
Lipocalin-2 Regulates Epidermal Growth Factor Receptor Intracellular Trafficking
By Lucie Yammine, Aniela Zablocki, William Baron, Fabiola Terzi, and Morgan Gallazzini
Excerpt from the article published in Cell Reports, volume 29, issue 7, P2067-2077.E6, November 12, 2019, DOI: https://doi.org/10.1016/j.celrep.2019.10.015
Editor’s Highlights
- Lcn2, also known as neutrophil gelatinase-associated lipocalin (NGAL), siderocalin, or uterocalin, is a 25-kDa secreted glycoprotein that belongs to the lipocalin superfamily and has been associated with a plethora of pathological conditions, such as cancers or chronic kidney disease (CKD).
- Lcn2 acts as an enhancer of Epidermal growth factor receptor (EGFR) recycling process and signaling.
- In breast cancer cells that the tumor-associated S2R (sigma-2 receptor)/Pgrmc1 (progesterone receptor membrane component 1) upregulates Lcn2 expression.
- S2R/Pgrmc1 activity correlates with an increase of EGFR levels at the plasma membrane. The implication of Lcn2/EGFR partnership in cancerous cells remains to be demonstrated.
Autors’ Highlights
- Lipocalin-2 (Lcn2) induces EGFR maintenance to the cell surface
- Lcn2 facilitates EGFR recycling upon TGF-α stimulation
- Cytosolic Lcn2 binds to EGFR in late endosomal compartments
- Lcn2 allows sustained EGFR activation leading to chronic kidney disease onset
Summary
Epidermal growth factor receptor (EGFR) activation and lipocalin-2 (Lcn2) expression are frequently observed in the same pathological contexts, such as cancers or chronic kidney disease (CKD). However, the significance of this association is unknown. Here, we describe the role of Lcn2 in regulating EGFR trafficking. We show that Lcn2 increases EGFR cell surface abundance and is required for transforming growth factor α (TGF-α)-induced EGFR recycling to the plasma membrane and sustained activation. Lcn2 binds to the intracellular domain of EGFR in late endosomal compartments and inhibits its lysosomal degradation. Consistently, Lcn2 enhances EGFR-induced cell migration after TGF-α stimulation. In vivo, Lcn2 gene inactivation prevents EGFR recycling to the plasma membrane in an experimental model of CKD. Remarkably, this is associated with a dramatic decrease of renal lesions. Together, our data identify Lcn2 as a key mediator of EGFR trafficking processes. Hence, therapeutic inhibition of Lcn2 may counteract the deleterious effect of EGFR activation.
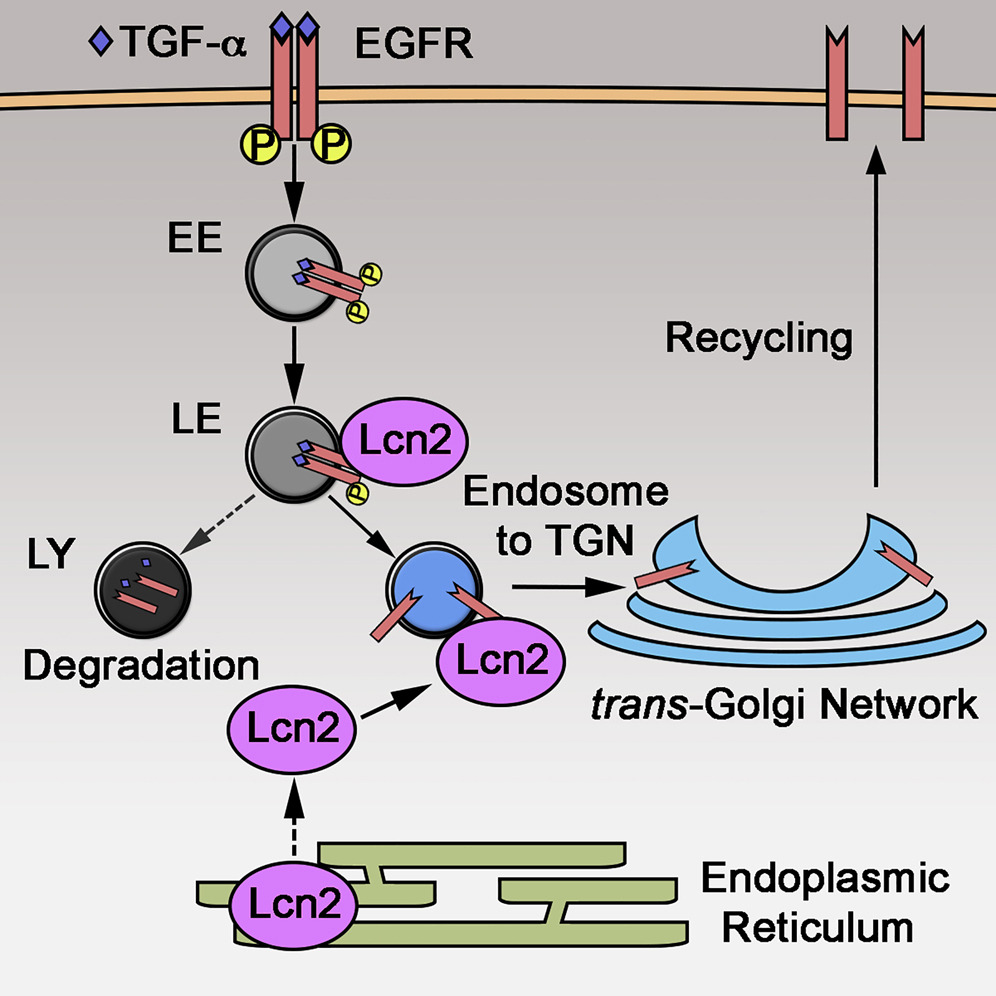
Introduction
Lcn2, also known as neutrophil gelatinase-associated lipocalin (NGAL), siderocalin, or uterocalin, is a 25-kDa secreted glycoprotein that belongs to the lipocalin superfamily (Flower et al., 1991). Lcn2 acts as a transporter of small hydrophobic substances and can sequester the siderophore-iron complex, limiting iron availability to bacteria and thus preventing the spread of infection (Goetz et al., 2002, Flo et al., 2004). In addition to its bacteriostatic role, Lcn2 expression has been associated with a plethora of pathological conditions, suggesting that it may have other functions outside infection (Chakraborty et al., 2012). In particular, Lcn2 overexpression has been reported to regulate cellular proliferation, migration, and differentiation in renal (Viau et al., 2010, Mori et al., 2005), vascular (Wang et al., 2015), cardiac (Buonafine et al., 2018), and cancer cells (Yang et al., 2009; Koh and Lee, 2015, Mongre et al., 2016, Ding et al., 2015, Iannetti et al., 2008). However, the molecular mechanisms underlying the role of Lcn2 in proliferation and migration are yet to be elucidated.
In the kidney, Lcn2 has been proposed as a promising biomarker of decreased kidney function (Bolignano et al., 2009), a hallmark of chronic kidney disease (CKD) onset. We have previously reported that the activation of epidermal growth factor receptor (EGFR) in a mouse model of CKD triggers the expression of Lcn2 and that Lcn2 gene deletion prevented the development of renal lesions by inhibiting the proliferative effect of EGFR (Viau et al., 2010). This observation is of particular interest because Lcn2 expression correlates with EGFR activity in other pathological contexts, such as breast (Leng et al., 2009), lung (Song et al., 2015), and prostate (Tung et al., 2013) cancers.
EGFR belongs to the ErbB receptor tyrosine kinase (RTK) family and is involved in a wide range of cellular processes, such as proliferation, growth, survival, and migration. EGFR can be activated by seven different ligands: EGF; transforming growth factor α (TGF-α); amphiregulin (AREG); Epigen (EPGN); heparin-binding EGF-like growth factor (HB-EGF); betacellulin (BTC); and epiregulin (EREG). The monomeric EGFR homo- or hetero-dimerizes with another member of the ErbB family upon ligand binding and transphosphorylates thereafter. This provides docking sites for adaptor proteins that will allow EGFR endocytosis and leads to the activation of intracellular signaling pathways, such as mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways. After internalization, ligand-bound EGFR is rapidly trafficked through the early endosome to the late endosome (Sorkin and Goh, 2009). Then, EGFR fate will depend on the ligand: EGF-bound EGFR will be targeted to lysosomal degradation, while TGF-α-bound EGFR will be recycled back to the plasma membrane (Roepstorff et al., 2009). Hence, EGFR pathway can be either transiently or constantly activated according to the nature of the ligand binding it. Interestingly, we have previously demonstrated that the expression of TGF-α is critically involved in CKD by stimulating EGFR-dependent cell proliferation (Laouari et al., 2012, Lautrette et al., 2005). Considering the role of EGFR over-activation in our previous study and in cancer, we hypothesized that Lcn2 expression in these pathological contexts may lead to a dysregulated activation of EGFR.
Here, we used in vitro and in vivo strategies to uncover the mechanism by which Lcn2 regulates EGFR activation. Lcn2 inactivation led to reduced EGFR-dependent signaling and increased EGFR lysosomal degradation. Lcn2 interacts with EGFR in late endosomal compartments after EGFR activation and endocytosis. This is relevant in vivo, where Lcn2 favors EGFR targeting to cellular membrane and over-activation in an experimental model of CKD. Together, our findings describe the mechanism by which Lcn2 modulates EGFR recycling process.
Results
EGFR Activation by Its Ligands Regulates Lcn2 Expression
To assess the role of Lcn2 in EGFR pathway, we used mIMCD-3 cells, an epithelial kidney cell line that spontaneously expresses Lcn2 (Viau et al., 2010). In order to better characterize our cell line, we first confirmed that mIMCD-3 cells responded to an EGF or TGF-α stimulation in terms of EGFR phosphorylation and downstream targets activation (e.g., ERK1/2 and AKT) in a dose-dependent manner (Figures S1A–S1E). Moreover, we showed that both EGF and TGF-α treatments were able to induce the expression of Lcn2 protein after 4 h of treatment (Figure 1A) and at least up to 24 h. This increase in Lcn2 protein abundance at 24 h was transcriptionally regulated, as we observed an increase in Lcn2 mRNA abundance after a 24-h EGF or TGF-α stimulation (Figure 1B). Because Lcn2 was first described as a secreted protein (Flower et al., 1991), we also showed that EGF and TGF-α tend to increase Lcn2 secretion as well between 4 h and 24 h of treatment (Figure 1C), although the differences are not statistically significant. Nevertheless, no effect in Lcn2 secretion was observed after 1 h, showing that Lcn2 secretion after EGFR activation is secondary to Lcn2 intracellular-induced expression. Then, we monitored EGFR expression upon stimulation in our cell line. Indeed, due to the high binding affinity of EGF to the receptor, an EGF-EGFR complex is more likely to undergo degradation, whereas a TGF-α-EGFR complex is keener to dissociation in the endosomes, allowing the receptor to be addressed back to the cell surface and the maintenance of its abundance (Roepstorff et al., 2009). As previously reported, we observed a decrease of EGFR protein abundance in EGF-stimulated cells, but not in TGF-α-stimulated ones, independently of ERBB1 mRNA expression (Figure 1D). These results suggest that EGF and TGF-α stimulation in mIMCD-3 induce Lcn2 mRNA and protein expression followed by an increased Lcn2 secretion.
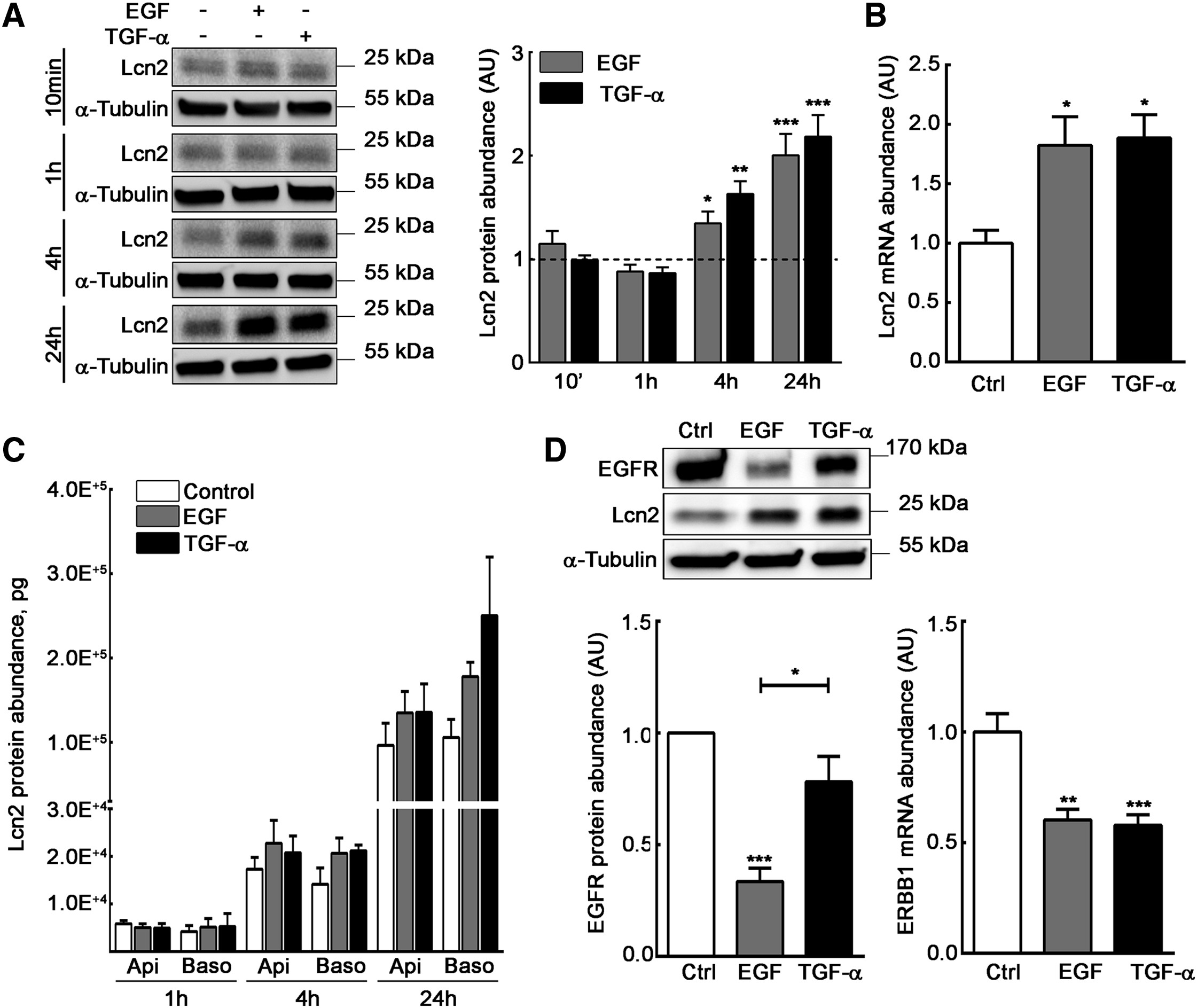
EGFR Activation Induces Lcn2 Expression and Subsequent Secretion
(A) EGF or TGF-α stimulated (10 nM) filter-cultured mIMCD-3 cells for 10 min, 1 h, 4 h, or 24 h. Immunoblots (left panel) and quantifications (right panel) of Lcn2 protein are shown. Dash line represents control condition value (n = 4).
(B) Filter-cultured mIMCD-3 cells were stimulated with EGF or TGF-α (10 nM) for 24 h. Quantifications show Lcn2 mRNA abundance (n = 6).
(C) Filter-cultured mIMCD-3 cells were stimulated or not (Control) with EGF or TGF-α (10 nM) for 1 h, 4 h, or 24 h. Apical and basolateral supernatants were collected and monitored by ELISA for Lcn2 protein secretion (n = 3).
(D) Filter-cultured mIMCD-3 cells were stimulated or not (Ctrl) with EGF or TGF-α (10 nM) for 24 h. Immunoblots and quantifications show EGFR protein abundance (left; n = 5) and ERBB1 mRNA abundance (right; n = 6).
Data are means ± SEM. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 versus control.
Lcn2 Promotes Cell Surface Expression of EGFR and Enhances Its Activation
Because we have previously described that Lcn2 expression regulates EGFR-induced phenotypes (Viau et al., 2010), we hypothesized that Lcn2 plays a role on EGFR early signaling, leading to its pathway regulation. Therefore, we studied the impact of Lcn2 knockdown on EGFR downstream signaling pathway. Interestingly, we observed a decrease of EGFR and ERK1/2 phosphorylation in sh-Lcn2 mIMCD-3 cells as compared to scramble mIMCD-3 cells when stimulated with either EGF or TGF-α (Figures 2A and 2B ). Because Lcn2 knockdown did not affect EGFR total protein abundance prior to stimulation (Figure S1F), we hypothesized that this effect may be due to a decreased availability of EGFR for its ligands. Indeed, in the absence of ligand, EGFR is internalized at the same rate as membrane turnover and is mainly recycled back thereafter (Herbst et al., 1994, Wiley, 2003). Because this steady-state EGFR recycling is regulated (Salazar and González, 2002), we postulated that Lcn2 might affect EGFR activation by its ligands by promoting its targeting to the cell surface and hence increasing its availability. To test this hypothesis, we performed a cell-surface biotinylation assay using the non-permeant sulfo-NHS-LC-biotin to label cell-surface proteins. We observed that Lcn2 knockdown decreased cell surface expression of EGFR but did not alter cell surface amount of Na,K-ATPase (Figure 2C). This demonstrates a specific effect on EGFR rather than a global effect on cell-surface protein content. Conversely, Lcn2 ectopic expression in Lcn2-naive cells induced a significant increase of EGFR cell surface abundance (Figure S1G). Hence, cells expressing Lcn2 exhibit increased EGFR abundance at the cell surface and are more prone to respond to EGFR ligands.
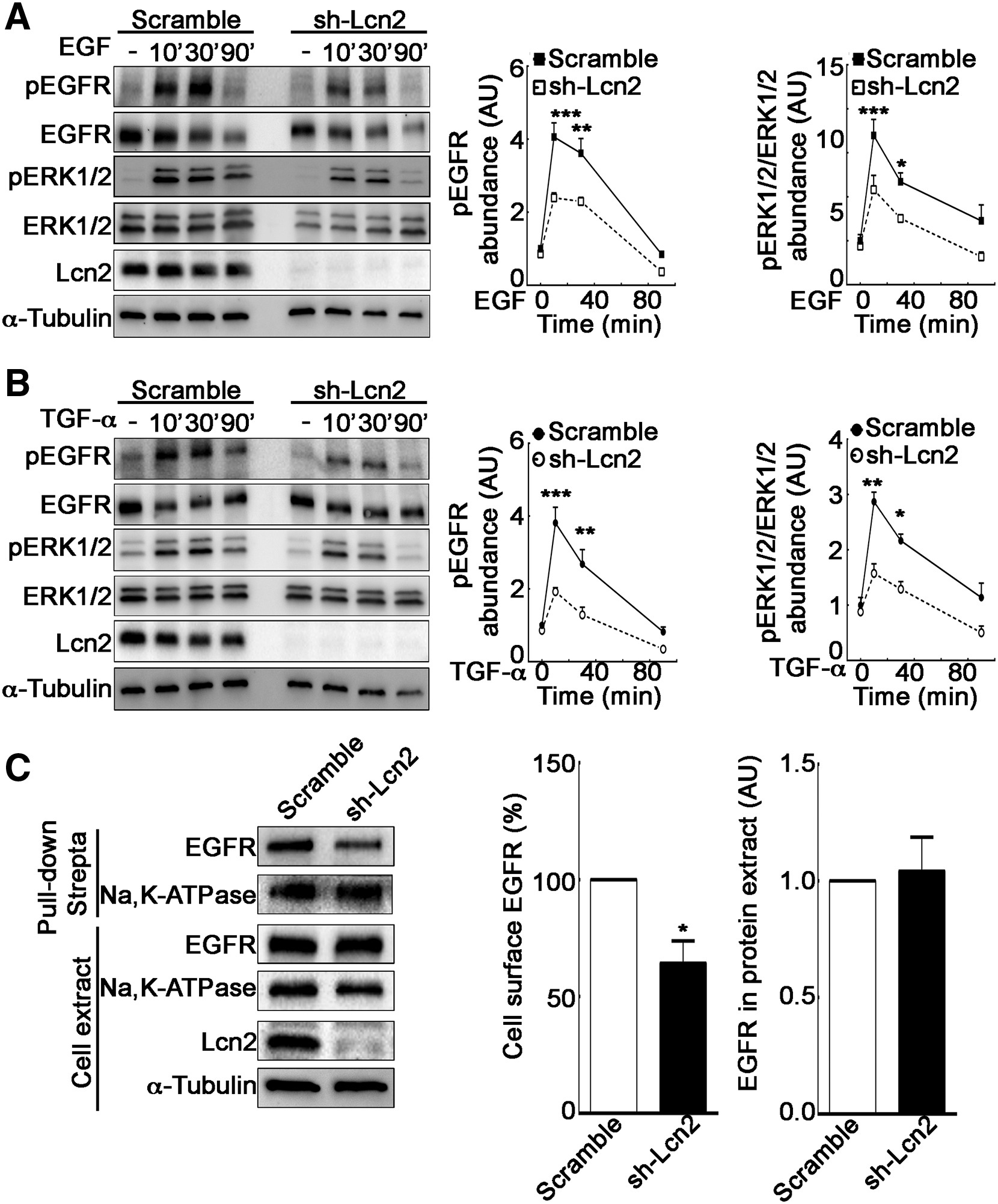
Lcn2 Facilitates EGFR Activation by Enhancing Its Cell Membrane Localization
(A and B) Filter-cultured sh-scramble or sh-Lcn2 mIMCD-3 cells were EGF (10 nM; A) or TGF-α stimulated (10 nM; B) for 10, 30, or 90 min. Representative immunoblot (left) of EGFR and ERK1/2 phosphorylation (pEGFR and pERK1/2, respectively) and quantification of pEGFR (middle) and pERK1/2 (right) over time (n = 3) is shown.
(C) sh-scramble or sh-Lcn2 mIMCD-3 cells were surface treated with EZ-Link Sulfo-NHS-LC-Biotin for 1 h prior to pull-down with Pierce Streptavidin Magnetic Beads. Representative immunoblot (left) and quantifications (right) of biotinylated EGFR in pull-down and total EGFR abundance in whole cell extract (n = 4) are shown.
Data are means ± SEM. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 versus scramble control.
Lcn2 Regulates EGFR Recycling upon TGF-α Stimulation
EGFR targeting to the plasma membrane is tightly linked to the regulation of its intracellular trafficking. To understand how Lcn2 modulates EGFR cell-surface expression, we tested whether Lcn2 regulates ligand-activated EGFR intracellular journey. Indeed, as observed in Figure 1D, EGF induces EGFR protein degradation while TGF-α promotes EGFR recycling back to the cell surface and hence the maintenance of EGFR protein abundance through time. We therefore postulated that, in this context, Lcn2 expression could be involved in EGFR recycling process in a TGF-α-specific manner. Accordingly, we observed that Lcn2 knockdown abolished EGFR protein maintenance upon stimulation with TGF-α for 24 h (Figure 3A). Furthermore, we observed a decrease in EGFR cell-surface labeling in TGF-α-stimulated sh-Lcn2 mIMCD-3 cells as compared to scramble cells (Figure 3B). Both scramble and sh-Lcn2 cell lines exhibit a decreased EGFR plasma membrane staining in response to EGF stimulation. This effect was independent of EGFR gene expression because no difference in ERBB1 mRNA abundance was observed between scramble and sh-Lcn2 mIMCD-3 cells after 24 h of TGF-α stimulation (Figure 3A). Importantly, this effect was specific to recycling-prone ligands, as we observed the same degradation of the receptor upon stimulation with EREG, another ligand known to induce EGFR recycling (Figure S2A). However, Lcn2 knockdown did not have any impact on EGFR abundance upon stimulation with EGF and HB-EGF, EGFR ligands known to induce the receptor degradation (Figures 3A and S2A). Knocking down Lcn2 using a distinct short hairpin RNA (shRNA) (sh2-Lcn2) confirmed EGFR degradation in TGF-α-stimulated cells in absence of Lcn2 (Figure S2B), demonstrating the specificity of Lcn2 loss on this phenomenon. This effect was not limited to kidney epithelial cells because it was also observed in A549 cells, a non-small-cell lung cancer cell line (Figure S2C). Of note, although we observed an increase of both protein and mRNA Lcn2 levels, regardless of the ligand in mIMCD-3 cells (Figures 1B, 3A, and S2A), in A549 cells, however, EGF or TGF-α stimulation induced a significant decrease in Lcn2 protein abundance (Figure S2C).
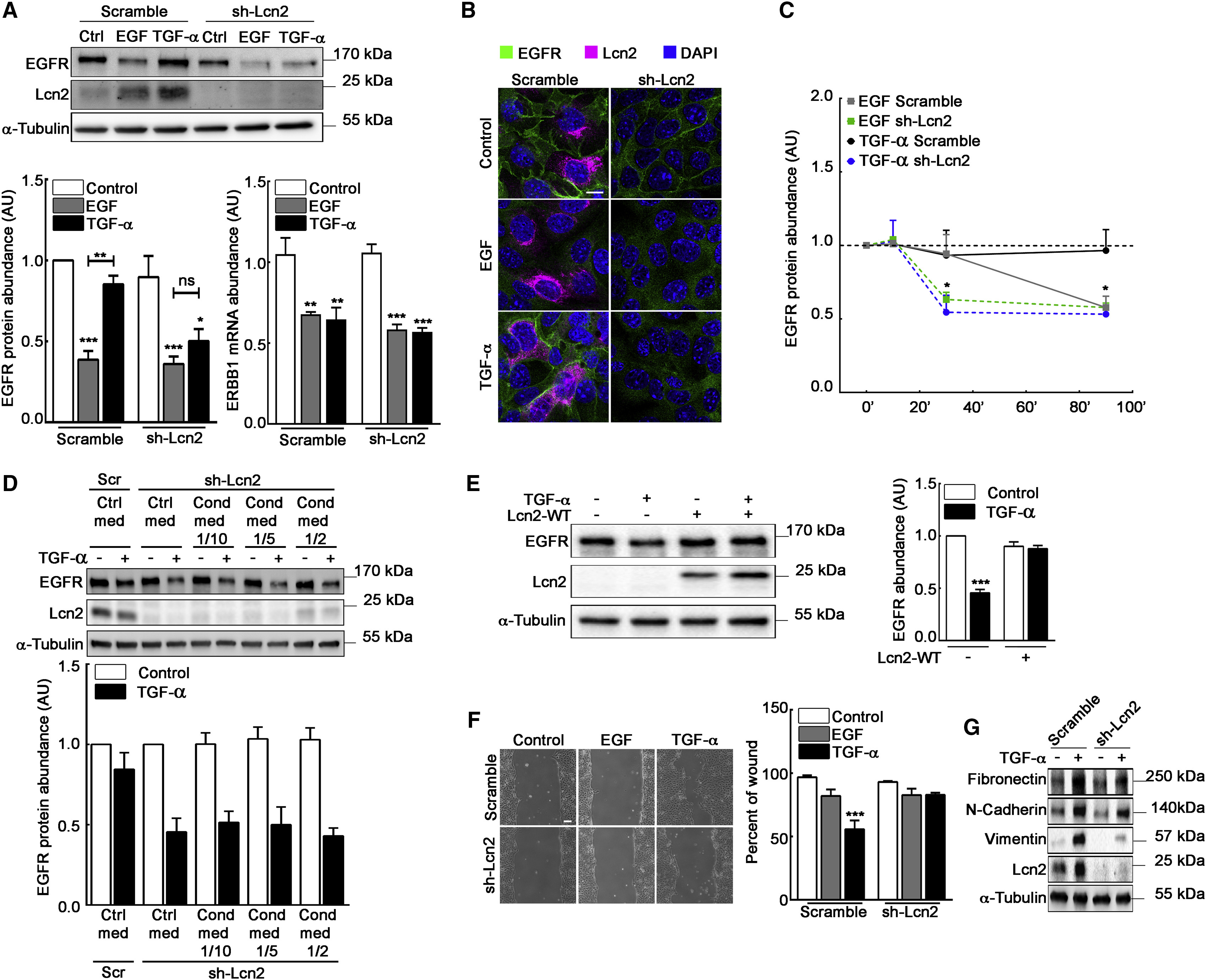
Lcn2 Modulates EGFR Abundance and TGF-α-Induced Phenotypes
(A) Filter-cultured sh-scramble and sh-Lcn2 mIMCD-3 cells were stimulated or not (Ctrl) with EGF or TGF-α (10 nM) for 24 h. Immunoblots (top) show EGFR and Lcn2 protein abundance and quantification show EGFR protein (bottom left) and ERBB1 mRNA (bottom right) abundance (n = 6).
(B) sh-scramble or sh-Lcn2 mIMCD-3 cells were EGF or TGF-α stimulated (10 nM) for 24 h. Immunofluorescence shows EGFR (green), Lcn2 (magenta), and nuclei counterstained with DAPI. Scale bar, 10 μm.
(C) sh-scramble and sh-Lcn2 mIMCD-3 cells were stimulated or not (Ctrl, dash line) with EGF or TGF-α (10 nM) for 10 min, 30 min, or 90 min. Quantifications show EGFR protein abundance over time (n = 4). Representative western blot is shown in Figure S2D.
(D) sh-Lcn2 mIMCD-3 cells were cultured with control media or with increased amount of mIMCD-3 cells conditioned media and stimulated or not with TGF-α (10 nM) for 2 h. sh-scramble mIMCD-3 cells cultured with control media and stimulated or not with TGF-α were used as a control for the effect of Lcn2 expression on EGFR protein abundance. Immunoblots (top) show EGFR and Lcn2 protein abundance and quantification (bottom) show EGFR protein abundance (n = 3).
(E) Empty vector (−) or Lcn2-WT (+) transfected sh-Lcn2 mIMCD-3 cells were TGF-α stimulated (10 nM) for 24 h. Representative immunoblot (left) of EGFR and Lcn2 protein and quantification (right) of EGFR abundance (n = 4) are shown.
(F) Wound healing assay on sh-scramble or sh-Lcn2 mIMCD-3 cells stimulated with EGF or TGF-α (10 nM) for 9 h. Scale bar, 100 μm. Quantification (right panel) of wound closure and percent of initial wound (n = 5) are shown.
(G) Filter-cultured sh-scramble or sh-Lcn2 mIMCD-3 were TGF-α stimulated (10 nM) for 24 h. Representative immunoblot of Fibronectin, N-Cadherin, Vimentin, and Lcn2 protein (n = 3) is shown.
Statistics source data are shown in Figure S3F. Data are means ± SEM. ∗∗p < 0.01; ∗∗∗p < 0.001 versus control.
EGFR protein degradation and maintenance upon stimulation are regulated mechanisms taking place shortly after EGFR internalization. Indeed, EGF-stimulated EGFR will be addressed to lysosomes for degradation while TGF-α-stimulated EGFR targets specific endosomes for recycling (Roepstorff et al., 2009). Thus, we hypothesized that Lcn2 may regulate EGFR trafficking journey. Therefore, we tested whether Lcn2 regulates EGFR recycling shortly after stimulation. Accordingly, Lcn2 knockdown decreased EGFR abundance as soon as 30 min after stimulation by TGF-α (Figures 3C and S2D), allowing us to hypothesize that this dramatic acceleration in EGFR degradation may rely on a role for Lcn2 in EGFR fate after endocytosis. We next wondered whether the secreted form of Lcn2 was responsible for the maintenance of EGFR protein abundance after TGF-α stimulation. To address this question, we treated sh-Lcn2 mIMCD-3 cells with conditioned medium obtained from cells secreting Lcn2 and incubated these cells with TGF-α. We demonstrated that addition of extracellular Lcn2 in the culture medium of Lcn2 knockdown cells failed to rescue EGFR protein abundance to the control level upon 2 h TGF-α stimulation (Figure 3D). The same results were obtained using a recombinant form of Lcn2 (Figure S2E). Of note, extracellular uptake of Lcn2 by sh-Lcn2 mIMCD-3 cells was rather weak because only a small amount of it was detected by western blotting. By contrast, rescuing Lcn2 expression by transfection of sh-Lcn2 mIMCD-3 cells did restore TGF-α-induced maintenance of EGFR to the control level (Figure 3E). No additive effect of Lcn2 ectopic expression has been observed in sh-scramble mIMCD-3 cells (Figure S3A). Interestingly, neither extracellular Lcn2 (recombinant or from conditioned medium) nor Lcn2 overexpression rescued EGF-induced degradation of EGFR (Figures S3B–S3D), confirming that Lcn2 regulates EGFR protein abundance only in a recycling prone ligand condition. Thus, Lcn2 regulates TGF-α-induced EGFR protein maintenance independently of its secretion.
It is known that TGF-α more potently induces cell migration than EGF in different cell types (Li et al., 2006, Andresen and Ehlers, 1998) and that this effect relies upon EGFR recycling after activation (McClintock and Ceresa, 2010). Thus, we performed a wound-healing assay on both scramble and sh-Lcn2 mIMCD-3 cells to test whether the absence of Lcn2 impacted TGF-α-induced phenotype. As expected, we observed that EGF failed to induce cell migration regardless of the presence of Lcn2 (Figure 3F). By contrast, Lcn2 knockdown prevented TGF-α-induced migration. This was associated with a decrease in several migration-related proteins abundance, such as Fibronectin, N-Cadherin, and Vimentin (Figures 3G and S3E). Collectively, these data strongly suggest a specific role of Lcn2 in EGFR intracellular trafficking and recycling upon TGF-α stimulation.
Lcn2 Facilitates EGFR Trafficking toward Recycling Compartment
To gain insight into the mechanism by which Lcn2 regulated EGFR protein maintenance upon TGF-α stimulation, we investigated its impact on EGFR lysosomal degradation (Opresko et al., 1995, Felder et al., 1990). Pre-treatment of sh-Lcn2 mIMCD-3 cells with chloroquine (Chloro), a lysosomal degradation inhibitor, completely blocked EGFR degradation, regardless of the ligand (Figure 4A). Consistent with these observations, we demonstrated a significant accumulation of EGFR in lysosomal compartments (Lamp2-positive structures) in TGF-α-stimulated sh-Lcn2 mIMCD-3 cells as compared to the scramble cell line (Figures 4B and S4A). As expected, EGF stimulation significantly increased EGFR staining in lysosomal compartments in both scramble and sh-Lcn2 mIMCD-3 cells (Figures S4B–S4D). Moreover, co-immunoprecipitation experiments demonstrated that, in absence of Lcn2, TGF-α-stimulated EGFR was more ubiquitinylated and bound to HRS (hepatocyte-growth-factor-regulated tyrosine kinase substrate), a protein involved in EGFR targeting to multivesicular bodies and lysosomal degradation (Hirano et al., 2006; Figure 4C). Together, these data indicate that, upon TGF-α stimulation, Lcn2 is critically involved in targeting EGFR toward recycling compartments rather than lysosomes.
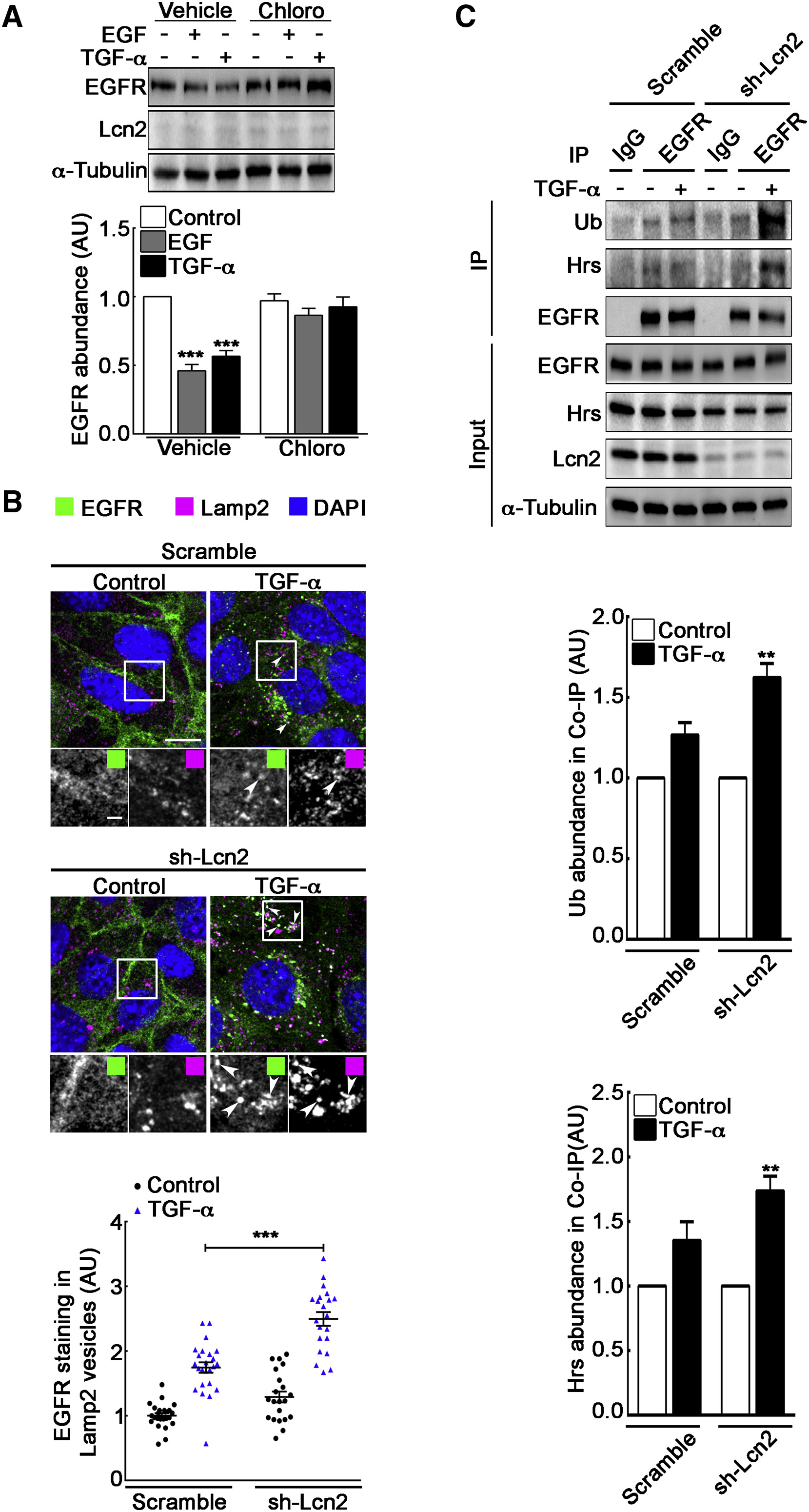
Lcn2 Inhibits EGFR Degradation in a TGF-α-Specific Manner
(A) sh-Lcn2 mIMCD-3 cells were treated without (vehicle) or with chloroquine 50 μM (Chloro) for 2 h prior to stimulation with EGF or TGF-α (10 nM) for 2 h. Representative immunoblot (top) of EGFR and Lcn2 protein abundance and quantification (bottom) of EGFR abundance (n = 5) are shown.
(B) sh-scramble or sh-Lcn2 mIMCD-3 cells were TGF-α stimulated (10 nM) for 1 h. Immunofluorescence shows EGFR (green), Lamp2 (magenta), and nuclei counterstained with DAPI. Scale bar, 10 μm; magnification scale bar, 2 μm. Graph shows a quantification of EGFR staining in Lamp2-positive vesicles (n = 3).
(C) sh-scramble or sh-Lcn2 mIMCD-3 cells were TGF-α stimulated (10 nM) for 15 min. Cell lysates were immunoprecipitated (IP) with immunoglobulin G (IgG) or anti-EGFR antibody. Representative immunoblot (top) of ubiquitinylated EGFR (Ub), Hrs, and EGFR protein abundance in IP and in whole cell lysate (input) is shown. Quantification of Ub (middle) and Hrs (bottom) in coIP with EGFR over stimulation (n = 3) is shown.
Data are means ± SEM. ∗∗p < 0.01; ∗∗∗p < 0.001 versus control.
Lcn2 Co-localizes with EGFR in Late Endosomal Compartments
Lcn2 is a protein synthesized in the endoplasmic reticulum (ER), transported in the Golgi apparatus, and then secreted (Figure S5A). To understand how Lcn2 could mediate EGFR trafficking, we co-stained EGFR and Lcn2 in TGF-α-stimulated mIMCD-3 cells at early time points. We observed an increasing number of co-localization events between the two proteins up to 30 min after stimulation (Figure 5A). Because this result suggested a possible association of Lcn2 to endosomal vesicles, we next co-stained Lcn2 with endosomal markers (Figure S5A). We observed that Lcn2 co-localized with the late endosomal marker Rab7 and with Rab9 vesicles, two proteins required for endosome to trans-Golgi network transport (Lombardi et al., 1993). By contrast, Lcn2 did not co-localize with Rab5-positive vesicles, a marker of the early endosomes (Figure S5A). Accordingly, live cell imaging, using total internal reflection fluorescence (TIRF) microscopy, in mIMCD-3 cells stably expressing Lcn2-Cherry and transfected with GFP-fused endosomal markers showed a dynamic co-staining of Lcn2 with Rab7 and Rab9, but not with Rab5, vesicles (Figure 5B; Videos S1, S2, and S3). Importantly, we demonstrated that Lcn2 and EGFR co-stained in Rab7- and Rab9-positive vesicles after TGF-α stimulation (Figures 5C and S5B). Together, these results suggest that a pool of Lcn2 protein localized at the late endosomal compartments may affect EGFR sorting upon TGF-α stimulation and facilitate its recycling to the cell surface via Rab9-positive vesicles.
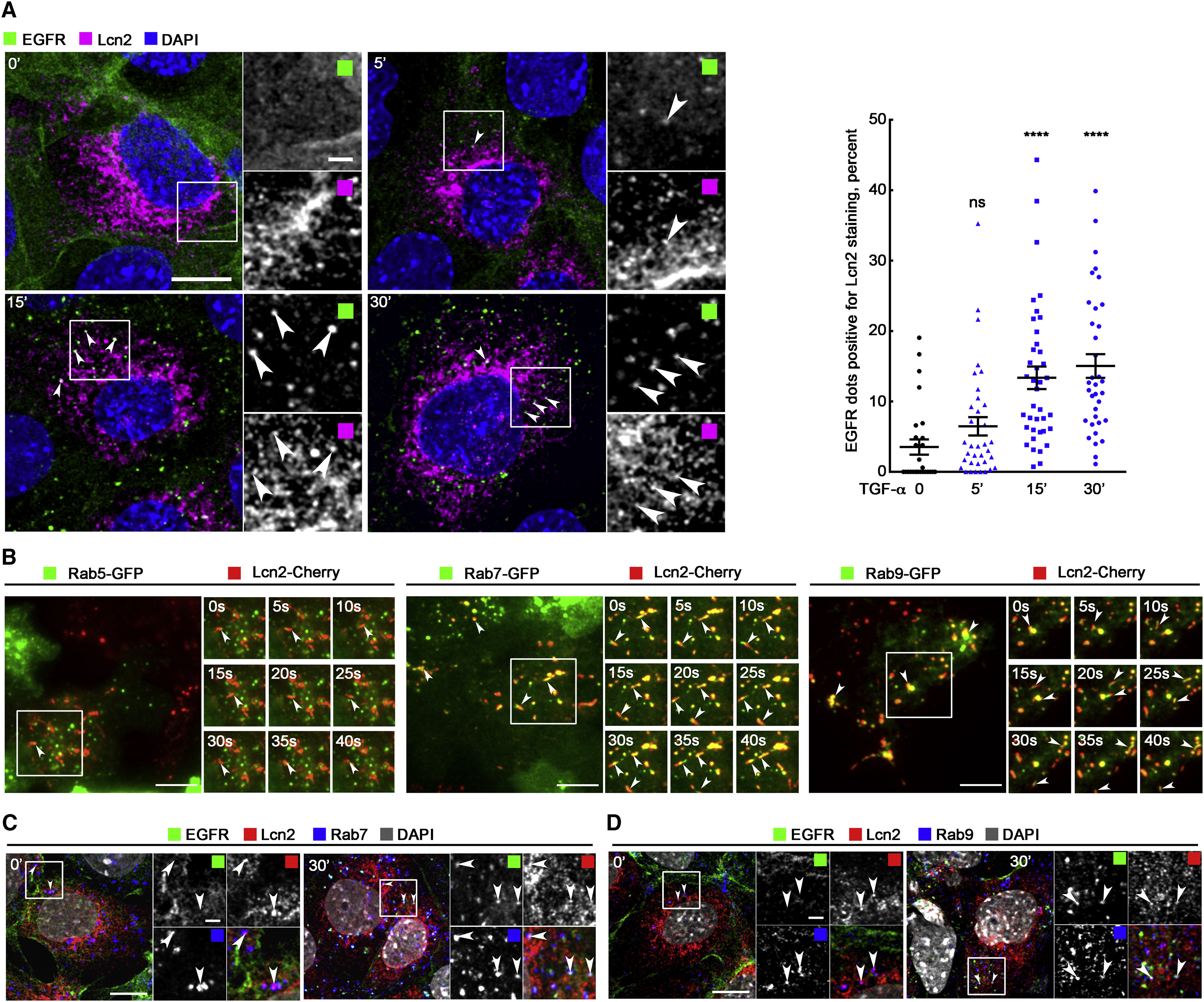
Lcn2 and EGFR Localize in the Same Compartments
(A) mIMCD-3 cells were TGF-α stimulated (10 nM) for 5, 15, and 30 min. Immunofluorescence (left) shows EGFR (green), Lcn2 (magenta), and nuclei counterstained with DAPI. Arrowheads point to the co-localization of EGFR and Lcn2. Scale bar, 10 μm; magnification scale bar, 2 μm. Graph (right) shows quantification of Lcn2 staining in EGFR-positive vesicles (in percent of total EGFR positive vesicles; n = 3). Data are means ± SEM. ∗∗∗∗p < 0.0001 versus control.
(B) mIMCD-3 cells stably expressing Lcn2-Cherry were transfected with Rab5-GFP (left), Rab7-GFP (middle), and Rab9-GFP (right) constructs. Real-time TIRF imaging shows Rabs-GFP (green) and Lcn2-Cherry (red) dynamic movements within 40 s. Arrowheads point to the co-localization of Rabs and Lcn2. Scale bars, 10 μm.
(C and D) mIMCD-3 cells were TGF-α stimulated (10 nM) for 30 min. Immunofluorescence shows EGFR (green), Lcn2 (red), Rab7 (blue; C), Rab9 (blue; D), and nuclei counterstained with DAPI (gray in merged images). Arrowheads point to the co-localization of EGFR, Rabs, and Lcn2. Scale bars, 10 μm; magnification scale bars, 2 μm.
TIRF Video Lcn2-mCherry and Rab5-GFP, Related to Figure 5
TIRF Video Lcn2-mCherry and Rab7-GFP, Related to Figure 5
TIRF Video Lcn2-mCherry and Rab9-GFP, Related to Figure 5
EGFR Activation Enhances Its Interaction with Lcn2
To provide further evidences regarding the molecular relationship between Lcn2 and EGFR, we performed immunoprecipitation experiments. We demonstrated a co-immunoprecipitation of endogenous Lcn2 with EGFR upon TGF-α stimulation in mIMCD-3 cells (Figure 6A). A similar result was also observed between ectopically expressed Lcn2 and EGFR in HEK293-transfected cells (Figure 6B). Interestingly, we observed Lcn2/EGFR association strengthened up between 10 and 30 min of TGF-α stimulation (Figures 6A and 6C), confirming our hypothesis that Lcn2 associates with EGFR in late endosomal compartments. Because EGFR is a transmembrane protein, Lcn2 might interact directly or indirectly either with its extracellular or intracellular domain. Using a biotin ligase (BirA) protein fusion approach (Roux et al., 2012), we generated a Lcn2-BirA fused protein to assess the proximity between Lcn2-BirA and full-length EGFR (FL), the extracellular domain of EGFR (ECD), or the intracellular domain of EGFR (ICD) (Figures 6D and 6E). We observed a biotinylation of both EGFR-FL and EGFR-ICD, but not of EGFR-ECD, indicating that Lcn2 is in close proximity with the intracellular domain of EGFR. These observations strongly suggest that Lcn2 forms a complex with EGFR, most likely toward its cytosolic domain.
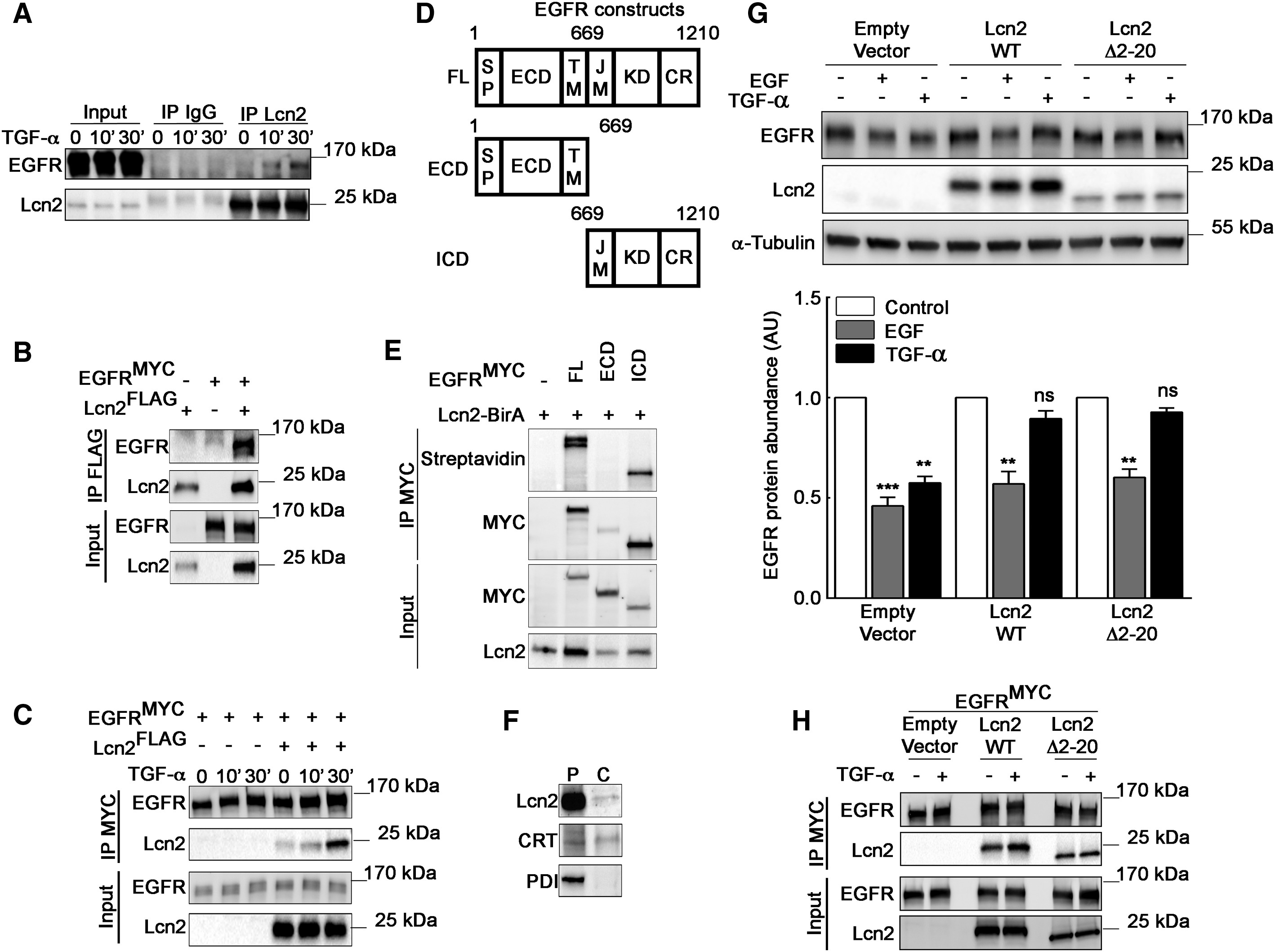
Lcn2 Interacts with EGFR in a Dynamic Manner
(A) mIMCD-3 cells were stimulated or not with TGF-α (10 nM) for 10 and 30 min. Cell lysates were immunoprecipitated (IP) with immunoglobulin G (IgG) or an anti-Lcn2 antibody. Representative immunoblot of EGFR and Lcn2 protein in IP and in whole-cell lysate (input) is shown.
(B) Anti-FLAG antibody immunoprecipitation (IP FLAG) of EGFRMYC– and Lcn2FLAG-transfected HEK293 cells lysate. Representative immunoblot of EGFR and Lcn2 protein in IP and in whole-cell lysate (input) is shown.
(C) Anti-MYC antibody immunoprecipitation (IP MYC) of EGFRMYC– and Lcn2FLAG-transfected HEK293 cells lysate stimulated with TGF-α (10 nM) for 10 and 30 min. Representative immunoblot of EGFR and Lcn2 protein in IP and in whole-cell lysate (input) is shown.
(D) A schematic representation of EGFR full-length (FL), EGFR extracellular domain (ECD), and EGFR intracellular domain (ICD), including EGFR signal peptide (SP), transmembrane (TM), and juxtamembrane (JM) regions; kinase domain (KD); and C-terminal region (CR).
(E) EGFRMYC full length, ECD, or ICD and Lcn2-BirA constructs were transiently expressed in HEK293 cells. Proteins were immunoprecipitated with anti-MYC antibody (IP MYC). Representative immunoblot of biotinylated proteins (streptavidin) and immunoprecipitated EGFR (MYC) in IP and EGFR (MYC) and Lcn2 abundance in cell lysate (input) is shown.
(F) mIMCD-3 cells were permeabilized with digitonin to determine cytosolic localization of endogenous Lcn2 protein. Representative immunoblots of permeabilized cells (P) and the cytosolic fraction (C) are shown. Calreticulin (CRT) (positive control) and protein disulfide-isomerase precursor (PDI) (negative control).
(G) Empty vector or Lcn2-WT or Lcn2 Δ2-20 transfected sh-Lcn2 mIMCD-3 cells were EGF or TGF-α stimulated (10 nM) for 2 h. Representative immunoblot (top) of EGFR and Lcn2 protein and quantification (bottom) of EGFR abundance (n = 3) is shown.
(H) Anti-MYC antibody immunoprecipitation (IP MYC) of EGFRMYC– and Lcn2-WTFLAG– or Lcn2-Δ2-20FLAG-transfected HEK293 cells lysate stimulated with TGF-α (10 nM) for 30 min. Representative immunoblot of EGFR and Lcn2 protein in IP and in whole-cell lysate (input) is shown.
Data are means ± SEM. ∗∗p < 0.01; ∗∗∗p < 0.001 versus control.
Interestingly, a recent report has shown that Lcn2 localizes to the nucleus and binds to the promoter of Twist1 (Wang et al., 2013), suggesting that a cytosolic pool of Lcn2 can translocate to the nucleus. To investigate whether cytosolic Lcn2 can be found in mIMCD-3 cells and be involved in EGFR trafficking, we performed subcellular fractionation using digitonin to selectively permeabilize the plasma membrane of our cells. We demonstrated that a fraction of Lcn2 could be found in the cytosol and therefore associates with the intracellular domain of EGFR (Figure 6F). Cytosolic localization of Lcn2 was also confirmed using another subcellular fractionation approach (subcellular fractionation kit; Figure S5C), as well as its previously described nuclear localization. Nevertheless, the relative abundance of Lcn2 remained unchanged in the different fractions and in the supernatant 2 h after TGF-α stimulation (Figures S5C and S5D). We next decided to reconstitute Lcn2 knockdown cells with a mutant form of Lcn2 baring a deletion of its signal peptide (Δ2-20 Lcn2), inducing the production of a cytosolic only form of Lcn2. This mutant is not secreted and migrates at a lower molecular weight than the wild-type Lcn2 in western blot due to the absence of glycosylation (Figure S5E). Interestingly, transfections of sh-Lcn2 mIMCD-3 cells with the wild-type or the Δ2-20 Lcn2 mutant showed a rescue of EGFR protein maintenance upon TGF-α stimulation (Figure 6G). No additive effect was observed when sh-scramble mIMCD-3 cells were transfected with Δ2-20 Lcn2 mutant (Figure S5F). Furthermore, we demonstrated that the Δ2-20 Lcn2 mutant co-immunoprecipitates with EGFR (Figure 6H). These results strongly suggest that a cytosolic form of Lcn2 is implicated in EGFR recycling process.
Lcn2 Expression in Damaged Kidneys Induces EGFR Localization to the Cell Membrane
Finally, we wondered whether our in vitro data are relevant in vivo. As a paradigm, we studied CKD progression, a pathological condition known to involve Lcn2 and a TGF-α-dependent EGFR activation (Laouari et al., 2012). Lcn2+/+ and Lcn2−/− mice were subjected to subtotal nephrectomy (Nx) in order to mimic CKD. We first confirmed that Nx led to the development of severe renal lesions in Lcn2+/+ mice, 2 months after Nx (Figure 7A). As previously described, the severity of renal lesions was markedly reduced in Lcn2−/− mice (Figure 7A). We then investigated EGFR and Lcn2 abundance and localization in kidney parenchyma. In control, sham-operated Lcn2+/+ and Lcn2−/− mice, we observed that EGFR localizes mainly to the cytoplasm of tubular epithelial cells (Figure 7B). In contrast, we failed to detect Lcn2 protein in these mice (Figures 7B and 7C). As expected, after Nx, the expression of Lcn2 was markedly increased in remnant kidneys of Lcn2+/+ mice (Figures 7B and 7C). This was associated with an increased localization of EGFR to the cell membrane of tubular cells (Figure 7B). This reinforcement was not due to an increase of EGFR abundance, as judged by the protein level (Figure 7C). Interestingly, Lcn2inactivation prevented such EGFR localization in Lcn2−/− Nx mice, suggesting that the beneficial effect of Lcn2 inactivation may be due to reduced EGFR activation (Figure 7B). Consistently, immunofluorescence experiments using anti-phospho-EGFR antibodies showed that EGFR phosphorylation and internalization occurred only in Lcn2+/+ Nx mice (Figure 7D). Interestingly, we observed a co-localization of Lcn2 with activated EGFR in damaged kidney of Lcn2+/+ Nx mice (Figure 7D). Together, these results indicate that Lcn2 regulates EGFR intracellular trafficking after Nx.
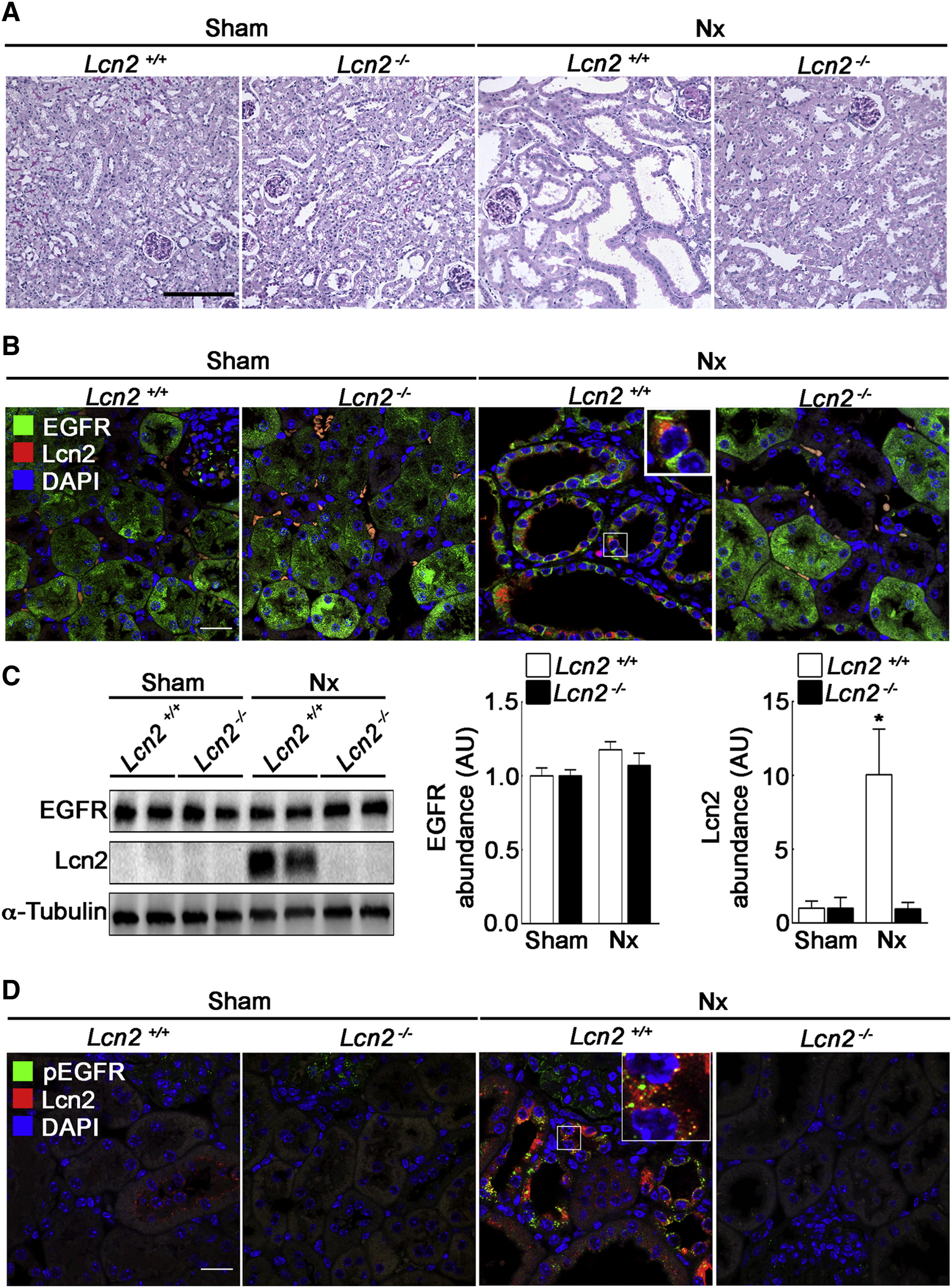
Lcn2 Modulates EGFR Localization in Damaged Kidneys
Lcn2+/+ or Lcn2−/− FVB/N mice were subjected to sham operation (sham) or nephron reduction (Nx).
(A) Morphology of kidney sections from Lcn2+/+ or Lcn2−/− sham and Nx mice. Scale bar, 100 μm.
(B) Immunofluorescence on kidney sections showing EGFR (green), Lcn2 (red), and nuclei counterstained with DAPI. Scale bar, 20 μm.
(C) Representative immunoblots (left) and quantifications of EGFR (middle) and Lcn2 (right) protein abundance in Lcn2+/+ or Lcn2−/− sham and Nx kidneys (n = 3 mice/group).
(D) Immunofluorescence on kidney sections showing activated EGFR (pEGFR; green), Lcn2 (red), and nuclei counterstained with DAPI. Scale bar, 20 μm.
Data are means ± SEM. ∗p < 0.05 versus sham.
Discussion
EGFR activation drives a large panel of signaling pathways regulating cellular functions and processes. EGFR over-activation can lead to uncontrolled cell proliferation responsible for a wide range of pathologies. Our group previously established that EGFR aberrant activation in kidney epithelial cells leads to renal lesions and progression toward end-stage renal disease (Laouari et al., 2012, Lautrette et al., 2005, Terzi et al., 2000). In this context, Lcn2 has been identified as an important mediator of EGFR-induced tubular cell proliferation (Viau et al., 2010). In this study, we establish that Lcn2 is a key modulator of EGFR intracellular trafficking. We demonstrate that, upon TGF-α stimulation, Lcn2 enhances EGFR recycling process and thus increases its downstream signaling pathways. We show that Lcn2 inactivation leads to lysosomal degradation of EGFR and limits EGFR-induced cellular migration. Importantly, Lcn2 seems to exert its effect on EGFR fate via its interaction with the intracellular domain of EGFR, and we identify a cytosolic fraction of Lcn2 that could be involved in this interaction. We were able to confirm our findings in an experimental model of CKD by observing a decreased targeting of EGFR to the tubular cell surface in Lcn2−/− mice as compared to Lcn2+/+ mice. Together, these data uncover Lcn2 as a key mediator of EGFR intracellular trafficking, leading to its sustained maintenance at the cell membrane and its availability for stimulation by TGF-α. This effect of Lcn2 on EGFR abnormal activation is responsible for the renal deterioration and progression toward CKD.
Our in vitro and in vivo data demonstrate that cell-autonomous Lcn2 expression will allow both increased abundance of EGFR to the cell surface and enhanced EGFR recycling to the plasma membrane upon TGF-α stimulation. Indeed, we demonstrate that only intracellular Lcn2 is able to facilitate EGFR recycling to the plasma membrane, as recombinant Lcn2 didn’t rescue EGFR abundance upon TGF-α stimulation in sh-Lcn2 cells. Furthermore, our data suggest the presence of a cytosolic fraction of Lcn2, which seems to be responsible for the interaction of Lcn2 with the intracellular domain of EGFR. Nevertheless, amino-acid sequence analysis of Lcn2 protein doesn’t identify by homology any putative direct binding site with EGFR. Likewise, the role of ligand-induced EGFR activation on the recruitment of Lcn2 is yet to be determined. Hence, the mechanism by which Lcn2 interacts directly or indirectly with EGFR remains to be elucidated. Lcn2 cytosolic localization implies that Lcn2 could escape the secretory pathway and retrotranslocate from the ER or the Golgi apparatus to the cytosol. Other proteins have been previously shown to retrotranslocate from the ER or the Golgi to the cytosol, like calreticulin or clusterin (Afshar et al., 2005, Nizard et al., 2007). Lcn2 cytosolic localization is of particular interest because it opens a wide range of putative cellular effects of Lcn2. Indeed, the majority of the reports regarding Lcn2 effect are mainly based on the role of the secreted form on iron transport and MMP9 stabilization (Chakraborty et al., 2012). Interestingly, Wang et al., 2013 recently demonstrate that, in hepatocellular carcinoma, Lcn2 can localize in the nucleus and effectively bind to the promoter of Twist1 to decrease its expression. This nuclear localization of Lcn2 suggests a translocation from a cytosolic pool of Lcn2, which is consistent with our data. Thus, Lcn2 subcellular localization could be subject to regulation. EGFR pathway activation has been reported to regulate protein localization. For instance, GOLM1 has been recently shown to escape from the Golgi apparatus after EGF stimulation to co-localize with EGFR in endosome-like structures (Ye et al., 2016). GOLM1 can then facilitate EGFR trafficking toward the plasma membrane. Hence, it is conceivable to propose that EGFR pathway activation may regulate Lcn2 localization and recruit Lcn2 during its journey within the endosomal compartment. Lcn2 has been recently reported as a phosphorylated protein (Weng et al., 2015); it will therefore be interesting to study the possible regulation of Lcn2 phosphorylation by EGFR.
Our results indicate that Lcn2 depletion induces an increased ubiquitinylated form of EGFR upon TGF-α stimulation and enhances its interaction with the endosomal sorting complexes required for transport (ESCRT) subunit Hrs. Ubiquitinylated EGFR and Hrs binding is the first step of a cascade of events leading to the ESCRT machinery recruitment, which is responsible for multivesicular bodies (MVBs) formation, resulting in EGFR lysosomal degradation (Schuh and Audhya, 2014). Because Lcn2 localizes at the late endosomes, it is tempting to propose that, by impairing Hrs/EGFR interaction and thus late endosome to MVB transition, Lcn2 prevents EGFR degradation when stimulated with recycling-prone ligands. It is worth noting that Lcn2 does not impact the first steps of EGFR intracellular trafficking, as it does not localize in the early endosomes. Hence, we hypothesize that Lcn2 may facilitate EGFR trafficking from the late endosomes to Rab9 endosomes, but not toward Rab4 or Rab11 recycling endosomes, because they originate from the early endosomes. Considering the lack of public data about Lcn2 interactome, the molecular mechanisms by which Lcn2 participates to these phenomena remain to be elucidated.
Our in vivo data show that Lcn2 expression during CKD induces EGFR localization to the renal tubular cell surface and thus allows a sustained activation of the receptor, leading to renal lesions. Other pathological conditions are associated with an unbalanced EGFR activation and intracellular fate responsible for migration and invasion phenotypes, such as breast, lung, and hepatic cancer (Al-Akhrass et al., 2017, Runkle et al., 2012, Ye et al., 2016). Interestingly, Lcn2 expression is also induced in these cancers (Leng et al., 2009, Ruiz-Morales et al., 2015, Wang et al., 2013, Yang et al., 2009). Considering that we demonstrate an impact of Lcn2 expression on EGFR-induced migration and on pro-epithelial-to-mesenchymal transition (EMT) markers recruitment and that Lcn2 can regulate EGFR recycling upon TGF-α stimulation in A549 cancer cell line, our findings indicate that the role of Lcn2 in EGFR recycling might be relevant to other pathological contexts, such as cancer. For instance, it has been described in breast cancer cells that the tumor-associated S2R (sigma-2 receptor)/Pgrmc1 (progesterone receptor membrane component 1) upregulates Lcn2 expression, which in turn promotes tumor formation (Mir et al., 2012). Furthermore, in this study, the authors demonstrate that S2R/Pgrmc1 activity correlates with an increase of EGFR levels at the plasma membrane. The implication of Lcn2/EGFR partnership in cancerous cells remains to be demonstrated.
In conclusion, our study demonstrates that Lcn2 acts as an enhancer of EGFR recycling process and signaling. This work is the first to dissect a precise intracellular role for Lcn2, as most Lcn2 literature focused on the secreted form of the protein. It unraveled possible cellular functions for Lcn2 in both physiological and physiopathological contexts. Hence, we propose Lcn2 as a promising therapeutic target able to counteract the deleterious effects of aberrant EGFR activation.