.4 (Xu and Lipscombe, 2001). The splice variants of Cav1.2 in arterial smooth muscle are more sensitive to DHPs than those in the myocardium (Liao et al., 2004; Cheng et al., 2009).
Because of the pathophysiological role of Cav1.2 in neurodegenerative disease, DHPs have been repurposed as a treatment for these diseases. DHPs have at least two advantages as drugs for CNS indications: safety and penetration of the blood–brain barrier (BBB). At therapeutic doses, no obvious side effects were observed for muscle function, hearing, CNS function, or insulin secretion, where LTCCs exert important functions (Levine et al., 2007). Several DHPs can cross the BBB in some species, including humans (Allen et al., 1983; Uchida et al., 1997). Intracerebral drug delivery methods have also improved recently (Patel et al., 2009; Lu et al., 2014).
Because of the known role of Cav1.2 in cognition and the imbalance in Ca2+ homeostasis found in AD, DHPs have been repurposed for AD treatment. In a survey of investigating the association between DHP or non-DHP calcium channel blocker and risk of developing AD or mortality, researchers found that the use of DHP did not reduce risk of AD but showed lower relative risk (Yasar et al., 2005). In vitro, nilvadipine, nitrendipine, and amlodipine reduced Aβ accumulation by affecting the production and clearance of Aβ. In vivo, nilvadipine and nitrendipine reduced Aβ deposition. In transgenic mouse models of AD (Tg APPsw (Tg2576) and Tg PS1/APPsw), chronic nilvadipine treatment resulted in lower Aβ levels and improved learning and spatial memory (Paris et al., 2011). These results suggest that some DHPs have significant benefits in the treatment of AD. Nilvadipine can also delay the degeneration of cognitive function in AD patients (Hanyu et al., 2007; Matsuda et al., 2008). Nitrendipine treatment reduced the risk of dementia by 55% in hypertensive patients compared with a control group (Forette et al., 2002). Since improvements in cognition are observed with non-DHP drugs like ACEI and thiazide (Bellew et al., 2004; Hanon and Forette, 2004; Fournier et al., 2009; Duron and Hanon, 2010), the protective effects of nivadipine and nitrendipine do not seem to be related to their antihypertensive effects. Although nilvadipine and nitrendipine have protective effects, their effectiveness depends on the severity of AD (Paris et al., 2011). After nilvadipine treatment, the very mild AD group showed less cognition decline whereas the moderate AD group showed greater cognition decline compared with their respective placebo-treated controls (Abdullah et al., 2020). This study suggests that AD severity affects the treatment results and nilvadipine may be restricted to patients with mild AD in the future.
The pathological mechanisms underlying PD are not yet clear. Symptomatic treatments are aimed at relieving deficits in motor symptoms and improving quality of life (Schulz et al., 2016; Obeso et al., 2017). Currently, pharmacotherapy includes dopamine mimetics (levodopa), synergists of levodopa (selegiline, carbidopa), dopamine receptor agonists (bromocriptine), dopamine-releasing drugs (amantadine), and anticholinergic drugs (trihexsyphenidyl). Neurosurgery and supportive treatments have been used clinically for many years (Oertel and Schulz, 2016; Schulz et al., 2016). However, none of these treatment methods can prevent or slow the progression of PD and the side effects of the treatments often limit the long-term benefits of symptomatic therapies. However, there are a few different drugs currently in preclinical trials. Because of LTCC-mediated Ca2+ load in SNc dopaminergic neurons, DHPs are considered for PD treatment. Studies have shown that isradipine has a significant neuroprotective effect on substantia nigral dopaminergic neurons in an MPTP-induced animal model of PD (Kupsch et al., 1995; Singh et al., 2016; Wang et al., 2017) and partially restores dopamine content in the striatum (Wang et al., 2017). Another DHP, nifedipine, was reported to improve apomorphine-induced rotation behavior in 6-OHDA-lesioned rats (Wang et al., 2012).
In humans, the ongoing phase III clinical study STEADY-PD is investigating the potential of the LTCC blocker isradipine for treatment of PD. Although the study showed that long-term treatment with immediate-release isradipine did not slow the clinical progression of early-stage PD, it did modestly decrease cumulative levodopa equivalent dose and the time needed for antiparkinsonian treatment (McFarthing and Simuni, 2019; Parkinson Study Group STEADY-PD III Investigators, 2020; Venuto et al., 2021). According to epidemiological studies (Becker et al., 2008; Ritz et al., 2010; Pasternak et al., 2012; Lee et al., 2014) and meta-analyses (Gudala et al., 2015; Lang et al., 2015; Mullapudi et al., 2016), patients treated with DHPs have a reduced risk of PD. Although DHPs have a history of safe use, the drug release time should be prolonged to avoid activation of the sympathetic nervous system, accompanied by reflex tachycardia and high cardiac oxygen consumption, flushing, hypotension, and headache (Carrara et al., 1994; Johnson et al., 2005). In some countries, extended-release formulations of isradipine are available and are already in phase II clinical trials in PD patients (Parkinson Study Group, 2013).
Other potential treatment strategies remain to be studied. Previous data show that the basal level of Cav1.2 in the hippocampus and cortex of ovariectomized APP/PS1 mice is significantly higher than that of wild-type mice. E2 or PPT could reverse this increased basal level of Cav1.2 by promoting the ubiquitination and degradation of Cav1.2 (Lai et al., 2019). Thus, ERα agonists (propylpyrazoletriol, dienestrol) may effectively alleviate the symptoms of AD. Hu et al. used a Tat-e9c peptide to compete for the Galectin-1 binding site on Cav1.2 and interfere with its ubiquitination and degradation (Hu et al., 2018), but whether Cavβ-derived peptides can be used to promote Cav1.2 degradation in the brain needs further study. The biggest concerns would be how to transport the peptide across the BBB and how to reduce the side effects in the cardiovascular system.
Conclusion
Cav1.2 plays important roles in the cardiovascular system, the CNS, and endocrine glands. In the brain, it mediates learning and memory, drug addiction, and neuronal development. Cav1.2 undergoes a variety of post-translational modifications, which are altered in neurodegenerative disease states. Recently identified modifications, such as S-nitrosylation, and their role in pathology require further study.
DHPs are widely prescribed for hypertension and myocardial ischemia and have been repurposed for use in neurodegenerative diseases including AD and PD. Several clinical trials show promising outcomes (summarized in Table 2). Although clinical studies have shown that DHPs have protective effects on neurodegenerative diseases, there are several issues with using DHPs to treat neurodegenerative diseases. First, achieving the requisite drug concentrations in the brain while avoiding fluctuations in blood pressure and cardiac function is a challenge. This may be addressed by the development of new drug-delivery strategies. Second, the relative lack of selectivity of DHPs is a big concern for their use in the CNS; unwanted effects may arise from antagonism of Cav1.3 channels. Furthermore, the universal expression of Cav1.2 may result in DHP side effects on normal brain functions. Further studies on channel modulation and more-specific methods of altering Cav1.2 channel function may lead to better therapeutic strategies for neurodegenerative diseases.