
Neuroprotective Effects of σ2R/TMEM97 Receptor Modulators in Neuronal Model of Huntington’s Disease

By Jing Jin, Nicolas Arbez, James J. Sahn, Yan Lu, Kathryn T. Linkens, Timothy R. Hodges, Anthony Tang, Robyn Wiseman, Stephen F. Martin, and Christopher A. Ross
Excerpt from the preprint published by ChemRxiv. Cambridge: Cambridge Open Engage; 2022; DOI 10.26434/chemrxiv-2022-l6xtv
This content has not been peer-reviewed.
Editor’s Highlights
- Huntington’s disease (HD) is a genetic neurodegenerative disease caused by an expanded CAG repeat in the human Huntingtin gene (HTT) that yields an expanded polyglutamine (polyQ) repeat in exon-1 of the human mutant huntingtin (mHTT) protein.
- Compounds binding to σ2R/TMEM97 protected cortical primary neurons from mHTT-induced toxicity as well as σ1R binding compounds.
- The results presented herein show for the first time that compounds modulating s2R/TMEM97 are neuroprotective in a HD cell model.
ABSTRACT:
Huntington’s disease (HD) is a genetic neurodegenerative disease caused by an expanded CAG repeat in the human Huntingtin gene (HTT) that yields an expanded polyglutamine (polyQ) repeat in exon-1 of the human mutant huntingtin (mHTT) protein. The presence of this polyQ repeat results in neuronal degeneration, and there is no cure nor treatment that modifies disease progression. Small molecules that bind selectively to the recently-cloned sigma 2 receptor (s2R), which is identical to TMEM97, have been shown to have neuroprotective effects in Alzheimer’s disease, traumatic brain injury and several other neurological disorders. To assess whether modulating pathways involving s2R/TMEM97 can provide protection in HD, we used an HD cell model to evaluate the effects of s2R/TMEM97 ligands on mHTT induced neuronal toxicity. We first synthesized a set of compounds designed to bind to s2R/TMEM97, and binding profiles (Ki) of these ligands were determined. Modulators with high selectivity for s2R/TMEM97 relative to s1R were then tested in our HD cell model. Primary cortical neurons were cultured in vitro for seven days and then co-transfected with either normal HTT construct (Htt N-586- 22Q/GFP) or mHTT construct (Htt-N586-82Q/GFP). Transfected neurons were treated with s2R/TMEM97 or s1R modulators for 48 h. After treatment, neurons were fixed and stained with Hoechst, and condensed nuclei were quantified to assess cell death in transfected neurons. Significantly, s2R/TMEM97 modulators reduce the neuronal toxicity induced by mHTT, and their neuroprotective effects are not blocked by NE-100, a selective s1R antagonist. These results indicate for the first time that s2R/TMEM97 modulators can protect neurons from mHTT induced neuronal toxicity, suggesting that targeting s2R/TMEM97 may provide a novel therapeutic approach to treat HD patients.
Graphical Abstract
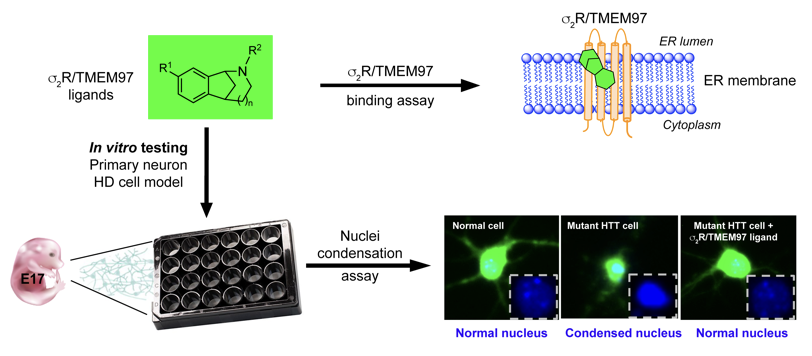
INTRODUCTION
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease caused by an expanded CAG repeat in the human Huntingtin gene (HTT) that yields an expanded polyglutamine (polyQ) repeat in exon-1 of the human mutant huntingtin (mHTT) protein.1 The mutant huntingtin protein (mHTT) preferentially affects striatum of HD patients, and the disease is manifested as uncontrolled chorea movement, cognitive decline and mood alterations that get progressively worse over time.2 Although some symptomatic treatments are available, there is no disease modifying treatment for HD, so there is an urgent need for neuroprotective therapies.3, 4
Small molecules have a rich history in drug discovery because of their ability to selectively target and inhibit or activate proteins involved in pathogenic pathways. In this context, compounds that bind to sigma receptors (σRs) are gaining prominence.5, 6 The sigma 1 receptor (σ1R), which shows no homology with any other mammalian protein, is located in the endoplasmic reticulum (ER) where it is enriched in the mitochondria-associated membrane subregion and is involved in calcium modulation.7, 8 Small molecules that bind to the σ1R have been shown to exhibit promising attributes in neurodegenerative and neurological disorders,9-12 and several σ1R ligands have been shown to have neuroprotective effects in animal models of HD,13-15 including pridopidine that is in human clinical trials.16-19
The sigma 2 receptor (σ2R), which is biochemically distinct from σ1R, was initially associated with cancer diagnosis and therapy,20-23 but more recently it has been implicated in neurological disorders.24, 25 The molecular identity of σ2R was an enigma from its discovery until a few years ago, when it was cloned and identified as the endoplasmic reticulum-resident transmembrane protein 97 (TMEM97),26 herein referred to as σ2R/TMEM97. Although the biological function of σ2R/TMEM97 is not well characterized, it is known to play a role in cholesterol trafficking and homeostasis,27 and it appears to be a binding partner of the lysosomal cholesterol transporter NPC1,28 a mutation in which results in Niemann-Pick disease type C. Small molecules that modulate σ2R/TMEM97 signaling show beneficial effects in different disease contexts, including cancer,29, 30 neuropathic pain,31 traumatic brain injury,32 alcohol use disorder,33, 34 and Alzheimer’s disease (AD).35-38 Moreover, a putative σ2R/TMEM97 antagonist is in Phase II clinical trials for treating AD.39
The finding that modulating σ2R/TMEM97 exhibits neuroprotection in several models of neurodegenerative disease prompted us to query whether compounds that bind to σ2R/TMEM97 might provide beneficial effects in an HD model. Toward testing this hypothesis, we evaluated a small panel of compounds with differing affinities and selectivities for σ2R/TMEM97 and σ1R in a primary neuron model of HD. These compounds include racemic AMA- 1127, DKR-1051, DKR-1677, UKH-1114, JJS-1678, BJM-1679, EES-1686, BEA-1687, MPC-1154, and HLJ- 1560 (Figure 1), some of which have been previously tested in other disease models.31, 32, 37 In this study, we used an HD cell model to assess the effects of these compounds upon mHTT-induced neuronal toxicity. Briefly, primary neurons were co-transfected with plasmid expression of a 586 N-terminal Htt polypeptide with either normal Q (Htt-N586-22Q) or expanded Q (Htt-N586-82Q) repeats and green fluorescent protein (GFP). Compounds that are selective for σ2R/TMEM97showed strong protective effects on mHTT-induced neuronal cell death as did several different compounds having σ1R selectivity. To exclude the possible involvement of σ1R modulation as a possible mechanism of action for the σ2R/TMEM97 compounds, the σ1R-selective antagonist, NE-100, was used. Notably, the protective effect of σ2R/TMEM97-selective compounds is not blocked by NE-100, clearly demonstrating that neuroprotection is not mediated by σ1R. These studies are the first to demonstrate that compounds that bind selectively to σ2R/TMEM97 are neuroprotective in an HD model, and they support further mechanistic studies of the function of σ2R/TMEM97 in mHTT protection as a possible new approach to treat HD.
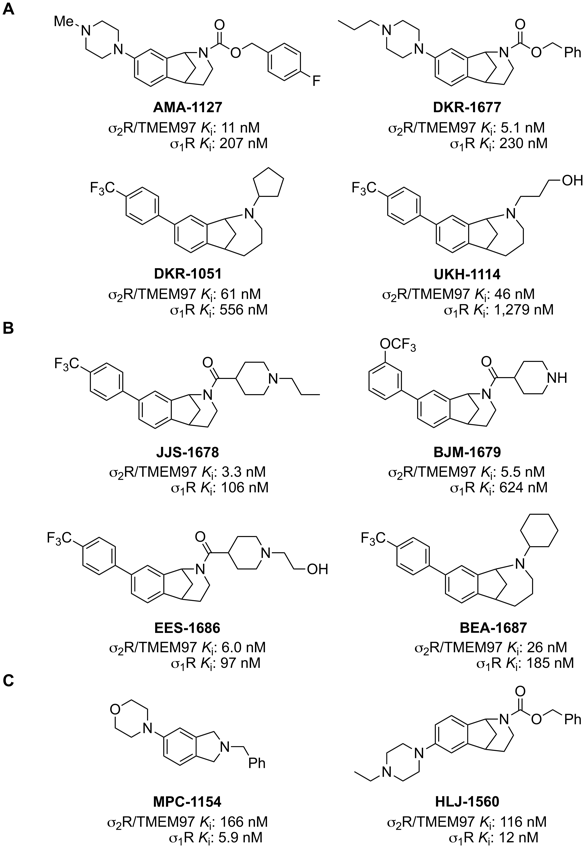
Structure of s2R/TMEM97-selective modulators and their binding affinities.
A. Structures of racemic B-norbenzomorphans (AMA-1127 and DKR-1677) and methanobenzazocines (DKR-1051 and UKH- 1114) that are selective modulators of s2R/TMEM97 and their binding affinities. Ki, for s2R/TMEM97 (rat) and σ1R (guinea pig). B. Structures of racemic B-norbenzomorphans (JJS-1678, BJM-1679, and EES-1686) and methanobenzazocines (BEA-1687) that are selective modulators of s2R/TMEM97 and their binding affinities. Ki, for s2R/TMEM97 (human) and σ1R (human). C. Structures of σ1R-selective modulators MPC-1154 and racemic HLJ-1560 and their binding affinities, Ki, for s2R/TMEM97 (rat) and σ1R (guinea pig).
…
RESULTS
Receptor binding profiles
The binding affinities (Ki) of all synthetic compounds for σ2R/TMEM97 and σ1R were determined at the Psychoactive Drug Screening Program. Prior to the identification of σ2R as TMEM97, Ki values were measured using σ2R sourced from rat PC12 cells and σ1R sourced from guinea pig brain, but subsequently σ2R/TMEM97 and s1R binding isotherms were determined using human protein obtained by transfection in HEK293T cells. Examination of the Ki values for AMA-1127, DKR-1051, DKR-1677 and UKH-1114, which were obtained using rat s2R/TMEM97 and guinea pig σ1R proteins, show that each of these compounds has high affinity and good selectivity for s2R/TMEM97 vs s1R (Figure 1A). Similarly, the Ki values for JJS-1678, BJM-1679, EES-1686 and BEA-1687, which were obtained using human s2R/TMEM97 and σ1R proteins, also display high affinity and good for s2R/TMEM97 vs s1R (Figure 1B). Examination of the structures of these s2R/TMEM97-selective compounds reveals that they belong to structural classes we have previously identified.40, 41, 43 Although the two molecular scaffolds differ by the presence of an extra methylene group in the methanobenzazocines DKR-1051 and UKH- 1114, two distinct chemotypes are also represented. Namely, AMA-1127 and DKR-1677 have a basic piperazine group on the aromatic ring of the B-norbenzomorphan core, whereas all of the other compounds have an aryl substituent on the parent molecular framework. We have also identified compounds that are selective for σ1R (guinea pig) vs s2R/TMEM97 (rat), and we used two of these, MPC-1154 and HLJ-1560 (Figure 1C), as controls. MPC-1154 represents a completely different class of compounds selective for σ1R,44 whereas HLJ-1560 differs from the s2R/TMEM97-selective ligands AMA-1127 and DKR-1677 by the orientation of the piperazine ring on the B-norbenzomorphan core.40, 41, 43
Compounds binding to σ2R/TMEM97 protected cortical primary neurons from mHTT induced toxicity as well as σ1R binding compounds.
To assess the possible neuroprotective effects of selective σ2R/TMEM97 modulators in the context of neurodegenerative processes associated with HD, we used a HD cell model that we have previously described.46 Briefly, primary cortical neurons were isolated from embryonic day 17 mouse brain and cultured in neurobasal medium for seven days. The neurons were then co-transfected with plasmids expressing HTT and GFP. Plasmids expressing 586 N-terminal amino acid of human huntingtin gene with either 22 polyglutamine (Htt-N586-22Q) or 82 polyglutamine (Htt-N586-82Q) repeats within exon 1were used. Parallel experiments were then performed in which neurons were simultaneously treated with σ2R/TMEM97 or σ1R modulators; vehicle treated neurons were
used as control. Forty-eight hours after transfection, neurons were fixed, and GFP + neurons with condensed nuclei were calculated. The effects of sigma receptor modulators on neuronal cell death were then evaluated. Representative images of neurons transfected with 22Q, 82Q and 82Q with s2R/TMEM97 modulators show that mHTT (82Q) transfected neurons had condensed nuclei, whereas and mHTT (82Q) treated with s2R/TMEM97 ligands AMA-1127, DKR-1051, UKH-1114, EES-1686 and BEA-1687 had relatively normal nuclei (Figure 2A and B). Similarly, mHTT (82Q) treated with the selective σ1R ligand HLJ-1560 also had relatively normal nuclei.
The effects of selective σ2R/TMEM97 modulators on mHTT neuronal toxicity were then assessed by a nuclear condensation assay as before (Figure 2C–H).46 Neurons were treated with σ2R/TMEM97 modulators at concentrations varying from 0.01–10 μM, and compounds having protective effects are AMA-1127 (Figure 2C), DKR-1015 (Figure 2D), UKH-1114 (Figure 2E), BJM-1679 (Figure 2F), EES-1686 (Figure 2G) and BEA-1687 (Figure 2H). These compounds provide significant neuroprotection at concentrations that range from a low of 10 nM for EES-1686 (Figure 2G) to a high of 5 μM for BJM-1679 (Figure 2F). Importantly, the observed neuroprotective effects were dose dependent with higher concentrations of the σ2R/TMEM97 modulator having greater protective effects (Figure 2C–H). Because these compounds have no effect on neurons transfected with Htt- N586-22Q, none appear to exhibit any intrinsic toxicity. Of the σ2R/TMEM97 modulators tested in these experiments, only DKR-1677 (Figure S1A) and JJS-1678 (Figure S1B) had no notable protective effect on neurons transfected with HTT-N586-82Q.
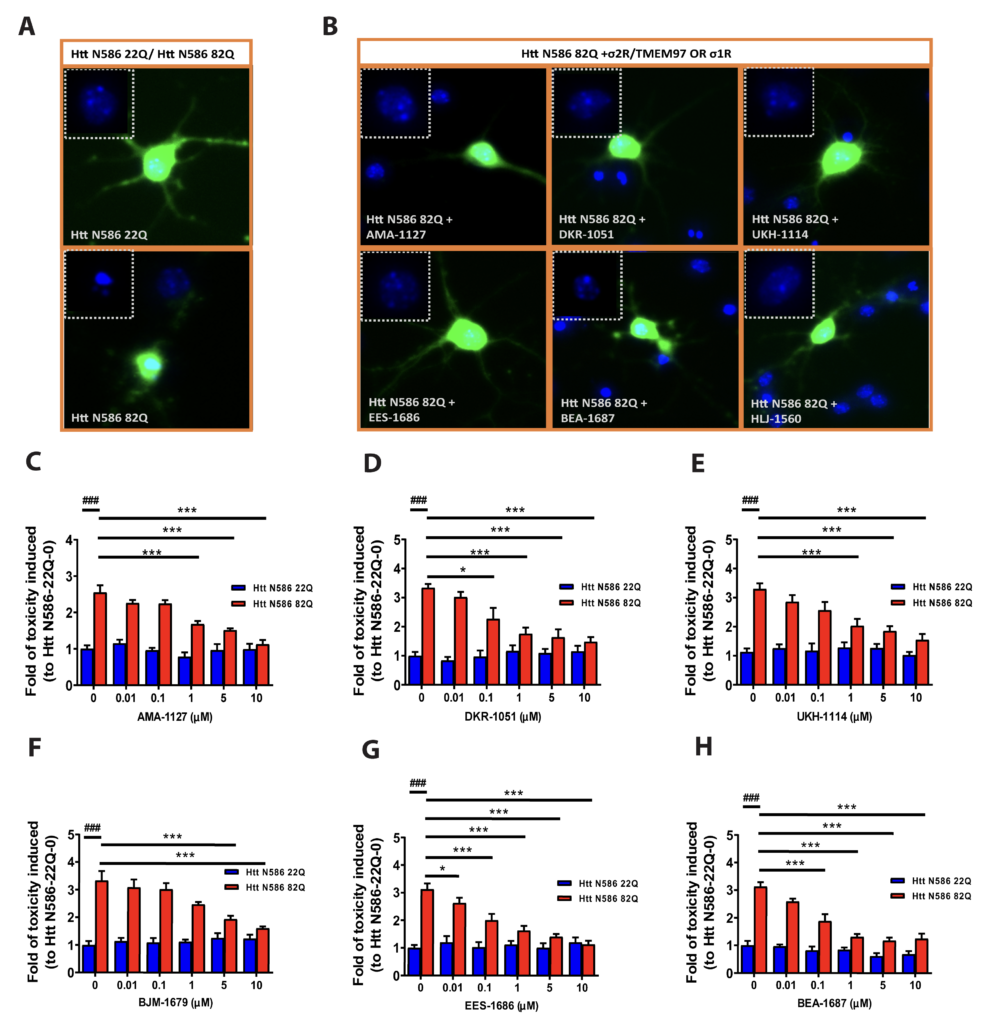
Neuroprotective effect of σ2R/TMEM97-selective modulators on mHTT induced toxicity.
Primary cortical neurons were co-transfected with either Htt-N586-22Q or Htt-N586-82Q and GFP. Four hours after transfection, neurons were treated with either σ1R or σ2R/TMEM97 modulators for 48 h,; whereupon neurons were fixed, and nuclei were stained with Hoechst. Cells with condensed nuclei were counted as dead cells. Only neurons transfected with plasmid were counted. A. Representative pictures for neurons transfected with Htt-N586- 22Q or Htt-N586-82Q and GFP. B. Representative pictures for neurons transfected with Htt-N586-82Q/GFP and treated with σ2R/TMEM97 or σ1R modulators. Neurons were treated with 1 μM of the indicated modulators. Insert boxes indicated viable cells with normal nucleic morphology. C-H. σ2R/TMEM97-selective modulators tested in HD cell model showing neuroprotection. These compounds are AMA-1127 (C), DKR-1051 (D), UKH-1114 (E), BJM-1679 (F), EES-1686 (G), and BEA-1687 (H). ### p<0.0001 vs Htt N586-22Q with 0. * p<0.05, *** p<0.0001 vs Htt N586-82Q with 0. n=6-8 independent experiments.
e also examined the effects of several selective σ1R modulators on neuron survival, and both MPC-1154 (Figure 3A) and HLJ-1560 (Figure 3B) protected neurons from mHTT induced cell toxicity at levels of 1 μM. The neuroprotective effects of these compounds are also dose dependent (Figure 3A, B).
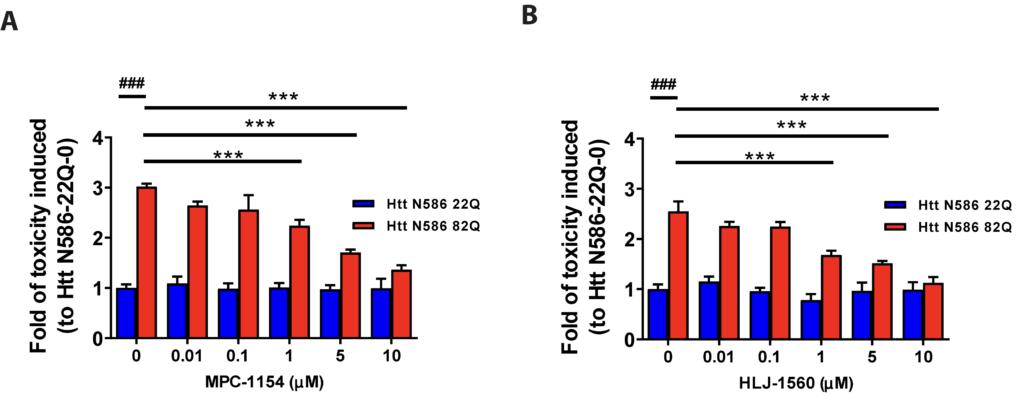
Neuroprotection effect of σ1R-selective modulators on mHTT induced toxicity.
Primary cortical neurons were co-transfected with either Htt-N586-22Q or Htt-N586-82Q and GFP. Four hours after transfection, neurons were treated with or without σ1R modulators at different concentrations. σ1R-selective modulators tested in HD cell model were as follows: MPC-1154 (A) or HLJ-1560 (B). ### p<0.0001 vs Htt N586-22Q with 0. *** p<0.0001 vs Htt N586-82Qwith 0. n=6-8 independent experiments.
NE-100 did not alter the effect of σ2R/TMEM97 binding compounds on mHTT toxicity.
The binding profiles of AMA-1127, DKR-1051, UKH-1114, BJM-1679, EES-1686, and BEA-1687 show that they are all selective for σ2R/TMEM97 vs σ1R and other CNS proteins (see Supporting Information), suggesting that their protective effect arises from modulating a pathway involving s2R/TMEM97. However, the function of s2R/TMEM97 is not well understood, and because there are no ligands that are confirmed s2R/TMEM97 antagonists, we used an established s1R antagonist, NE-100, to block any s1R pathway that might be operative.48, 49 NE-100 had no effect on the protective attributes of the DKR- 1015, UKH-1114, EES-1686 and BEA-1687, whereas NE-100 treatment abolished the neuroprotective effects of the selective s1R modulators HLJ-1560 (Figure 4) and MCP-1154 (Figure S2). These results support the hypothesis that compounds that selectively bind to s2R/TMEM97 mitigate mHTT-induced neuronal toxicity by a pathway that is distinct from modulating s1R.
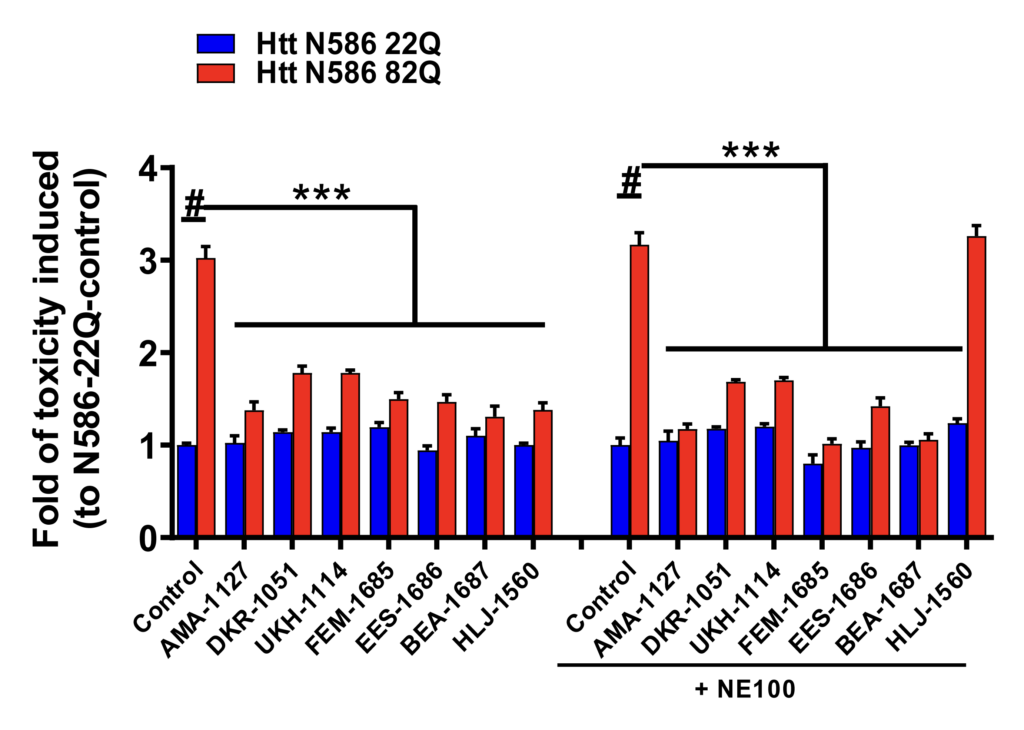
Specificity of σ2R/TMEM97 modulators.
To further explore the specificity of σ2R/TMEM97 modulators, selective σ1R antagonist, NE-100 was used in the primary cortical neurons treated σ1R or σ2R/TMEM97 modulators. Primary cortical neurons were co-transfected with either Htt-N586-22Q or Htt-N586-82Q and GFP. Four hours after transfection, neurons were treated with modulators with or without a pretreatment with 10 μM of NE-100. After 48 h, neurons were fixed and nuclei were stained. We included one σ1R-selective modulator, HLJ- 1560, as a positive control, which its effect will be blocked by NE-100. Cell death were quantified using a nuclei condensation assay. NE-100 abolished the protective effect of the σ1R modulator, HLJ-1560, but it did not influence the effects of σ2R/TMEM97 modulators. *** p<0,0001 vs Htt N586-82Q, # p<0,001 vs Htt N586-22Q. n=4-6 independent experiments.
DISCUSSION
HD is a devastating disease for which there is neither a cure nor an approved treatment that slows, stops or reverses its progression. HD patients typically develop symptoms at mid-adulthood, and the associated disabilities worsen over time ending in death within 10-20 years following the onset of symptoms.50-52 Research using cell and animal models has achieved significant progress in understanding the etiology and pathology of HD, but effective treatments have been elusive. HD is caused by mutation in HTT gene that encodes mutant huntingtin (mHTT) with expanded polyglutamine repeats (>37Q).51, 52 Mutant HTT is specifically toxic to striatal medium spiny neurons and causes neuronal death in the striatum.50-52 The mechanism of neuronal death includes mHTT-related transcriptional dysregulation, neurotrophic factor deficit, abnormal mitochondrial function, energy and cholesterol metabolic abnormalities, and impaired protein degradation.53 Although numerous attempts toward discovering agents that can reduce or reverse mHTT toxicity have been unsuccessful, genetic modification of mHTT expression is a promising technique, albeit one limited by central delivery and lack of specificity.54, 55
The results detailed herein show that compounds that selectively target s2R/TMEM97 significantly protect neurons in primary cultures from toxicity induced by mHTT. The neuroprotective effects of the s1R ligands MPC- 1154 and HLJ-1560 are not entirely surprising because s1R agonists have been shown to be neuroprotective in HD animal models,13-15 and the s1R agonist pridopidine is in clinical trials for treating HD.16-18 However, the present findings are the first to demonstrate that s2R/TMEM97 ligands are neuroprotective in an HD model. In particular, AMA-1127, DKR-1051, UKH-1114, EES-1686 and BEA-1687, which bind with high affinity and selectivity to s2R/TMEM97, protect neurons from mHTT-induced toxicity in a dose-dependent manner at concentrations as low as 10 nM for EES-1686. It is notable that some of the compounds tested in these studies also display beneficial effects in models of other neurological conditions. For example, DKR-1051 and UKH-1114 relieve mechanical hypersensitivity in an animal model of neuropathic pain.31 Somewhat surprisingly, DKR-1677, which reduces axonal degeneration in a blast model of traumatic brain injury (TBI) and improves survival of cortical neurons and oligodendrocytes in the controlled cortical impact injury model of TBI,32 has no neuroprotective activity in this HD model, suggesting that mechanisms of neuroprotection in TBI are different from those in HD.
We wanted to confirm that binding of AMA-1127, DKR-1051, UKH-1114, EES-1686 and BEA-1687 to s2R/TMEM97 was responsible for their protective attributes using a s2R/TMEM97 antagonist. However, the functional activity of s2R/TMEM97 has not been fully elucidated, so there is no generally accepted assay available exclude the possibility that s1R binding was responsible for their protective effects. Toward this end, we treated neurons with AMA-1127, DKR-1051, UKH-1114, EES-1686 and BEA-1687 together with the known s1R HLJ-1560 served as a positive s1R control. NE-100 blocked the protective effect of the s1R binding compound HLJ-1560, but it did not block the effects of any of the s2R/TMEM97 binding compounds. These results exclude the possibility that the observed protective effects of AMA-1127, DKR-1051, UKH-1114, EES-1686 and BEA-1687 arise from binding to the s1R. In view of the exceptional selectivity of these compounds for s2R/TMEM97 relative to about 45 GPCRs, transmembrane proteins, and neurotransmitter transporters (see Supporting Information), it seems highly likely that modulation of s2R/TMEM97 protects neurons from mHTT-induced toxicity.
In summary, the results presented herein show for the first time that compounds modulating s2R/TMEM97 are neuroprotective in a HD cell model. EES-1686, the most potent compound studied, is a promising lead compound for advancing to in vivo experiments. Although further studies are needed, these investigations suggest that targeting s2R/TMEM97 may be a novel therapeutic strategy for developing HD treatments.