
New Pharmacological Strategies against Pancreatic Adenocarcinoma: The Multifunctional Thiosemicarbazone FA4
By Dario P. Anobile, Mauro Niso, Adrian Puerta, Stephanie M. Fraga Rodrigues, Francesca S. Abatematteo, Amir Avan, Carmen Abate, Chiara Riganti, and Elisa Giovannetti
Excerpt from the article published in Molecules 2022, 27, 1682. DOI: https://doi.org/10.3390/molecules27051682
Editor’s Highlights
- Pancreatic ductal adenocarcinoma (PDAC) represents about 95% of all types of pancreatic cancer and is characterized by aggressive biological behavior and dismal prognosis.
- The sigma-2 (σ2) receptor mRNA TMEM97 is expressed in both primary pancreatic cancer cell cultures, at significantly higher values compared to the normal human pancreatic ductal epithelial HPDE cells.
- Since σ2 receptor activity is known to trigger apoptosis in PDAC cells.
- A σ2 receptor agonist, induced a higher percentage of apoptotic cells than gemcitabine and showed relevant antiproliferative activity with half maximal inhibitory concentration (IC50) values ranging from 0.701 to 0.825 μM.
Abstract
A new sigma-2 (σ2) receptor ligand (FA4) was efficiently synthesized and evaluated for cytotoxic, proapoptotic, and antimigratory activity on pancreatic ductal adenocarcinoma (PDAC) primary cell cultures, which restrained the aggressive and chemoresistant behavior of PDAC. This compound showed relevant antiproliferative activity with half maximal inhibitory concentration (IC50) values ranging from 0.701 to 0.825 μM. The cytotoxic activity was associated with induction of apoptosis, resulting in apoptotic indexes higher than those observed after exposure to a clinically relevant concentration of the gemcitabine, the first-line drug used against PDAC. Interestingly, FA4 was also able to significantly inhibit the migration rate of both PDAC-1 and PDAC-2 cells in the scratch wound-healing assay. In conclusion, our results support further studies to improve the library of thiosemicarbazones targeting the σ-2 receptor for a deeper understanding of the relationship between the biological activity of these compounds and the development of more efficient anticancer compounds against PDAC.
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) represents about 95% of all types of pancreatic cancer and is characterized by an aggressive biological behavior and dismal prognosis. The global burden of PDAC has more than doubled over the past 25 years, and it is predicted to become the second cancer-related death reason in 2040 [1,2].
Five-year survival rates have just reached about 10%, but therapeutic options are limited, and often patients develop resistance to the standard treatments based on gemcitabine [3].
In the attempt to identify new vulnerabilities to overcome chemoresistance, the so-called sigma (σ-1 and σ-2) receptors have attracted special interest, because they are overexpressed in several tumors, including PDAC, compared to healthy tissues, and their ligands trigger cancer cell apoptosis by pleiotropic mechanisms, including intracellular calcium oscillation, endoplasmic reticulum stress, and alteration of mitochondrial polarization [4,5]. The σ1 receptor has been defined as a pluripotent modulator mainly residing in the mitochondria-associated ER membrane (MAM) where it exerts its functions through protein–protein interactions [6]. The σ2 receptor has only recently been identified as the TMEM97 protein. Its crystal structure has been resolved, helping in the understanding of the ligand–protein interaction and paving the way for more in-depth investigations of the protein biological functions [7,8]. Of note, 20-(S)-hydroxycholesterol has been identified as an endogenous interactor of σ2 receptor/TMEM97 [9].
Recently, Niso and collaborators have evaluated the anticancer activity of a series of σ2 receptor agonists belonging to the thiosemicarbazone family (including the FA4 compound) in immortalized cell lines, both in vitro and in vivo. Of note, these ligands were also able to induce apoptosis in the human PDAC cell lines AspC1, MiaPaCa2, and PANC-1 in the low micromolar range [10]. σ-2 receptor agonists have been shown to induce apoptosis with limited toxicities in different preclinical models and, thus, are a promising possibility to trigger apoptosis or autophagy when chemoresistance to traditional therapies is developed [11,12].
Starting from this basis, our work aimed to further validate the effects of the better-performing thiosemicarbazone, namely FA4, in patient-derived primary cell cultures (PDAC-1 and PDAC-2), which are ex vivo cell populations recovered directly from fresh surgically resected samples from PDAC patients. These cells have the advantage of preserving the most important patient features because they are cultured in vitro for a few passages, in order to maintain the genetic hallmarks of the primary originator tumors [13]. Moreover, bioinformatic studies conducted in our laboratory highlighted that the gene coding for the σ-2 receptor was overexpressed in the PDAC-1 and PDAC-2 cells compared to the normal immortalized pancreatic ductal cells HPDE. These models were used to further establish the cytotoxic and proapoptotic effects of FA4, as well to investigate its inhibitory effects on cell migration.
Remarkably, we found that in both PDAC1 and PDAC2, the IC50 of the FA4 compound was lower than the one reported in cancer immortalized cell lines, suggesting that the compound also exerts a good cytotoxic activity in primary cell cultures. This effect was associated with apoptosis induction, which was comparable to the proapoptotic effects of gemcitabine, a drug commonly used in the treatment of PDAC [14].
As for the second objective of our study, FA4 has been shown to exert antimigratory effects and could, therefore, provide new opportunities for therapy against the pro-invasive and metastatic behavior of PDAC [15].
2. Results
2.1. TMEM97 mRNA Expression in Pancreatic Cancer Primary Cells
Our next generation sequencing (NGS) transcriptomic data showed that the σ-2 receptor mRNA TMEM97 was expressed in both primary pancreatic cancer cell culture, at significantly higher values compared to the normal human pancreatic ductal epithelial HPDE cells (Figure 1). These results were in line with previous findings, demonstrating a higher expression of the σ2 receptors on the plasma membranes of PDAC cell lines compared to HPDE cells (Supplemental Figure S1) [10]. Of note, the highest Fragments per Kilobase Million (FPKM) value was measured in PDAC-2 cells (Figure 1), which originated from the most clinically aggressive tumor [16].
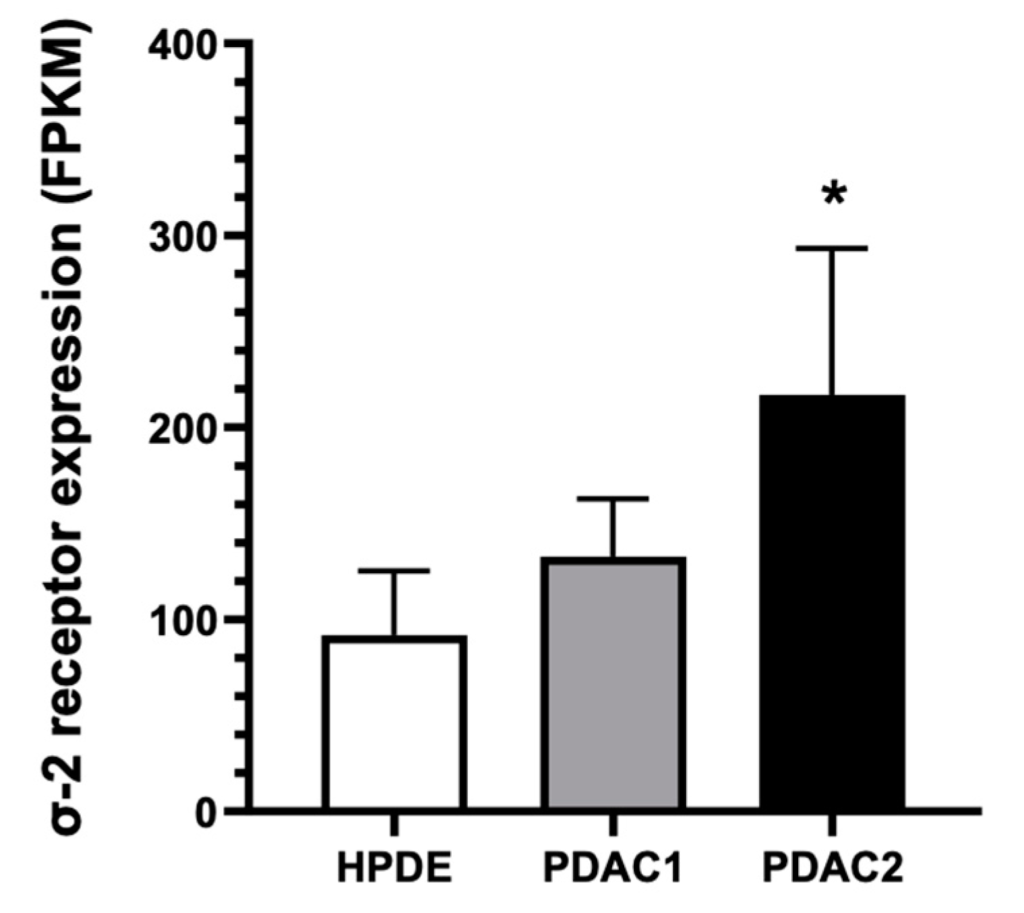
Two primary pancreatic cancer cell cultures (PDAC-1 and PDAC-2) originated from patients undergoing surgical resection for PDAC showed significantly higher mRNA expression levels of the σ2 receptor compared to HPDE cells. Columns represent the mean values obtained from three independent experiments; bars represent the SD. * p < 0.05 compared to HPDE cells.
2.2. FA4 Synthesis
FA4 was obtained according to a polypharmacology strategy, following other σ2 receptor-targeting thiosemicarbazones (e.g., MLP44 and PS3) that had shown promising antitumor properties in a panel of immortalized human PDAC cells and in a murine (KP02) tumor model [17,18]. All these thiosemicarbazones were designed with the aim to bind σ-2 receptors, that are overexpressed in a number of cancers [19,20], and chelate metals (in particular iron and copper ions) in order to obtain synergic effects by adding the effects of the alteration of the redox state of cells due to metal chelation to the cytotoxic properties proper of the σ2 agonists [21].
FA4, which was designed based on the σ2 reference agonist siramesine, demonstrated a superior anticancer activity in a panel of PDAC cells in comparison with its congeners MLP44 and PS3, that only differ in the basic moiety, with the (Z)-N,N-dimethyl-2-(2-oxoindolin-3-ylidene)hydrazinecarbothioamide unchanged (Figure 2).
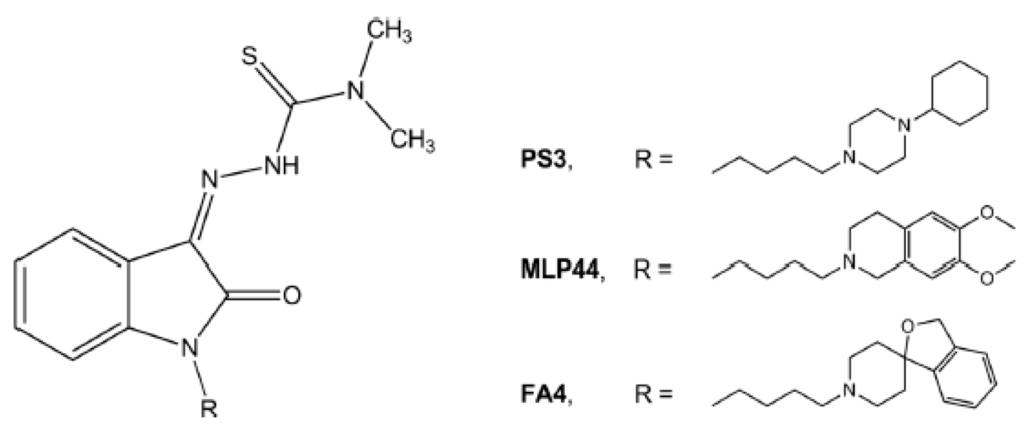
Chemical structures of the thiosemicarbazones (MLP44, PS3, and FA4) targeting the σ2 receptor.
The results obtained in commercial PDAC cell lines [10] highlighted how the basic moiety 3H-spiro[isobenzofuran-1,4′-piperidine] of FA4, which differs from the 6,7-dimethoxytetrahydroisoquinoline in MLP44 or the 1-cyclohexylpiperazine in PS3, may drive this increased activity. While MLP44 and PS3 underwent a deconstruction approach in order to evaluate how the chelating moiety impacts on the σ receptor affinity and overall activity, this was not investigated for FA4. Indeed, the removal of the thiosemicarbazone moiety importantly reduced the cytotoxic activity of MLP44 and PS3 [10]. Thus, FA4 was deconstructed by removing the thiosemicarbazone moiety, resulting in compound MT8 that appears like a simplified isomer of the siramesine, in which the butyl linker is in position-1 rather than position-3 of the indole ring, devoid of the p-fluorophenyl portion.
According to a previously reported procedure [17], the synthesis of compound MT8 was obtained by nucleophilic substitution of the spiropiperidine system on the 1-(4-chlorobutyl)-1H-indole portion (Scheme 1).
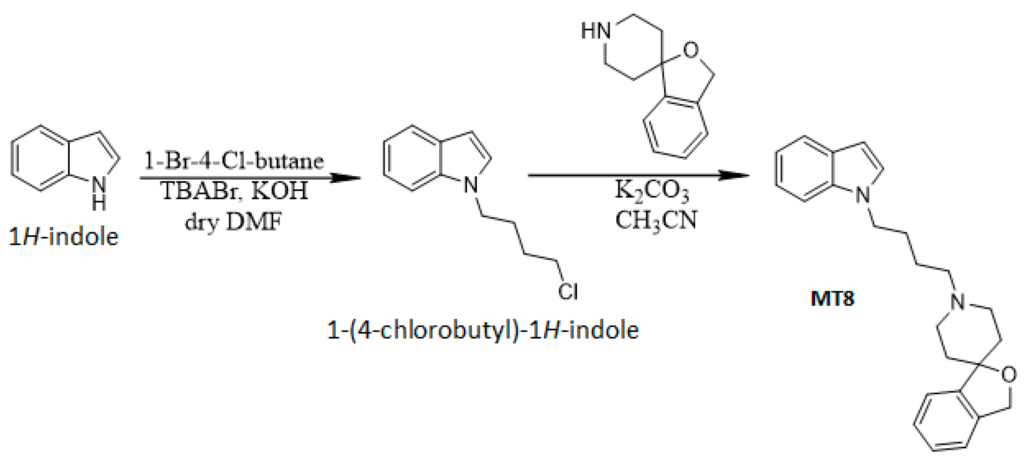
Schematic representation of the MT8 synthesis.
The compounds underwent radioligand binding assays on σ1 and σ2 receptors, according to the previously adopted protocols [17,18]. While the affinity for the σ2 receptor was subnanomolar (Ki = 0.43 nM, Table 1), for MT8,the affinity for the σ-1 receptor was in the two-digit nanomolar range (Ki = 27.3 nM, Table 1): this compound was a moderately selective high-affinity σ-2 receptor ligand, with an improved profile compared to siramesine that, from our perspective, did not discriminate between the two σ subtypes (Ki = 10.5 nM for σ1 and Ki = 12.6 nM for σ2 receptors, respectively) [22].
Table 1. σ receptor binding affinities and PANC-1 cell line viability of FA4 and MT8.
| Binding Assay | Cytotoxicity | ||
|---|---|---|---|
| Ki ± SD a (nM) | IC50 ± SD a (μM) | ||
| Compound | σ1 | σ2 | PANC-1 |
| MT8 | 27.3 ± 2.6 | 0.43 ± 0.1 | 19.1 ± 1.3 |
| FA4 | 51.3 ± 2.5 b | 15.8 ± 3.4 | 3.01 ± 0.9 c |
| Siramesine | 10.5 ± 1.3 d | 12.6 ± 1.4 | >50 |
| DTG | 29.3 ± 4.3 b | 29.5 ± 3.3 | >50 |
| (+)-pentazocine | 3.9 ± 0.3 | nd | >50 |
These data also suggest how the thiosemicarbazone portion on this scaffold was slightly detrimental for the interaction with the σ2 binding site, although FA4 retained an optimal binding profile in the nanomolar range (Ki = 15.8 nM for the σ1 and Ki = 51.3 nM for the σ2 receptors, respectively, Table 1). MT8 was evaluated for its anticancer properties in the PANC-1 cell line, in which FA4 activity appeared promising, both in vitro and in vivo [10]. MT8 had an appreciable cytotoxic effect in the low micromolar range (IC50 = 19.1 μM), but it was six-fold less potent than FA4.
The cytotoxicity of MT8 was also evaluated in other cancer cell lines, such as the lung adenocarcinoma A549 cells, the breast adenocarcinoma MCF7 cells, and the hepatocarcinoma HepG2 cells, displaying an IC50 = 4.29 μM in A549, IC50 = 6.02 μM in MCF7, and IC50 = 3.20 μM in HepG2 cells. These data highlight the higher aggressiveness of PANC-1 cells, in which MT8 displayed the lowest potency. On the other hand, the data also confirmed the need for a strategy with a greater impact on pancreatic tumors. From this perspective, the multitarget FA4 appeared to be a promising candidate and was selected to be further investigated in the primary PDAC cell cultures of the present study.
2.3. Antiproliferative Activity
Cell viability upon exposure to FA4 was measured with the Sulforhodamine B (SRB) assay. FA4 caused a concentration-dependent inhibition of proliferation (Figure 3), with IC50 values of 0.825 ± 0.006 and 0.701 ± 0.059 μM in the PDAC-1 and PDAC-2 cells, respectively (Table 2). Lastly, we performed additional experiments to evaluate the in vitro cytotoxicity of FA4 against normal HPDE cells and normal fibroblast Hs27 cells. The results of these studies allowed us to calculate the selectivity index (SI, calculated as IC50 non-tumor cells/IC50 tumor cells), which were 11 and 7 in HPDE and Hs27 cells, respectively. Interestingly, FA4 had SI values similar to the values measured for gemcitabine, as reported in previous studies in the same PDAC primary cells [23].
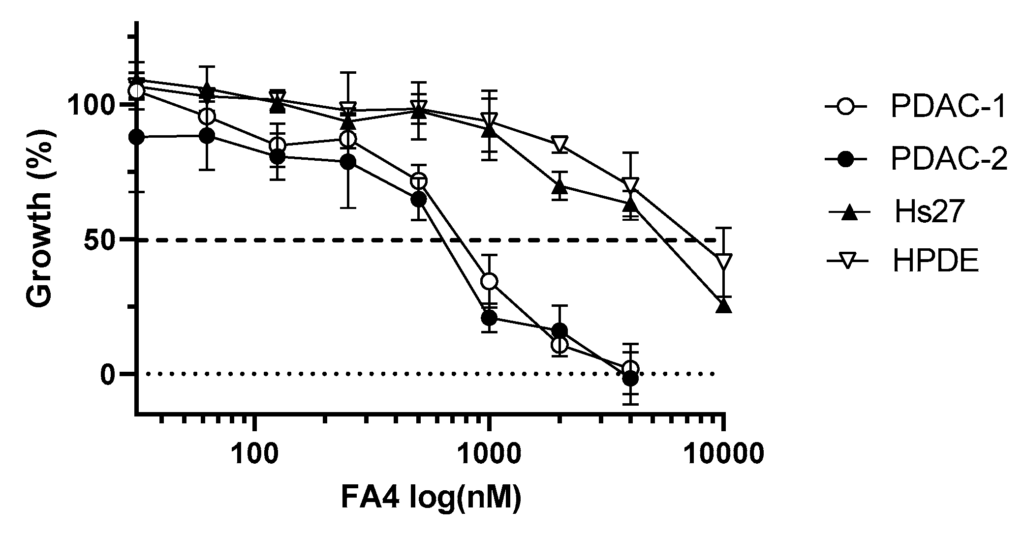
Representative dose-dependent inhibition of cell growth by FA4 in the PDAC primary cell lines PDAC-1 and PDAC-2 and in the HPDE and Hs27 cells. Cell viability was determined using the SRB assay after 72 h of treatment with increasing concentrations of FA4. Data points represent mean percentage of cell growth ± SD of three independent experiments.
| Cell Line | Cytotoxicity |
|---|---|
| IC50 ± SD (μM) | |
| PDAC1 | 0.825 ± 0.006 |
| PDAC2 | 0.701 ± 0.059 |
| HPDE | 8.069 ± 0.302 |
| Hs27 | 5.551 ± 0.333 |
Of note, the IC50 values of FA4 in PDAC-1 and PDAC-2 cells were lower than the values reported in human commercial PDAC cell lines, namely BxPC3, AspC1, MiaPaCa2, and PANC-1, where they ranged between 0.88 μM and 3.01 μM [10].
IC50 values ± SD were determined by the graphical interpolation of the dose–response curves reported in Figure 3.
2.4. Apoptosis Induction
Previous studies reported that the modulation of σ2 receptor activity contributes to the initiation of the apoptotic process [4,10]. Therefore, we evaluated the effect of FA4 on the induction of apoptosis in the PDAC primary cultures. To this aim, we measured the externalization of the plasma membrane phosphatidylserine, a reliable marker of cell apoptosis, which was quantified by the measurement of the fluorescence of annexin-V by spectrophotometric assay and microscopy. After 24 h of treatment with 10 μM FA4, a significant increase in the portion of apoptotic cells was observed compared to untreated control cells. The highest apoptotic index was measured in the PDAC-2 cells (17.6 ± 3.6, compared to 15.5 ± 4.9 in the PDAC-1 cells).
Remarkably, the percentages of apoptotic cells after treatment with FA4 were significantly higher compared to the number of cells that underwent apoptosis after treatment with gemcitabine at 10 μM (Figure 4), indicating that FA4 had superior anticancer activity.
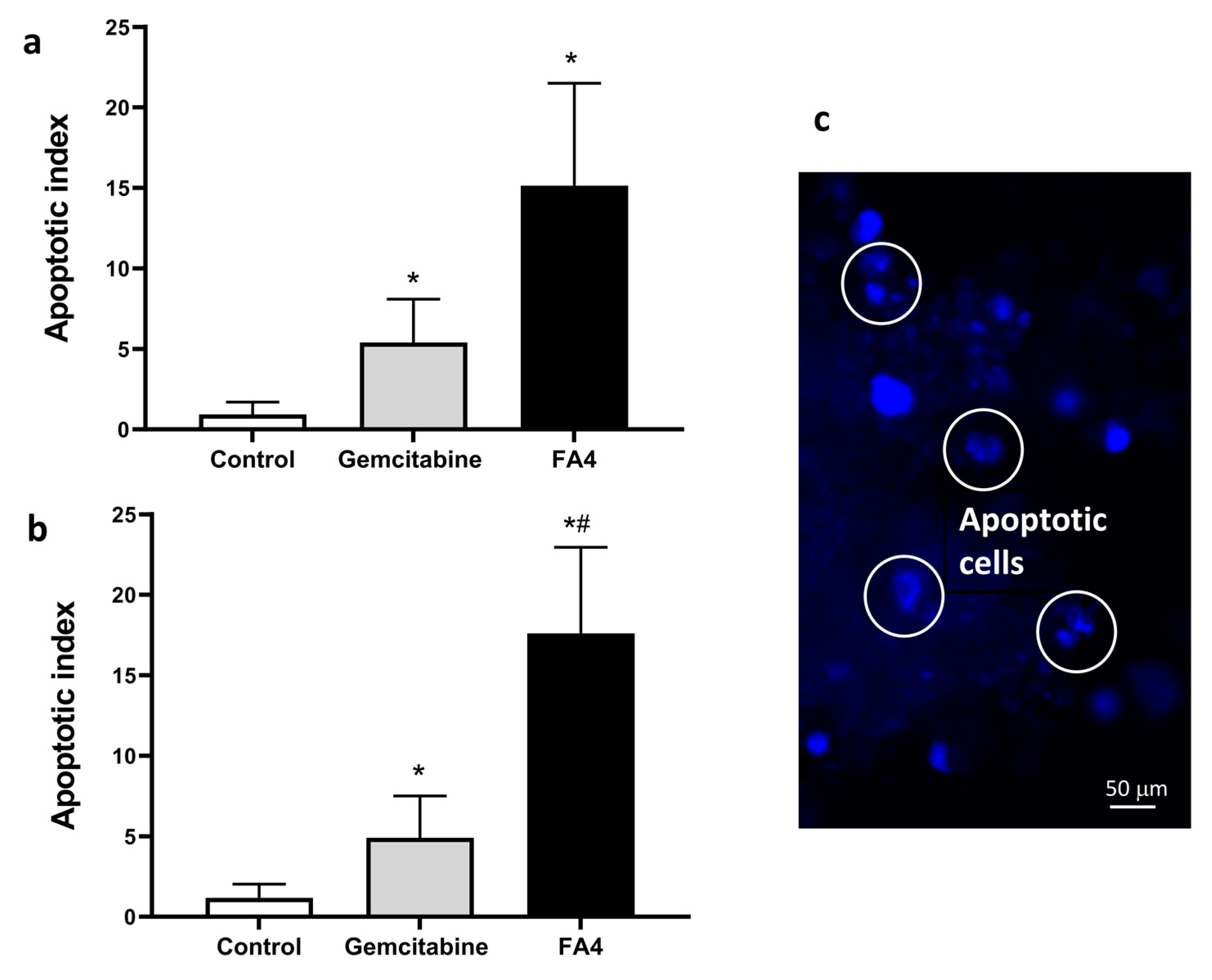
Effects of FA4, on apoptosis induction in PDAC-1 (a) and PDAC-2 (b) pancreatic cancer cells. The apoptotic index was calculated by evaluating the annexinV-FITC fold change compared to control cells after 24 h treatment. Gemcitabine was used as a positive control. Columns represent the mean; bars represent the SD. (c) Representative pictures of PDAC-2 cells with apoptotic features as assessed by bisbenzimide staining after treatment with FA4. * p < 0.05, significantly different compared with control; # p < 0.05, significantly different compared with gemcitabine.
2.5. Migration Assay
Cancer cell migration is a key factor contributing to the aggressiveness of PDAC. It is therefore important to find new compounds that can counteract or stop this process [24]. To evaluate the effect of FA4 on cell migratory properties, a migration assay was performed by incubating cells for 24 h with FA4. As reported in Figure 5 and Supplemental Figure S2, the compound was able to inhibit migration over 24 h with respect to the control and had an antimigratory activity superior to previous data with gemcitabine in these PDAC cell cultures [25,26].
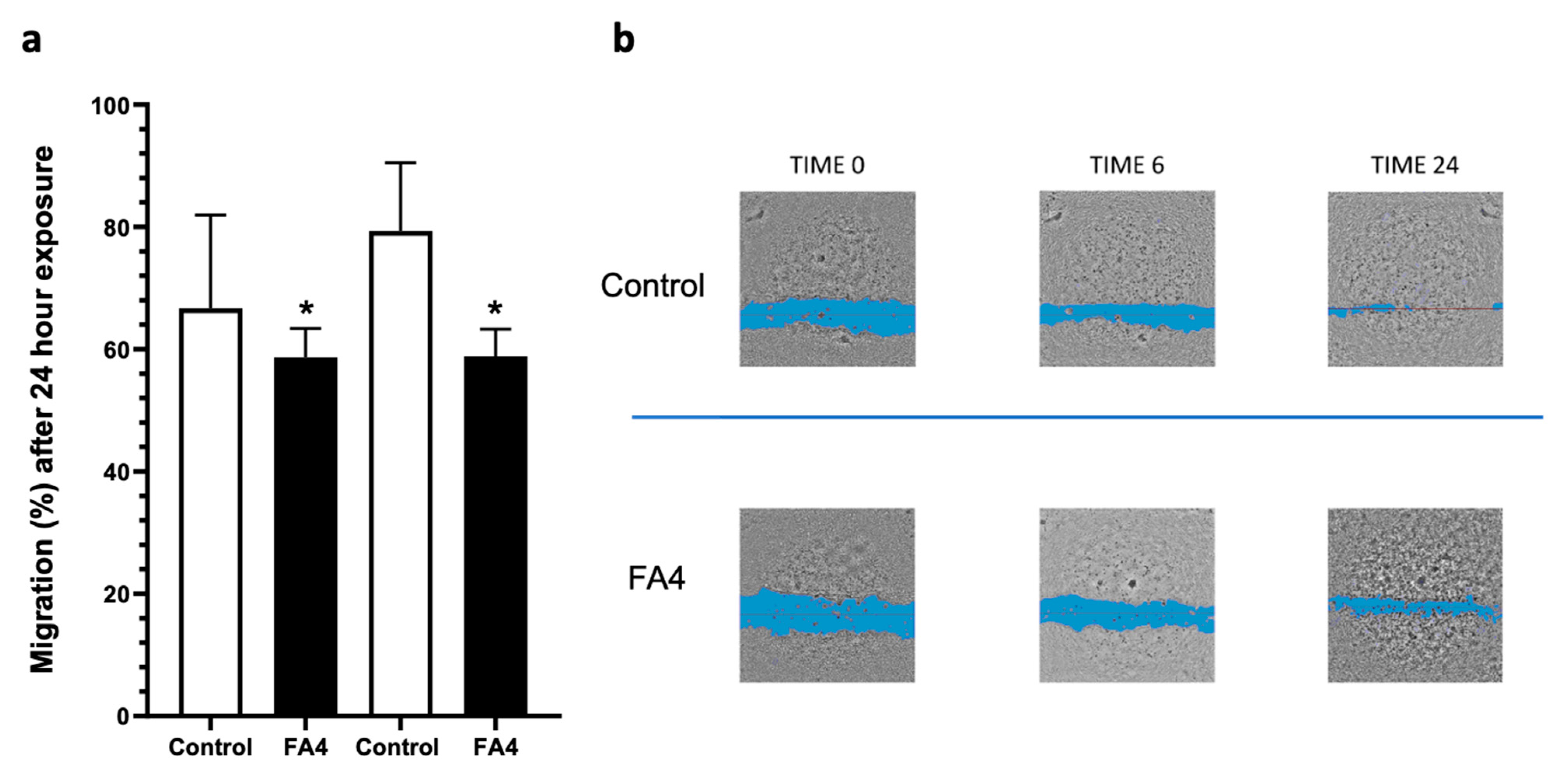
Results of the migration assay performed by incubating confluent cells after the scratch with FA4 10 µM in the PDAC-1 and PDAC-2 cells (a), and representative pictures taken at different time points (0, 6, and 24 h) in PDAC-2 cells (b). Data are derived from three independent experiments. Columns represent the mean values obtained from three independent experiments in treated and untreated (control) cells; bars represent the SD; asterisks indicate the statistical significance compared to the control. * p < 0.05.
3. Discussion
PDAC is a highly aggressive tumor, and therapeutic options are limited by constitutive and acquired resistance to treatment. In the search for a novel way to improve this dismal scenario, σ2 receptors have recently demonstrated encouraging results both in vitro and in vivo, showing the efficacy of novel compounds such as FA4 especially in PDAC cells resistant to gemcitabine [10]. To further define the potentiality of this promising drug, we studied the activity of FA4 in primary patient-derived cell cultures. These PDAC preclinical models, mimicking the molecular complexity of the original PDAC tumors, represent an improvement for the experimental testing of anticancer agents and might reduce their failure in follow-up clinical trials [16].
First, we found that the σ2 receptor/TMEM97 gene was significantly upregulated compared to normal pancreatic epithelial cells, particularly in patient-derived cell lines with particular aggressiveness [16]. This observation prompted us to investigate how σ2 receptor-targeting agents performed compared to gemcitabine, currently used as first-line therapy, with a special focus on advanced and chemorefractory tumors, where the efficacy of gemcitabine is low [3].
Following a multi-targeting drug synthesis approach, we designed a series of σ2 receptor agonists [21]. A preliminary screening on different commercial PDAC cell lines, indicated FA4, based on the modification of the structure of σ2 reference agonist siramesine, as the lead compound in terms of cytotoxicity [10]. By further deconstructing the siramesine structure, we produced a second congener, MT8, devoid of the thiosemicarbazone portion. Although MT8 had a higher affinity for the σ2 receptor than FA4, its cytotoxic profile was worse in the PANC-1 cell line, while it showed a good cytotoxic profile (in the same range as FA4) in other cancer cell lines. This result suggests that the σ-2 engagement by MT8 could be responsible for part of the cytotoxicity effects, but that the presence of the thiosemicarbazone moiety in FA4 importantly improved its cytotoxicity in PDAC cells. These data are of particular interest because they were obtained in PANC-1 cells, which are typically resistant to the activity of gemcitabine [27].
Prompted by these results, we narrowed our focus to FA4 and compared its biological effects with those of gemcitabine in primary PDAC samples. First, dose–response assays to measure cell viability indicated a good antitumor effect on PDAC cell and a higher IC50 on non-transformed cells, such as pancreatic epithelial cells and fibroblasts. Second, the SI was comparable for FA4 and gemcitabine [23]. Together, these results indicated that FA4has a potentially good therapeutic window.
Since σ2 receptor activity is known to trigger apoptosis in PDAC cells [4,10], we next tested the proapoptotic effect of FA4 in our primary PDAC cells. Interestingly, FA4 induced a higher percentage of apoptotic cells than gemcitabine. We considered that FA4 had stronger anticancer activity but the same SI of gemcitabine; hence, our results indicate that FA4 was a stronger and safer anti-PDAC agent, at least in vitro. FA4 was also safe in PDAC xenografts [10], suggesting that it could be noteworthy of further investigation in patient-derived xenografts (PDXs) and in a clinical setting. Furthermore, FA4 also decreased the migration of PDAC cells, revealing another important property in the treatment of PDAC, a cancer known for its high invasiveness [24].
These results prompt further studies on the multiple effects of FA4 on primary patient-derived cell cultures and PDXs and on the pharmacological effects of a co-incubation of FA4 with the drugs that represent the actual standard of care for PDAC, such as gemcitabine. Other future work will help to better clarify the pharmacokinetics and pharmacodynamics of what we hope will be a new drug in the fight against such aggressive tumor.