
PERK, Beyond an Unfolded Protein Response Sensor in Estrogen-Induced Apoptosis in Endocrine-Resistant Breast Cancer
By Ping Fan and V. Craig Jordan
Excerpt from the article published in Molecular Cancer Research, 1 February 2022; 20 (2): 193–201. https://doi.org/10.1158/1541-7786.MCR-21-0702
Editor’s Highlights
- Sigma-1 receptor (Sig1R), an endoplasmic reticulum-resident membrane protein, stabilizes IP3R and highly interacts with the chaperone protein GRP78 to regulate Ca2+ exchange at MAMs.
- Overexpression of Sig1R suppresses PRK-like endoplasmic reticulum kinase (PERK) activation.
- PERK functions as a double-edged sword in therapy for ER-positive breast cancer, depending on the duration and intensity of stress.
- PERK drives invasion and metastasis of breast cancer undergoing epithelial-to-mesenchymal transition.
- PERK is a component enriched in mitochondria-associated endoplasmic reticulum membranes (MAMs) that further sensitizes mitochondria to stress onset in the endoplasmic reticulum.
- MAMs attract wide attentions due to their functional importance in organelle communication at membrane contact sites
- Sig1R may regulate mitochondrial dynamics by interacting with mitochondrial tethering protein mitofusin 2 (MFN2) at membrane contact sites.
- The biological implications of MAMs in controlling apoptosis may shed light on how to precisely modulate molecular interactions, particularly between PERK and MAM-tethering proteins, for the improvement of breast cancer therapy.
Abstract
The discovery of 17β-estradiol (E2)–induced apoptosis has clinical relevance. Mechanistically, E2 over activates nuclear estrogen receptor α that results in stress responses. The unfolded protein response (UPR) is initiated by E2 in the endoplasmic reticulum after hours of treatment in endocrine-resistant breast cancer cells, thereby activating three UPR sensors—PRK-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1α (IRE1α), and activating transcription factor 6 (ATF6) with different functions. Specifically, PERK plays a critical role in induction of apoptosis whereas IRE1α and ATF6 are involved in the endoplasmic reticulum stress-associated degradation (ERAD) of PI3K/Akt/mTOR pathways. In addition to attenuating protein translation, PERK increases the DNA-binding activity of NF-κB and subsequent TNFα expression. In addition, PERK communicates with the mitochondria to regulate oxidative stress at mitochondria-associated endoplasmic reticulum membranes (MAM). Furthermore, PERK is a component enriched in MAMs that interacts with multifunctional MAM-tethering proteins and integrally modulates the exchange of metabolites such as lipids, reactive oxygen species (ROS), and Ca2+ at contact sites. MAMs are also critical sites for the initiation of autophagy to remove defective organelles and misfolded proteins through specific regulatory proteins. Thus, PERK conveys signals from nucleus to these membrane-structured organelles that form an interconnected network to regulate E2-induced apoptosis. Herein, we address the mechanistic progress on how PERK acts as a multifunctional molecule to commit E2 to inducing apoptosis in endocrine-resistant breast cancer.
Introduction
Endocrine therapies, through targeting of estrogen receptor (ER) with selective ER modulators (SERM) or preventing synthesis of 17β-estradiol (E2) by aromatase inhibitors (AI), are standards of care for ER-positive patients with breast cancer (1, 2). However, acquired resistance to the endocrine therapies is a major challenge in treatment of these patients (3, 4). Paradoxically, E2/ERα induces apoptosis in endocrine-resistant breast cancer in vitro and in vivo (5–9). This discovery has clinical relevance (10) to treat AI-resistant breast cancer in clinical trials with 30% benefit rate (11) and to interpret the reduction of breast cancer incidence for postmenopausal women taking E2 alone as hormone replacement therapy (HRT; ref. 12). Nevertheless, breast cancer incidence increases in postmenopausal women taking E2 plus medroxyprogesterone acetate (MPA) as classical HRT (12) because MPA has glucocorticoid activity that blocks E2-induced apoptosis (13). Our publications have demonstrated that glucocorticoids suppress E2-induced inflammatory response, thereby inhibiting E2-induced apoptosis in endocrine-resistant breast cancer (13–15). Thus, further investigation of the mechanisms underlying E2-induced apoptosis will find more factors that blunt E2-induced apoptosis and more ways to improve the therapeutic effects of E2-induced apoptosis.
Long-term endocrine therapies suppress the proliferative activity of ERα, which also alter interactions between ERα and other lipid metabolism- or inflammation-associated transcription factors, such as peroxisome proliferator-activated receptor γ (PPARγ) and NF-κB in nucleus (15–17). Therefore, an E2-deprived state causes metabolic disorders and inflammatory stress (14, 16, 18, 19) that render endocrine-resistant breast cancer cells overexpressing PPARγ and constitutively activating NF-κB (17, 20). In these settings, E2 initiates apoptosis through over activation of nuclear ERα that subsequently triggers stress responses, including endoplasmic reticulum stress, oxidative stress, and inflammatory stress responses in endocrine-resistant breast cancer cells (15–17, 20–25). Among these stress responses, E2 quickly activates the unfolded protein response (UPR) in the endoplasmic reticulum after hours of treatment in endocrine-resistant cells, thereby activating three sensors—PRK-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1 alpha (IRE1α), and activating transcription factor 6 (ATF6) with different functions (Fig. 1; refs. 21, 23). Specifically, PERK plays a critical role in induction of apoptosis whereas IRE1α and ATF6 are involved in the endoplasmic reticulum stress-associated degradation (ERAD) of PI3K/Akt/mTOR–associated pathways after E2 treatment (21, 23). In addition to attenuating translation, sustained activation of PERK deteriorates stress responses in mitochondria and activates NF-κB/TNFα axis, ultimately determining cell fate (20, 21, 24). The key role of NF-κB in E2-induced apoptosis creates new opportunities to modulate its DNA-binding activity through other nuclear transcription factors, such as glucocorticoid receptor (GR; ref. 15) and PPARγ (17), thereby affecting E2-induced apoptosis.
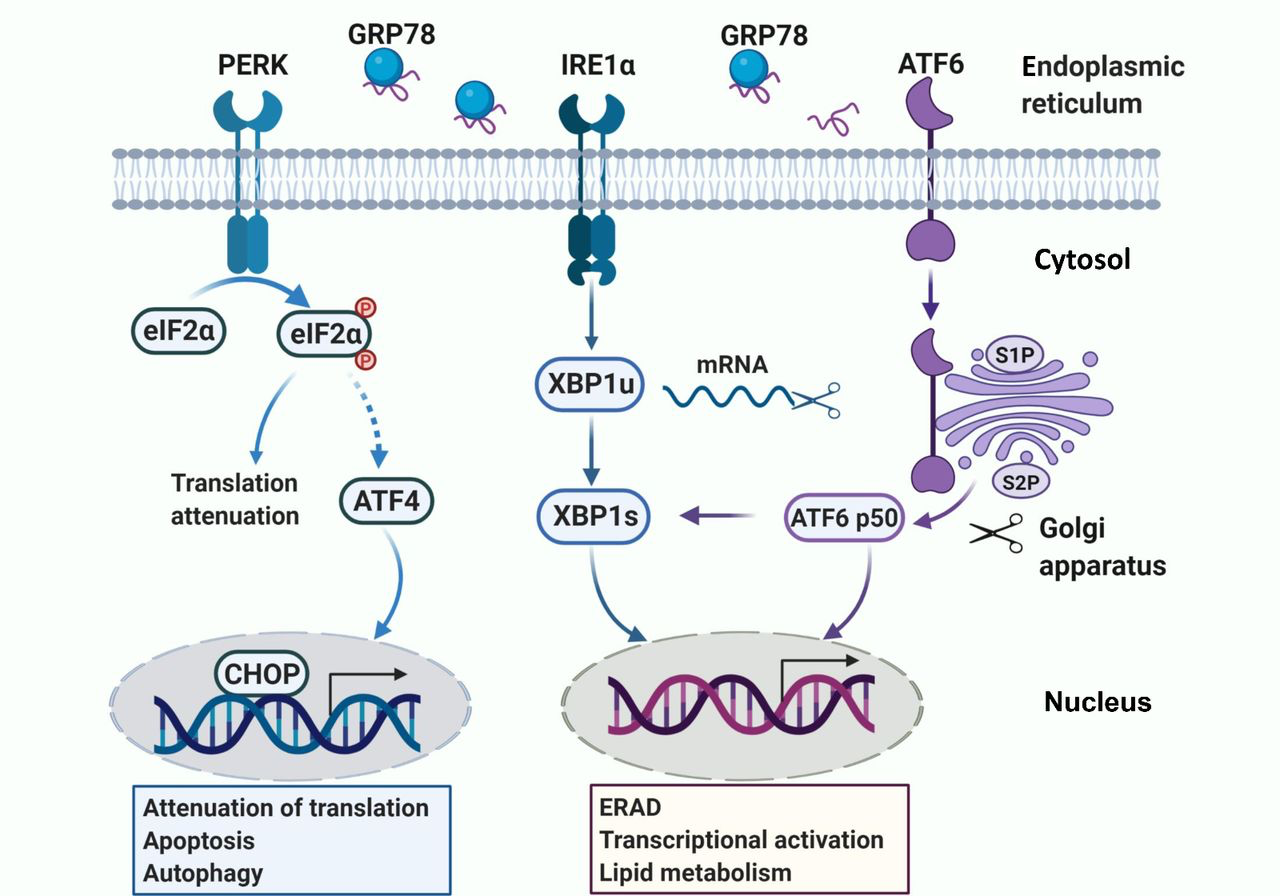
Activation of three sensors of UPR with different functions. E2 treatment quickly activates UPR in endocrine-resistant breast cancer cells. Then, three sensors are activated with different functions. PERK phosphorylates eIF2α to attenuate protein translation. Given that the PERK activation is persistent, ATF4 is selectively activated and the downstream pro-apoptotic protein CHOP is induced. IRE1α splices XBP1 mRNA to spliced XBP1 (s-XBP1) that regulates genes expression to promote protein folding and induces endoplasmic reticulum stress-associated degradation (ERAD). ATF6 is cleaved by S1P and S2P after translocation to the Golgi apparatus upon stress. Active ATF6p50 is then released to increase the transcription activity of XBP1 and regulate ERAD.
Furthermore, PERK kinase communicates with the mitochondria to regulate oxidative stress at mitochondria-associated endoplasmic reticulum membranes (MAM; ref. 26). PERK is a component enriched in MAMs that interacts with multifunctional MAM tethering proteins and integrally modulates the exchange of metabolites such as lipids, reactive oxygen species (ROS), and Ca2+, at contact sites (27). Particularly, the transfer of ROS and Ca2+ determines cell fate (28). In addition, the MAMs are also the contact sites for the initiation of specific forms of autophagy [endoplasmic reticulum-phagy (ER-phagy) and mitophagy] to remove damaged or defective portions of organelle mediated by many specific receptor proteins (25). Apparently, multiple molecules are undergoing dynamic and active communications at MAMs with the initial purpose to maintain the normal biological functions of these two organelles. Persistent stress that disturbs the homeostasis at MAM interfaces leads to apoptosis. Herein, we describe the mechanistic progress on how PERK acts as a multifunctional molecule to commit E2 to inducing apoptosis in endocrine-resistant breast cancer.
E2 Over Activates Nuclear ERα to Induce Stress-Associated Apoptosis
Unlike the traditional apoptotic mechanism caused by cytotoxic chemotherapy with cell-cycles arrest (29), cells that undergo apoptosis induced by E2 simultaneously activate proliferation with an increased S phase of cell cycle (21, 22, 29). Thus, identifying the mechanisms underlie E2-induced apoptosis is necessary. Antiestrogens, tamoxifen and ICI 182,780 completely block E2-induced apoptosis in endocrine-resistant breast cancer cells (21, 29), suggesting that E2 induces apoptosis through ER (21, 29). There are two ERs: ERα and ERβ that control E2-regulated process in women. The function of both ERs in E2-induced apoptosis has been identified by siRNAs specific for ERα and ERβ (14). Only the loss of ERα prevents E2-induced apoptosis, demonstrating that E2 induces apoptosis via ERα (14). Both non-genomic and genomic pathways of ERα are activated by E2 in these resistant cells despite apoptosis occurring after E2 therapy (16, 21). The synthetic macro-compound estrogen-conjugated dendrimer (EDC) contributes to distinguishing the functions of these two pathways in E2-induced apoptosis (30), which specifically activates non-genomic pathway of ERα mediated by tyrosine kinase c-Src (Fig. 2; refs. 21, 30). Notably, EDC does not induce apoptosis in endocrine-resistant breast cancer cells, instead, it increases cell proliferation (21). These findings demonstrate that E2 induces apoptosis mainly through nuclear ERα.
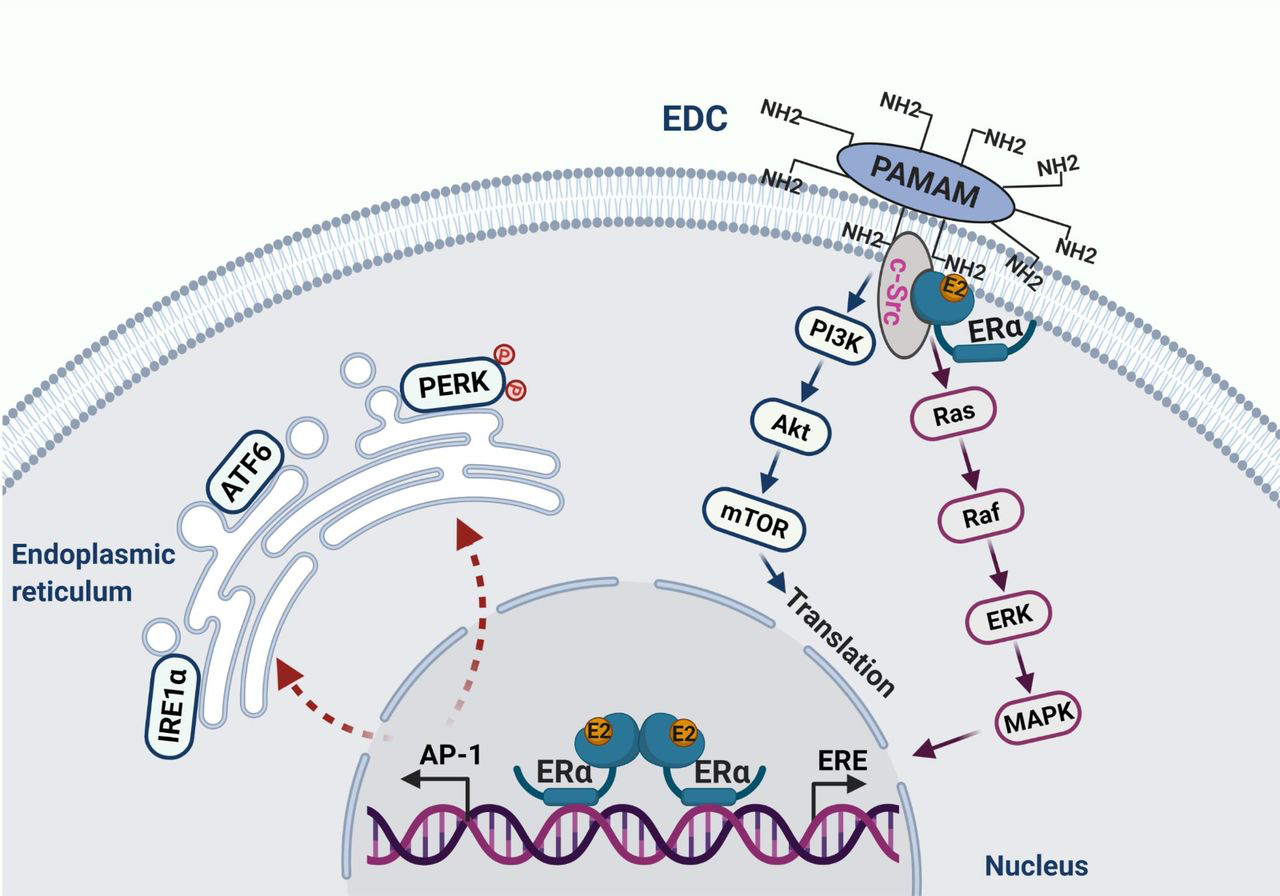
E2 initiates apoptosis through over activation of nuclear ERα. The macromolecule EDC specifically activates non-genomic pathways of ERα mediated by c-Src, which stimulates the proliferation of endocrine-resistant breast cancer cells. In the nucleus, E2 activates classical transcription pathway ERE, which is involved in cellular proliferation. Simultaneously, E2 consistently activates the tethering pathway of ERα, particularly AP-1 family members. This leads to stress responses in the endoplasmic reticulum.
c-Src plays a critical role in the mediation of non-genomic pathway of EDC (21, 30). The proliferative action of EDC helps us to exclude the mechanism of c-Src participates in E2-induced apoptosis via non-genomic pathway of ERα (Fig. 2). Compelling evidence indicates that c-Src mediates multiple signaling pathways of ERα in breast cancer cells (21, 31, 32). We observed that c-Src mediates E2-induced stress responses that result in apoptosis after E2 therapy (21). Therefore, inhibition of tyrosine kinase activity of c-Src blocks E2-induced apoptosis (21, 33). In addition, c-Src is involved in ubiquitin-dependent proteolysis of ERα and affects ERα’s transcriptional activity (32). As a result, inhibition of c-Src increases ERα expression levels and activates classical ERα transcription pathways with increasing estrogen-responsive element (ERE) activity in these endocrine-resistant breast cancer cells (21, 33) in spite of blocking E2-induced apoptosis (21, 33). Enhancement of ERE activities after blockade of apoptosis by the c-Src inhibitor suggests that the classical transcription pathway of ERα is not involved in E2-induced apoptosis (21). Similarly, the PPARγ agonist increases ERE-regulated pS2 expression whereas it inhibits E2-induced oxidative stress (17). Andruska and colleagues (34) reported the supportive observation that treatment with ERα biomodulator BHPI induces apoptosis in several ERα-positive endocrine-resistant models due to the excessive activation of stress responses even though BHPI suppresses classical ERα-regulated transcription. Different from E2, BHPI strongly activates plasma membrane enzyme phospholipase Cγ (PLCγ), producing inositol 1,4,5-triphosphate (IP3), which opens inositol trisphosphate receptor (IP3R) calcium channels, rapidly depleting Ca2+ stores in the endoplasmic reticulum. This leads to activation of UPR (34).
All of these results suggest that activation of classical ERα-dependent transcription pathways occurs separately from the initiation of stress responses to induce apoptosis in endocrine-resistant breast cancer cells (17, 21, 34). Further investigation demonstrates that E2 persistently activates transcription activity of ERα through the tethering pathways, such as activator protein-1 (AP-1) family members in endocrine-resistant breast cancer cells (15, 16), which differs from the transient activation of AP-1 family members in endocrine-sensitive breast cancer cells (15, 16). The difference in activating AP-1 family members between endocrine-sensitive breast cancer cells and endocrine-resistant breast cancer cells suggests that long-term endocrine therapies alter the function of ERα in response to E2. These observations raise the possibility that the tethering pathways activated by ERα play a role in triggering E2-induced apoptosis (15, 16). Along with the tethering pathways, E2 constantly activates AP-1–associated Jun aminoterminal kinase (JNK) for the original purpose to promote growth while simultaneously causing stress responses (15, 16, 22, 35). When the stress accumulates persistently, it will result in apoptosis (21, 35, 36).
Multiple Functions of PERK in the Activation of Apoptotic Cascades
Compelling findings have uncovered molecular mechanisms underlying E2-induced apoptosis via accumulation of stress, including endoplasmic reticulum stress, oxidative stress, and inflammatory stress responses (16, 21, 37). Among these stress responses, UPR is the first one initiated by E2 in endoplasmic reticulum after hours of treatment (21, 23). E2 activates three sensors of UPR with differential functions in endocrine-resistant breast cancer cells: PERK phosphorylates eIF2α to attenuate protein translation whereas IRE1α and ATF6 mainly mediate ERAD of PI3K/Akt/mTOR pathways (23). Specifically, sustained activation of PERK, but not IRE1α and ATF6, plays a key role in mediation of E2-induced apoptosis (21, 23). Consistently, the ERα biomodulator BHPI exerts different actions on PERK and IRE1α/XBP-1 during the induction of apoptosis. It persistently activates PERK but suppresses IRE1α/XBP-1 expression due to the functional inhibition of ERα, suggesting that activation of PERK is sufficient to induce apoptosis (34). In support of this, another research group reported that PERK and IRE1 have different functions in determining cell fate (38, 39). They used chemical-genetic strategies to individually activate these two sensors in HEK293 cells. Their results demonstrated that sustained PERK signaling promotes apoptosis, whereas the equivalent duration of IRE1 signaling enhances cell proliferation (38, 39). Inhibition of IRE1 activity is effective at preventing MYC-driven breast cancer development (40). Thus, differential functions of three sensors of UPR result in co-occurrence of apoptosis and proliferation in endocrine-resistant breast cancer cells after E2 therapy (21, 22, 29).
All of the findings described above also emphasize the importance of PERK in mediation of apoptosis (20, 21, 34). Generally, sustained activation of PERK phosphorylates eIF2α, which induces activation of ATF4 and the downstream pro-apoptotic protein CHOP (Fig. 3). Nevertheless, this apoptotic effect of PERK is not solely dependent on the phosphorylation of eIF2α (20). Persistent activation of PERK leads to mitochondrial fragmentation, enhanced Ca2+ overload, increased ROS production, and overexpressed BH3-only proteins (Fig. 3; refs. 20, 21, 27). Furthermore, PERK participates in the regulation of inflammatory responses, particularly involving in the induction of TNFα expression by E2. TNFα is expressed in a delayed pattern that peaks after 72 hours of E2 treatment and relies on PERK for activation of NF-κB (Fig. 3; refs. 20, 21, 24). PERK phosphorylates stress-associated transcription factor STAT3 and promotes its translocation to the nucleus. The STAT3 facilitates activation of NF-κB and induction of TNFα expression, ultimately leading to apoptosis (20, 24). The PERK/NF-κB/TNFα axis has been identified to play a critical role in inducing apoptosis after E2 therapy (20, 24). The key role of NF-κB in E2-induced apoptosis creates new opportunities to modulate its DNA-binding activity through other nuclear transcription factors, such as GR (15) and PPARγ (17), thereby affecting E2-induced apoptosis (13, 15, 17). As a result, investigators are testing novel therapeutic strategies to determine how to precisely modulate UPR to improve the therapeutic effects of E2-induced apoptosis on endocrine-resistant breast cancer.
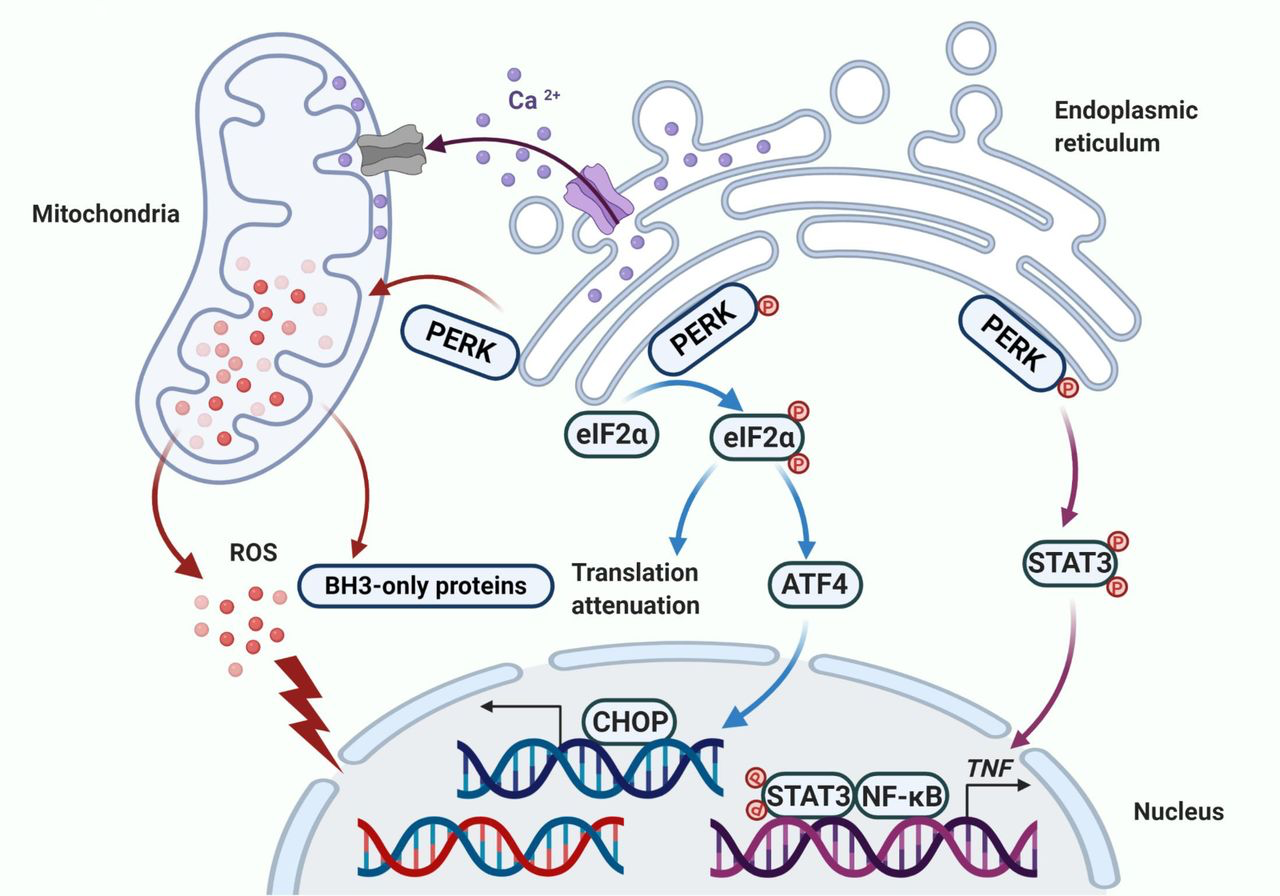
PERK activates apoptotic cascades in multiple ways. Apoptosis is ultimately induced by sustained PERK activation after E2 therapy in endocrine-resistant breast cancer cells. The classical way that PERK induces apoptosis is by activating ATF4 and the downstream pro-apoptotic protein CHOP. In addition, PERK regulates the function of the mitochondria and results in the release of ROS and overexpression of BH3-only proteins. Finally, the extrinsic apoptotic pathway is activated by PERK that is mediated by STAT3 to increase the DNA-binding activity of NF-κB and subsequently induce TNFα expression.
PERK Is Enriched in MAMs and Interacts with MAM-Tethering Proteins to Regulate Metabolite Transfer
Two major metabolic organelles—the endoplasmic reticulum and mitochondria—are involved in E2-induced apoptosis in endocrine-resistant breast cancer (21, 22). UPR activation occurs before the oxidative stress after E2 therapy with the initial purpose of maintaining the homeostasis (21, 22). Nevertheless, the mitochondria are highly sensitive to different cellular insults, including unfolded proteins, Ca2+, lipids, and ROS in the endoplasmic reticulum (41). The exchange of metabolites such as Ca2+, lipids, and ROS, from the endoplasmic reticulum to the mitochondria results in mitochondrial dysfunction and determines the final cell fate (41). How the endoplasmic reticulum communicates stress over to the mitochondria after E2 therapy in endocrine-resistant breast cancer cells remains unclear.
The result that inhibition of PERK activity completely blocks oxidative stress and expression of BH-3–only proteins induced by E2 demonstrates that PERK functions as a crucial UPR sensor to mediate the cross-talk between the endoplasmic reticulum and mitochondria (20, 21, 26, 27, 42). In addition to accumulation of unfolded proteins (21, 42), imbalance of metabolites, such as Ca2+, lipids, and ROS can activate PERK in the lumen of the endoplasmic reticulum (21, 31, 34, 43, 44). Sustained activation of PERK causes mitochondrial dysfunction (20, 21, 26, 27). Of note, PERK is a component enriched in MAMs (26) that further sensitizes mitochondria to stress onset in the endoplasmic reticulum (41). Recently, the MAMs attract wide attentions due to their functional importance in organelle communication at membrane contact sites (45, 46), which are tightly tethered by many multifunctional proteins that integrally regulate lipid metabolism, Ca2+ signaling, and ROS homeostasis (Fig. 4; refs. 45, 46). Among these proteins, PERK interacts with the mitochondrial tethering protein mitofusin 2 (MFN2; refs. 27, 47) to stabilize endoplasmic reticulum–mitochondrial contacts (27). This structural effect of PERK on MAMs appears to be dependent on the PERK cytosolic kinase domain, but does not occur through PERK downstream signaling of eIF2α (27). Apart from being a tethering protein, MFN2 regulates the function of PERK, which participates in mitochondrial fusion, inflammation, and efficient uptake of Ca2+ at contact sites from the endoplasmic reticulum to the mitochondria (47–49). Similarly, the endoplasmic reticulum-localized tethering protein, phosphofurin acidic cluster sorting protein 2 (PACS2) recruits chaperone protein calnexin (CNX) to regulate Ca2+ signaling by stabilizing MAMs structure and affecting the function of ion channels (50, 51). Particularly, two ion channels, IP3R on the endoplasmic reticulum luminal membrane and voltage-dependent anion-selective channel localized to the mitochondria outer membranes, are key to the transfer of Ca2+ (Fig. 4; ref. 41). Sigma-1 receptor (Sig1R), an endoplasmic reticulum-resident membrane protein, stabilizes IP3R and highly interacts with the chaperone protein GRP78 to regulate Ca2+ exchange at MAMs (52). Notably, overexpression of Sig1R suppresses PERK activation (52). In addition, Sig1R may regulate mitochondrial dynamics by interacting with MFN2 at contact sites (53). Thus, the interaction of these tethering proteins with each other renders MAMs the hubs for Ca2+ signaling communicated between the endoplasmic reticulum and mitochondria (54).
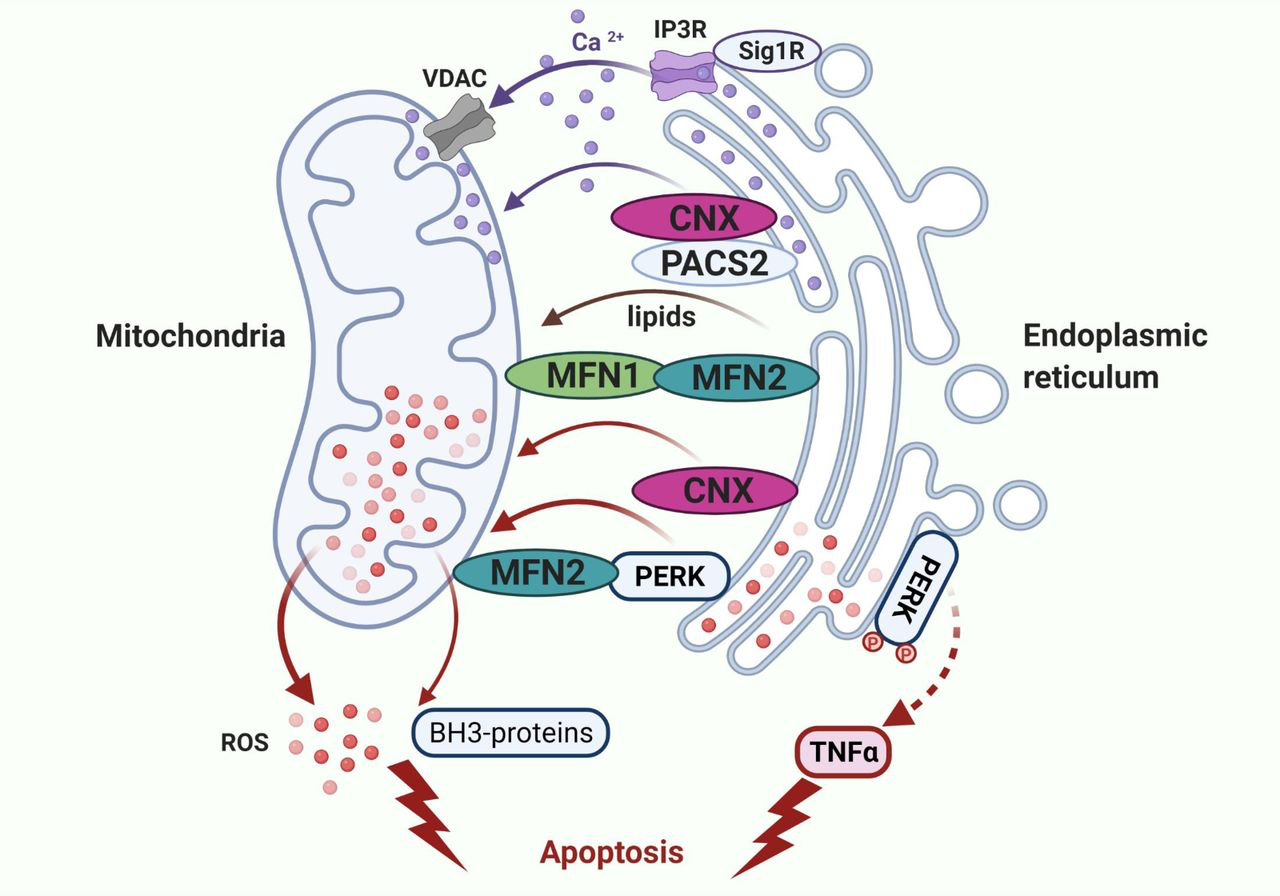
Integral regulation of metabolite exchange at MAMs. Multiple MAM-tethering proteins, including PERK, participate in the transfer of metabolites such as ROS and Ca2+ at MAMs. Briefly, both MFN1 and MFN2 are mitochondrial fusion proteins. PERK physically interacts with MFN2 to stabilize MAMs structure and regulate metabolite exchange. Two ion channels, inositol trisphosphate receptor (IP3R) and voltage-dependent anion-selective channel (VDAC), are key to the transfer of Ca2+. Sigma-1 receptor (Sig1R) stabilizes IP3R and regulates Ca2+ exchange between two organelles. Similarly, phosphofurin acidic cluster sorting protein 2 (PACS2) recruits chaperone protein calnexin (CNX) to regulate Ca2+ signaling by affecting the function of ion channels.
In addition to the role of these proteins in regulation of Ca2+ signaling, the accumulation of ROS at MAMs is a critical factor controlling the Ca2+ exchange at MAM-interface that chemically modifies ion channels (55). Like the mitochondria, the endoplasmic reticulum is a major source of ROS derived from oxidative protein folding that forms disulfide bonds in secretory proteins to stabilize their structures (56–58). The endoplasmic reticulum oxidoreductase 1α is a major enzyme required for the formation of disulfide bonds in secretory proteins that results in an increase in ROS productions (56, 57). In addition, the chaperone protein CNX interacts with NADPH oxidase 4 (Nox4) to regulate redox status within the endoplasmic reticulum (51). Given that ROS accumulate in MAMs, they oxidize the calcium ion channel IP3R that leads to the release of calcium flux from the endoplasmic reticulum and exposure of the mitochondria to high concentrations of Ca2+ (51, 59). Indeed, constitutive Ca2+ release to the mitochondria plays an essential role in maintaining the normal cell bioenergetics (60). When stress in the endoplasmic reticulum is persistent, depletion of Ca2+ from the endoplasmic reticulum to the mitochondria that results in mitochondrial fragmentation and initiates intrinsic apoptotic pathways (28, 61, 62). Therefore, MAMs not only provide a conduit for the transfer of metabolites such as lipids, ROS, and Ca2+ between organelles, but also integrally regulate biological functions to determine cell fate. Notably, the exchange of ROS and Ca2+ at MAMs is critical for controlling apoptosis (28).
The MAMs Are Contact Sites for the Initiation of Autophagy
Both the endoplasmic reticulum and mitochondria undergo stress responses after E2 therapy in endocrine-resistant breast cancer (21, 22). These two organelles are highly dynamic, constantly reshaping their membrane structures to maintain functional homeostasis (40). Autophagy is implicated to be an important way to maintain organelle integrity, recycling cellular macromolecules and damaged organelles through the lysosomal pathway under nutrient or energy deficiency for the survival of cells (63–65). Nevertheless, excessive autophagy can promote cell death (66). It is well documented that autophagy is regulated by PI3K/Akt/mTOR pathways that are sensitive to nutrient and energy deficiency (67, 68). These pathways are activated by E2 in the first 48 hours of treatment (15, 21, 22), contributing to the hyperproliferation of endocrine-resistant breast cancer cells (21, 22). Subsequently, the UPR sensors IRE1α and ATF6 mediate ERAD of PI3K/Akt/mTOR–associated pathways after 72 hours of E2therapy (15, 23). Suppression the activity of nutrient sensor mTOR after E2 therapy is sufficient to induce autophagy (68, 69). Simultaneously, the energy stress sensor, adenosine monophosphate (AMP)–activated protein kinase (AMPK) is activated to regulate protein translation and apoptosis (21, 69). The balance between mTOR and AMPK is a fundamental mechanism of cellular adaptation to stress (21, 69).
Furthermore, both endoplasmic reticulum stress and oxidative stress can selectively induce autophagy to mitigate the respective stress responses through specific forms of endoplasmic reticulum-phagy (ER-phagy) and mitophagy (70–72). Recently, many selective ER-phagy- and mitophagy-regulatory proteins are identified to functionally remodel the endoplasmic reticulum and mitochondria during nutrient or energy stress (71, 73–78). For instance, endoplasmic reticulum-resident proteins such as FAM134B, SEC62, and TEX264 function as receptors to initiate ER-phagy (Fig. 5; refs. 73–76). Similarly, mitochondrial proteins such as FUNDC1, NIX, and BNIP3 serve as mitophagy receptors to remove dysfunctional organelles via autophagy (Fig. 5; refs. 76–78). Most of these proteins contain LC3-interacting region (LIR) that interacts with the autophagy membrane protein LC3 or other autophagy genes for the initiation of ER-phagy and mitophagy (Fig. 5; refs. 72–79). Remarkably, MAMs are also critical interfaces for recruiting the autophagy machinery to remove defective portions of organelles and recycle the major components of membrane structures (25). Specifically, MAMs proteins such as MFN2, IP3R, CNX, and PACS2 interact with autophagy regulatory proteins to selectively remove the defective portions of organelles (71, 80, 81). In parallel, the autophagy regulatory proteins AMPK and mTORC2 translocate to MAMs upon stress, and mTORC2 phosphorylates these MAM-associated proteins to regulate the dynamics of membrane structures at contact sites (80, 81). Beyond its connections to the mitochondria, the endoplasmic reticulum widely contacts the endosome system, Golgi apparatus, and plasma membrane, forming an interconnected network among these organelles (82–85). Therefore, multiple pathways, including ERAD, the ubiquitin-proteasome system, and autophagy are activated to remove misfolded proteins and damaged organelles from endocrine-resistant breast cancer cells after E2 therapy (17, 21–23, 86).
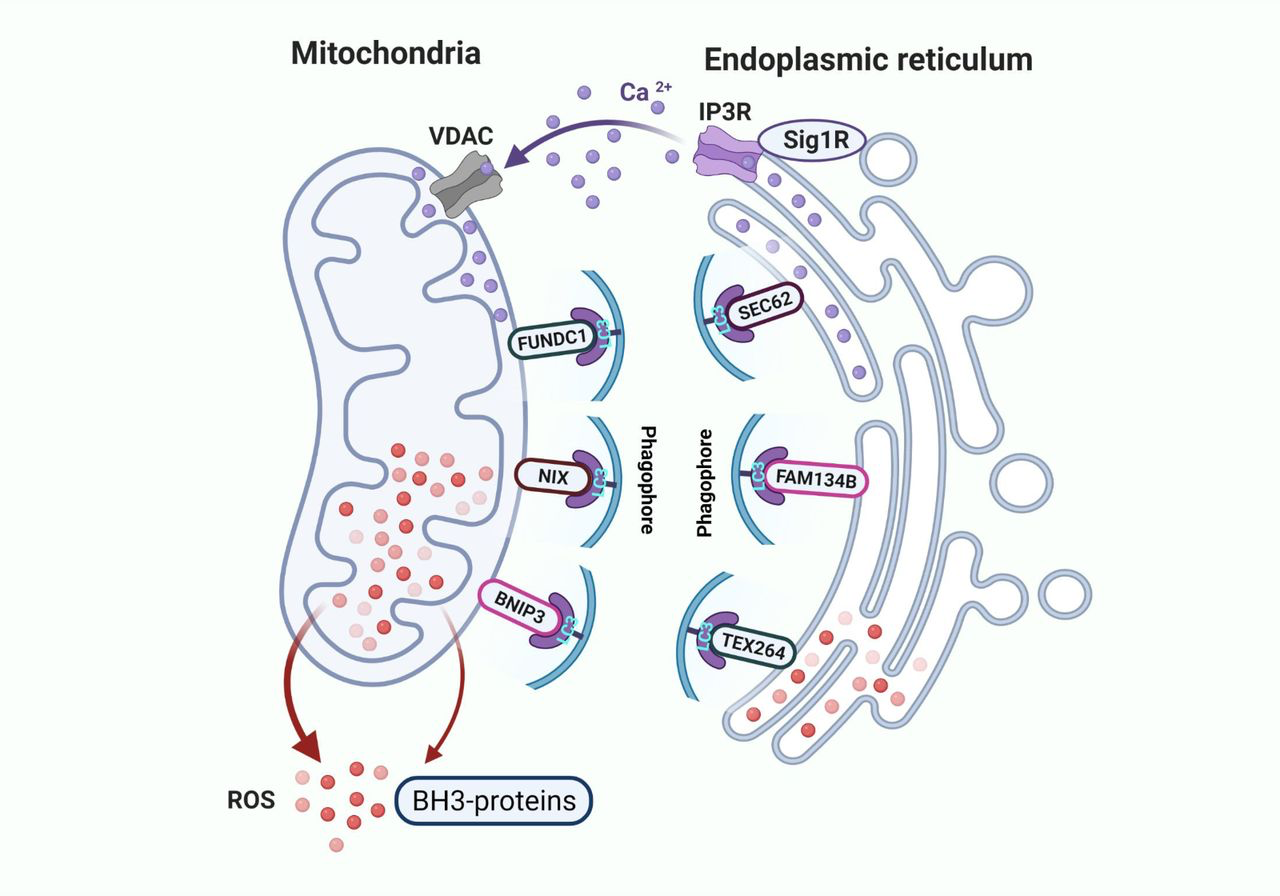
Initiation of autophagy at MAMs. MAMs are critical contact sites for the initiation of ER-phagy and mitophagy. Multiple specific mitochondria- and endoplasmic reticulum-regulatory proteins are involved in the removal of defective or damaged organelles. For instance, endoplasmic reticulum-resident proteins such as FAM134B, SEC62, and TEX264 function as receptors to initiate ER-phagy. Similarly, mitochondrial proteins such as FUNDC1, NIX, and BNIP3 serve as mitophagy receptors to remove dysfunctional organelles. Particularly, most of these proteins contain LC3-interacting region (LIR) that interact with autophagy membrane protein LC3 to form phagophores.
Therapeutic Potentials of Multifunctional PERK and Future Challenges
The discovery of E2-induced apoptosis has clinical relevance, which benefits for patients with breast cancer after long-term endocrine therapies as well as for menopausal women taking E2 alone as HRT (10–12). In these settings, E2 response is altered, displayed as over activation of nuclear ERα, which subsequently triggers stress responses in the endoplasmic reticulum and mitochondria in endocrine-resistant breast cancer cells (15–17, 20–24, 25). In this process, UPR conveys the nuclear signals to organelles. Before apoptosis, rapid activation of PERK and its downstream eIF2α signaling indicates that accumulation of unfolded proteins is the initial type of stress in the endoplasmic reticulum after hours of E2 therapy (20, 21). How nuclear ERα activation quickly generates large amounts of unfolded proteins is under investigation. The distinct characteristic of persistent activation of tethering pathways after E2 therapy is one of the potential mechanisms that produce short half-life proteins such as c-Fos in endocrine-resistant breast cancer cells (15, 16).
Ultimately, apoptosis is due to sustained PERK activation by E2 therapy, resulting in the induction of apoptotic cascades such as the NF-κB/TNFα axis and oxidative stress (20, 21, 23, 33). Recent findings demonstrated that PERK is enriched in MAMs and regulates the exchange of metabolites such as ROS and Ca2+ between two major organelles, emphasizing the implied role of PERK in cell fate determination (26, 27). Nevertheless, our observations provide new evidence that the basal mitochondrial conditions after endocrine therapies affect the biological function of PERK to regulate redox status. For instance, the long-term E2-deprived (LTED) breast cancer cell lines MCF-7:5C and MCF-7:2A are derived from the same parental endocrine-sensitive MCF-7 cells (15, 22). The difference between them is that MCF-7:2A cells have a stronger antioxidant system in the mitochondria than do MCF-7:5C cells (15, 22). This makes MCF-7:2A cells undergo apoptosis later than MCF-7:5C cells after E2 therapy (15, 22) despite the fact that E2 activates PERK similarly in two cell lines (21, 22). Of note, PERK functions as an antioxidant in MCF-7:2A cells but an oxidative stress inducer in MCF-7:5C cells (21), ultimately leading to opposite biological effects of the PERK inhibitor on these two LTED cell lines. How basal mitochondrial conditions affect these molecular functions remain unclear (17). It is very likely that PERK differentially coordinates with other oxidative stress-responsive molecules such as Nrf2 and NF-κB to modulate redox homeostasis, depending on the cellular context (17). All of these results complicate the function of PERK in the regulation of metabolites transfer in MAMs, demonstrating a reciprocal connection between PERK and the mitochondria.
In addition, we cannot exclude the pro-survival activities of PERK during E2-induced apoptosis because E2-induced apoptosis and proliferation co-occur in endocrine-resistant breast cancer cells (21, 22, 29). PERK functions as a double-edged sword in therapy for ER-positive breast cancer, depending on the duration and intensity of stress (21, 22, 29, 87). Recent findings demonstrate that PERK drives invasion and metastasis of breast cancer undergoing epithelial-to-mesenchymal transition (88, 89). Feather of PERK is a challenge for the therapeutic purpose.
Collectively, the findings described herein demonstrate that PERK widely interacts with a variety of molecules, including MAM-tethering proteins and apoptotic and/or metabolic transcription factors, to convey signals between organelles and nucleus, beyond functions as a classical UPR sensor (20–24, 27). Upon stress, MAM formation increases and provides important interfaces for the dynamic communications between the endoplasmic reticulum and mitochondria to determine cell fate (61). The biological implications of MAMs in controlling apoptosis may shed light on how to precisely modulate molecular interactions, particularly between PERK and MAM-tethering proteins, for the improvement of breast cancer therapy.