
Lysosomal exocytosis releases pathogenic α-synuclein species from neurons in synucleinopathy models
By Ying Xue Xie, Nima N. Naseri, Jasmine Fels, Parinati Kharel, Yoonmi Na, Diane Lane, Jacqueline Burré, and Manu Sharma
Excerpt from the article published in Nature Communications 13, 4918, 22 August 2022, DOI: https://doi.org/10.1038/s41467-022-32625-1
Editor’s Highlights
- The release of pathogenic aggregates of the neuronal protein α-Synuclein (αSyn) into the extracellular space instigate the neuron-to-neuron transmission and spread of αSyn pathology in synucleinopathies including Parkinson’s disease.
- Multiple cellular pathways have linked PD and αSyn aggregation with lysosomal storage disorders.
- The aggregates/fibrils of αSyn could form inside the acidified lumen of lysosomes, e.g., from monomers taken up via chaperone-mediated autophagy or macroautophagy of synaptic vesicles
- The accumulation of pathogenic αSyn species in neuronal lysosomes, and pinpoint to lysosomal exocytosis as a pathway of release for these pathogenic αSyn species into the extracellular milieu.
- This pathway, at first sight, appears to have relevance to the release of undegraded aggregates of other proteins as well that transmit pathology from cell-to-cell.
Abstract
Considerable evidence supports the release of pathogenic aggregates of the neuronal protein α-Synuclein (αSyn) into the extracellular space. While this release is proposed to instigate the neuron-to-neuron transmission and spread of αSyn pathology in synucleinopathies including Parkinson’s disease, the molecular-cellular mechanism(s) remain unclear. To study this, we generated a new mouse model to specifically immunoisolate neuronal lysosomes, and established a long-term culture model where αSyn aggregates are produced within neurons without the addition of exogenous fibrils. We show that neuronally generated pathogenic species of αSyn accumulate within neuronal lysosomes in mouse brains and primary neurons. We then find that neurons release these pathogenic αSyn species via SNARE-dependent lysosomal exocytosis. The released aggregates are non-membrane enveloped and seeding-competent. Additionally, we find that this release is dependent on neuronal activity and cytosolic Ca2+. These results propose lysosomal exocytosis as a central mechanism for the release of aggregated and degradation-resistant proteins from neurons.
Introduction
Cytoplasmic aggregates of the synaptic protein α-synuclein (αSyn) are a characteristic feature of multiple neurodegenerative diseases termed “synucleinopathies”, including Parkinson disease (PD). Over the course of these age-linked diseases, αSyn assembles into amyloid-type β-sheet-rich aggregates, depositing as Lewy bodies and/or Lewy neurites within neurons1,2,3. The neuro-anatomical propagation of αSyn pathology is highly stereotyped, and has been defined well-enough to be used in staging PD—originating in the medulla and olfactory bulb, then advancing to the midbrain and basal forebrain, followed by “spread” to the neocortex4,5. This observation, combined with detection of extracellular αSyn in human cerebrospinal fluid6, led to the original conjecture that pathogenic species of αSyn may be transmitted neuron-to-neuron via prion-like permissive templating. Subsequently, in PD patients who had received fetal neuron transplants, appearance of Lewy pathology in the non-diseased grafted neurons also pointed to the transmission of αSyn pathology from the surrounding PD-affected neurons7,8. These observations were followed by animal studies which confirmed either neuron-to-neuron transmission of αSyn pathology9, or induction of intraneuronal αSyn pathology by an inoculum of extracellular αSyn fibrils10,11.
However, unclarity persists in how the cytosolic aggregates are conveyed into the extracellular milieu: Proposed pathway(s) include secretion of αSyn monomers, oligomers and/or larger aggregates inside extracellular vesicles—such as exosomes12,13,14, or microvesicles15—which have been categorized collectively as “unconventional secretion”. Other proposed mechanisms include synaptic vesicle release16, or exocytosis of other types of vesicles17,18,19, exophagy20, membrane translocation21, or trafficking of lysosome-associated aggregates through tunneling nanotubes22,23. Yet, these studies have not identified a neuronal organelle that first accumulates and then secretes the pathogenic αSyn aggregates generated within the neurons, resulting in the current mechanistic gap.
Interestingly, PD is associated with mutations in the glucocerebrosidase gene, which cause Gaucher disease, the most common lysosomal storage disorder24,25. In addition, multiple cellular pathways have linked PD and αSyn aggregation with lysosomal storage disorders26,27.
Here, we show that neuronally-produced pathogenic αSyn species accumulate within the neuronal lysosomes in mouse brains and in primary neurons, and that these seeding-competent species are then released into the extracellular space via SNARE-dependent lysosomal exocytosis.
Results
Pathogenic species of αSyn accumulate within the lysosomes of transgenic αSynA53T mouse brains
Rapid neurodegeneration and synapse loss in cysteine string protein-α knockout mice (CSPα−/−) is rescued by transgenic expression of human αSyn carrying the familial PD mutation A53T (Tg-αSynA53T)28. However, beyond 5 months, the rescued Tg-αSynA53T/CSPα−/− mice begin exhibiting neurodegeneration due to the transgenic overexpression of αSynA53T. Curiously, we found that Tg-αSynA53T/CSPα−/− mice have accelerated accumulation of the pathogenic versions/species of αSyn compared to Tg-αSynA53T/CSPα+/− mice, detectable by antibodies against phospho-Ser129 αSyn, filamentous αSyn, and amyloid-type folds (Supplementary Fig. S1). Meanwhile, there was no change in monomeric αSyn levels (Supplementary Fig. S1). CSPα+/− mice have normal CSPα function29. This suggested that brains of Tg-αSynA53T/CSPα−/− mice are collecting αSyn aggregates faster in the absence of CSPα.
Loss-of-function mutations in CSPα cause the lysosomal storage disorder adult-onset neuronal ceroid lipofuscinosis (ANCL) or Kufs disease30,31,32,33. Specifically, loss of CSPα function leads to progressive accumulation of lysosomes containing undegraded material such as lipofuscin/residual bodies in ANCL patient brains30, in CSPα−/− mouse brains29, in Drosophila models carrying the ANCL mutations34, and in ANCL patient-derived induced neurons35. Tg-αSynA53T/CSPα−/− mice also accrue lysosomal proteins such as LAMP1 and cathepsin-L, as well as ATP5G—a mitochondrial protein characteristically stored in lysosomes of CSPα loss-of-function models and ANCL patient neurons (Supplementary Fig. S1). The enhanced accumulation of lysosomal contents in Tg-αSynA53T/CSPα−/− brains, together with the exaggerated buildup of αSyn aggregates in these brains, plus the association of lysosomal dysfunction and lysosomal storage disorders with PD24,25,26,27, led us to investigate whether neuronally-produced αSyn aggregates deposit within the lysosomes.
We next aimed to determine whether αSyn aggregates accumulate within lysosomes, even in the absence of CSPα-/--driven lysosomal pathology. Thus, we tested whether aged Tg-αSynA53T mice, which display severe αSyn-driven pathology, have αSyn aggregates within their lysosomes.
We found pathogenic αSyn species, but not monomeric αSyn, in dextranosomes (heavy lysosomes loaded with dextran to enhance isolation-purity) isolated from the brains of 6-month-old Tg-αSynA53T mice (Fig. 1). These lysosomal fractions precisely coincided with pathogenic αSyn species and had highly enriched lysosomal proteins as well as cathepsin-D activity, with negligible contamination from other organelle protein-markers or mitochondrial and peroxisomal enzyme activities (Fig. 1). This experiment indicates that isolated intact lysosomes from aged Tg-αSynA53T mice contain pathogenic αSyn species.
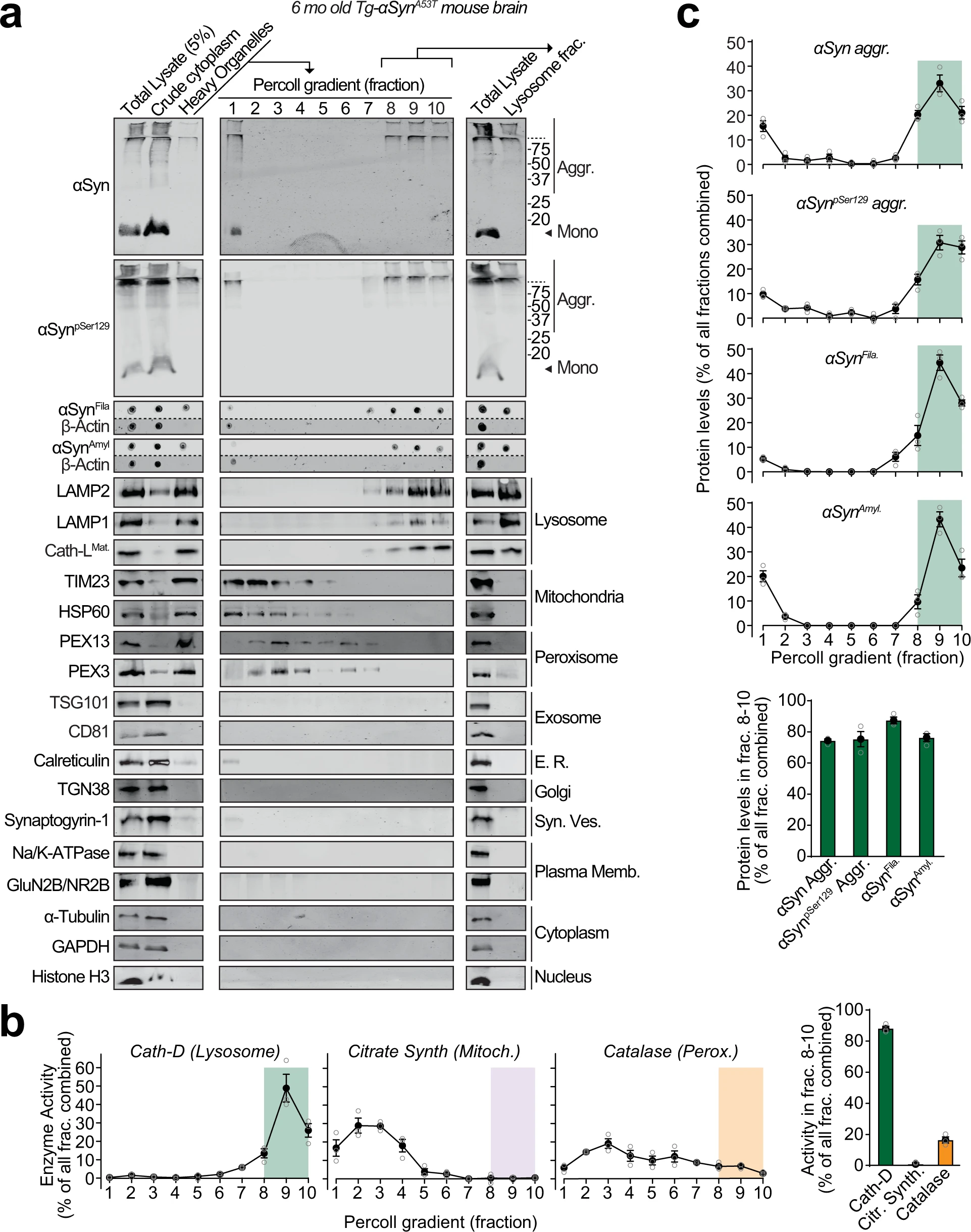
Pathogenic αSyn aggregates accumulate within lysosomes of aged Tg-αSynA53Tmice.
Lysosomes were isolated from 6-month-old Tg-αSynA53T mouse brains via Percoll gradient centrifugation of the heavy organelle fraction—which contained peroxisomes, heavy lysosomes loaded in vivo with dextran-70, and mitochondria swollen ex vivo by CaCl2. a Lysosome (dextranosome) enrichment was determined by immunoblotting for markers of indicated organelles, compared to the respective levels in the total lysate input. Membrane-matched dot blots (β-Actin = loading control) are separated by dashed lines. b Left panels—Activities of enzymes contained within lysosomes (cathepsin-D), mitochondria (citrate synthase), and peroxisomes (catalase) were measured, testing for isolation of intact organelles. Right panel—Summary graph of enzyme activity present in the combined “lysosomal fractions” (fractions 8–10). c Top panels—Levels in each Percoll gradient fraction of pathogenic αSyn species: Aggregated (αSyn Aggr), aggregates phosphorylated at Ser129 (αSynpSer129 Aggr), filamentous (αSynFila), and amyloid-type (αSynAmyl). Bottom panel—Summary graph of these αSyn species present in the combined “lysosomal fractions” (fractions 8–10). (n = 3). All data are shown as means ± SEM, where “n” represents mouse brains.
Pathogenic species of αSyn accumulate within the lumen of neuronal lysosomes
The lysosomes studied above are a mixture of lysosomes from all brain cells. To isolate and study lysosomes specifically from neurons, we generated transgenic mice expressing HALAMP1Myc—late-endosomal/lysosomal protein LAMP1 epitope-tagged on its luminal domain with tandem 2xHA, and on its cytoplasmic tail with 2xmyc—driven by the neuron-specific synapsin-I promoter (Fig. 2a–d and Supplementary Fig. S2a–d), enabling affinity-isolation of neuronal late-endosomes/lysosomes from mouse brains. The Tg-HALAMP1Mycmice were then crossed to the Tg-αSynA53T mice to generate Tg-αSynA53T/Tg-HALAMP1Mycprogeny (Supplementary Fig. S3a–g). Immunoisolation experiments from aged Tg-αSynA53T/Tg-HALAMP1Myc mouse brains indicated that pathogenic versions of αSyn co-isolate with neuronal late-endosomes/lysosomes (Fig. 3a). We found that ~10% of the SDS-resistant αSyn aggregates, and ~20% of the more mature αSyn aggregates (filamentous and amyloid-type) reside within neuronal late-endosomes/lysosomes (Fig. 3a). No monomeric αSyn resided in neuronal late-endosomes/lysosomes (Fig. 3a). Together with the dextranosome-isolation experiments (Fig. 1), this result indicates that a non-trivial portion of pathogenic αSyn species in the brain is found within neuronal lysosomes.
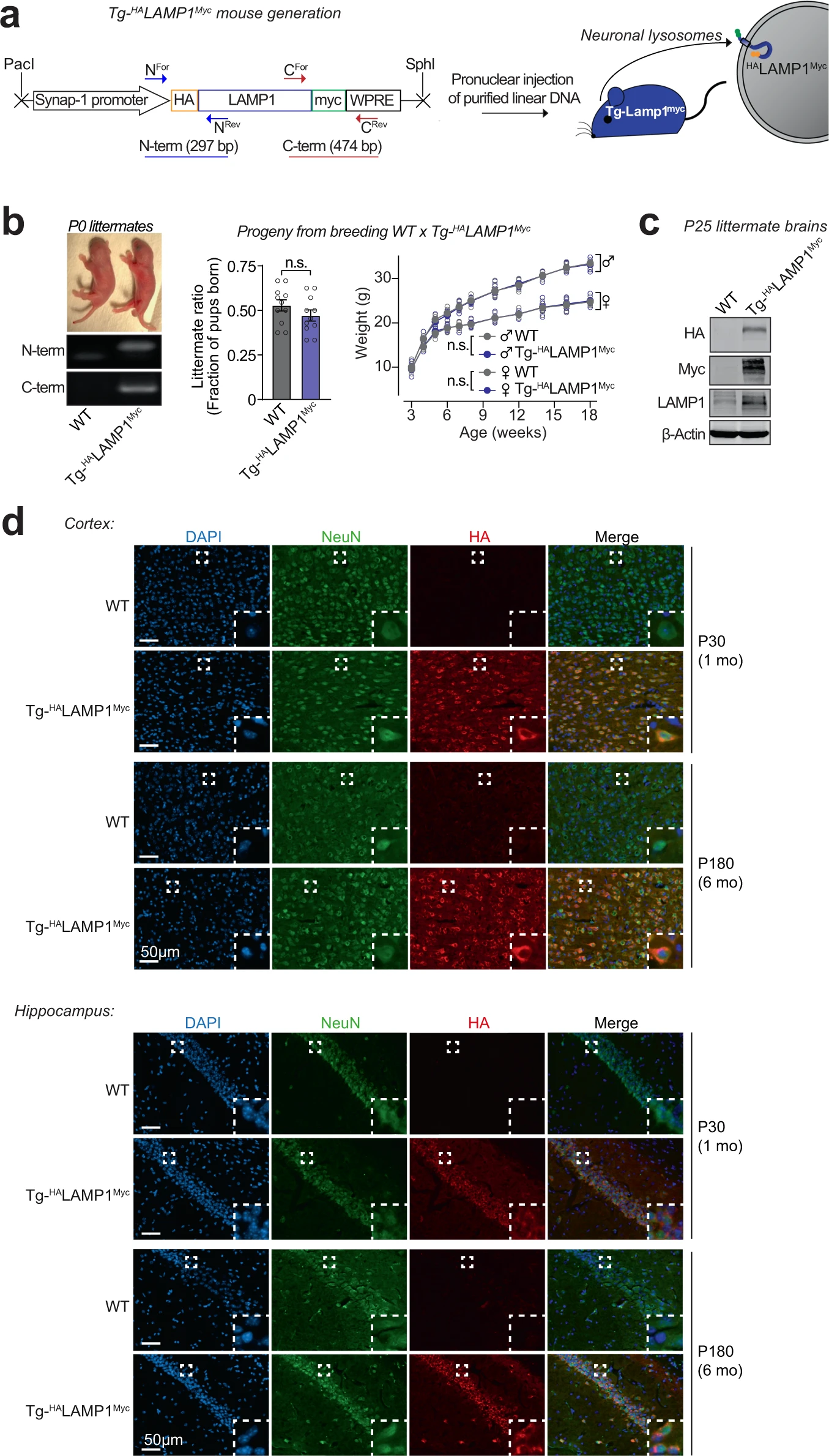
Development of a transgenic mouse model (NeuLyso-Tag mouse) to isolate neuronal lysosomes from brains.
a A lentiviral vector expressing HALAMP1Myc via the neuron-specific synapsin-1 promoter was used to generate Tg-HALAMP1Myc (or “NeuLyso-Tag”) mice via pronuclear injection of linearized DNA comprising the synapsin-1 promoter and the HALAMP1Myc cDNA. b Tg-HALAMP1Myc mice were PCR-genotyped using primer-pairs at the N- and the C-terminal ends of the HALAMP1Myc cDNA. Single insertion-site for the transgene was suggested by equal numbers of WT and Tg- HALAMP1Myc pups born per litter (n = 11 litters). There was also no effect of the transgene on birth-ratio and growth (indicated by weight gain; n = 5 per group) of the Tg- HALAMP1Myc mice compared to WT mice. c Immunoblots show that HALAMP1Myc protein is detectable in mouse brains by immunoblots against the N-terminal HA tag and the C-terminal myc tag (representative of n = 7 littermates of each genotype). d Histological colocalization of anti-HA immunofluorescence with NeuN staining in the cortex and hippocampus shows that HALAMP1Myc construct is expressed in the neurons of Tg-HALAMP1Mycmouse brains at 1 and 6 months of age (representative of >10 stained sections from n = 3 littermates of each genotype). All data represent means ± SEM. n.s. not significant, by paired 2-tailed Student’s t test for littermate ratio and by RM 2-way ANOVA for weight gain over age (each mouse matched over age). Note:The FSW-HALAMP1Myc lentiviral vector is described and tested in Supplementary Fig. S2, and further characterization of the Tg- HALAMP1Myc mice is included in Supplementary Fig. S3.
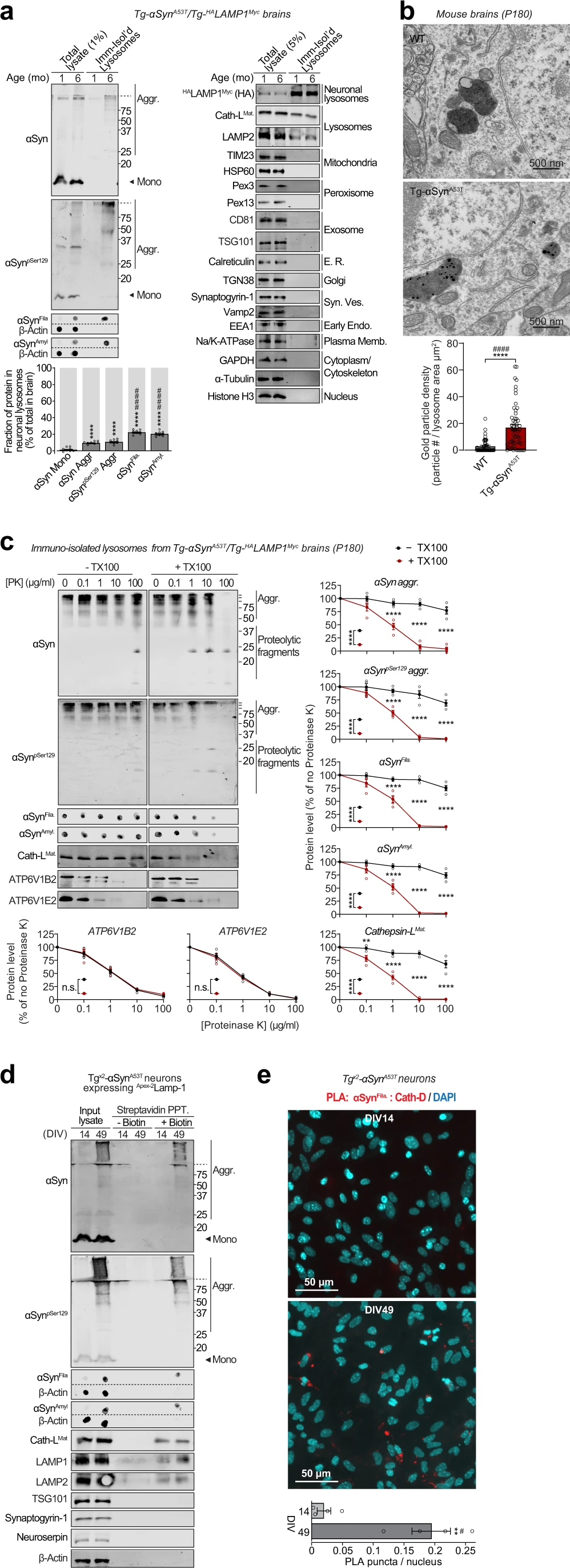
Pathogenic αSyn species accumulate within neuronal lysosomes in mouse brains and primary neurons.
a Neuronal late-endosomes/lysosomes immunoisolated from the brains of 1- and 6-month-old Tg-αSynA53T/Tg-HALAMP1Myc mice using anti-myc antibodies were analyzed by immunoblotting for levels of αSyn, αSynpSer129, αSynFila, and αSynAmyl (Mono monomer, Aggr aggregates). The fraction of these αSyn species residing within neuronal lysosomes at 6 months of age (bottom graph) was back-calculated from the fraction of each αSyn species immunocaptured (αSyn species immunocaptured/total input), normalized to the fraction of neuronal late-endosomes/lysosomes captured—indicated by the fraction of HALAMP1Myc captured (HA blot immunocaptured/total input). Membrane-matched dot blots (β-Actin = loading control) are separated by dashed lines, and immunoblots against markers of lysosomes and of other organelles are also shown (n = 8). b Electron micrographs showing increased αSyn aggregate-labeling within the lysosomal lumen in 6-month-old Tg-αSynA53T brains (2 lysosomes shown with 12 and 5 gold particles) compared to WT brains (2 lysosomes shown with 1 possible and 0 gold particles). (n = 50 lysosomes for WT and n = 55 lysosomes for Tg-αSynA53T). c Lysosomes isolated from the brains of 6-month-old Tg-αSynA53T/HALAMP1Myc mice (as in a) were subjected to on-bead limited proteolysis using the indicated concentrations of proteinase K (PK) in the absence or presence of 0.1% Triton X-100. Immunoblots for αSyn (αSyn, αSynpSer129, αSynFila, and αSynAmyl), mature cathepsin-L (Cath-LMat; lysosome lumen), and vATPase subunits ATP6V1B2 and ATP6V1E2 (lysosomal proteins exposed on the cytosolic side) (n = 4 brains, isolation, and proteolysis). d Tgx2-αSynA53T primary cultures lentivirally expressing a synapsin-1 promoter-driven Apex-2LAMP1 construct were subjected to proximity-labeling with biotin at 14 or 49 days in vitro (DIV). Labeled proteins were precipitated on streptavidin-coated magnetic beads and immunoblotted for the indicated pathogenic αSyn species, as well as for the marker proteins cathepsin-L (lysosomes), LAMP1 and LAMP2 (late-endosomes/lysosomes), TSG101 (exosomes), synaptogyrin-1 (synaptic vesicles), neuroserpin (non-lysosomal/constitutively secreted neuronal protein), and β-actin (cytosol). Membrane-matched dot blots (β-Actin = loading control) are separated by dashed lines. (representative of n = 3). e Tgx2-αSynA53T primary cultures were subjected to in situ proximity ligation assay (PLA) at either DIV14 (top) or at DIV49 (bottom), with primary antibodies against α-SynFila and cathepsin-D (Cath-D). Quantification of PLA puncta per nucleus is shown, performed identically between images from DIV14 and 49 (n = 4 cultures and PLA experiments; data from 3 images averaged for each n). Data represent means ± SEM. Each “n” corresponds to independently aged mouse littermates in a, lysosomes in b, independent mouse brains, immunoisolation and proteolysis experiments in c, independent primary neuron cultures in d, and independent pups, cultures, and PLA experiments in e. ****P < 0.0001 by 1-way ANOVA with Dunnett multiple-comparison correction and ####P < 0.0001 by non-parametric Kruskal–Wallis test with Dunn’s multiple-comparison adjustment; all tests comparing levels of each αSyn aggregate-type to αSyn monomer in a; ****P < 0.0001 by unpaired 2-tailed Student’s t test and ####P < 0.0001 by non-parametric Mann–Whitney test in b; 2-way ANOVA with Bonferroni multiple comparisons post-test (compared at each PK concentration) in c; and **P < 0.01 by unpaired 2-tailed Student’s t test and #P < 0.05 by non-parametric Mann–Whitney test in e.
Electron microscopy showed at the ultrastructural level that αSyn aggregates were localized within the lysosomal lumen of cortical neurons in 6-month-old Tg-αSynA53T mouse brains (Fig. 3b). This was further corroborated by limited proteolysis of immunoisolated lysosomes from 6-month-old αSynA53T/Tg-HALAMP1Myc mice, where αSyn aggregates were resistant to proteinase K digestion, only in the absence of a detergent, similar to the lysosome-luminal enzyme cathepsin-L, and in contrast to the lysosomal vATPase subunits that are exposed on the cytosolic side of lysosomal membrane (Fig. 3c). Altogether, these experiments localize αSyn aggregates inside the lysosomal lumen.
To understand the fate of these lysosomal αSyn aggregates, we developed a neuronal model which accumulates pathogenic αSyn species. Compared to Tg-αSynA53T mice, homozygous Tg-αSynA53T mice (Tgx2-αSynA53T), which express twice the amount of transgenic αSynA53T, have shorter lifespans, earlier onset of neuromuscular pathology, and begin accumulating pathogenic αSyn species earlier—by only 6 weeks of age (Supplementary Fig. S4a–f). Accordingly, long-lived primary neurons from Tgx2-αSynA53T mice also accrued pathogenic αSyn species by 6 weeks, including aggregates detectable by antibodies against αSyn fibrils and amyloid-type conformations (Supplementary Fig. S5a, b), which were Triton X-100 insoluble, extractable in SDS, and disrupted by urea (Supplementary Fig. S5c).
Importantly, we further verified the accumulation of these pathogenic αSyn aggregates in lysosomes using a second independent immunoisolation approach. Isolation of lysosomes from Tgx2-αSynA53T neurons using the HA-tagged lysosomal protein TMEM192 (previously characterized in36,37) captured αSyn aggregates within neuronal lysosomes at DIV49, but not earlier at DIV35 and DIV21 (Supplementary Fig. S6). No signal for these pathological αSyn species was detected in the lysosomes isolated from DIV49 αSyn knockout (αSyn−/−) neurons.
We then applied two independent proximity-labeling techniques to specifically mark and track the pathogenic αSyn species located within neuronal lysosomes. First, we targeted Apex-2 to the lysosomal lumens of Tgx2-αSynA53T primary neurons. We lentivirally expressed a synapsin-1 promoter-driven construct, comprised of Apex-2 fused to the luminal domain of a truncated version of LAMP1 (Apex-2LAMP1) (Supplementary Fig. S7a–c), which proximity-labels the luminal contents of neuronal late-endosomes/lysosomes with biotin. In these Tgx2-αSynA53T neurons expressing Apex-2LAMP1, we found that pathogenic αSyn species as well as lysosomal proteins (typified by cathepsin-L) were biotinylated (Fig. 3d). Second, we used an in situ proximity ligation assay, and found that filamentous αSyn aggregates co-localized with cathepsin-D, i.e., within the lysosomal lumen of Tgx2-αSynA53T primary neurons (Fig. 3e). Together, these results show that pathogenic αSyn species accumulate within the lumen of neuronal lysosomes.
Pathogenic αSyn species are secreted from neurons via SNARE-dependent lysosomal exocytosis
Release of αSyn from neurons, particularly the seeding-competent pathogenic versions of αSyn, is considered a key step in the spatial progression of synucleinopathies38, and αSyn has been documented in extracellular fluids of PD patients6. We also found pathogenic species of αSyn in the cerebrospinal fluid of aged Tg-αSynA53T mice (Fig. 4a). Importantly, this extracellular pool of αSyn aggregates is not enveloped in membranes, as they were not protected from proteinase K digestion (Fig. 4a), and their proteolytic susceptibility was unaffected by the presence of detergent, akin to the lysosomal luminal protein cathepsin-L and neuroserpin—a non-lysosomal secreted protein (Fig. 4a). This was contrary to the well-protected contents of exosomes, typified by TSG101, which becomes susceptible to proteolysis only in the presence of detergent (Fig. 4a).
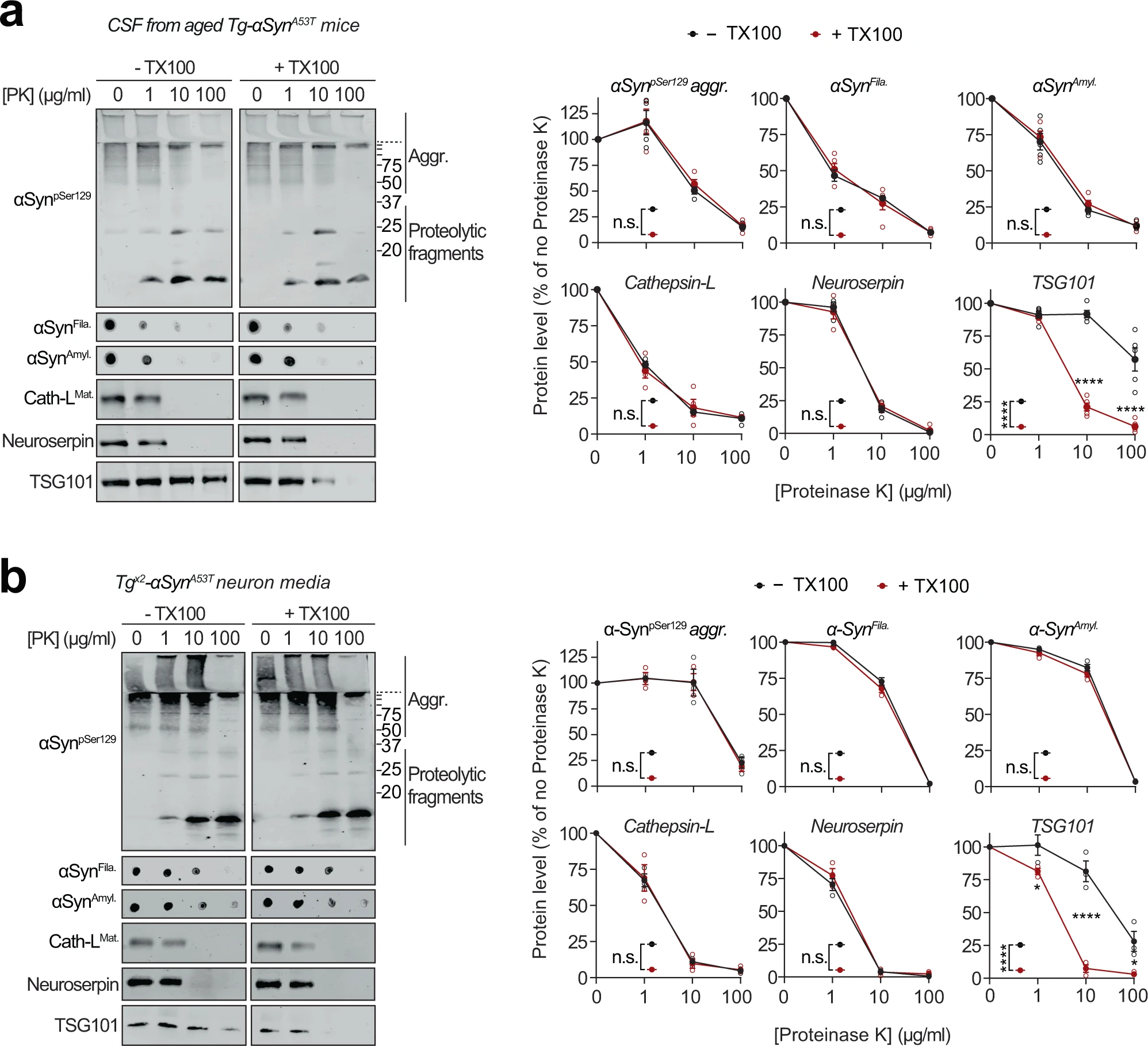
Extracellular αSyn species in cerebrospinal fluid and in primary neuron media are not membrane-enveloped.
a Cerebrospinal fluid (CSF) collected from 6-month-old Tg-αSynA53T mice were subjected to limited proteolysis using indicated concentrations of proteinase K (PK) in the absence or presence of 0.1% Triton X-100. Immunoblots for αSyn protein levels (αSynpSer129, αSynAmyl, and αSynFila), mature cathepsin-L (Cath-LMat), TSG101, and neuroserpin (n = 5 for αSynAmyl, αSynFila, TSG101; n = 4 for αSynpSer129, Cath-LMat). b Media collected from Tgx2-αSynA53T neuron cultures at DIV49 was subjected to limited proteolysis in presence or absence of 0.1% Triton X-100 and analyzed as in a (n = 3). All data represent means ± SEM. Each ‘n’ is an independent collection (CSF or medium) and proteolysis experiment. n.s. not significant; *P < 0.05; ****P < 0.0001 by 2-way ANOVA with Bonferroni multiple comparisons post-test (compared at each PK concentration).
The pathogenic αSyn species were also detected in the medium of Tgx2-αSynA53T neurons collected at DIV49, but were not yet present at DIV35 or in the media of DIV49 αSyn−/− and WT neurons (Supplementary Fig. S5d). This extracellular pool of αSyn aggregates, similar to that in cerebrospinal fluid, was not protected from proteinase K digestion (Fig. 4b), indicating that it is not membrane-enveloped. Additionally, proteolytic susceptibility of these αSyn aggregates, as well as that of cathepsin-L and neuroserpin were unaffected by the presence of detergent (Fig. 4b). In contrast, membrane-enveloped exosomal contents were well-protected, typified by TSG101, which became susceptible to proteolysis only in the presence of detergent (Fig. 4b). Notably, we found no evidence of neuronal death as a potential source of extracellular αSyn aggregates (Supplementary Fig. S5e, f).
Accumulation of the pathogenic αSyn species inside lysosomes, together with their un-enveloped presence in the extracellular space, prompted us to investigate whether lysosomes are directly releasing the αSyn aggregates contained inside them.
We thus collected the medium from Tgx2-αSynA53T primary neurons, in which the lysosomal contents had been biotinylated via neuron-specific expression of Apex-2LAMP1 (as in Fig. 3band Supplementary Fig. S7). We found that lysosomal contents, typified by cathepsin-L, as well as pathogenic αSyn species, could be precipitated from the medium of Tgx2-αSynA53Tneurons via their biotin-modification (Fig. 5a). In contrast, exosomal contents, typified by TSG101, were not biotinylated/precipitated. This result points to lysosomal exocytosis—SNARE-dependent fusion of lysosomes with the plasma membrane—as the likely mechanism for the exit of αSyn aggregates from neurons.
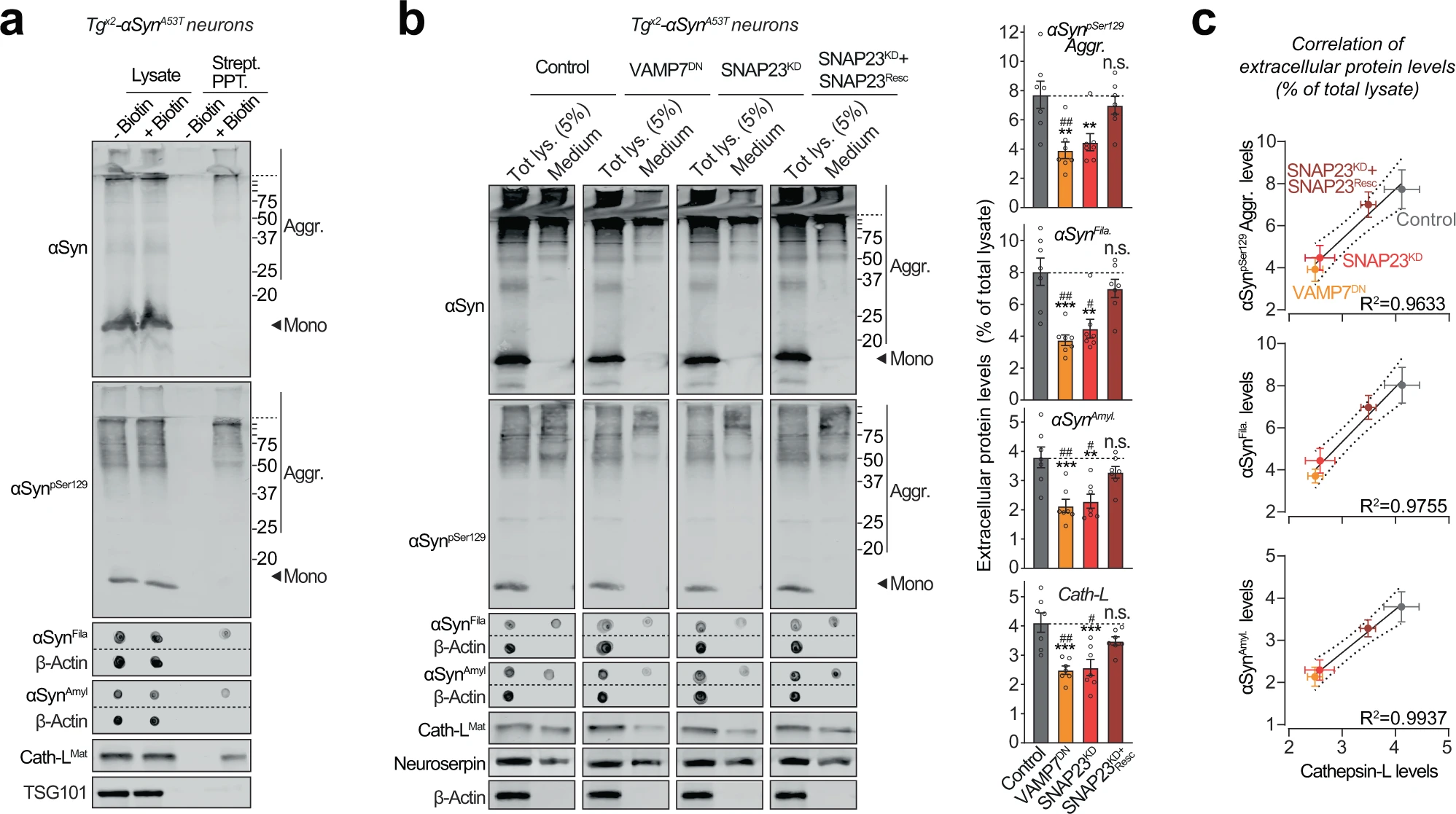
Pathogenic αSyn species are secreted from neurons via SNARE-dependent lysosomal exocytosis.
a Tgx2-αSynA53T primary neurons lentivirally expressing a synapsin-1 promoter-driven Apex-2LAMP1 (infected on DIV7) were subjected to proximity-labeling on DIV 47. Biotinylated proteins released into the media during 48 h (by DIV49) were precipitated on streptavidin-magnetic beads and immunoblotted for the indicated αSyn species (αSyn, αSynpSer129, αSynAmyl, and αSynFila), lysosome-luminal protein cathepsin-L, and exosome luminal protein TSG101. Strept. PPT. streptavidin precipitate. Membrane-matched dot blots (β-Actin = loading control) are separated by dashed lines. (representative of n = 3). b Media collected over 2 days (DIV 47–49) from Tgx2-αSynA53T primary neurons lentivirally expressing control (GFP), GFP-VAMP7DN fragment, SNAP23KD shRNA, and SNAP23KD shRNA plus knockdown-resistant SNAP23 rescue-construct (infected on DIV7) were immunoblotted for the indicated αSyn species (αSyn, αSynpSer129, αSynAmyl, and αSynFila), cathepsin-L (Cath-LMat; late-endosome/lysosome-luminal), neuroserpin (non-lysosomally/constitutively secreted from neurons), and β-actin (cytoplasmic). Membrane-matched dot blots (β-Actin = loading control) are separated by dashed lines. For quantification (graphs on the right), protein level in the medium was normalized to its levels in total cellular lysate (5% loaded) (n = 7). c Correlation between the levels of pathogenic αSyn species and the levels of cathepsin-L released in the medium, upon the indicated manipulation of SNARE-dependent lysosomal exocytosis; derived from data shown above in b. Linear regression is shown with dotted lines depicting the 95% confidence intervals, and Pearson’s correlation coefficient is indicated on the bottom right of each graph. (n = 7). All data represent means ± SEM. Each “n” corresponds to a separate mouse litter used for a batch of neuronal culture and infection. In b n.s. not significant; **P < 0.01; ***P < 0.001 by 1-way ANOVA with Dunnett multiple-comparison correction compared to control; and #P < 0.05; ##P < 0.01; by non-parametric Kruskal–Wallis test with Dunn’s multiple-comparison adjustment compared to control.
SNARE-dependent lysosomal exocytosis is triggered by cytosolic calcium39, which in turn is modulated by neuronal activity and/or exchange with organellar calcium stores. We first pharmacologically modulated neuronal activity in Tgx2-αSynA53T neurons, by using TTX or CNQX/APV to inhibit activity, and K+ or bicuculline methiodide to enhance activity. Release of αSyn aggregates as well as that of cathepsin-L directly correlated with neuronal activity (Supplementary Fig. S8a). Next, we inhibited the release of calcium from the endoplasmic reticulum using dantrolene/2-APB, or inhibited the influx of extracellular calcium using YM-58483, in Tgx2-αSynA53T neurons (Supplementary Fig. S8b). Both strategies reduced the release of αSyn aggregates and cathepsin-L into the medium (Supplementary Fig. S8b). Neither the manipulation of neuronal activity nor the reduction of cytoplasmic calcium caused neuron death (Supplementary Fig. S8c, d). These pharmacological experiments further suggest that release of pathogenic αSyn species from neurons is mediated via calcium-regulated SNARE-dependent lysosomal exocytosis.
To test this hypothesis directly, we modulated the SNARE proteins responsible for lysosome-to-plasma membrane fusion. First, we identified VAMP7 as the prominent v-SNARE in immunoisolated lysosomes (Supplementary Fig. S9a). We then screened for the Qb-type t-SNAREs which interact and thus co-immunoprecipitate with VAMP7. SNAP23 was the clearest Qb/t-SNARE candidate (Supplementary Fig. S9a). Based on these results and prior studies validating them40, we tested four strategies to disrupt lysosomal exocytosis: shRNA knockdown of either SNAP23 (SNAP23KD) or VAMP7 (VAMP7KD), and overexpression of dominant-negative fragments of either SNAP23 (SNAP23DN) or VAMP7 (VAMP7DN). In wild-type primary neurons, the release of cathepsin-L was used to signal lysosomal exocytosis, opposed to neuroserpin—a non-lysosomal secreted protein. VAMP7KD and SNAP23DN had no significant effect on lysosomal exocytosis, but VAMP7DN severely reduced it by ~80% and SNAP23KD partially reduced it by ~40%, with no effect on cell viability (Supplementary Fig. S9b–d). Therefore, we applied the latter two strategies to disrupt lysosomal exocytosis next.
Importantly, in Tgx2-αSynA53T neurons, both VAMP7DN and SNAP23KD strategies reduced the release of pathogenic αSyn species into the medium, and the effect of SNAP23KD was rescued by overexpression of a knockdown-resistant version of wild-type SNAP23 (Fig. 5b). Moreover, the release of pathogenic αSyn species was significantly correlated with the change in lysosomal exocytosis, measured as cathepsin-L released into the medium (Fig. 5b, c). No β-actin was detectable in the medium (Fig. 5b). These results suggest that SNARE-dependent exocytosis of lysosomes is essential and rate-determining for the release of pathogenic αSyn species from neurons.
αSyn species released via lysosomal exocytosis can seed aggregation of purified recombinant αSyn and of endogenous αSyn in neurons
Pathogenic spread requires that the released αSyn is able to template or “seed” the assembly of monomeric αSyn into amyloid-type aggregates.
To test whether the pathogenic αSyn species exocytosed from neurons are seeding-competent, we performed in vitro aggregation of purified recombinant myc-tagged wild-type αSyn (Supplementary Fig. S10a), in the presence of extracellular media collected from neuron cultures generated either from wild-type mice, or from Tgx2-αSynA53T mice with or without lentiviral expression of VAMP7DN fragment (Fig. 6a–c). Aggregation kinetics, measured by amyloid-binding fluorescent dye K114 (Fig. 6a) or Thioflavin-T (Fig. 6b), were enhanced by medium from Tgx2-αSynA53T neurons, compared to the medium from wild-type neurons. Suppressing lysosomal exocytosis in the Tgx2-αSynA53T neurons, via VAMP7DNexpression, diminished the aggregation-promoting effect of the Tgx2-αSynA53T medium (Fig. 6a, b). When myc-αSyn aggregation kinetics were measured by quantitative immunoblotting for either disappearance of myc-tagged αSyn (myc-αSyn) monomers or for the appearance of filamentous and amyloid-type aggregates (shown in αSynFila and αSynAmylblots), we again found that compared to the wild-type neuron medium, aggregation was augmented by the Tgx2-αSynA53T medium, and the expression of VAMP7DN fragment diminished this effect (Fig. 6c). Notably, the effect of wild-type neuron medium on myc-αSyn aggregation rate was indistinguishable from the effect of unconditioned medium and αSyn−/−neuron medium (Supplementary Fig. S10b). In all the assays, a lag-time of nearly 2 weeks is apparent before the appearance of amyloid signal with the wild-type medium (Fig. 6a–c). Importantly, this lag is shortened or eliminated by the secreted aggregates in Tgx2-αSynA53Tmedium (Fig. 6a–c), with a reduced effect when lysosomal exocytosis is suppressed by VAMP7DN fragment (Fig. 6a–c).
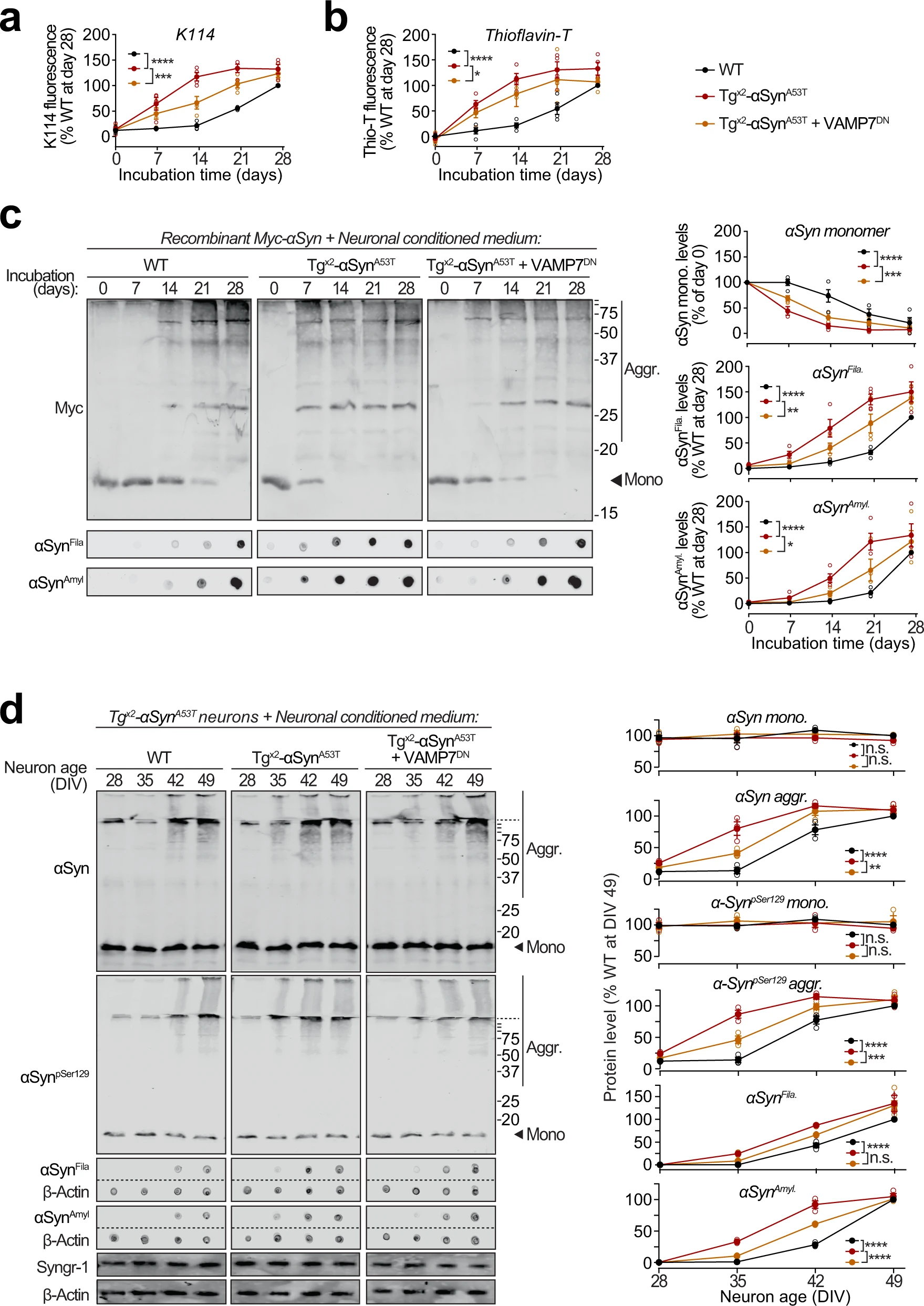
Seeding of aggregation of recombinant and neuronally expressed αSyn by pathogenic αSyn species exocytosed from neurons.
a–c Recombinant purified myc-tagged αSyn (myc-αSyn) was shaken at 37 °C in presence of concentrated extracellular medium from mouse cortical neuron cultures collected over 48 h (DIV 47–49), generated either from wild-type (WT) mice, or from Tgx2-αSynA53T mice with or without lentiviral expression of VAMP7 dominant-negative fragment (VAMP7DN; infected at DIV7). Aggregation of myc-αSyn was analyzed at the indicated days of incubation by the following assays: a Congo-red derivative, amyloid-binding dye K114 fluorescence at 390/535 nm (n = 4). b Amyloid-binding dye Thioflavin-T fluorescence at 450/485 nm (n = 4). c Quantitative immunoblotting for the myc epitope-tag, where aggregation is measured as disappearance of monomeric myc-αSyn (top; n = 4); dot-blotting for filamentous myc-αSyn aggregates using αSynFila antibody (middle; n = 4); and dot-blotting for amyloid-type myc-αSyn aggregates using αSynAmyl A11 antibody (bottom; n = 4). d Concentrated media collected over 48 h (DIV 47–49) from WT neurons, or from Tgx2-αSynA53Tneurons with or without lentiviral expression of VAMP7DN was added to the medium of Tgx2-αSynA53T neurons at DIV14 and removed 48 h later by replacing the medium. Neurons were harvested at indicated times, and αSyn aggregation was measured by immunoblotting, and normalized to synaptogyrin-1 (Syngr-1) levels. Membrane-matched dot blots (β-Actin = loading control) are separated by dashed lines. (n = 3). All data represent means ± SEM, where each “n” is an independent media collection and aggregation experiment. n.s. not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 by 2-way ANOVA.
Moreover, in Tgx2-αSynA53T primary neurons, appearance of αSyn aggregates was accelerated by media from Tgx2-αSynA53T neurons, but not from wild-type neuron media (Fig. 6d). Importantly, media from Tgx2-αSynA53T neurons expressing the VAMP7DN fragment—which inhibits lysosomal exocytosis, had a significant reduction in this effect (Fig. 6d).
These results substantiate that αSyn species exocytosed by lysosomes are capable of amyloid-nucleation via permissive templating, as well as spreading this seeding activity into live neurons.
Discussion
Release of αSyn aggregates from neurons is thought to transmit neuropathology to other yet-unaffected neurons. Here, we find that pathogenic αSyn species accumulate within neuronal lysosomes in mouse brains and in primary neurons, and are then released from neurons via SNARE-dependent lysosomal exocytosis (Fig. 7).
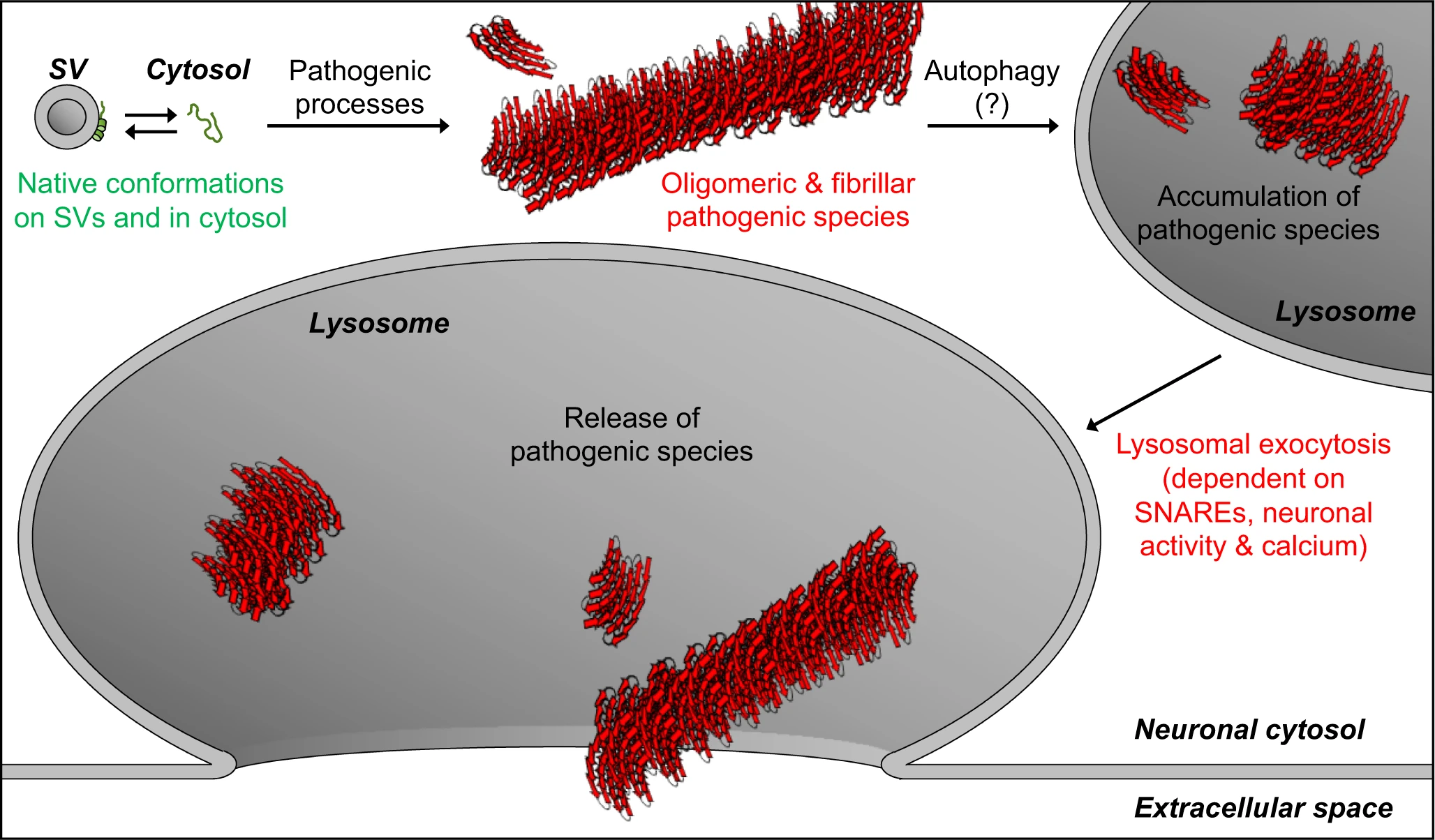
Model of our findings: Lysosomal exocytosis releases pathogenic αSyn species from neurons.
Native αSyn is expressed as a natively unstructured monomer in the cytosol, in equilibrium with an α-helical conformer upon binding the highly curved synaptic vesicle (SV) membrane. Pathogenic processes oligomerize αSyn into β-sheet-rich amyloid-type aggregates of various sizes. We demonstrate here that these larger pathogenic species, generated in the neuronal cytosol, accumulate in neuronal lysosomes over time, possibly due to their high structural and metabolic stability. SNARE-dependent lysosomal exocytosis releases these non-membrane-enveloped aggregates, and this release is upregulated by neuronal activity and cytosolic Ca2+.
It is important to note that for αSyn pathology to transmit from neuron-to-neuron, the lysosomal exocytosis mechanism explains the following observations, which underscore that the extracellular pool of pathogenic αSyn is likely not membrane-enveloped: (a) the stark pathology and rapid propagation caused by inoculated pre-formed fibrils10,11,41,42,43,44, (b) the efficacy of humoral and cellular immunotherapy against αSyn (reviewed in ref. 45), (c) the efficacy of passive immunization using antibodies against αSyn epitopes (reviewed in ref. 45), and (d) much of the extracellular αSyn has been found as non-enveloped6,46,47, while a minority of αSyn is contained in extracellular vesicles or exosomes12,18,20. However, other proposed means of αSyn aggregate-transmission might simultaneously contribute to pathological propagation in synucleinopathies38.
Key gaps still remain in how the pathogenic αSyn species are targeted and trafficked to lysosomes, as well as in the fate of extracellular αSyn aggregates following their release from neurons:
The aggregates/fibrils of αSyn could form inside the acidified lumen of lysosomes48, e.g., from monomers taken up via chaperone-mediated autophagy or macroautophagy of synaptic vesicles, especially if their degradation is delayed due to reduced or disrupted lysosomal activity49. Yet, we found no presence of αSyn monomers within isolated lysosomes, possibly due to their rapid degradation by lysosomal proteases, making this possibility less likely. Macroautophagy of oligomers and/or larger aggregates from the cytoplasm, termed “aggrephagy”, is a more likely mechanism, following from the well-recorded ability of autophagosomes to encapsulate cytoplasmic structures including aggregates50, and target them to lysosomes.
Once released from the neurons, the fate of extracellular/interstitial αSyn aggregates also remains unclear, and can follow multiple possible scenarios: uptake into microglia51, astrocytes52, and/or oligodendrocytes53, and/or drainage via glymphatics54,55 or blood flow6,56. However, for the extracellular αSyn aggregates to cause the stereotypic “spread” of pathology via a prion-like etiology, other neurons will have to take up the released aggregates, and this process remains even less understood than the glial uptake or clearance scenarios.
The results presented here provide evidence of the accumulation of pathogenic αSyn species in neuronal lysosomes, and pinpoint to lysosomal exocytosis as a pathway of release for these pathogenic αSyn species into the extracellular milieu. This pathway, at first sight, appears to have relevance to the release of undegraded aggregates of other proteins as well that transmit pathology from cell-to-cell (reviewed in ref. 38), and follow-up studies will be needed to test this speculation.