
Neuroinflammation is independently associated with brain network dysfunction in Alzheimer’s disease

By Fangda Leng, Rainer Hinz, Steve Gentleman, Adam Hampshire, Melanie Dani, David J. Brooks, and Paul Edison
Excerpt from the article published in Molecular Psychiatry, 06 December 2022. DOI: https://doi.org/10.1038/s41380-022-01878-z
Editor’s Highlights
- Neuroinflammation has been established as an important component in Alzheimer’s pathology.
- Alzheimer’s dementia is increasingly understood as a consequence of brain network dysfunction.
- Neuroinflammation is an independent predictor of structural and functional network disruption, while the amout of Aβ in Aβ+ population was not linearly associated with brain connectivity changes.
- Aβ deposition initiates neuroinflammation, which then directly lead to structural and functional brain network damage, while also promoting hyperphosphorylated tau formation and aggregation
- At the very early stage of the Alzheimer’s disease, a stronger response of anti-inflammatory microglia is associated with better outcome.
- In the later stages, the detection of β-sheeted fibrillary amyloid by PET, might already mark the failure of the protective response, followed by a deleterious inflammatory response.
Abstract
Brain network dysfunction is increasingly recognised in Alzheimer’s disease (AD). However, the causes of brain connectivity disruption are still poorly understood. Recently, neuroinflammation has been identified as an important factor in AD pathogenesis. Microglia participate in the construction and maintenance of healthy neuronal networks, but pro-inflammatory microglia can also damage these circuits. We hypothesised that microglial activation is independently associated with brain connectivity disruption in AD. We performed a cross-sectional multimodal imaging study and interrogated the relationship between imaging biomarkers of neuroinflammation, Aβ deposition, brain connectivity and cognition. 42 participants (12 Aβ-positive MCI, 14 Aβ-positive AD and 16 Aβ-negative healthy controls) were recruited. Participants had 11C-PBR28 and 18F-flutemetamol PET to quantify Aβ deposition and microglial activation, T1-weighted, diffusion tensor and resting-state functional MRI to assess structural network and functional network. 11C-PBR28 uptake, structural network integrity and functional network orgnisation were compared across diagnostic groups and the relationship between neuroinflammation and brain network was tested in 26 Aβ-positive patients. Increased 11C-PBR28 uptake, decreased FA, network small-worldness and local efficiency were observed in AD patients. Cortical 11C-PBR28 uptake correlated negatively with structural integrity (standardised β = −0.375, p = 0.037) and network local efficiency (standardised β = −0.468, p < 0.001), independent of cortical thickness and Aβ deposition, while Aβ was not. Network structural integrity, small-worldness and local efficiency, and cortical thickness were positively associated with cognition. Our findings suggest cortical neuroinflammation coincide with structural and functional network disruption independent of Aβ and cortical atrophy. These findings link the brain connectivity change and pathological process in Alzheimer’s disease, and suggest a pathway from neuroinflammation to systemic brain dysfunction.
Introduction
The pathological hallmarks of Alzheimer’s disease are extracellular β-amyloid (Aβ) plaques and intraneuronal neurofibrillary tangles (NFT) consisting of tau protein. The ‘amyloid cascade hypothesis’ posits initial Aβ aggregation leads to intracellular tau phosphorylation, tangle formation and neuronal dysfunction [1]. Both Aβ and NFT are associated with neuroinflammation in the form of activated microglia, which might mediate their neurotoxicity [2]. Genome-wide association studies (GWAS) have found that innate immune-related genes such as TREM2 (Triggering Receptor Expressed on Myeloid Cells 2) are associated with the risk of sporadic AD, independent of apolipoprotein E4 status [3]. It is now well-accepted that neuroinflammation plays a significant role in the process of neurodegeneration [4].
Brain network disruption is considered responsible for cognitive impairment in AD [5]. Diffusion tensor imaging (DTI) and resting-state functional MRI (rs-fMRI) have enabled evaluation of brain network structural integrity and functional organisation in vivo [6, 7]. Recent studies have revealed disruption of structural and functional connectivity early in the Alzheimer’s trajectory and they are associated with cognitive impairment [8, 9].
However, the pathological events disrupting structural and functional brain networks in the Alzheimer’s spectrum remain to be clarified. While there is imaging and pathological evidence that tau deposition is associated with white-matter degeneration [10, 11], the influence of amyloid deposition on brain connectivity in Alzheimer’s disease remains controversial [12, 13]. More importantly, the influence of neuroinflammation on brain connectivity needs further evaluation, given its close relationship with Alzheimer’s pathologies and the supportive function of homoeostatic microglia for neurones in healthy human brain [14]. We hypothesised that levels of microglial activation are responsible for network structural integrity damage and functional organisation disruption in AD. To test the hypothesis, we analysed the association between 11C-PBR28 uptake and the brain’s structural and functional network measures in Aβ-positive patients while controlling for amyloid deposition and grey-matter integrity.
…
Results
Demographics
14 AD, 12 MCI and 16 HCs were included in the current study, with their demographics summarised in Table 1. The mean ages (±SD) of the AD and MCI patients were 73.4 ± 7.7 and 75.8 ± 7.6, which were older than HCs (63.1 ± 8.2). The mean MMSE were 23.0 ± 3.6, 28.0 ± 1.5 and 28.7 ± 1.7 for the AD, MCI and HCs, respectively.
| AD | MCI | HC | ||
|---|---|---|---|---|
| Cases | 14 | 12 | 16 | – |
| Age | 73.7 ± 7.7a | 75.8 ± 7.6b | 63.1 ± 8.2 | 0.00012** |
| Gender (Male/Female) | 8/6 | 6/6 | 7/9 | 0.74 |
| IQ | 114 ± 16.9 | 118 ± 8.1 | 115 ± 9.4 | 0.68 |
| NART | 12.2 ± 10.3 | 10.7 ± 7.1 | 12.6 ± 7.5 | 0.83 |
| MMSE | 23.0 ± 3.6a | 28.0 ± 1.5 | 28.7 ± 1.7 | <0.0001** |
| Rey Copy | 25.6 ± 10.8 | 32.2 ± 4.3 | 34.5 ± 2.8 | 0.022* |
| Rey Imm | 5.2 ± 4.6a | 9.8 ± 6.3 | 18.2 ± 11.5 | 0.004* |
| Rey Del | 3.2 ± 4.2a | 10.5 ± 6.6 | 18.0 ± 7.5 | <0.0001** |
| WLM Imm | 15.4 ± 9.8a | 30.0 ± 10.5b | 45.2 ± 8.5 | <0.0001** |
| WLM Del | 3.8 ± 6.3a | 13.1 ± 8.2b | 28.4 ± 6.2 | <0.0001** |
| Hopkins Imm | 9.8 ± 8.8a | 18.3 ± 6.8b | 26.2 ± 8.6 | 0.00016** |
| Hopkins Del | 1.6 ± 1.9a | 6.7 ± 9.1b | 12.4 ± 7.1 | 0.003* |
| Hopkins RI | 5.0 ± 3.8a | 7.9 ± 3.9b | 11.4 ± 0.9 | <0.0001** |
| LNS | 3.6 ± 2.4a | 6.3 ± 3.0a | 10.71 ± 3.20 | <0.0001** |
| Semantic Fluency | 11.3 ± 3.2a | 14.7 ± 4.2b | 23.8 ± 8.6 | <0.0001** |
| Verbal Fluency | 35.6 ± 11.9a | 38.7 ± 13.5b | 49.2 ± 12.9 | 0.038* |
| HADS Anxiety | 6.8 ± 3.8 | 6.0 ± 3.9 | 5.2 ± 3.5 | 0.58 |
| HADS Depression | 5.6 ± 4.3 | 4.0 ± 2.4 | 3.2 ± 3.6 | 0.28 |
Demographics of the study cohort.
Alzheimer’s disease (AD), Mild cognitive impairment (MCI), and healthy control (HCs)
SD standard deviation, NART National Adult Reading Test, MMSE Mini-Mental State Examination, Rey Rey–Osterrieth complex figure test, Imm immediate recall, Del delayed recall, WLM Wechsler Memory Scale–Logical Memory test, Hopkins Hopkins Verbal Learning Test, RI Recognition Index, LNS Letter-Number Sequencing, HADS Hospital Anxiety and Depression Scale.
*p < 0.05; **p < 0.001 in ANOVA.
aSignificant difference between Alzheimer’s dementia patients and healthy controls in post hoc comparisons (p < 0.05).
bSignificant difference between mild cognitive impairment patients and healthy controls in post hoc comparisons (p < 0.05).
11C-PBR28 PET
Voxel-wise analysis confirmed increased 11C-PBR28 uptake in bilateral superior and middle frontal gyri, bilateral frontal pole, bilateral middle and inferior temporal gyri, bilateral fusiform gyri, bilateral lateral occipital gyri, left hippocampus, left parahippocampal gyrus, and left angular gyrus in AD patients (corrected for age and gender, Fig. 1A). In Aβ+ MCI patients, elevated 11C-PBR28 uptake was seen in right inferior and middle temporal gyri, right temporal pole, right fusiform gyrus and right parahippocampal gyrus (corrected for age and gender, Fig. 1B). ROI analysis localised elevated 11C-PBR28 uptake in medial temporal lobe in AD patients compared to HCs (1.09 ± 0.11 versus 1.01 ± 0.07, p = 0.029). No significant difference was found between the MCI patients (1.04 ± 0.08) and HCs. TSPO genotypes did not show significant influence on SUVR values at both ROI level and voxel level.
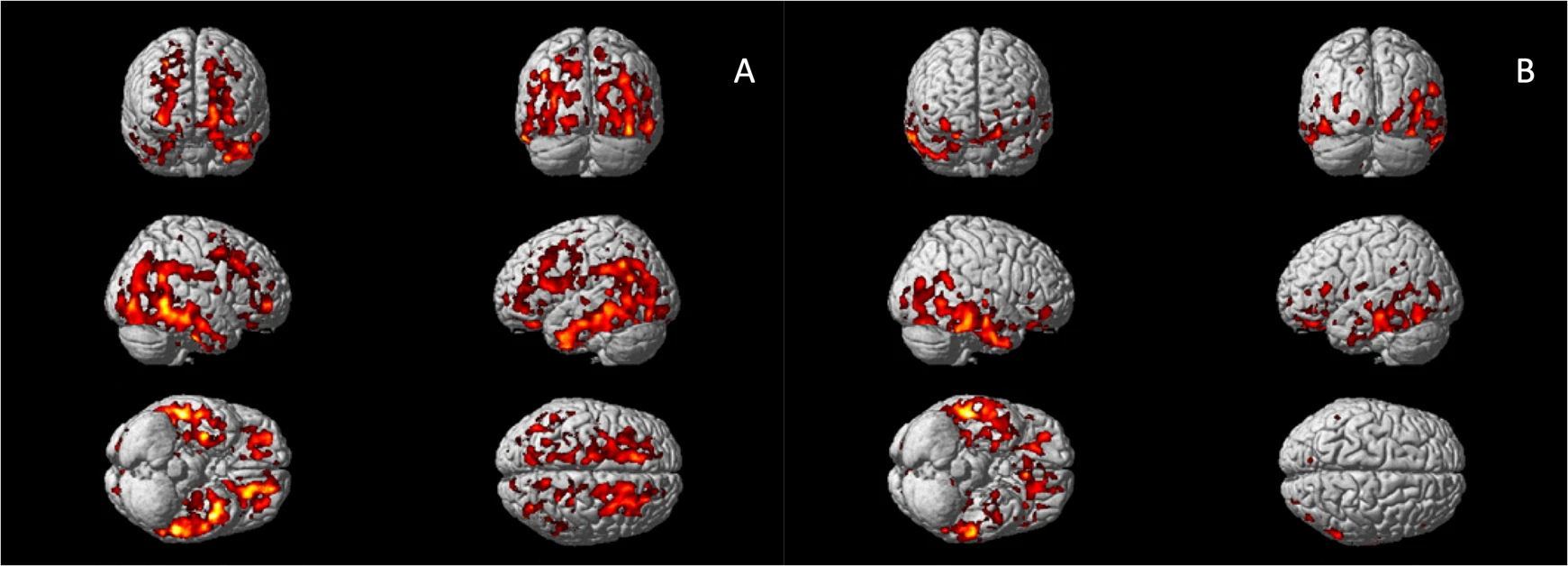
Voxel-wise group comparisons of 11C-PBR28 PET images.
A Increased 11C-PBR28 uptake ratio in the Alzheimer’s dementia patients compared to healthy controls; (B) Increased 11C-PBR28 uptake ratio in the Aβ+ mild cognitive impairment patients compared to healthy controls.
Cross-group comparisons of network metrics and cortical thickness
The first PC of FA values (PC1, 40.3% variance explained) did not differ across the groups, while PC2 (9.7% variance explained) of the AD patients was significantly lower than HCs (p = 0.003, age and gender corrected). The FA PC2 axis distinguished AD and HCs with good separation (Supplementary Fig. 2). Loading weights revealed that all TOIs had positive weights on FA PC1 while long association fibres had higher weights on PC2, suggesting FA PC1 as measure of overall white-matter integrity and FA PC2 as a marker of association fibres, where disease-specific changes might happen (Supplementary Fig. 2). Voxel-wise comparison showed decreased FA in the right inferior longitudinal fasciculus (ILF) and right superior longitudinal fasciculus (SLF) in the AD patients compared to HCs (corrected for age and gender, Fig. 2A).
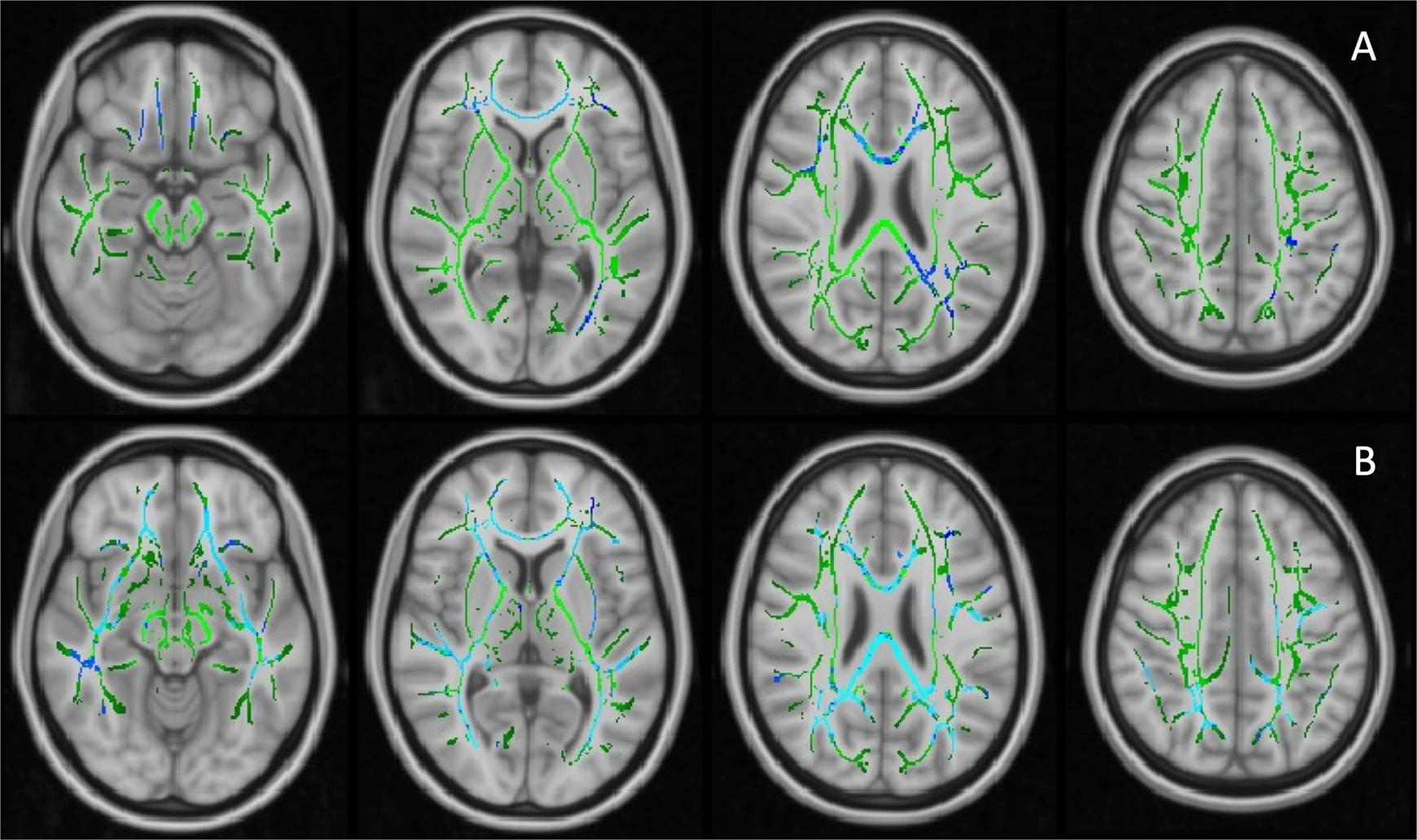
Voxel-wise analysis of FA values on TBSS skeletons.
A Decreased FA values in Alzheimer’s dementia patients compared to healthy controls, age and gender adjusted; (B) Significant negative relationship between 11C-PBR28 uptake (11C-PBR28 PC1) and FA values in Aβ+ patients, corrected for age, gender, cortical thickness and 18F-flutemetamol uptake. Significant clusters (FDR corrected) are shown in blue (negative association); TBSS skeleton is shown in green.
Functional network small-worldness (SW) and local efficiency (Eloc) were significantly decreased in the AD patients, while global efficiency (Eg) was not (1.47 ± 0.25 versus 1.71 ± 0.31, 0.72 ± 0.03 versus 0.75 ± 0.02, and 0.52 ± 0.02 versus 0.51 ± 0.02, p = 0.029, 0.003, and 0.34, respectively Fig. 3A–C). AUC of SW and Eloc were also lower in AD patients (0.69 ± 0.08 versus 0.74 ± 0.09, and 0.339 ± 0.005 versus 0.343 ± 0.004, p = 0.062 and 0.074, respectively, Supplementary Fig. 3A–C).
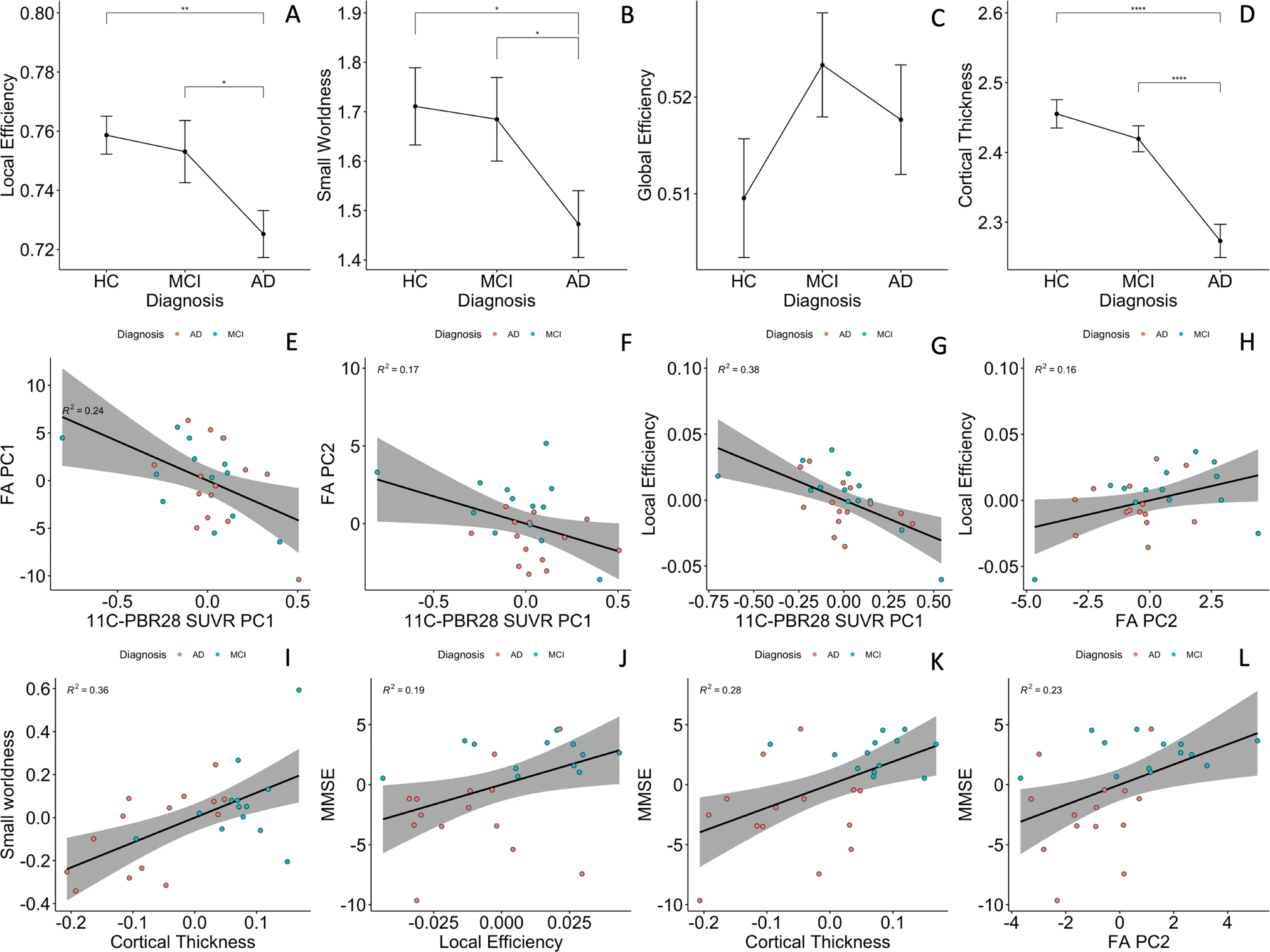
Group comparisons and partial correlations.
A–C Group comparisons of functional network topology metrics found reduced small-worldness and local efficiency in patients with Alzheimer’s dementia compared to healthy controls; (D) patients with Alzheimer’s disease had significantly lower cortical thickness compared to healthy controls and mild cognitive impairment participants. E, F Cortical 11C-PBR28 uptake has significant negative correlations with PC1 and PC2 of FA values in Aβ+ patients; (G) cortical 11C-PBR28 uptake has significant negative correlation with local efficiency; (H) FA PC2 positively correlates with local efficiency; (I) cortical thickness positively correlates with small-worldness; (J–L) FA PC2, local efficiency and cortical thickness positively correlate with mini-mental state examination scores. AD Alzheimer’s dementia, MCI mild cognitive impairment, HC healthy controls, PC principal component, FA fractional anisotropy. *p < 0.05 in pairwise comparisons; ****p < 0.0001 in pairwise comparisons.
AD patients had decreased average cortical thickness (2.27 ± 0.09 mm) compared to HCs (2.46 ± 0.08 mm, p < 0.0001), but MCI patients (2.42 ± 0.07 mm) did not show significant cortical thinning (p = 0.21, Fig. 3D).
Influence of cortical neuroinflammation and amyloid deposition on network structural integrity in the Alzheimer’s continuum
In Aβ+ patients, cortical thickness had a positive association with FA PC2 (standardised β = 0.48, 95% CI 1.37–0.85, p-perm = 0.027, corrected for age and gender), while 18F-flutematemol PC1 was not associated with either of the FA PCs, suggesting that association fibre integrity is quantitatively associated with grey-matter preservation, but not Aβ plaque load. 11C-PBR28 PC1 was negatively associated with both FA PC1 and FA PC2 (standardised β = −0.44, 95% CI: 0.11–0.13, p-perm = 0.018; standardised β = −0.375, 95%CI: −0.75 to −0.01, p-perm = 0.037, corrected for cortical thickness in addition to age and gender, Fig. 3E, F). To test whether microglial activation had stage-dependent relationship with brain structural network integrity at MCI and AD stages, we also performed same linear analysis in the two groups separately, and the results suggested same direction of association (Supplementary Materials Fig. 6).
To further explore the influence of inflammation and Aβ in different cortices on structural network integrity, SCCA was performed between PET and FA measures Negative correlation was found between 11C-PBR28 and FA values (λ = 0.70, r = 0.62, p-perm = 0.034), with 11C-PBR28 uptake in bilateral frontal, parietal and posterior cingulate cortices having non-zero weights. Canonical correlation between 18F-flutematemol and FA was not significant (λ = 0.50, r = 0.43, p-perm = 0.53), reinforcing the above findings that neuroinflammation, but not amount of Aβ in the cortex is associated with white-matter damage in Alzheimer’s disease. Interestingly, when both 11C-PBR28 and 18F-flutematemol were included, 18F-flutematemol uptake information did not improve the model fits under different level of sparsity constraints (Supplementary Fig. 5). Under heavy LASSO regularisation (λ < 0.50), none of the 18F-flutematemol ROIs was assigned non-zero weight. Even under light regularisation, the 18F-flutematemol ROIs had minimal canonical weights in the model (Table 2), and the overall model performance was less satisfactory compared with the model with 11C-PBR28 only (λ = 0.70, r = 0.60, p-perm = 0.064).
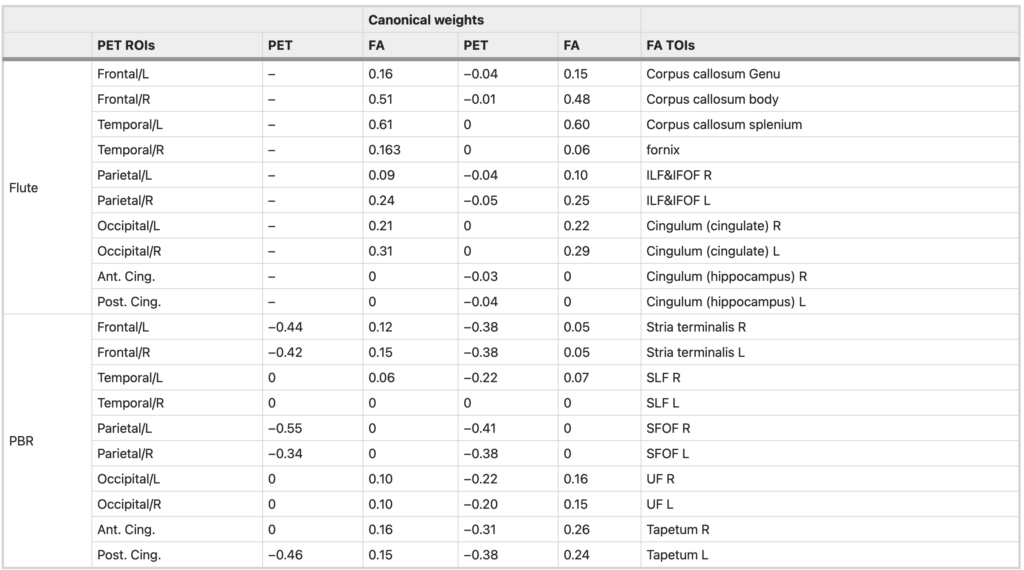
Canonical correlation between microglial activation/amyloid deposition and FA values.
Canonical weights in correlation between PET tracer uptakes and FA values in SCCA analysis. The first two columns show canonical correlation between 11C-PBR28 PET SUVR and FA values; the last two columns show canonical correlation between 11C-PBR28, 18F-flutematemol SUVR and FA values. The lasso penalty (λ) was 0.70 in both SCCA analysis presented in the table. Negative weights on tracer uptake measures and positive weights on FA values indicate negative relationship between the two set of variables.
ILF inferior longitudinal fasciculus, IFOF inferior fronto-occipital fasciculus, SLF superior longitudinal fasciculus, SFOF superior fronto-occipital fasciculus, UF uncinate fasciculus, L left, R right, Ant anterior, Post posterior, Cing cingulate, Flute 18F-flutemetamol regional uptake, PBR 11C-PBR28 regional uptake.
A voxel-wise regression analysis, which included age, gender, cortical thickness, 18F-flutemetamol and 11C-PBR28 uptake, supported the SCCA findings. 11C-PBR28 PC1 was associated with decreased FA in the forceps major, right inferior fronto-occipital fasciculus (IFOF) and ILF in Aβ+ patients (Fig. 2B).
Altogether, the above findings suggested a negative influence of cortical neuroinflammation on brain structural network integrity, which is independent of grey-matter atrophy and Aβ deposition in AD.
Influence of cortical neuroinflammation and amyloid deposition on network functional organisation in the Alzheimer’s continuum
In the Aβ+ patients, 11C-PBR28 PC1 negatively correlated with local efficiency (standardised β = −0.468, 95% CI: −0.846 to −0.018, p-perm < 0.001, cortical thickness corrected in addition to age and gender, Fig. 3G), but not global efficiency nor small-worldness. AUC of network Eloc was also negatively associated with 11C-PBR28 PC (standardised β = −0.432, 95% CI: −0.775 to −0.160, p-perm = 0.045). 18F-flutematemol PC did not correlate with the network topology metrics. Similar results was found in AD and MCI groups in sub-group analyses (Supplementary Fig. 6). The above findings suggest that cortical neuroinflammation is associated with disruption of brain functional network organisation, in particular the effective processing of information within locally interconnected subnetworks.
Network structural integrity, functional network organisation and cognition
FA PC2 had significant positive association with local efficiency (standardised β = 0.44, 95% CI: 0.08–0.79, p-perm = 0.047, cortical thickness corrected in addition to age and gender, Fig. 3H), while cortical thickness correlated with small-worldness (standardised β = 0.61, 95% CI: 0.21–1.01, p-perm = 0.001, PC2 corrected in addition to age and gender, Fig. 3I). Similar results were obtained using Eloc and SW AUC in regression analysis (standardised β = 0.45, 95%CI: 0.05–0.85 p-perm = 0.021 and standardised β = 0.56, 95% CI: 0.16–0.96 p-perm = 0.005, Supplementary Fig. 3). These findings suggested that effective functional brain network organisation is dependent on both grey-matter and structural network integrity.
FA PC2, local efficiency and cortical thickness positively correlated with MMSE (standardised β = 0.44, 95%CI: 0.04–0.84, p-perm = 0.025; standardised β = 0.52, 95% CI: 0.22~0.82, p-perm = 0.031; and standardised β = 0.60, 95% CI: 0.29–0.92, p-perm = 0.0004, respectively, Fig. 3J–L). Local efficiency AUC also marginally correlated with MMSE (standardised β = 0.39, 95% CI: −0.007–0.79, p-perm = 0.052, Supplementary Fig. 3). The above findings supported the idea that brain structural network integrity, effective functional network organisation and grey-matter preservation are the basis of cognition.
As there was a potential outlier in the above association analyses (Fig. 3), we have identified the outlier (a 75-year-old MCI patient) and re-examined the above findings. The results remain similar to the original ones (Supplementary Material 9, Supplementary Fig. 7), supporting the above inferences.
We have also examined whether age have confounded the previous findings. We found that in both healthy volunteers and the combined cohort (diagnostic groups considered), age did not have significant effect on the metrics we used in the study (probably due to moderate sample size). To further ensure the above inferences were not confounded by age, we have also performed a 5:5:5 exact match of HC, MCI and AD patients by age (within 1 year), and observed similar results (Supplementary Material 10).
Direct effect and indirect effect of neuroinflammation on functional network
As neuroinflammation was associated with both network structural integrity and functional organisation, we evaluated whether the effect of neuroinflammation on functional network was mediated by structural integrity. A linear mediation analysis with 11C-PBR28 and 18F-flutematemol PCs, FA PC2 and age as independent variables, Eloc as dependent variable and FA PC2 as mediator showed a significant average direct effect of 11C-PBR28 PC on local efficiency (95% quasi-Bayesian credible interval −0.25 to −0.06, p = 0.002) and a marginal average causal mediation effect via FA PC2 (95% quasi-Bayesian credible interval −0.12 to −0.001, p = 0.066). The above analysis suggested that neuroinflammation may have both direct and indirect negative influence (via structural network damage) on brain functional organisation disruption.
Discussion
This study evaluated the influence of neuroinflammation and Aβ deposition on brain structural and functional network in Alzheimer’s disease using a cross-sectional multimodal imaging dataset. Our findings suggest that neuroinflammation, but not Aβ deposition, directly influences the effective functioning of brain networks, which in turn is responsible for cognitive impairment in AD. By adjusting for amyloid deposition in the linear model, we were able to illustrate that microglial activation had a unique contribution to (putatively subsequent) network disruption, which is not dependent on the quantity of amyloid deposition. The current findings have bridged our understanding of macroscopic brain connectivity change and microscopical pathological processes in AD, and suggested a pathway from neuroinflammation to cognitive impairment via network disruption.
Neuroinflammation has been established as an important component in Alzheimer’s pathology [26]. Neuroimaging studies have found that microglial activation is associated with lower baseline cognition and faster cognitive decline [27, 28]. At early MCI stage, neuroinflammation, but not NFT, was found to correlate with Aβ deposition [29], while in AD patients tau pathology was more related to neuroinflammation [30], suggesting a possible mediation role of neuroinflammation. This directional flow from neuroinflammation to tau aggregation is further supported by post-mortem pathological and genetic analyses [31].
Alzheimer’s dementia is increasingly understood as a consequence of brain network dysfunction. rs-fMRI studies have reported impaired network connectivity and abnormal network topology in AD and MCI patients [32,33,34,35,36]. In our study, we confirmed the presence of significantly decreased small-worldness and network segregation (Eloc) in the AD patients, implying a disrupted brain functional network for information processing.
The basis of the functional network is inevitably structural network [37], and white-matter degeneration is an early feature of AD [9]. We found decreased microstructural integrity in association fibres in AD patients, which correlated with cognition, consistent with the literature [38]. More interestlingly, Aβ and hyperphosphorylated tau may propagate in a prion-like manner via the white-matter tracts [39, 40], highlighting the important position of brain structural and functional connectivity in AD pathegenesis.
Recently, it has been reported that anterior temporal neuroinflammation is associated with longitudinal cognitive decline in Aβ+ patients [28], and that an independent component of brain neuroinflammation is associated with abnormal functional connectivity within and between brain networks [41]. Given the current evidence, we further investigated the influence of neuroinflammation on brain structural and functional network’s overall organisation. We have demonstrated that neuroinflammation is an independent predictor of structural and functional network disruption, while the amout of Aβ in Aβ+ population was not linearly associated with brain connectivity changes. On the one hand, the lack of linear association between amyloid deposition and structural/functional network disruption might reflect non-linear relationships [42]. One the other hand, it may suggest that once the disease process has been initiated, the quantity of Aβ deposition no longer directly influcences brain network and cognitive decline [1]. Indeed, this hypothesis could explain the controversial results from clinical trials targeting Aβ in the symptomatic stage of Alzheimer’s disease [43].
During brain development, microglia contribute to neurogenesis, axon outgrowth, synapse modelling and myelination [44]. In adulthood, microglia have dynamic and direct interactions with neuronal network structures [45] and are involved in the maintenance and repair of myelin [44]. In AD brains, activated microglia undergo phenotypic changes and migrate to Aβ plaques and NFTs [46]. Activated microglia can cause direct damage to oligodendrocytes and axons through ROS production (reactive oxygen species), matrix metalloproteinases and pro-inflammatory cytokines [47]. Accumulation of activated microglia has also been reported to co-locate with damaged axonal initial segments in a mouse model of AD [48]. Furthermore, an ex vivo experiment has demonstrated that activated microglia can induce neuritic beading of axons, inhibit mitochondrial function and axonal transport through N-methyl-D-aspartate (NMDA) receptor signalling [49]. The preclinical evidence above explains the biological basis of how neuroinflammation could induce structural and functional network disruption.
Our findings, together with previous evidence, suggests a framework of systemic brain damage in Alzheimer’s continumm, where Aβ deposition initiates neuroinflammation, which then directly lead to structural and functional brain network damage, while also promoting hyperphosphorylated tau formation and aggregation. The tau pathology, in turn, is associated with further network damage as well as cortical atrophy [10, 50]. Structural network and functional network disruption, as well as cortical atrophy, eventually result in cognitive impairment (Fig. 4). It is possible that the initial activation of microglia by other inflammatory factors such as trauma, infection and systemic inflammation, contributes to future impairment of brain connectivity, formation of tau pathology, and progression of the disease [4, 51]. Recently, a ground breaking multicentre study by Pascoal et al. have demonstrated that microglial activation may propagate together with tau pathology in AD, suggesting that the prion-like network-based tau propagation is associated with basline neuroinflammatory network [51]. Together with previous evidence, it is worth considering whether microglial activation at downstream regions, caused by amyloid deposition and various pathological factors, could pave the way for the prion-like propagation of tau protein or local dissemination of tau pathology.
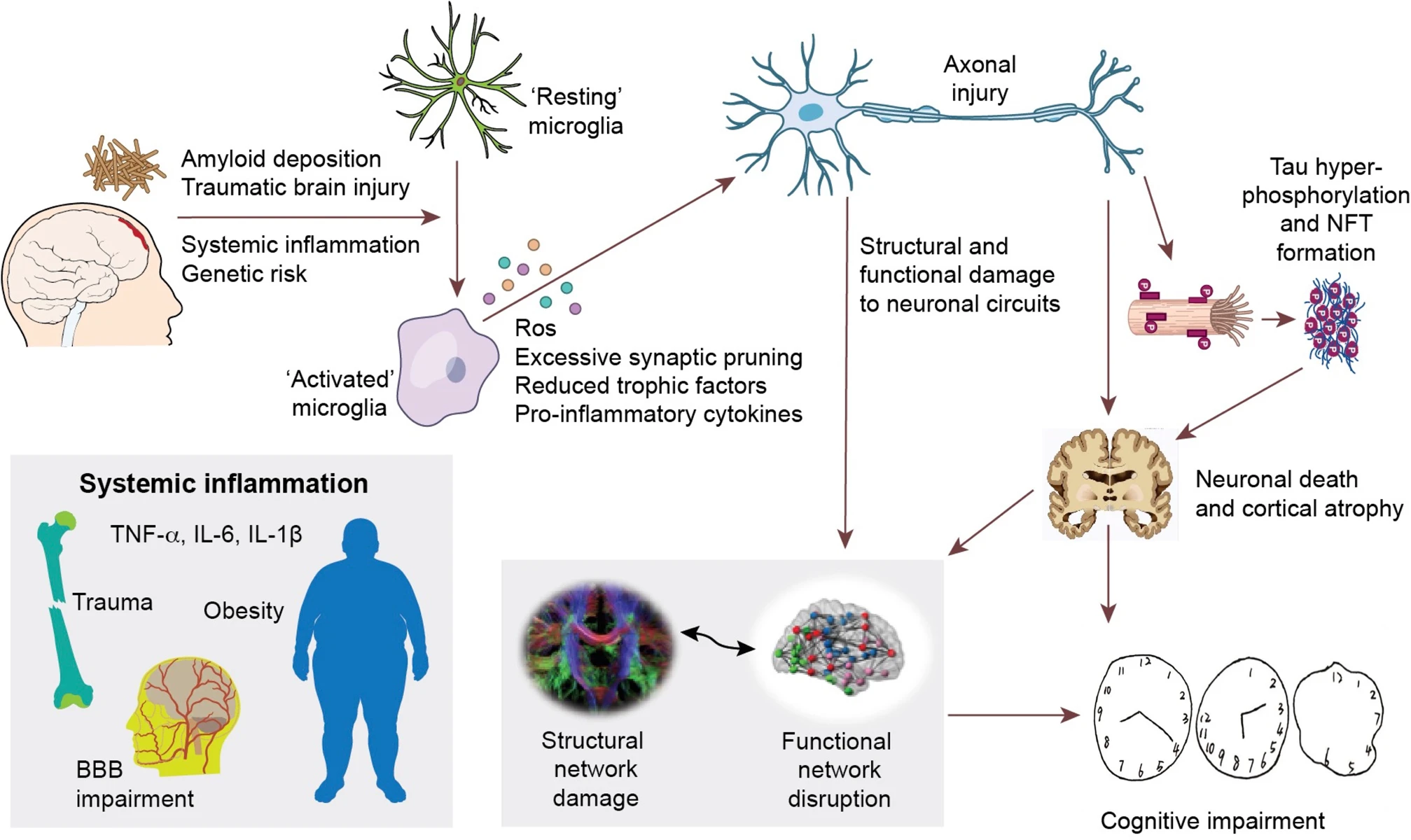
Hypothetical framework of pathological events leading to structural and functional network impairment and cognitive decline in Alzheimer’s disease.
The initial amyloid deposition, together with other inflammatory risk factors (including systemic inflammation, traumatic brain injuries, and genetic risk factors, etc.) initiate neuroinflammation, which could cause neuronal damage via production of reactive oxygen species, pro-inflammatory cytokines, reduced trophic support and direct synaptic pruning. These aforementioned events could then directly damage neuronal circuits and cause abnormal neuronal activity, resulting in disruption of structural and functional brain connectivity. On the other hand, these events could also facilitate the propagation of hyperphosphorylated tau species and formation of intracellular neurofibrillary tangles (NFT). NFT formation further causes neuronal dysfunction, axonal destabilisation and neuronal death, hence cortical atrophy and network dysfunction. This cascade of events eventually leads to cognitive impairment in Alzheimer’s disease.
While there is evidence that at the very early stage of the disease, a stronger response of anti-inflammatory microglia is associated with better outcome [52, 53], in the later stages, the detection of β-sheeted fibrillary amyloid by PET, might already mark the failure of the protective response, followed by a deleterious inflammatory response.
The limitations of the current study are: (1) its cross-sectional nature and thus causal relationships need validation by longitudinal studies; (2) The age difference between healthy volunteers and cognitively impaired patients were rather large, and may have influenced the cross-group comparisons, dispite the efforts made to correct for the matter using linear regression mothods. The mismatch of age could be due to the fact that healthy volunteers were recruited early on in the current study, and a more balanced recruitment is recommended for future studies. (3) The hypothetical framework between neuroinflammation, tau aggregation and downstream network damage and cortical atrophy also needs to be further tested, especially the how inflammation may interact with the tau propagation and how the two factors may influence brain network together.
In conclusion, our findings reinforce the theory that a network failure underlies cognitive impairment in AD, and is compatible with the framework in which neuroinflammation, initiated by amyloid pathology, plays a significant upstream role in causing structural and functional connectivity impairment as the disease progresses. Therefore, it is essential to consider: (1) the inclusion of neuroinflammation in the current ‘amyloid-tau-neurodegeneration’ framework and (2) targeting neuroinflammation in the symptomatic stage of Alzheimer’s disease to preserve brain networks and delay disease progression.